

Abstract
The prolyl isomerase Pin1 binds to the phosphorylated Ser/Thr-Pro motif of target proteins and enhances their cis-trans conversion. This report is the first to show that Pin1 expression in pancreatic β cells is markedly elevated by high-fat diet feeding and in ob/ob mice. To elucidate the role of Pin1 in pancreatic β cells, we generated β-cell–specific Pin1 KO (βPin1 KO) mice. These mutant mice showed exacerbation of glucose intolerance but had normal insulin sensitivity. We identified two independent factors underlying impaired insulin secretion in the βPin1 KO mice. Pin1 enhanced pancreatic β-cell proliferation, as indicated by a reduced β-cell mass in βPin1 KO mice compared with control mice. Moreover, a diet high in fat and sucrose failed to increase pancreatic β-cell growth in the βPin1 KO mice, an observation to which up-regulation of the cell cycle protein cyclin D appeared to contribute. The other role of Pin1 was to activate the insulin-secretory step: Pin1 KO β cells showed impairments in glucose- and KCl-induced elevation of the intracellular Ca2+ concentration and insulin secretion. We also identified salt-inducible kinase 2 (SIK2) as a Pin1-binding protein that affected the regulation of Ca2+ influx and found Pin1 to enhance SIK2 kinase activity, resulting in a decrease in p35 protein, a negative regulator of Ca2+ influx. Taken together, our observations demonstrate critical roles of Pin1 in pancreatic β cells and that Pin1 both promotes β-cell proliferation and activates insulin secretion.
Keywords: cell proliferation, diabetes, insulin secretion, pancreatic islet, prolyl isomerase, salt inducible kinase
Introduction
Pancreatic β cells secrete insulin in response to elevated glucose concentrations and to control glucose homeostasis (1, 2). In pancreatic β cells, glucose is metabolized to produce ATP via oxidative phosphorylation, and the increase in intracellular ATP levels induces closure of the ATP-sensitive potassium channel, leading to Ca2+ influx through voltage-dependent calcium channels and subsequent exocytosis of insulin granules (3, 4). In states of insulin resistance in peripheral tissues such as the liver and muscles under obese conditions, the mass of pancreatic β cells becomes enlarged to meet the demand for more insulin to be supplied to peripheral tissues (5–7). Both impaired glucose-induced insulin secretion and the β-cell mass reduction are regarded as major causes of type 2 diabetes mellitus (1). However, the molecular mechanisms underlying the aforementioned functions of pancreatic β cells have not as yet been fully elucidated.
On the other hand, prolyl isomerase Pin1 was initially identified as a regulator of mitosis that raises the cyclin D content (8). Pin1 requires the motif including Ser(P)/Thr(P)-Pro for binding with substrates, and to date, many reports have revealed Pin1 to have major involvement in the pathogenesis of cancer and Alzheimer's disease (9–11). In addition, interestingly, the Pin1 expressions in metabolic organs were found to be highly up-regulated by high-fat diet feeding and under hyperglycemic conditions (12–15). Indeed, Pin1 binds to and modulates numerous important proteins that are key to the regulation of glucose and lipid metabolism, including IRS-1, Crtc2, and AMPK (12, 16, 17). Thus, Pin1 contributes to the regulation of gluconeogenesis, adipogenesis, fibrosis, fatty acid oxidation, and bone differentiation as well as of the onset of metabolic syndromes, such as obesity and nonalcoholic steatohepatitis (12, 16–20). Clinical studies have shown serum Pin1 concentrations to be highly correlated with the progression of metabolic syndromes (21, 22).
In this study, we first identified increased expression of Pin1 in the pancreatic β cells of obese mice, which is reportedly associated with increased insulin secretion and enlarged β-cell mass, which are mechanisms compensating for insulin resistance. To elucidate the patho-physiological significance of increased Pin1 expression in pancreatic β cells, we generated β-cell–specific Pin1 KO mice, which exhibited impaired insulin secretion and glucose intolerance. This is the first study to demonstrate the critical roles of Pin1 in pancreatic β cells, especially β-cell proliferation and the insulin-secretory step.
