

Abstract
Megakaryoblastic leukemia (MKL)/serum-response factor (SRF)-mediated gene transcription is a highly conserved mechanism that connects dynamic reorganization of the actin cytoskeleton to regulation of expression of a wide range of genes, including SRF itself and many important structural and regulatory components of the actin cytoskeleton. In this study, we examined the possible role of MKL/SRF in the context of regulation of profilin (Pfn), a major controller of actin dynamics and actin cytoskeletal remodeling in cells. We demonstrated that despite being located on different genomic loci, two major isoforms of Pfn (Pfn1 and Pfn2) are co-regulated by a common mechanism involving the action of MKL that is independent of its SRF-related activity. We found that MKL co-regulates the expression of Pfn isoforms indirectly by modulating signal transducer and activator of transcription 1 (STAT1) and utilizing its SAP-domain function. Unexpectedly, our studies revealed that cellular externalization, rather than transcription of Pfn1, is affected by the perturbations of MKL. We further demonstrated that MKL can influence cell migration by modulating Pfn1 expression, indicating a functional connection between MKL and Pfn1 in actin-dependent cellular processes. Finally, we provide initial evidence supporting the ability of Pfn to influence MKL and SRF expression. Collectively, these findings suggest that Pfn may play a role in a possible feedback loop of the actin/MKL/SRF signaling circuit.
Keywords: actin, cell migration, cytoskeleton, profilin, serum-response factor (SRF), MKL, SAP-domain
Introduction
Dynamic remodeling of actin cytoskeleton is an essential feature for many physiological processes. This is thought to depend partly on the gene expression program orchestrated by the serum-response factor (SRF),3 a ubiquitously expressed and highly conserved transcription factor. SRF binds to the CArG (CC(A/T)6GG) consensus sequence found in the control regions of a wide array of genes, including SRF itself and many involved in regulating actin cytoskeletal, adhesion, and contractility functions (e.g. actin, myosin, vinculin, filamin, integrin, calponin, ARP3, and cofilin) (1). Depending on the target gene, SRF-dependent gene expression is potently stimulated by two broad classes of transcriptional cofactors: myocardin and ternary complex factors. The myocardin family transcriptional co-activators include myocardin (expressed exclusively in cardiac and smooth muscle cells) and the broadly expressed two myocardin-related transcription factors, namely MRTF-A (also known as MAL or MKL1 (megakaryoblastic leukemia-1)) and MRTF-B (or MKL2). Depending on the context, the two isoforms of MKL either exhibit functional redundancy or have unique functions (2).
Contrasting the constitutively active characteristic of myocardin, MKL function is highly signal-responsive. The MKL-SRF signaling axis is tightly regulated by the state of actin polymerization in cells. Under the basal condition, MKL binds to G-actin, and this interaction holds MKL inactive by promoting its cytoplasmic sequestration. Actin polymerization in response to extracellular signals liberates MKL from G-actin allowing nuclear accumulation of MKL and SRF activation. Therefore, MKL/SRF signaling is considered to be a key pathway that connects dynamic reorganization of actin cytoskeleton to gene expression control. Because transcriptional targets of SRF include structural and regulatory components of the actin cytoskeletal system, and MKL and SRF can influence each other's expression, a complex feedback loop exists between MKL-SRF signaling and actin polymerization in cells (3–6).
Profilins (Pfns) belong to a class of small 14–17-kDa actin-binding proteins found in virtually every eukaryotic organism. The two main isoforms of Pfn (Pfn1 and Pfn2) are encoded by distinct genes located at different genomic loci. Pfn1 (the most widely studied form) is ubiquitously and abundantly expressed in all cell types with the exception of skeletal muscle cells. In vertebrates, Pfn2 is highly enriched in the brain but is also expressed in low amounts (generally at a much lower level than Pfn1) in other cell types. Because of their abilities to (a) promote nucleotide exchange (ADP-to-ATP) on actin and (b) to stimulate actin polymerization driven by various actin-assembly factors through direct interaction, Pfn isoforms play key roles in regulating actin dynamics and actin cytoskeletal remodeling in cells. Pfn has been functionally linked to many actin-dependent cellular functions ranging from cell migration to cytokinesis, vesicle trafficking, and transcription/splicing control of genes (7, 8).
How expression of Pfn is regulated in cells is poorly understood. There is evidence in the literature that SRF can bind to a conserved intronic region of the PFN1 gene (9, 10), although the functional relevance of this interaction remained unexplored. Given SRF's prominent role in the regulation of cytoskeletal gene expression and MKL being an important cofactor for SRF activation, in this study we investigated whether MKL plays any role in the regulation of Pfn expression. We herein demonstrate that even though Pfn1 and Pfn2 are encoded by distinct genes, intracellular levels of these two isoforms can be co-regulated by a common mechanism that indirectly involves the action of MKL but does not appear to be dependent on its SRF-related activity. Unexpectedly, our studies reveal that cellular externalization rather than the transcription of Pfn1 is affected by perturbations of MKL thus highlighting a novel mechanism of regulation of the intracellular level of Pfn1. Finally, we provide initial evidence that the reverse phenomenon may also be true, i.e. Pfn could have the ability to modulate MKL and SRF (a transcriptional target of the MKL/SRF unit) expression. These findings suggest a possible feedback system between MKL and Pfn, adding another complexity in the current model of actin/MKL/SRF signaling.
Results
MKL is an important upstream regulator of Pfn expression
To explore the potential role of MKL in Pfn regulation, we first performed gene silencing of MKL in HEK-293 (a human embryonic kidney epithelial cell line) and MDA-MB-231 (MDA-231, a human breast cancer cell line). In both cell lines, knockdown of MKL1 alone significantly down-regulated the expression of SRF (this is consistent with MKL's ability to promote SRF activity and in turn stimulate SRF expression) and concomitantly reduced the levels of Pfn1 and Pfn2 (Fig. 1A; the knockdown efficiency of MKL1 in these cell lines ranged from 70 to 80%). On average, Pfn1 expression was reduced by 60 and 50% in HEK-293 and MDA-231 cells, respectively, in response to MKL1 knockdown; the corresponding reductions in Pfn2 expression in these two cell lines were 70 and 50%, respectively (at least in MDA-231 cells, co-silencing of MKL2 expression did not reduce Pfn1 expression any further, data not shown). Pfn down-regulation in HEK-293 cells in response to MKL1 knockdown was also verified by a second siRNA (MKL1 siRNA#2) targeting a different region of MKL1 mRNA (supplemental Fig. S1). To further determine whether MKL1 depletion down-regulates the expression of one Pfn isoform downstream of the other, we performed isoform-specific knockdown of Pfn in both HEK-293 and MDA-231 cells. Consistent with the previous findings in normal mammary epithelial cells and breast cancer cell lines by another group (11), we also found that selective knockdown of Pfn1 had a negligible effect (10–20% change, not statistically significant) on the expression of Pfn2 and vice versa in either HEK-293 or MDA-231 cells (supplemental Fig. S2). These data argue against the regulation of one Pfn isoform downstream of the other, and they likely suggest that MKL1 depletion leads to down-regulation of Pfn1 and Pfn2 in a coordinated fashion through a common upstream mechanism.
Figure 1.

Effect of loss-of-function of MKL on the expression of Pfn isoforms in HEK-293 and MDA-231 cells. A, immunoblot analyses of MKL1, SRF, Pfn1, and Pfn2 expression in HEK-293 and MDA-231 cells 72 h after transfection with either MKL1 or control (cont) siRNA. The bar graphs show quantification (mean ± S.D.) of changes in Pfn1 and Pfn2 expression in response to MKL1 knockdown (data summarized from at least three experiments; *, p < 0.05; **, p < 0.01). B–D, immunoblot analyses of Pfn1 and Pfn2 expression in MDA-231 cells in response to either overnight (O/N ∼16 h) LatB (B) or 3 h LatB (C) or 24 h CCG-1423 treatment (D). LatB and CCG-1423 were used at 5 μm concentration. LatB experiments were performed with cultures in complete growth media. For CCG-1423 experiments, cells were serum-starved for 4 h before treating with CCG-1423 in complete growth media. The bar graphs show the mean ± S.D. values of the fold-changes in Pfn1 and Pfn2 expression with respect to the corresponding control conditions (data summarized from at least three experiments; **, p < 0.01). GAPDH blots serve as the loading control.
To further corroborate the gene silencing results, we next investigated whether Pfn expression is susceptible to pharmacological loss of MKL function. G-actin plays a key role in negative regulation of MKL/SRF signaling because G-actin binding restrains MKL in the cytoplasm by inhibiting its nuclear import as well as promoting its nuclear export. Accordingly, treating cells with the actin-depolymerizing drug latrunculin B (LatB) is a commonly used technique to induce cytoplasmic accumulation of MKL and inhibit SRF activation (LatB increases the cellular G-actin level and stabilizes the G-actin–MKL complex in the cytoplasm) (3, 4). Consistent with the effects of gene silencing of MKL1, overnight (16–18 h), LatB treatment also led to a dramatic (∼65%) co-reduction of Pfn isoforms in MDA-231 cells (Fig. 1B). We also performed a short-term (3 h) treatment of LatB in MDA-231 cells, which also showed a trend of Pfn down-regulation, although the extent of Pfn down-regulation was more prominent in the case of overnight LatB treatment (Fig. 1C). Because LatB experiments were not feasible in weakly adherent HEK-293 cells (LatB treatment even for a short duration caused detachment of these cells), to extend our observations in another cell type, we performed similar experiments in HmVEC-1, an immortalized human microvascular endothelial cell (EC) line. We saw similar down-regulation of Pfn1 and Pfn2 expression after either overnight or a short duration (3 h) exposure to LatB (supplemental Fig. S3, A and B). Consistent with LatB's effects, co-silencing of MKL1 and MKL2 (as MKL2 is abundantly expressed in EC; the mean knockdown efficiency of MKL isoforms was ∼85%) also resulted in dramatic reduction in the expression of the two Pfn isoforms (Pfn1 by 76% and Pfn2 by 80%) along with SRF in HmVEC-1 cells (supplemental Fig. S3C).