Results
Pin1 protein in pancreatic islets was elevated by high-fat diet feeding and in ob/ob mice
Eight-week-old mice were fed a high-fat diet (60% fat, 8% carbohydrate, and 25% protein) or a normal diet (5% fat and 23% protein) for 20 weeks, and pancreatic islets were then isolated and subjected to immunoblotting using anti-Pin1 antibody. The Pin1 protein level in high-fat diet-fed mice was increased to ∼3-fold that of the controls (Fig. 1A). Similarly, Pin1 expression levels in the islets from ob/ob mice were increased, to ∼2.7 times the level in control lean mice (Fig. 1B). These mice reportedly showed the phenotype of hyperinsulinemia with β-cell mass enlargement, which compensates for obesity-related insulin resistance (6, 23).
Figure 1.

Pin1 expressions are increased in islets from obese mice. A, islets were prepared from mice fed a normal diet or a high-fat diet for 20 weeks (n = 4). B, islets were isolated from 10-week-old male wild-type mice or ob/ob mice. Pin1 protein levels in the islets were demonstrated by immunoblot (IB) analysis (n = 4). C and D, glucose tolerance and insulin tolerance tests were performed, using three groups of mice (n = 5). E, islets were isolated from 8-week-old male mice. The islets were then incubated with HKR buffer containing 3 or 20 mm glucose for 1 h. F, islet areas were measured to allow comparisons among the three groups. **, p < 0.01; ***, p < 0.001; n.s., not significant. Representative data from three independent experiments are shown. Error bars, S.E.
Deficiency of Pin1 in pancreatic β cells drives glucose intolerance
To investigate the roles of Pin1 in pancreatic β cells, we generated pancreatic β-cell–specific Pin1 knock-out mice (βPin1 KO mice) by mating Pin1 flox mice with mouse insulin promoter-Cre transgenic (MIP-Cre)2 mice. Recent reports have demonstrated that the MIP-Cre or the rat insulin promoter (RIP)-Cre gene may impact glucose metabolism (25, 26). Therefore, we initially assessed whether the MIP-Cre gene affects glucose or insulin tolerance, as compared with that in C57BL/6J (wild type) or Pin1 flox mice. Neither glucose nor insulin tolerance tests showed any differences among the three groups (Fig. 1, C and D). In addition, the areas of pancreatic β-cell or glucose-induced insulin release were unaffected by insertion of the MIP-Cre or the flox gene (Fig. 1, E and F). Hence, we concluded that the inserted MIP-Cre gene had no effect on glucose metabolism and selected Pin1 flox mice as a control. Immunoblotting with anti-Pin1 antibody clearly showed almost complete absence of Pin1 protein in the islets of these βPin1 KO mice, whereas Pin1 expressions in other tissues were unchanged (Fig. 2A). The Pin1 mRNA level in islets of βPin1 KO mice was also markedly decreased, as compared with that in the controls (Fig. 2B). To investigate the cell type–specific expressions of Pin1 in islets, we performed double immunostaining with anti-Pin1 and either anti-glucagon or anti-insulin antibodies. Pin1 expression was ubiquitously observed in numerous cell types in islets of the control (Pin1 f/f) mice (top panels of Fig. 2, C and D). In contrast, in the βPin1 KO mouse islets, Pin1 expressions were absent from the β-cell areas stained with anti-insulin antibody (bottom panel of Fig. 2C), whereas Pin1 expressions were still detectable in the glucagon-positive α-cell areas (bottom panel of Fig. 2D). Next, we carried out experiments to investigate the effect of ablating Pin1 in β cells on blood glucose homeostasis. Whereas there was no difference in fasting blood glucose levels between control and βPin1 KO mice, βPin1 KO mice had higher values in the refed state than the control (Fig. 2E). Glucose tolerance test (GTT) results indicated elevated serum glucose concentrations at 15 and 30 min in βPin1 KO, as compared with the control Pin1 flox (Fig. 2F). In accordance with the GTT results, elevations of the serum insulin concentration at 15 and 30 min in control mice were significantly smaller than those in the control Pin1 flox mice (Fig. 2G).