Although LatB is widely used to suppress MKL/SRF signaling, because LatB's action is not specific to MKL, we wanted to further confirm the sensitivity of Pfn expression to a more direct and selective strategy of MKL inhibition. In unstimulated (serum-starved) cells, MKL rapidly shuttles back and forth between the nuclear and the cytoplasmic compartments, and serum stimulation blocks the nuclear export of MKL thus effectively promoting nuclear accumulation of MKL (4). Nuclear import of MKL can be blocked by CCG-1423, a small molecule inhibitor that binds to the actin-binding RPEL domain of MKL and interferes with its interaction with the nuclear import factor importin α/β complex (12, 13). It is now a widely used pharmacological agent to block MKL/SRF signaling. Therefore, we next studied the effect of CCG-1423 treatment on Pfn expression in serum-starved MDA-231 cells. Consistent with the outcome of the previous loss-of-function experiments (gene silencing and LatB treatment) of MKL, we observed a dramatic >80% depletion of both Pfn1 and Pfn2 expression within 24 h of CCG-1423 treatment in MDA-231 cells (Fig. 1D). On a quantitative measure, the extent of Pfn down-regulation upon either LatB or CCG-1423 was greater than that elicited by MKL knockdown; this was not entirely surprising because knockdown of MKL was not 100% in our experiments.
Based on the results of the foregoing loss-of-function experiments, we next asked whether forced elevation of MKL is capable of increasing the cellular level of any of the Pfn isoforms. We initially performed pilot experiments to examine the changes in Pfn expression in response to graded overexpression of wild-type (WT) MKL1 as a FLAG-tagged fusion protein in HEK-293 cells. In our first set of pilot experiments, the amount of MKL1 plasmid for a 35-mm culture dish was titrated from 0.25 to 1 μg. Even with this modest range of plasmid dosage, we were able to achieve an estimated ∼10–30-fold increase in MKL1 levels compared with the vector control group. Under these conditions, we observed co-elevation of Pfn isoforms (the ranges of Pfn1 and Pfn2 increases were 1.8–2- and 2.2–2.8-fold, respectively; these values represent the mean fold-changes based on two independent pilot experiments) (supplemental Fig. S4A). In follow-up pilot experiments, we titrated down MKL1 plasmid dosage, which showed that even 3.5-fold overexpression of MKL1 is sufficient to elicit elevation of Pfn1 and Pfn2 expression by 3.6- and 3.1-fold, respectively (these values represent the mean fold-changes based on two independent pilot experiments) (supplemental Fig. S4B). Note that because MKL2 level was not considered in these fold-change calculations, the overall fold-change in MKL is somewhat overestimated. Importantly, if one takes into account the increase of both Pfn isoforms, the total Pfn level increase on an absolute scale is not trivial. Although MKL1-dependent Pfn induction did not quantitatively correlate with the level of rise of MKL (we speculate that this could be due to an internal adaptive feedback mechanism between MKL/SRF signaling and Pfn, limiting excessive intracellular levels of Pfn in cells), the general observation was nonetheless qualitatively consistent with the results from MKL1 knockdown experiments. To statistically validate these results in HEK-293 cells and to further extend these findings in MDA-231 cells, we performed WT MKL1 overexpression in these two cell types and analyzed Pfn expression (in these and all subsequent experiments, we followed a transfection protocol of 1 μg of plasmid in a 35-mm culture dish). We confirmed that MKL1 overexpression led to concomitant elevation of SRF and Pfn expression in both HEK-293 and MDA-231 cells (Fig. 2A). On an average, Pfn1 expression increased by 2.3- and 1.7-fold in HEK-293 and MDA-231 cells, respectively; the corresponding increases in Pfn2 expression in these two cell lines were ∼2.5- and 2.4-fold, respectively. Because these results were based on transient transfection experiments where transfection efficiency is typically around 70–80%, these fold-change values were likely underestimated to some extent. These overexpression experimental data taken together with the findings from the foregoing loss-of-function experiments suggested that MKL is an upstream regulator of Pfn expression.
Figure 2.

Effect of MKL1 overexpression on Pfn expression. A, immunoblot analyses of MKL1, SRF, Pfn1, and Pfn2 expression 48 h after transfection with either empty vector (EV) or FLAG-tagged wild-type (WT) MKL1 in HEK-293 and MDA-231 cells (images of MKL1 bands were acquired at a very low exposure (0.05 s) to prevent saturation of the MKL1 overexpression lane signal, which prevented the endogenous MKL1 band from being detected). B, schematic of MKL structure (RPEL, three actin-binding regions that also has basic rich B2 region containing nuclear localization signal); B1, basic region that has a second nuclear localization signal and SRF-binding site; Q, glutamine-rich domain; SAP, DNA-binding domain; LZ, leucine zipper (dimerization) domain; TA, transcriptional activation domain). C, HEK-293 cells expressing various FLAG-tagged MKL1 constructs (WT, ΔN100 (lacks the first 100 amino acids), ΔB1 (internal deletion of amino acids 222–237)) were immunostained with anti-FLAG (green) antibody and DAPI (red) and scored for % cells for subcellular localization of FLAG-MKL1 as summarized in the graph below (N, exclusively nuclear; C, exclusively cytoplasmic; N/C, localized in both cytoplasmic and nuclear compartments; n indicates the number of cells analyzed in each group). D, anti-FLAG immunoblot analyses of nuclear (nucl) versus cytoplasmic (cyto) fractions prepared from HEK-293 cells 48 h after transfection with the indicated FLAG-tagged MKL constructs. Histone-H3 and tubulin blots serve as loading controls for nuclear and cytoplasmic fractions, respectively. E, total extracts of HEK-293 cells transfected with the above constructs were analyzed by immunoblotting for expression of Pfn1, Pfn2, and GAPDH (loading control); the anti-FLAG blot shows comparable expression levels of the various MKL1 constructs. The bar graph shows the average ± S.D. of the fold-changes in Pfn expression with respect to the corresponding control transfection condition (data summarized from three experiments; **, p < 0.01; *, p < 0.05). The electrophoretic mobility shift of ΔN100-MKL1 was not apparent in this blot as these samples were run on a high percentage (15%) gel.
As both LatB and CCG-1423 are known to functionally inhibit MKL by impairing its nuclear localization, we asked whether MKL-dependent induction of Pfn can be also prevented by a molecular strategy that is capable of reducing nuclear accumulation of MKL. The basic-rich B1 domain of MKL1 contains one of the two nuclear localization signal sequences of MKL1 (a schematic representation of MKL1 with its different domains is shown in Fig. 2B), and previous studies have shown that deletion of the entire B1 domain prominently reduces the ability of MKL1 to localize in the nucleus. Conversely, upon deletion of the N-terminal actin-binding RPEL domain, MKL cannot be sequestered in the cytoplasm by the action of actin monomers, and therefore it exhibits a higher propensity to localize in the nucleus and activate gene transcription in a constitutive manner (i.e. even in the absence of any serum stimulation) (14, 15). We next overexpressed either WT-MKL1 or mutants of MKL1 that lack either the entire B1 domain (ΔB1-MKL1) or the first 100 amino acids containing the RPEL domain (ΔN100-MKL1, note that the last RPEL domain ends at amino acid 89 and that ΔN100 deletion does not interfere with any other functional domains of MKL1 as the next known domain starts at amino acid 219) in HEK-293 cells and analyzed the changes in Pfn expression. Immunofluorescence and subcellular fractionation studies confirmed reduced nuclear localization of ΔB1-MKL1 compared with the other two variants of MKL1 as expected (Fig. 2, C and D). Immunoblot analyses of total cell extracts showed that deletion of the B1 domain blocks MKL1's ability to induce Pfn isoforms when overexpressed in HEK-293 cells (Fig. 2E; overexpression of ΔN100-MKL1 mutant resulted in Pfn elevation similar to the effect of WT-MKL1). These data are consistent with the requirement of nuclear localization of MKL1 for its ability to promote Pfn expression in cells. However, we cannot rule out an alternative explanation (not mutually exclusive) that the B1 domain of MKL1 might mediate molecular interactions that are important for Pfn regulation.
MKL regulates Pfn expression through its SAP-domain function
Given the parallel between the changes in SRF and Pfn expression upon various perturbations of MKL as seen in previous experiments and that deletion of the B1 domain (which is also responsible for SRF binding) abrogates MKL's ability to induce Pfn, we next asked whether MKL-dependent modulation of Pfn expression is through its effect on SRF activation. We initially analyzed the effect of silencing of SRF on Pfn expression in various cell lines. Surprisingly, transient knockdown of SRF in HEK-293, MDA-231, and HmVEC-1 cells did not elicit any significant change in the basal level of expression of either of the two Pfn isoforms (the average knockdown efficiencies of SRF in HEK-293, MDA-231, and HmVEC-1 cells were 70, 70, and 90%, respectively) (Fig. 3A; supplemental Fig. S3C). However, because SRF knockdown was not complete, we could not completely rule out the possibility that some low levels of residual SRF activity in SRF-silenced cells may be sufficient to maintain Pfn expression. We also examined Pfn levels in previously generated sublines of mouse 3T3 fibroblasts that stably expressed either control or SRF-shRNA (two independent SRF-shRNA clones (1 and 2) were analyzed). Down-regulation of transcriptional activity of SRF in these two clones of SRF-depleted fibroblasts was previously confirmed by dramatic attenuation of serum-induced transcription of SRF target genes, including EGR2 and CTGF) (16). We estimated the knockdown efficiency of SRF to be equal to ∼97 and ∼70% in clones 1 and 2, respectively. Pfn1 level was essentially unaffected by stable knockdown of SRF in 3T3 cells (Fig. 3B; note that we were not able to detect Pfn2 expression in these fibroblasts presumably because of general low abundance of Pfn2 in most non-neuronal cells). Although the MKL1 promoter has SRF-binding sites (10), interestingly, neither stable nor transient knockdown of SRF led to any noticeable reduction in the basal expression level of MKL1 (Fig. 3, A and B, and supplemental Fig. S3). These data suggest that at its endogenous level of activation, SRF is likely dispensable for regulating the basal expression level of MKL1 (which also explains why Pfn expression was unchanged upon SRF depletion). These SRF knockdown studies in multiple cell lines provided a preliminary indication that SRF may not play a key role in controlling at least the basal expression level of Pfn. In complementary experiments, although overexpression of SRF led to elevated Pfn levels in both HEK-293 and MDA-231 cells, SRF overexpression was also associated with an elevated MKL1 level in these cell lines thus suggesting that hyperactivation of SRF can stimulate MKL expression (Fig. 3C). We proposed that SRF overexpression-induced elevation of Pfn expression is an indirect effect of MKL up-regulation rather than SRF being a direct downstream mediator of MKL-dependent induction of Pfn. To test this postulate, we next investigated the effect of disrupting SRF-activating function on MKL's ability to stimulate Pfn level in HEK-293 cells. Mutagenesis studies revealed that Lys-237, Tyr-238, His-239, and Tyr-241 residues located in the B1 domain of MKL1 are critical for its SRF interaction and that alanine substitution on these residues abrogates MKL1–SRF interaction and MKL1-mediated SRF activation (15). Therefore, we overexpressed either WT MKL1 or a triple-point mutant version of MKL1 (K237A/Y238A/H239A, denoted as MKL13p(mut-B1)) in HEK-293 cells, and we analyzed the changes in Pfn expression, the results of which are shown in Fig. 3D. Overexpression of WT-MKL1 but not MKL13p(mut-B1) led to increased SRF expression, which confirmed the inability of this MKL1 mutant to activate SRF. However, MKL13p(mut-B1) was able to elevate Pfn expression and to an extent comparable with that seen in WT-MKL1 overexpressers. These results argue against SRF being a direct downstream mediator of MKL1-dependent regulation of Pfn.
Figure 3.

MKL regulates Pfn expression likely through an SRF-independent SAP domain-directed function. A, immunoblot analyses of MKL1, SRF, Pfn1, Pfn2, and GAPDH (loading control) expression in HEK-293 and MDA-231 cells 72 h after transient transfection with either control or SRF-siRNA (bar graph shows quantification (mean ± S.D.) of immunoblot data summarized from three independent experiments; NS, not significant). B, left, immunoblot analyses of SRF, Pfn1, and GAPDH (loading control) expression in control versus SRF-shRNA-expressing mouse 3T3 fibroblasts (two independent SRF shRNA stable clones (#1, #2) are shown); data are representative of two independent experiments; right, immunoblot analyses of MKL1 expression in control versus SRF-shRNA (#1) expressers of 3T3 cells. C, immunoblot analyses of SRF, MKL1, Pfn1, and Pfn2 expression in HEK-293 and MDA-231 cells 48 h after transfection with either SRF overexpression vector or empty vector (EV) as control. Images of SRF bands were acquired at a very low exposure (0.05 s) to prevent saturation of the SRF overexpression lane signal, which prevented the endogenous SRF band from being detected. The bar graph shows the mean ± S.D. values of the fold-changes in Pfn1 and Pfn2 expression with respect to the corresponding empty vector control transfection condition. D and E, immunoblot analyses of lysates of HEK-293 cells showing the effects of overexpression of either WT versus 3p(mut-B1) mutant form of MKL1 (D) or WT versus various deletion mutants form of MKL1 (E) on Pfn expression (3p(mut-B1), K237A/Y238A/H239A; ΔSAP = deletion of amino acids 343–378; ΔTA = a truncated form consisting of the first 630 amino acids); all MKL1 constructs are FLAG-tagged. The bar graphs show the mean ± S.D. values of the fold-changes in Pfn1 and Pfn2 expression with respect to the corresponding empty vector control transfection conditions. All data are summarized from three independent experiments; **, p < 0.01; *, p < 0.05; NS, not significant). GAPDH blots serve as the loading control.
A previous study demonstrating differential gene expression and phenotypic distinctions between MKL and SRF knock-out megakaryocytes suggested that MKL can have SRF-independent transcriptional activity in cells (17). It has been shown that MKL can transcriptionally regulate gene expression in breast cancer cells in an SRF-independent manner that is dependent on its SAP-domain (a homology domain that is found in SAF-A/B, Acinus, and PIAS proteins and is thought to facilitate DNA binding of MKL) function (note that some of the MKL-regulated genes require both SAP- and SRF-related function) (18). Because our findings suggested that MKL likely regulates Pfn expression without a direct involvement of SRF, we wondered whether SAP-domain-directed transcriptional function of MKL1 might be important in the context of Pfn regulation. To address this question, we next investigated the effect of deletion of either the SAP or the transcriptional activation (TA) domain on MKL1's ability to promote Pfn expression in HEK-293 cells. We found that upon deletion of either the SAP (ΔSAP-MKL1) or the transcriptional activation (ΔTA-MKL1; this construct is completely impaired in transcriptional activity) domain, MKL1 was unable to induce Pfn elevation when overexpressed in HEK-293 cells (Fig. 3E). These data are consistent with a scenario in which MKL regulates Pfn expression through its SAP-domain-directed transcriptional activity.
MKL's regulation of Pfn expression is STAT1-dependent
Our next goal was to search for alternative pathways that might link SAP-domain function of MKL to Pfn regulation. A previous study had shown that conditional deletion of the MKL1 gene in mouse mammary gland is associated with alterations in the transcript levels of certain STAT (signal transducer and activator of transcription) pathway-associated genes (19). There is evidence of STAT3's ability to be recruited to the promoter and transcriptionally regulate the expression of Pfn1 in rat aortic endothelial cells specifically in response to diabetic condition-mimicking stimuli (20). STAT has been shown to bind to the promoter of chickadee, the Drosophila homolog of Pfn1 (note that unlike in mammals, Drosophila has only one STAT isoform that is structurally similar to STAT3/STAT5) (21). Based on these findings, we explored whether there could be a potential connection between MKL, STAT, and Pfn. To test this, we first examined the effects of knockdown and overexpression of MKL1 on the expression levels of two major STAT isoforms, namely STAT1 and STAT3, in various cells. We found that in all three cell lines (HEK-293, MDA-231, and HmVEC-1), STAT1 expression was reduced upon silencing of MKL1 either alone or in conjunction with MKL2 (as done in HmVEC-1 cells) (Fig. 4A; supplemental Fig. S3C). STAT1 down-regulation in response to MKL1 knockdown was also verified by a second siRNA (MKL1 siRNA#2) targeting a different region of MKL1 mRNA (supplemental Fig. S1). STAT3 expression also followed a similar trend in response to MKL knockdown, although STAT3 down-regulation in MDA-231 cells was not as pronounced as observed in the other two cell lines (Fig. 4A; supplemental Fig. S3C). Consistent with these knockdown experiment results, overexpression of WT-MKL1 led to elevation of both STAT1 and STAT3 levels in HEK-293 cells (mean fold-changes: ∼2.1 (STAT1) and ∼2.5 (STAT3)) and MDA-231 (mean fold-changes: ∼1.5 (STAT1) and ∼2.5 (STAT3)) (Fig. 4B). Together, these results demonstrate that MKL can modulate the expression level of certain STAT family members. As in the case of Pfn, silencing SRF had no impact on the expression of either of the two STAT isoforms in any of the three cell lines (supplemental Figs. S3 and S5), and furthermore, deletion of either the SAP or the TA domain blocked MKL1's ability to up-regulate STAT isoforms when overexpressed in HEK-293 cells (Fig. 4C). Collectively, these data suggest that MKL modulates STAT expression in a SAP-domain function-directed manner.
Figure 4.

MKL promotes the expression of STAT isoforms through its SAP domain function. A and B, immunoblot analyses of HEK-293 and MDA-231 extracts showing the effects of either knockdown (A) or overexpression (B) of MKL1 on STAT1 and STAT3 expression (lysates were prepared 72 and 48 h after siRNA and plasmid transfections, respectively). The bar graphs show the mean ± S.D. values of fold-changes in STAT isoforms associated with MKL1 perturbations. C, STAT1 and STAT3 immunoblot analyses of HEK-293 extracts following overexpression of either WT form or the indicated deletion mutants of MKL1. The bar graphs show the mean ± S.D. values of the fold-changes in STAT1 and STAT3 expression with respect to the corresponding control transfection condition. All data are summarized from three experiments (**, p < 0.01; *, p < 0.05, NS, not significant; EV, empty vector). GAPDH blots serve as the loading control.