Figure 2.

Deletion of Pin1 in pancreatic β cells reduced insulin secretion and exacerbated glucose intolerance. A, expressions of Pin1 protein in various tissues of control and βPin1 KO mice. B, Pin1 mRNA levels in isolated islets from control and βPin1 KO mice (n = 5). C and D, immunostaining with anti-insulin, anti-glucagon, or anti-Pin1 antibody in pancreatic sections from control and βPin1 KO mice. E, blood glucose levels in fasted and 1-h re-fed states of 10-week-old male mice fed chow diets (n = 6–9). F, glucose tolerance tests on normal diet-fed mice. Glucose (2 g/kg) was intraperitoneally injected into 8-week-old male mice after they had been fasted overnight (n = 6). G, plasma insulin levels after stimulation with glucose. Mice were fasted overnight and then injected with glucose solution (2 g/kg) (n = 6). H, insulin tolerance tests on 7-week-old male mice fed a normal diet. Insulin solution (0.75 IU/kg) was intraperitoneally injected into the mice after they had been fasted for 4 h (n = 6–8). I, glucose tolerance tests on mice fed the HFHS diet for 14 weeks (n = 6). J, insulin tolerance tests on mice fed the HFHS diet for 15 weeks (n = 6). *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant. Representative data from two (A, C, and D) or three independent experiments (B and E–J) are shown. IB, immunoblotting; Error bars, S.E.
Clearer results were obtained from the GTT after feeding of a diet containing both high fat and high sucrose (HFHS) for 14 weeks. Markedly reduced glucose clearance from blood in the βPin1 KO mice, as compared with the control mice, was observed (Fig. 2I). In contrast, there were no differences in the lowering of serum glucose levels in response to insulin between the two groups of mice, regardless of whether the normal or the HFHS diet had been given to these mice (Fig. 2, H and J). This series of experimental results indicated impaired glucose-induced insulin secretion and normal insulin sensitivity in the βPin1 KO mice.
Islet and β-cell areas are reduced in βPin1 KO mice
To investigate the cause of reduced insulin secretion in βPin1 KO mice, as demonstrated by GTT, the β-cell mass sizes were compared between βPin1 KO and control mice. The average pancreatic islet size as well as the ratios of β-cell mass to that of the entire pancreas in sections from βPin1 KO mice were revealed to be significantly smaller than those in control mice (Fig. 3, A and B). However, insulin contents per equal amount of protein in the islets isolated from the two groups did not differ (Fig. 3C). To elucidate whether proliferation or apoptosis is involved in this β-cell mass difference, anti-Ki67 and TUNEL staining were performed. Cell proliferation, as shown by anti-Ki67-positive staining in islets from βPin1 KO mice, was significantly decreased as compared with that in control mice (Fig. 3D), whereas the ratios of apoptotic cells did not differ significantly (Fig. 3E). In addition, elimination of the Pin1 gene had no effects on the gene expression levels of key factors, such as Ins1, PDX1, Glut2, and NeuroD, known to be involved in the maturation or differentiation of pancreatic β cells (Fig. 3F). Taking these observations together, the difference in β-cell mass might be attributable to cell proliferation changes rather than to alterations in apoptosis or differentiation. Thus, we next examined the expression levels of cell cycle-related genes, such as cyclin D1, cyclin D2, CDK2, and CDK4. Whereas their mRNA levels were unaltered (data not shown), the protein expression level of cyclin D1 in the Pin1 KO islets was reduced by ∼50% (Fig. 3G), which is in good agreement with a previous report showing a relationship between the expression levels of Pin1 and cyclin D1 (27).
Figure 3.