We next investigated the effects of knockdown and overexpression of these two variants of STAT on Pfn expression. First, in HEK-293 cells, knockdown of only STAT1 (but not STAT3) resulted in prominent 60% down-regulation of Pfn isoforms (Fig. 5A). Conversely, Pfn isoforms were coordinately elevated (mean values of increase of Pfn1 and Pfn2 equal to 2.5- and 3-fold, respectively) upon overexpression of only STAT1 but not STAT3 (Fig. 5B). Although we estimated 10–15-fold increase of STAT1 or STAT3 expression in our overexpression experiments, judging from the immunoblot data in supplemental Fig. S4, it is possible that even a modest level of STAT1 increase may be sufficient to induce Pfn elevation. In these cells, perturbation of STAT1 did not affect the expression of STAT3 and vice versa (Fig. 5, A and B). In fact, even when STAT3 expression was forcibly knocked down, STAT1 overexpression was still able to result in Pfn elevation in HEK-293 cells (Fig. 5C). We also extended some of these findings in HmVEC-1 and MDA-231 cells. In these two cell lines, knockdown of either STAT1 or STAT3 caused prominent down-regulation of Pfn isoforms (supplemental Fig. S6, A and B). Depending on the cell type and the STAT variant, reduction of the Pfn1 and Pfn2 levels ranged from 50 to 90%. However, in these two cell lines, knockdown of one STAT variant also led to concomitant down-regulation of the other variant (it was not surprising because there is evidence of cross-talk between STAT1 and STAT3 signaling in a context-dependent manner (22, 23)). STAT1 knockdown-induced reduction of Pfn1 and Pfn2 expression in HmVEC-1 cells was also verified by a second STAT1 siRNA (STAT1 siRNA#2) targeting a different region of mRNA (supplemental Fig. S7A). Conversely, forced overexpression of either STAT1 or STAT3 led to co-elevation of Pfn isoforms (by 2–2.7-fold (mean) depending on the Pfn isoforms) in MDA-231 cells (note that STAT1 overexpression also promoted STAT3 expression and vice versa) (supplemental Fig. S6C). Collectively, these results clearly demonstrated the importance of STAT in the regulation of Pfn expression. Although HEK-293 cell data suggest that STAT1 alone is sufficient to promote Pfn expression, we do not rule out the possibility of STAT3 as a collaborator in a cell type-specific context.
Figure 5.

MKL promotes Pfn expression through modulating STAT1 in HEK-293 cells. A and B, immunoblot analyses of Pfn expression in HEK-293 cells following either knockdown (A) or overexpression (B) of STAT variants (lysates were prepared 72 and 48 h after siRNA and plasmid transfections, respectively). C, immunoblot analyses of HEK-293 extracts showing the effect of STAT1 overexpression with or without STAT3 knockdown on Pfn expression. D, immunoblot analyses of HEK-293 extracts showing the effect of FLAG-MKL1 overexpression with or without STAT1 knockdown on Pfn expression (siRNA transfection was performed 24 h prior to plasmid transfection). All bar graphs accompanying the immunoblots show the mean ± S.D. values of the fold-changes in Pfn expression with respect to the corresponding control transfection condition (all data summarized from three experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant; EV, empty vector). GAPDH blots serve as the loading control.
Given the ability of MKL to simultaneously elevate the expression levels of STAT1 and Pfn isoforms, we further asked whether STAT1 plays any role in MKL-dependent regulation of Pfn. To test this, we overexpressed WT-MKL1 in control versus STAT1-siRNA transfected cells and analyzed the changes in Pfn expression. We found that silencing STAT1 expression prevented MKL1-induced elevation of both Pfn1 and Pfn2 in HEK-293 cells (Fig. 5D; this result was further verified by the use of STAT1 siRNA#2, see supplemental Fig. S7B) suggesting that MKL-mediated regulation of Pfn is STAT1-dependent.
MKL regulates Pfn expression in a post-transcriptional manner
To gain insight into the mode of regulation of Pfn by MKL, we analyzed the mRNA levels of Pfn in control versus MKL1 siRNA-transfected HEK-293 cells by quantitative RT-PCR. Although silencing of MKL1 elicited a 60% decrease in the expression of Pfn1 at the protein level, surprisingly, we found no significant difference in the mRNA level of Pfn1 between control and MKL1 knockdown HEK-293 cells (Fig. 6A). We detected a small ∼20% difference in the average Pfn2 mRNA level between the control and MKL1 knockdown groups, which also clearly did not correlate with the robust 70% change in the corresponding protein expression. With knockdown using MKL1 siRNA#2, we did not detect any significant change in the mRNA level of either Pfn1 or Pfn2 in HEK-293 cells (Fig. 6A). Similarly, the mRNA levels of Pfn isoforms were also not significantly affected when STAT1 expression was knocked down (supplemental Fig. S8). These data suggest that Pfn expression is altered upon MKL1 knockdown in a post-transcriptional manner. We further asked whether altered protein stability might be responsible for MKL1-dependent changes in Pfn expression. However, blocking either the proteasomal (by MG-132 treatment)- or lysosomal (by ammonium chloride treatment)-mediated protein degradation pathway failed to reverse MKL1 knockdown-induced down-regulation of Pfn expression (Fig. 6, B and C). These experimental observations tend to further rule out the possibility of accelerated protein degradation accounting for Pfn down-regulation upon loss of MKL1 function.
Figure 6.

Loss-of-function of MKL1 down-regulates Pfn expression by promoting its extracellular release rather than its transcription. A, quantitative RT-PCR analyses show the mean ± S.D. values of the fold-changes in Pfn1 and Pfn2 mRNA levels after knockdown of MKL1 (data shown for two different MKL1 siRNAs) relative to control-siRNA transfectants of HEK-293 cells (data summarized from three independent experiments for each siRNA with three technical replicates/group; *, p < 0.05; NS, not significant). B, immunoblot analyses of MKL1, Pfn1, Pfn2, p27Kip1, and GAPDH (loading control) expressions in HEK-293 cells 72 h after transfection with the indicated siRNAs and following treatment with either 5 μm MG-132 or DMSO (vehicle (veh) control) for 12 h. p27Kip1, a cell cycle protein that is rapidly turned over by proteasomal degradation, shows elevation upon MG-132 treatment serving as a positive control in these experiments (data representative of three experiments). C, immunoblot analyses of MKL1, Pfn1, LC3, and GAPDH (loading control) expressions in HEK-293 cells 72 h after transfection with the indicated siRNAs and being treated with either 10 mm NH4Cl (blocks lysosomal degradation pathway) or vehicle control for 12 h. LC3, an autophagy-related protein that is subjected to lysosomal degradation, shows elevation upon NH4Cl treatment serving as a positive control in these experiments (data representative of three experiments). The bar graphs in B and C show the mean ± S.D. values of the fold-changes in Pfn isoforms (relative to control) associated with the indicated conditions. D–F, representative immunoblot analysis of Pfn1 level in the conditioned media prepared from HmVEC (D) and MDA-231 (E) cells following overnight treatment with either vehicle (veh) or LatB or CCG-1423. CD63, a well-known marker of extracellular vesicles, including exosomes, was used as a loading control for conditioned media samples in HmVEC-1 experiments. Coomassie staining of the SDS-PAGE of conditioned media derived from MDA-231 cells confirmed equal loading. F summarizes the quantification of fold-changes of Pfn1 (n = 3 experiments; **, p < 0.01). G, representative immunoblot analyses of Pfn1 along with the other indicated proteins in cellular extracts versus the conditioned media prepared from MDA-231 cells following transfection with either control or MKL1 siRNA (note that MKL1, STAT1, and SRF were not detected in the conditioned media). The numbers in parentheses represent the individual fold-changes in the released Pfn1 content in each of the two experiments with the mean fold-change equaling ∼4-fold.
Although Pfn is mainly an intracellular protein, a previous study reported the presence of Pfn1 along with several other cytoskeletal proteins in exosomes (24). Pfn1 has been also detected in the secretome of pancreatic cancer cells (25) and serum (26). These observations suggest that a certain fraction of cellular Pfn1 may be externalized into the extracellular milieu. Therefore, to explore whether loss-of-function of MKL could affect the intracellular level of Pfn by somehow impacting its externalization, we next analyzed the Pfn1 and Pfn2 levels in the conditioned media of HmVEC-1 and MDA-231 cells following overnight treatment of either LatB or CCG-1423. For both cell types, we observed a significantly elevated level (depending on the cell type and the treatment, the mean fold-increase ranged from 3- to ∼5-fold) of Pfn1 in the conditioned media concomitant with the expected decrease in the corresponding cellular fractions in response to either LatB or CCG-1423 treatment (Fig. 6, D–F). In alignment with these results, knockdown of MKL1 expression also led to a prominent increase in the level of externalized Pfn1 in the conditioned media of MDA-231 cells (Fig. 6G). Interestingly, we also detected GAPDH and tubulin in the conditioned media in our experiments; this is consistent with the previously reported findings of the presence of these two proteins in the exosomes (27, 28). Unfortunately, because of general low abundance of Pfn2 in non-neuronal cells, we were not able to detect Pfn2 in the conditioned media by immunoblot analyses. We also performed live-dead staining of HmVEC-1 and MDA-231 cells for either CCG-1423 treatment or MKL knockdown settings, which showed 97–99% cell viability under these experimental conditions (supplemental Fig. S9). Note that CCG-1423 reduced cell proliferation (accounting for ∼15–20% lower overall cell number compared with the DMSO-treated group) and spreading. A more obvious phenotypic change elicited by CCG-1423 compared with MKL knockdown was not entirely surprising for at least two reasons. First, MKL knockdown was not complete by siRNA treatment. Second, there is evidence of at least one other cellular target (MICAL) of CCG-1423 besides MKL (29), and therefore the biological effect of CCG-1423 likely extends beyond its action on MKL. Nonetheless, no biologically significant difference in cell viability between the different experimental settings strongly suggests that our observation of a substantial increase in the released amount of Pfn1 upon loss-of-function of MKL is not a result of compromised cell viability.
In complementary experiments, we further analyzed the effect of overexpression of WT-MKL1 on the mRNA level and the externalized content of Pfn1. Consistent with the results of the knockdown experiments, overexpression of MKL1 also did not significantly change the mRNA level of either Pfn1 or Pfn2 in HEK-293 cells (supplemental Fig. S10A). Furthermore, overexpression of MKL1 was associated with an average of a 40% reduction (based on two independent experiments) in the level of Pfn1 in the conditioned media concomitant with the expected elevation of its intracellular level in MDA-231 cells (supplemental Fig. S10B). Based on these collective findings, we conclude that MKL plays an important role in somehow regulating the cellular retention of at least Pfn1.
Pfn may also be an important determinant of MKL/SRF expression
Consistent with Pfn1's important role in facilitating actin polymerization in cells, Treisman and co-workers (3, 5) had previously shown that overexpressing Pfn1 in fibroblasts promotes nuclear localization of MKL and transcriptional activity of SRF, at least as measured in a transfection-based reporter assay, and these effects are dependent on Pfn1's ability to interact with actin. To further extend these observations, we next studied the effect of perturbations of Pfn on MKL and SRF (as SRF is a transcriptional target of SRF itself) levels in MDA-231 cells. We first found that co-depletion of Pfn isoforms led to a significant reduction in both MKL1 and SRF levels in MDA-231 cells (Fig. 7A). Consistent with these observations, we also found overexpression of Pfn1 alone resulted in elevation of both MKL1 (∼2.75-fold) and SRF levels (by ∼1.5-fold) in MDA-231 cells (Fig. 7B). Note that even though MKL1 level was elevated, Pfn2 expression did not appear to be affected (Fig. 7B, the possible reason is addressed under “Discussion”). Based on our initial evidence supporting Pfn1's ability to influence MKL/SRF expression along with our foregoing results of Pfn1 being modulated downstream of MKL, a feedback loop between MKL/SRF, Pfn1, and dynamic control of actin polymerization integrating the action of STAT is possible as schematically hypothesized in Fig. 7C.
Figure 7.

Initial evidence of sensitivity of MKL and SRF expression to perturbations of Pfn. A, immunoblot analyses of MDA-231 extracts showing the effect of co-depletion of Pfn isoforms (via transfection of pooled siRNAs targeting Pfn1 and Pfn2) on MKL1 and SRF levels. The bar graph summarizes the quantification (mean ± S.D.) of the fold-changes of MKL and SRF (n = 3 experiments; *, p < 0.05). B, immunoblot analyses of MDA-231 extracts showing the effect of transient overexpression of Pfn1 (cloned into GFP-IRES backbone vector) on MKL1, SRF, and Pfn2 levels (cells transfected with the GFP-IRES backbone vector served as a control group). The bar graphs show the mean ± S.D. values of the fold-changes in MKL1, SRF, and Pfn2 expressions with respect to the corresponding control transfection condition (n = 3 experiments; *, p < 0.05). GAPDH blots serve as the loading control. EV, empty vector. C, hypothetical model of actin/MKL/Pfn/SRF signaling circuit. This model integrates the current findings of Pfn being regulated downstream of MKL in an SRF-independent manner through STAT and our initial evidence supporting Pfn's ability to also modulate MKL and in turn SRF expression, thus possibly enabling a positive feedback loop. SRF activation can either elicit a feedforward (through promoting actin polymerization, MKL expression) action amplifying the response or a negative feedback action (through elevating G-actin level) thus dampening the response beyond a certain limit (dashed lines with question marks indicates mechanisms unknown).
Loss of MKL1 promotes breast cancer cell motility through down-regulating Pfn expression
Finally, to explore a possible functional connection between MKL1 and Pfn, we next asked whether Pfn plays any role in MKL-mediated regulation of actin-dependent cellular events such as cell migration. Specifically, we investigated the effect of MKL1 knockdown on random motility of MDA-231 cells without or with transient overexpression of Pfn1. We found that knockdown of MKL1 reduced the basal level of Pfn1 as expected (Fig. 8A) and increased the average speed of MDA-231 cells by ∼1.9-fold (Fig. 8B; this result was further confirmed by the use of a second MKL1 siRNA (MKL siRNA#2) (supplemental Fig. S11)). This is consistent with our previously published finding of enhanced motility of MDA-231 cells upon Pfn1 knockdown, a phenotype that was related to altered phosphoinositide signaling (30). Hypermigratory phenotype of MDA-231 cells upon MKL1 depletion was completely reversed when Pfn1 expression was forcibly elevated thus suggesting that down-regulation of Pfn1 expression contributed to enhanced motility of MKL1-silenced MDA-231 cells (Fig. 8B). In the absence of MKL1 knockdown, transient overexpression of Pfn1 also reduced the average speed of MDA-231 cells, which is consistent with our previously published findings related to the effect of stable overexpression of Pfn1 on MDA-231 cell motility (31). These data provided a proof-of-principle of MKL's ability to impact cellular phenotypes through modulating Pfn expression.
Figure 8.
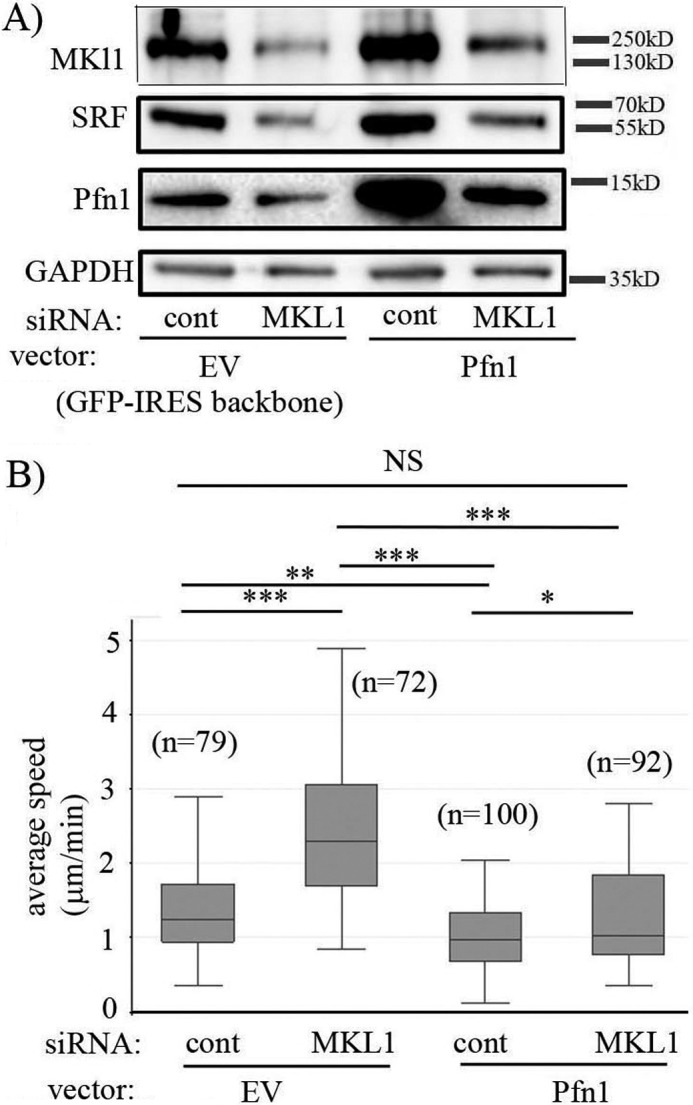
Loss-of-function of MKL1 promotes MDA-231 cell motility through down-regulating Pfn1 expression. A, immunoblot analyses of MKL1, SRF, Pfn1, and GADPH (loading control) expression in MDA-231 cells co-transfected with the indicated siRNAs (control versus MKL1) and overexpression vectors (GFP-IRES backbone (EV) versus GFP-IRES-Pfn1). B, box-whisker plot summarizing the average speed of migration of these four groups of cells (transfected cells were identified by GFP fluorescence) in random-motility assays. In the plot, the middle line, the upper and lower hinges of the box represent the median, 75th and 25th percentile of data, and the whiskers represent the maximum and minimum values (data summarized from three experiments; n, number of cells analyzed in each group pooled from all experiments; *, p < 0.05; **, p < 0.01; ***, p < 0.001; NS, not significant).
Discussion
Although Pfns are important control elements of actin polymerization, very little is known about how their expression is controlled in cells. In this study, we report several novel findings. First, we show that despite their different genomic locations, the two main cellular isoforms of Pfn can be co-regulated through a common signaling pathway involving the action of the transcriptional co-activator MKL; importantly, this mechanism is generalizable across different cell types. However, contrasting the conventional paradigm of transcriptional control of cytoskeletal genes by the MKL/SRF axis, our studies suggest an unconventional SRF-independent mode of action of MKL indirectly linking to a post-transcriptional regulation of Pfn isoforms. With further evidence of MKL1-dependent changes in the extracellular release of at least Pfn1, we show for the first time that the cellular level of Pfn can be modulated at the level of its externalization. In light of these findings, the functional significance of SRF's binding to an intronic region of the PFN1 gene as originally discovered by Miano and co-workers (9) still remains unclear (it is possible that SRF may act as a distal transcription factor regulating the expression of some genes around the Pfn locus). Although SRF is not a direct downstream mediator of the MKL-dependent modulation of Pfn, our SRF-overexpression studies suggest that SRF hyperactivation could lead to elevated Pfn levels in cells indirectly through increasing MKL expression; whether this occurs in any physiological scenario remains to be seen. Second, we identify STAT1 as an important upstream regulator of Pfn expression and further show that MKL regulates Pfn expression indirectly by controlling the level of STAT1 via its SAP-domain (a putative DNA-binding domain of MKL that has been implicated in SRF-independent transcriptional control of MKL (18, 32)) function. SAP-domain-directed transcriptional regulation by MKL is recently gaining attention; however, the key downstream players in this process remain to be identified. Because STAT isoforms are regulated by the SAP-domain activity of MKL, it further raises the possibility of a STAT-centric gene expression control downstream of SRF-independent SAP-domain activity of MKL that can extend beyond and above the context of regulation of Pfn. Third, consistent with the molecular evidence of MKL's role in Pfn regulation, we demonstrate for the first time the feasibility of a pharmacological strategy to trigger Pfn depletion using CCG-1423, a small molecule that has already shown significant promise in preclinical studies either as an anti-fibrotic agent (33) or a possible therapeutic agent in type II diabetes (34). Given the association of increased Pfn expression with certain cancers and vascular pathology (11, 26, 35–40), this study now opens up the possibility of CCG-1423 as a potential pharmacological agent to reverse Pfn-associated aberrant cellular phenotypes in disease conditions. Therefore, our findings may have a potential translational significance. Fourth, similar to MKL's ability to indirectly regulate Pfn levels, we provide initial evidence of Pfn being a possible determinant of the expressions of MKL and SRF. Finally, we provide evidence of a functional connection between MKL and Pfn in the context of regulation of cell motility suggesting that MKL-dependent modulation of cellular phenotype is Pfn-dependent.