Pin1 deficiency diminished pancreatic β-cell mass. Pancreatic sections were prepared from 10-week-old male mice fed normal chow (n = 5). A and B, islet areas and β-cell mass were measured for comparison between control and βPin1 KO mice. C, insulin contents in islets. Proteins were extracted from islets of 12-week-old male mice. Insulin contents were measured by the ELISA method and then adjusted by protein levels (n = 4–5). D and E, Ki67-positive cells were counted as an indicator of cell proliferation. TUNEL-positive cells were counted to allow calculation of apoptosis rates. F, expression levels of markers of mature islets. RNA was extracted from islets of 8-week-old male mice. Expressions of mRNA were measured by real-time PCR (n = 5). G, reduction of cyclin D1 protein in response to Pin1 gene ablation. Cyclin D1 protein levels in islets were assessed by immunoblotting (IB) (n = 6–7). *, p < 0.05; **, p < 0.01; ***, p < 0.001; n.s., not significant. Representative data from one of three independent experiments are shown. Error bars, S.E.
Pin1 is essential for hypertrophy of β cells in mice fed the HFHS diet
Insulin resistance in peripheral tissues is well known to drive the compensatory hypertrophy of pancreatic β cells. As shown in Fig. 1, Pin1 expression in islets is significantly increased in ob/ob and high-fat diet fed mice. In addition, taking the smaller β-cell mass in βPin1 KO mice into consideration, we can reasonably speculate that Pin1 contributes to the compensatory proliferation of β cells in islets. To assess this possibility, mice fed the HFHS diet for 20 weeks were prepared. Feeding of the HFHS diet increased both β-cell areas and the ratio of β cells to the entire pancreatic area in control mice (right panels of Fig. 4, A and B). In contrast, no such hypertrophy of β cells in response to HFHS diet feeding was observed in the βPin1 KO mice (left panels of Fig. 4, A and B). In addition, staining with the anti-Ki67 antibody revealed Ki67-positive cells to be increased in control mice but not in βPin1 KO mice (Fig. 4C), whereas there was no significant difference in the number of TUNEL-positive cells between the two groups (Fig. 4D). Thus, HFHS-induced β-cell hypertrophy might be attributable to increased cell proliferation but not to diminished apoptosis.
Figure 4.

Pin1 is essential for compensatory hypertrophy of pancreatic β-cell mass. A–D, compensatory hypertrophy of β-cell mass was suppressed in βPin1 KO mice. Both control and βPin1 KO mice were fed the HFHS diet for 20 weeks (n = 6–7). A and B, islet areas and β-cell masses were measured for comparison between control and βPin1 KO mice. C, Ki67-positive cells were counted as an indicator of cell proliferation. D, TUNEL positive cells were counted to allow calculation of apoptosis ratios. *, p < 0.05; **, p < 0.01; n.s., not significant. Representative data from three independent experiments are shown. Error bars, S.E.
Pin1 gene deletion in pancreatic β cells impairs insulin secretion
Furthermore, the ratios of secreted insulin per total insulin content were compared between the islets isolated from βPin1 KO and control Pin1 flox mice, employing stimulation with glucose or KCl. It was revealed that the ratio of secreted insulin to the entire cellular insulin content of the βPin1 KO mouse islets was significantly lower than the ratio in the control islets, regardless of whether the stimulation was with high glucose or KCl (Fig. 5, A and B). Therefore, in addition to its role in cell proliferation, it was suggested that Pin1 plays an important part in the insulin-secretory step. To determine the impaired step, contents of ATP, accumulation of which is the first step leading to insulin release, were measured. Intracellular ATP concentrations did not differ between the islets prepared from Pin1 flox versus those from βPin1 KO mice (Fig. 5C).
Figure 5.