Some of the details of how MKL regulates the Pfn level by controlling STAT expression are still unclear. It was previously shown that conditional knock-out of MKL1 in the mammary gland is associated with transcriptional up-regulation of SOCS3 (suppressor of cytokine signaling 3), a key negative regulator of JAK-STAT signaling (19). Therefore, it is possible that MKL regulates STAT expression by modulating SOCS3 transcription (either directly or indirectly). An alternative possibility is that MKL may directly regulate STAT transcription. At least it has been reported that SRF can bind to the STAT3 promoter (10), and MKL1 and STAT3 can physically interact and transactivate gene expression in breast cancer cells (41). It is not known whether other STAT members can directly interact with MKL1 or at least co-complex with MKL1–STAT3. Because STAT family members can homo- and heterodimerize to regulate the expressions of their own and other STATs, it is also possible that the MKL1–STAT interaction may play a role in regulating STAT expression. Although there is evidence of STAT3's ability to directly bind to the promoter and stimulate the transcription of the Pfn1 gene in rat aortic endothelial cells, STAT3 recruitment to the Pfn1 promoter was shown to occur only in response to a certain stimulus (such as oxysterol treatment as demonstrated in that study) (20). These findings suggest that STAT3 has the potential to transcriptionally enhance Pfn expression under certain conditions, but it is not one of the main regulators of Pfn under basal conditions. Potential roles of other STAT family members in the context of Pfn regulation have not been explored previously. Our studies show that STAT1 can promote Pfn expression without requiring STAT3. Our RT-PCR experiments revealed no appreciable changes in Pfn mRNA levels upon either MKL1 or STAT1 knockdown (this likely explains why alteration in Pfn1 expression was also never reported in any of the previous microarray studies involving either knock-out or a dominant-negative mutant of MKL1 (18, 19)). However, we do not exclude the possibility of a transcriptional component of Pfn gene regulation in a hyperactivated STAT scenario. Although beyond the scope of this work, we need to address in the future how the loss of MKL function ultimately leads to increased cellular release of Pfn1 and what the mode of release of Pfn1 is. Clearly, exosomal release of Pfn1 will be an interesting avenue to pursue in the future.
It was shown previously that overexpression of Pfn1 can promote nuclear localization of MKL1 and increase SRF activation, and this effect of Pfn1 on MKL/SRF signaling requires its actin-binding function (3, 5). MKL and Pfn1 can also compete for G-actin binding (6). Therefore, Pfn1 likely promotes MKL/SRF activation via inhibiting MKL–actin complex formation through direct competition with MKL and/or promoting actin polymerization. The present findings of MKL1's positive regulation of Pfn1 through STAT1, and our initial evidence supporting Pfn1's ability to promote MKL and in turn SRF expression, raise the possibility of a feedback loop between MKL/SRF, Pfn1, and dynamic control of actin polymerization integrating the action of STAT (Fig. 7C). Although further studies are needed to examine this hypothetical feedback model in detail and extend the generality of such a model across various cell types, a few points are still worth addressing with regard to this model. First, according to this feedback model, increased expression and/or nuclear localization of MKL1 induced by Pfn1 overexpression would be expected to cause an increase in Pfn2 expression, but this was not observed in our studies. This may be caused by a possible negative feedback signal from hyperactivated SRF somehow dampening the autoregulatory loop (as illustrated in Fig. 7C). An alternative explanation could be that because all of our overexpression experiments were done in serum-containing culture where MKL is predominantly nuclear, additional contribution from Pfn1 overexpression in terms of regulating MKL localization may be inconsequential. Second, our data showing LatB-induced Pfn depletion (also consistent with the proposed schematic model) raises an interesting possibility that Pfn may be regulated in a G-actin-sensitive manner. Future studies investigating the effects of F-actin-stabilizing drugs or perturbation of other actin-polymerization regulatory proteins on Pfn expression will shed further insight into this process. Third, Pfn's ability to regulate MKL/SRF (a major transcriptional system) suggests that we should consider additional pathways by which Pfn may regulate actin-dependent processes such as cell migration beyond the traditional model of Pfn's role in regulating actin polymerization at the leading edge as conceived in the literature. In fact, a recent proteomic study by Moens and co-workers and our group revealed that overexpression of Pfn1 in MDA-231 cells is associated with alterations in the expression of many proteins that have been functionally linked to cytoskeletal regulation, adhesion, proliferation, and survival (42). Of particular interest, cofilin1, an SRF target gene (9) and a major regulator of actin cytoskeleton dynamics and cell motility, was shown to be one of the differentially expressed proteins between control and Pfn1-overexpressing cells.
Finally, it was previously shown that co-depletion of MKL isoforms (MKL1 and MKL2) leads to faster migration of strongly adherent cells (such fibroblasts) but has an opposite effect on the motility of weakly adherent transformed epithelial cells and breast cancer cells, including MDA-231 cells (43). Inhibition of MDA-231 motility upon co-depletion of MKL1 and MKL2 was attributed to impairment in cell–substrate adhesion. Our studies demonstrate that MDA-231 cells actually become hypermotile if expression of only one of the isoforms of MKL (MKL1 in our case) is selectively suppressed, a phenotype that is related to down-regulation of Pfn1 expression. Although we do not have sufficient evidence yet to propose a generalized hypothesis, it is possible that there may be an optimum range of MKL activity that is most productive for migration of certain types of cells (such as MDA-231 cells). Future structure–function studies dissecting the role of various functional domains of MKL on cell migration will likely yield mechanistic insights into this phenomenon.
Experimental procedures
Cell culture and reagents
HEK-293 and MDA-MB-231 (MDA-231) cells were cultured as described previously (44). HmVEC-1 cells (ATCC, CRL-3243) were cultured in MCDB131 (Life Technologies, Inc.) growth medium (10% (v/v) FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 10 ng/ml EGF, 1 μg/ml hydrocortisone, 10 mm l-glutamine). Generation and maintenance of sublines of 3T3 fibroblasts stably expressing either control or SRF-shRNA have been described previously (16). Latrunculin B, CCG-1423, and MG-132 were purchased from Cayman Chemicals (Ann Arbor, MI), Santa Cruz Biotechnology (Dallas, TX), and Sigma, respectively.
Plasmids and siRNAs
Wild-type and deletion constructs of MKL1 (FLAG-tagged) have been previously described (14). FLAG-tagged STAT1α construct was kindly provided by Dr. Jennifer Grandis (University of California, San Francisco). SRF and FLAG-tagged STAT3 plasmids were obtained through Addgene. Pfn1 cDNA was subcloned into pIRES2-AcGFP backbone vector (Clontech) at the XhoI- and BamHI-cloning sites. The MKL13p(mut-B1) mutant was generated on the FLAG-MKL1 plasmid using the following primers: K237A (sense, 5′-GAAGAAGCTCGCGTACCACCAGT-3′), K237A and Y238A (sense, 5′-GAAGAAGCTCGCGGCCCACCAGT-3′), and K237A, Y238A, and H239A (sense, 5′-GCTCGCGGCCGCCCAGTACATCC-3′). Plasmid DNA transfections for HEK-293 and MDA-231 cells were done using XtremeGENE HP (Roche Diagnostics, Basel, Switzerland) and Lipofectamine 3000 (Life Technologies, Inc.) transfection reagents, respectively, according to the manufacturers' instructions. For knockdown of genes, MKL1 siRNA (Santa Cruz Biotechnology, Dallas, TX, sc-43944; Thermo Fisher Scientific, Waltham, MA, 133259 (#2)), MKL2 siRNA (Santa Cruz Biotechnology, sc-61074), SRF siRNA (Santa Cruz Biotechnology, sc-36563), STAT1 siRNA (Santa Cruz Biotechnology, sc-44123; Thermo Fisher Scientific, s279 (#2)), STAT3 siRNA (Santa Cruz Biotechnology, sc-29493), Smartpool Pfn1 siRNA (GE Dharmacon, Lafayette, CO; M-012003-01-0005), or Smartpool Pfn2 siRNA (Santa Cruz Biotechnology, sc-78482) were transfected using either Transfection Reagent 1 (Dharmacon) or Transfection Reagent 2 (Dharmacon) for HEK-293/HmVEC-1 or MDA-231 cells, respectively. As control, Smartpool non-targeting siRNA (D-001206-13-05, Dharmacon) was transfected in cells. All siRNA transfection was performed at a 100 nm concentration. The targeting sequences of SRF-shRNAs have been described previously (16).
Subcellular fractionation
Cells were extracted from 10-cm dishes at 80% confluence for 15 min in Buffer A (10 mm HEPES, 10 mm KCl, 0.1 mm EDTA, 0.1% Nonidet P-40, 1 mm DTE, protease and phosphatase inhibitors, pH 7.9). Samples were centrifuged at 5000 rpm for 10 min, and the supernatant was collected as the cytoplasmic fraction. The cell pellet was washed and centrifuged (5000 rpm, 5 min) three times with Buffer A. The pellet was then resuspended in Buffer B (20 mm HEPES, 0.4 mm NaCl, 1 mm EDTA, 10% glycerol, 1 mm DTE, 0.1% SDS, protease and phosphatase inhibitors, pH 7.9) and vigorously vortexed for 1 h at 4 °C. Samples were centrifuged at 13,000 rpm for 10 min, and the supernatants were collected as the nuclear fractions. Purity of the cytoplasmic and nuclear fractions was assessed by tubulin and histone-H3 immunoblots.
Conditioned media preparation
Conditioned media were collected from the culture dish following overnight incubation of cells in serum-free media. The collected media were filtered (0.45 μm size) and concentrated using a 10-kDa cutoff filter. The concentrate was reconstituted with 2× Laemmli sample buffer and boiled before being analyzed by gel electrophoresis.
Immunostaining
Cells expressing various FLAG-MKL constructs were washed with PBS, fixed with 3.7% formaldehyde for 15 min, and permeabilized with 0.5% Triton X-100 for 5 min. Following blocking with 10% goat serum in PBS at room temperature for 30 min, cells were incubated with a monoclonal FLAG antibody (Sigma; 1:100) diluted in 5% goat serum for 1 h at room temperature. After washing cells two times with 0.02% Tween 20 and twice with PBS, cells were incubated with FITC-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA) for 1 h. Cells were again washed with 0.02% Tween 20 and PBS, two times each, before mounting with the mounting medium with DAPI (Sigma). Slides were imaged using a ×20/0.4 NA objective on an Olympus IX71 inverted microscope. Quantification of FLAG-MKL1 localization was performed by overlaying green fluorescence (FLAG-MKL1) and DAPI fields. Cells were scored based on the presence of FLAG-MKL1 either completely within the nucleus, completely outside of the nucleus, or present in both compartments. The percentage of cells within each compartment was calculated for data representation.
Cell viability assay
HmVEC-1 and MDA-231 cells were stained using a live/dead assay kit (source: Life Technologies, Inc.) following the manufacturer's protocol. Stained cells were imaged with a ×4/0.13 NA objective.
Immunoblotting
Equal number of cells for different experimental groups were seeded in the wells of 6-well plate 16–18 h before the day of extraction (cells reached ∼70–80% confluency at the time of extraction). Cell lysates were prepared by 80–90 μl of a modified RIPA buffer (25 mm Tris-HCl, pH 7.5, 150 mm NaCl, 1% (v/v) Nonidet P-40, 5% (v/v) glycerol, 1 mm EDTA, 50 mm NaF, 1 mm sodium pervanadate, protease inhibitors supplemented with 6× sample buffer diluted to 1× with the final SDS concentration in the lysis buffer were equivalent to 2%). For immunoblotting, 10–25 μl of lysate (MDA-231, HEK-293: 10–15 μl; HmVEC: 25 μl) was loaded onto SDS-polyacrylamide gel. Immunoblotting conditions for the various antibodies were as follows: monoclonal Pfn1 (Abcam (ab124904), Cambridge, MA, 1:3000); monoclonal GAPDH (Bio-Rad (G9545), 1:3000); monoclonal p27Kip1 (BD Biosciences (610241), 1:2000); polyclonal MKL1 (Santa Cruz Biotechnology (sc-32909), 1:500); polyclonal MKL2 (Santa Cruz Biotechnology (sc-98989), 1:500); polyclonal SRF (Santa Cruz Biotechnology (sc-335), 1:500); polyclonal Pfn2 (Santa Cruz Biotechnology (sc-100955), 1:500); monoclonal STAT1 (Cell Signaling (9172P), Danvers, MA, 1:1000); monoclonal STAT3 (Cell Signaling (4904P), 1:1000); monoclonal LC3 (Cell Signaling (12741T), 1:1000); monoclonal tubulin (Sigma (T9026), 1:3000); polyclonal CD63 (Systems Biosciences (EXOAB-CD63A-1), 1:1000); polyclonal histone-H3 (Bioss Antibodies (bs-0349R), 1:100); and monoclonal FLAG (Sigma (F3165), 1:3000).
mRNA extraction/quantitative RT-PCR
Total RNA was extracted from cultured cells using the RNeasy mini kit (Qiagen) according to the manufacturer's instructions. Complementary DNA (cDNA) was synthesized from 1 μg of RNA using the Quantitect reverse transcription kit (Qiagen) following the manufacturer's instructions. Each PCR was prepared with 50 ng of cDNA, 12.5 μl of SYBR Select Master Mix (Thermo Fisher Scientific), 1 μm (final concentration) forward and reverse primers, and water for a total volume of 25 μl. Thermal cycling and data analysis were performed using the StepOne Plus Real-Time PCR System and StepOne Software (Applied Biosystems) to detect quantitative mRNA expression of Pfn1, Pfn2, and GAPDH (endogenous control). The primer sequences for GAPDH were 5′-CGGAGTCAACGGATTTGGTCGTAT-3′ (sense) and 5′-AGGCTTCTCCATGGTGGTGAAGAC-3′ (antisense). The primer sequences for Pfn1 were 5′-CGAGAGCAGCCCCAGTAGCAGC-3′ (sense) and 5′-ACCAGGACACCCACCTCAGCTG-3′ (antisense). The primer sequences for Pfn2 were 5′-TGTCGGCAGAGCTGGTAGAGTCTT-3′ (sense) and 5′-GCAGCTAGAACCCAGAGTCTCTCAA-3′ (antisense). The PCR cycling conditions for GAPDH, Pfn1, and Pfn2 were 95 °C (30 s), 55 °C (30 s), and 72 °C (1 min) for a total of 35 cycles.
Migration assay
Single cell random migration assay with MDA-231 cells was performed according to our published protocol (45).
Quantification and statistics
Immunoblots were quantified using ImageJ to calculate and normalize band area and intensity (band intensities of proteins of interest were normalized with respect to their loading controls). Quantitative RT-PCR data were generated using StepOne Software (Applied Biosystems) and expressed as mean ± S.D. Analysis of variance was performed to determine statistical significance for the differences in the mean between the control and the experimental groups, and a p value less than 0.05 was considered statistically significant.
Author contributions
M. J. and D. G. were involved in design and execution of experiments and preparing the manuscript. N. C. was involved in execution of experiments. R. P. was involved in generation of various MKL constructs and intellectual contributions to the study. P. R. was involved in the overall planning and execution of the study and preparation of manuscript.
Supplementary Material
This work was supported in part by National Institutes of Health Grant 2R01CA108607 (to P. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.

This article contains supplemental Figs. S1–S11.
- SRF
- serum-response factor
- MKL
- megakaryoblastic leukemia
- Pfn
- profilin
- LatB
- latrunculin B
- EV
- empty vector
- DTE
- dithioerythritol
- TA
- transcriptional activation
- EC
- endothelial cell.
References
- 1. Olson E. N., and Nordheim A. (2010) Linking actin dynamics and gene transcription to drive cellular motile functions. Nat. Rev. Mol. Cell Biol. 11, 353–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Pipes G. C., Creemers E. E., and Olson E. N. (2006) The myocardin family of transcriptional co-activators: versatile regulators of cell growth, migration, and myogenesis. Genes Dev. 20, 1545–1556 [DOI] [PubMed] [Google Scholar]
- 3. Miralles F., Posern G., Zaromytidou A. I., and Treisman R. (2003) Actin dynamics control SRF activity by regulation of its co-activator MAL. Cell 113, 329–342 [DOI] [PubMed] [Google Scholar]
- 4. Vartiainen M. K., Guettler S., Larijani B., and Treisman R. (2007) Nuclear actin regulates dynamic subcellular localization and activity of the SRF cofactor MAL. Science 316, 1749–1752 [DOI] [PubMed] [Google Scholar]
- 5. Sotiropoulos A., Gineitis D., Copeland J., and Treisman R. (1999) Signal-regulated activation of serum-response factor is mediated by changes in actin dynamics. Cell 98, 159–169 [DOI] [PubMed] [Google Scholar]
- 6. Posern G., Miralles F., Guettler S., and Treisman R. (2004) Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF co-activator MAL. EMBO J. 23, 3973–3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ding Z., Bae Y. H., and Roy P. (2012) Molecular insights on context-specific role of profilin-1 in cell migration. Cell Adh. Migr. 6, 442–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Witke W. (2004) The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 14, 461–469 [DOI] [PubMed] [Google Scholar]
- 9. Sun Q., Chen G., Streb J. W., Long X., Yang Y., Stoeckert C. J. Jr., and Miano J. M. (2006) Defining the mammalian CArGome. Genome Res. 16, 197–207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Esnault C., Stewart A., Gualdrini F., East P., Horswell S., Matthews N., and Treisman R. (2014) Rho-actin signaling to the MRTF co-activators dominates the immediate transcriptional response to serum in fibroblasts. Genes Dev. 28, 943–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mouneimne G., Hansen S. D., Selfors L. M., Petrak L., Hickey M. M., Gallegos L. L., Simpson K. J., Lim J., Gertler F. B., Hartwig J. H., Mullins R. D., and Brugge J. S. (2012) Differential remodeling of actin cytoskeleton architecture by profilin isoforms leads to distinct effects on cell migration and invasion. Cancer Cell 22, 615–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Evelyn C. R., Wade S. M., Wang Q., Wu M., Iñiguez-Lluhí J. A., Merajver S. D., and Neubig R. R. (2007) CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol. Cancer Ther. 6, 2249–2260 [DOI] [PubMed] [Google Scholar]