Ablation of the Pin1 gene impairs the insulin release response. A and B, measurement of insulin release in isolated islets stimulated with 20 mm glucose for 1 h or with 45 mm KCl for 30 min. HKR buffer containing 3 mm glucose was used as a control. Supernatants were collected, and insulin contents were measured by the ELISA method. The rates of insulin release were calculated by adjusting for total insulin contents (n = 4–5). C, measurement of intracellular ATP levels in the isolated islets. Isolated islets were stimulated with 3 or 20 mm glucose for 15 min. Then ATP levels were measured. D, Min6 cells were loaded with Fura-2 for 30 min. After being washed with PBS, intracellular Ca2+ changes were measured. High glucose was added 1 min after starting the measurement (n ≥ 25 cells). E, regulation of insulin release by SIK2. F, decreased insulin release in response to pan-SIK inhibitor treatment. Isolated islets were incubated with 1 μm HG-9-91-01 overnight and then stimulated with 20 mm glucose for 1 h (n = 4–5). G, the association between Pin1 and SIK2. Both FLAG-SIK2 and S-tagged Pin1 were overexpressed in 293T cells. Next, the cells were immunoprecipitated (IP) with FLAG beads, and binding proteins were eluted employing FLAG peptide. H, Pin1 endogenously binds to SIK2. Proteins were extracted from Min6 cells and then immunoprecipitated with Pin1 antibody. Finally, proteins in the samples were detected employing SIK2 antibody. *, p < 0.05; **, p < 0.01; n.s., not significant. Representative data from three independent experiments are shown. IB, immunoblotting; AUC, area under the curve; error bars, S.E.
Next, the effect of Pin1 on high glucose–induced Ca2+ influx using MIN6 insulinoma cells was examined. Two adenoviruses expressing Pin1 shRNA reduced Pin1 expression, and Min6 lacking Pin1 showed reduced Ca2+ influx in response to high-glucose stimulation (Fig. 5D).
Pin1 associates with SIK2
We subsequently searched for Pin1 binding partners, which would affect the steps between ATP synthesis and Ca2+ influx leading to insulin secretion. Fig. 5D shows the role of salt-inducible kinase 2 (SIK2) in insulin release through p35 phosphorylation. In brief, the phosphorylation of p35 by SIK2 results in the induction of its polyubiquitination and degradation, thereby nullifying suppression of the voltage-dependent calcium channel (VDCC) by CDK5 (28) (Fig. 5E). Indeed, it was confirmed that pan-SIK inhibitor treatment reduces high glucose–induced insulin secretion in isolated islets (Fig. 5F).
We speculated that Pin1 might be involved in the SIK2–CDK5–VDCC pathway and examined the associations of Pin1 with p35, CDK5, and SIK2. FLAG-tagged p35, CDK5, and SIK2 were each overexpressed with S-tagged Pin1 in 293T cells, and anti-FLAG tag antibody immunoprecipitates were immunoblotted with anti-S-tag antibody. As shown in Fig. 5F, this experiment revealed that S-tag Pin1 binds with FLAG-SIK2 (Fig. 5G) but not with either FLAG- p35 or CDK5 (data not shown). The endogenous interaction between Pin1 and SIK2 was also detected in Min6 cells (Fig. 5H).
Subsequently, we focused on identifying the Pin1 domain required for the association with SIK2. Pin1 has two domains, WW and PPIase. We prepared GST alone and GST fusion proteins containing the WW domain only, the PPIase domain only, and full-length Pin1, whereas S-tagged SIK2 was overexpressed in 293 cells. Because GST fusion proteins containing full-length Pin1 and that with only the WW domain, but not that containing the PPIase domain, bound to S-tagged SIK2, the WW domain of Pin1 was revealed to be responsible for the association with SIK2 (Fig. 6A).
Figure 6.

Pin1 binds to both Ser-179–Pro and Thr-420–Pro sites of SIK2 via its WW domain. A, Pin1 associates with SIK2 through the WW domain. FLAG-tagged SIK2 were overexpressed in 293T cells, and cell lysates were prepared. The cell lysates were incubated with GST protein-conjugated beads, and the proteins were then immunoblotted (IB) with FLAG antibody. B, two Ser/Thr-Pro sites of SIK2 are essential for Pin1 binding. All Ser/Thr-Pro sites in SIK2 were substituted with alanine. Then one of the Ser/Thr residues was restored. Cell lysates including SIK2 mutants were incubated with GST protein-conjugated beads. C, GST–Pin1 was incubated with wild-type SIK2 or the SIK2 mutant in which both Ser-179 and Thr-420 had been switched to alanine. Representative data from one of three independent experiments are shown. CBB, Coomassie Brilliant Blue.
On the other hand, SIK2 contains 11 Ser(P)/Thr-Pro motifs, which are required for the association with Pin1. We generated cDNAs encoding the mutated SIK proteins, of which all Ser/Thr-Pro sites except for the one indicated site were substituted with Ala-Pro. Wild-type and mutated SIK2 proteins were overexpressed in 293 cells. These cell lysates were subjected to the pulldown assay with GST–Pin1 proteins. The wild-type and mutant SIK2 in which Ser-179 or Thr-420 was intact were found to be associated with GST–Pin1 (Fig. 6B). In addition, the SIK2 mutant in which both the Ser-179 and the Thr-420 site had been substituted with Ala showed no capacity to interact with Pin1 (Fig. 6C). These results indicate that SIK2 possesses two portions, one containing Ser-179 and the other Thr-420, that are necessary for the association with Pin1.
Pin1 enhances SIK2 activity, leading to p35 degradation and Ca2+ influx
Because the two binding sites of SIK2 with Pin1 exist in or near the kinase domain, we investigated the effects of their associations with Pin1 on the kinase activity of SIK2 by measuring the p35 phosphorylation level at Ser-91. It was confirmed that wild-type p35, but not the mutated p35 in which Ser-91 had been replaced with Ala, was phosphorylated by SIK2 in vitro (Fig. 7A). Co-incubation with Pin1 strongly enhanced the SIK2-induced phosphorylation of p35 in vitro, as compared with either p35 or SIK2 alone (Fig. 7B). The phosphorylation of p35 at the Ser-91 site reportedly enhances its degradation through polyubiquitination (28). Therefore, we investigated the effect of Pin1 on the p35 protein expression level. When both p35 and wild-type Pin1 were overexpressed in 293T cells, the amount of p35 protein was dramatically decreased (Fig. 7C). However, p35 expression was unaffected by the W34A Pin1 mutant, which is unable to bind substrates, or by the K63A Pin1 mutant, which is an inactive form of PPIase, indicating that the isomerase activity of Pin1 is essential for the degradation-promoting effect on p35 (Fig. 7C). To confirm that the decrease in p35 protein in response to Pin1 depends on SIK2, we used an S91A p35 mutant, which cannot be phosphorylated by SIK2. Pin1 overexpression had no effect on the expression of this mutant, indicating that Pin1 decreased p35 protein via regulation of SIK2 (Fig. 7D).
Figure 7.

SIK2 is activated by the association with Pin1 and is involved in the insulin secretion pathway. A, phosphorylation of p35 at Ser-91 by SIK2. SIK2 protein was incubated with GST–p35 WT or GST–p35 S91A for 30 min, and the level of phospho-p35 was then examined by Western blotting (IB). B, up-regulation of SIK2 activity by Pin1. GST–p35 protein was reacted with SIK2, and SIK2 binding with Pin1 was allowed to proceed for 30 min. Phosphorylation levels of p35 were determined by Western blotting. C, Pin1 decreases p35 expression, depending on the isomerase activity of Pin1. Both FLAG-p35 and S-tag Pin1 were transfected into 293T cells. The p35 expression levels were determined by Western blotting. W34A is the Pin1 mutant form that is unable to bind substrates, and K63A is the inactive form of PPIase. D, the reductions in p35 proteins in response to Pin1 depend on SIK2 activity. Both S-tag Pin1 and FLAG-SIK2 were transfected into 293T cells. The SIK2 S91A mutant is not phosphorylated by SIK2. E, Min6 cells were infected with adenovirus coding shLacZ or shPin1. After 72 h, samples were prepared, and total p35 levels were determined by Western blotting. F, islets were isolated from both control and βPin1 KO mice. The p35 protein expression levels were investigated using Western blotting. G, isolated islets were incubated with DMSO or 50 μm olomoucine for 2 h. The cells were then incubated in the presence of 20 mm glucose for 1 h (n = 4–5). **, p < 0.01; n.s., not significant. Shown are representative data from three independent experiments. Error bars, S.E.