- 13. Hayashi K., Watanabe B., Nakagawa Y., Minami S., and Morita T. (2014) RPEL proteins are the molecular targets for CCG-1423, an inhibitor of Rho signaling. PLoS One 9, e89016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muehlich S., Wang R., Lee S. M., Lewis T. C., Dai C., and Prywes R. (2008) Serum-induced phosphorylation of the serum-response factor co-activator MKL1 by the extracellular signal-regulated kinase 1/2 pathway inhibits its nuclear localization. Mol. Cell. Biol. 28, 6302–6313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zaromytidou A. I., Miralles F., and Treisman R. (2006) MAL and ternary complex factor use different mechanisms to contact a common surface on the serum-response factor DNA-binding domain. Mol. Cell. Biol. 26, 4134–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lewis T. C., and Prywes R. (2013) Serum regulation of Id1 expression by a BMP pathway and BMP responsive element. Biochim. Biophys. Acta 1829, 1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smith E. C., Thon J. N., Devine M. T., Lin S., Schulz V. P., Guo Y., Massaro S. A., Halene S., Gallagher P., Italiano J. E. Jr., and Krause D. S. (2012) MKL1 and MKL2 play redundant and crucial roles in megakaryocyte maturation and platelet formation. Blood 120, 2317–2329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gurbuz I., Ferralli J., Roloff T., Chiquet-Ehrismann R., and Asparuhova M. B. (2014) SAP domain-dependent Mkl1 signaling stimulates proliferation and cell migration by induction of a distinct gene set indicative of poor prognosis in breast cancer patients. Mol. Cancer 13, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sun Y., Boyd K., Xu W., Ma J., Jackson C. W., Fu A., Shillingford J. M., Robinson G. W., Hennighausen L., Hitzler J. K., Ma Z., and Morris S. W. (2006) Acute myeloid leukemia-associated Mkl1 (Mrtf-a) is a key regulator of mammary gland function. Mol. Cell. Biol. 26, 5809–5826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Romeo G. R., and Kazlauskas A. (2008) Oxysterol and diabetes activate STAT3 and control endothelial expression of profilin-1 via OSBP1. J. Biol. Chem. 283, 9595–9605 [DOI] [PubMed] [Google Scholar]
- 21. Shields A. R., Spence A. C., Yamashita Y. M., Davies E. L., and Fuller M. T. (2014) The actin-binding protein profilin is required for germline stem cell maintenance and germ cell enclosure by somatic cyst cells. Development 141, 73–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ho H. H., and Ivashkiv L. B. (2006) Role of STAT3 in type I interferon responses. Negative regulation of STAT1-dependent inflammatory gene activation. J. Biol. Chem. 281, 14111–14118 [DOI] [PubMed] [Google Scholar]
- 23. Qi Y. F., Huang Y. X., Wang H. Y., Zhang Y., Bao Y. L., Sun L. G., Wu Y., Yu C. L., Song Z. B., Zheng L. H., Sun Y., Wang G. N., and Li Y. X. (2013) Elucidating the crosstalk mechanism between IFN-γ and IL-6 via mathematical modelling. BMC Bioinformatics 14, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Théry C., Boussac M., Véron P., Ricciardi-Castagnoli P., Raposo G., Garin J., and Amigorena S. (2001) Proteomic analysis of dendritic cell-derived exosomes: a secreted subcellular compartment distinct from apoptotic vesicles. J. Immunol. 166, 7309–7318 [DOI] [PubMed] [Google Scholar]
- 25. Grønborg M., Kristiansen T. Z., Iwahori A., Chang R., Reddy R., Sato N., Molina H., Jensen O. N., Hruban R. H., Goggins M. G., Maitra A., and Pandey A. (2006) Biomarker discovery from pancreatic cancer secretome using a differential proteomic approach. Mol. Cell. Proteomics 5, 157–171 [DOI] [PubMed] [Google Scholar]
- 26. Caglayan E., Romeo G. R., Kappert K., Odenthal M., Südkamp M., Body S. C., Shernan S. K., Hackbusch D., Vantler M., Kazlauskas A., and Rosenkranz S. (2010) Profilin-1 is expressed in human atherosclerotic plaques and induces atherogenic effects on vascular smooth muscle cells. PLoS One 5, e13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jella K. K., Yu L., Yue Q., Friedman D., Duke B. J., and Alli A. A. (2016) Exosomal GAPDH from proximal tubule cells regulate ENaC activity. PLoS One 11, e0165763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hosseini-Beheshti E., Pham S., Adomat H., Li N., and Tomlinson Guns E. S. (2012) Exosomes as biomarker enriched microvesicles: characterization of exosomal proteins derived from a panel of prostate cell lines with distinct AR phenotypes. Mol. Cell. Proteomics 11, 863–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lundquist M. R., Storaska A. J., Liu T. C., Larsen S. D., Evans T., Neubig R. R., and Jaffrey S. R. (2014) Redox modification of nuclear actin by MICAL-2 regulates SRF signaling. Cell 156, 563–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bae Y. H., Ding Z., Das T., Wells A., Gertler F., and Roy P. (2010) Profilin1 regulates PI(3,4)P2 and lamellipodin accumulation at the leading edge thus influencing motility of MDA-MB-231 cells. Proc. Natl. Acad. Sci. U.S.A. 107, 21547–21552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zou L., Jaramillo M., Whaley D., Wells A., Panchapakesa V., Das T., and Roy P. (2007) Profilin-1 is a negative regulator of mammary carcinoma aggressiveness. Br. J. Cancer 97, 1361–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Asparuhova M. B., Ferralli J., Chiquet M., and Chiquet-Ehrismann R. (2011) The transcriptional regulator megakaryoblastic leukemia-1 mediates serum-response factor-independent activation of tenascin-C transcription by mechanical stress. FASEB J. 25, 3477–3488 [DOI] [PubMed] [Google Scholar]
- 33. Johnson L. A., Rodansky E. S., Haak A. J., Larsen S. D., Neubig R. R., and Higgins P. D. (2014) Novel Rho/MRTF/SRF inhibitors block matrix-stiffness and TGF-β-induced fibrogenesis in human colonic myofibroblasts. Inflamm. Bowel Dis. 20, 154–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jin W., Goldfine A. B., Boes T., Henry R. R., Ciaraldi T. P., Kim E. Y., Emecan M., Fitzpatrick C., Sen A., Shah A., Mun E., Vokes V., Schroeder J., Tatro E., Jimenez-Chillaron J., and Patti M. E. (2011) Increased SRF transcriptional activity in human and mouse skeletal muscle is a signature of insulin resistance. J. Clin. Invest. 121, 918–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cui X. B., Zhang S. M., Xu Y. X., Dang H. W., Liu C. X., Wang L. H., Yang L., Hu J. M., Liang W. H., Jiang J. F., Li N., Li Y., Chen Y. Z., and Li F. (2016) PFN2, a novel marker of unfavorable prognosis, is a potential therapeutic target involved in esophageal squamous cell carcinoma. J. Transl. Med. 14, 137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ding Z., Joy M., Bhargava R., Gunsaulus M., Lakshman N., Miron-Mendoza M., Petroll M., Condeelis J., Wells A., and Roy P. (2014) Profilin-1 downregulation has contrasting effects on early vs. late steps of breast cancer metastasis. Oncogene 33, 2065–2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Janke J., Schlüter K., Jandrig B., Theile M., Kölble K., Arnold W., Grinstein E., Schwartz A., Estevéz-Schwarz L., Schlag P. M., Jockusch B. M., and Scherneck S. (2000) Suppression of tumorigenicity in breast cancer cells by the microfilament protein profilin 1. J. Exp. Med. 191, 1675–1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karamchandani J. R., Gabril M. Y., Ibrahim R., Scorilas A., Filter E., Finelli A., Lee J. Y., Ordon M., Pasic M., Romaschin A. D., and Yousef G. M. (2015) Profilin-1 expression is associated with high grade and stage and decreased disease-free survival in renal cell carcinoma. Hum. Pathol. 46, 673–680 [DOI] [PubMed] [Google Scholar]
- 39. Tang Y. N., Ding W. Q., Guo X. J., Yuan X. W., Wang D. M., and Song J. G. (2015) Epigenetic regulation of Smad2 and Smad3 by profilin-2 promotes lung cancer growth and metastasis. Nat. Commun. 6, 8230. [DOI] [PubMed] [Google Scholar]
- 40. Romeo G., Frangioni J. V., and Kazlauskas A. (2004) Profilin acts downstream of LDL to mediate diabetic endothelial cell dysfunction. FASEB J. 18, 725–727 [DOI] [PubMed] [Google Scholar]
- 41. Liao X. H., Wang N., Liu L. Y., Zheng L., Xing W. J., Zhao D. W., Sun X. G., Hu P., Dong J., and Zhang T. C. (2014) MRTF-A and STAT3 synergistically promote breast cancer cell migration. Cell. Signal. 26, 2370–2380 [DOI] [PubMed] [Google Scholar]
- 42. Coumans J. V., Gau D., Poljak A., Wasinger V., Roy P., and Moens P. D. (2014) Profilin-1 overexpression in MDA-MB-231 breast cancer cells is associated with alterations in proteomics biomarkers of cell proliferation, survival, and motility as revealed by global proteomics analyses. Omics 18, 778–791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leitner L., Shaposhnikov D., Mengel A., Descot A., Julien S., Hoffmann R., and Posern G. (2011) MAL/MRTF-A controls migration of non-invasive cells by upregulation of cytoskeleton-associated proteins. J. Cell Sci. 124, 4318–4331 [DOI] [PubMed] [Google Scholar]
- 44. Gau D., Veon W., Zeng X., Yates N., Shroff S. G., Koes D. R., and Roy P. (2016) Threonine 89 is an important residue of profilin-1 that is phosphorylatable by protein kinase A. PLoS One 11, e0156313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bae Y. H., Ding Z., Zou L., Wells A., Gertler F., and Roy P. (2009) Loss of profilin-1 expression enhances breast cancer cell motility by Ena/VASP proteins. J. Cell. Physiol. 219, 354–364 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.