Next, we examined whether Pin1 knockdown influences the expression of endogenous p35 protein and found that it was increased by Pin1 knockdown (Fig. 7E). In isolated islets from Pin1 flox MIP-Cre mice, an increase in p35 protein was observed, as compared with islets from control mice (Fig. 7F).
The p35 protein works in partnership with CDK5, and the p35–CDK5 complex suppresses Ca2+ entry via VDCC. Taking into consideration that Pin1 ablation enhances p35–CDK5 activity, it is reasonable to suggest that Pin1 deficiency might well attenuate Ca2+ influx induced by high glucose, as shown in Fig. 5D. Finally, if the impairment of insulin release in Pin1 KO islets depends upon up-regulation of CDK5 activity via increased p35 protein, CDK5 inhibitor treatment would presumably reverse the reduction of insulin secretion. Therefore, we examined the effect of olomoucine, a CDK5 inhibitor, on insulin release. Treatment with 50 μm olomoucine blocked the inhibition of insulin secretion normally seen with Pin1 deficiency (Fig. 7G). Taking these results together, we can reasonably assume that Pin1 enhances SIK2 activity and, thereby, promotes both p35 degradation and Ca2+ influx.
Discussion
Diabetes mellitus is an increasingly common disease, characterized by hyperglycemia due to deficient insulin action, which in turn is due to insulin resistance and/or insufficient insulin secretion (1–3). Obesity or overnutrition appears to be the most common cause of insulin resistance, for which increased insulin secretion with an enlarged pancreatic β-cell mass compensates during the period through glucose intolerance development to the early stage of type 2 diabetes mellitus (29, 30). This is the first report, to our knowledge, demonstrating the role of Pin1 in pancreatic β-cell functions. Our results also strongly suggest the involvement of Pin1 in the mechanism underlying increased insulin secretion in the state of obesity-related insulin resistance.
First, the Pin1 expression level in pancreatic β cells was shown to be markedly elevated in high-fat diet-fed and ob/ob mice, both reportedly showing hyperinsulinemia. On the other hand, βPin1 KO mice showed impaired insulin secretion, leading to glucose intolerance. Based on both sets of results, we recognized the important role of Pin1 in pancreatic β cells. Regarding the mechanism underlying impaired insulin secretion in βPin1 KO mice, two independent factors were identified. One role of Pin1 in pancreatic β cells is to increase the β-cell mass by up-regulating cell proliferation, which is supported by the mass of β cells in βPin1 KO mice being significantly smaller than that in the controls, and enlargement in response to HFHS feeding failed to occur. Previous reports revealed that Pin1 binds to and activates ∼30 proteins potentially involved in cell proliferation, including cyclin D1, Myc, β-catenin, and so on (27, 31–33). In this study, we showed that Pin1 ablation in islets reduced cyclin D1 protein, which is consistent with a previous report showing that Pin1 associates with cyclin D1 and enhances the stability of its protein. Indeed, cyclin D1 reportedly plays important roles in cell proliferation and hypertrophy in pancreatic islets (34, 35). On the other hand, although β-catenin, one of the known Pin1 binding partners, is also considered to contribute to cell growth in islets, no reduction of β-catenin in Pin1-deficient islets was observed in this study (data not shown). Thus, it is reasonable to assume that up-regulation of cyclin D1 protein is involved, at least playing a contributory role, in Pin1-induced β-cell proliferation. However, we cannot rule out the possibility that more factors exist that are involved in cell proliferation via the association with Pin1.
The other role of Pin1 is to activate the insulin-secretory step. It was revealed that not only glucose- but also KCl-induced intracellular Ca2+ concentration elevation and insulin secretion were impaired in Pin1 KO β cells, despite there being no difference in the cellular ATP level or insulin content per equal amount of protein. Thus, we attempted to identify the protein that binds to Pin1 and affects regulation of the intracellular Ca2+ concentration in β cells. We found that SIK2 binds to Pin1, which leads to enhancement of its kinase activity. SIK2, which belongs to the AMP-activated protein kinase family, functions as a key player in insulin secretion while serving a gluconeogenesis-regulating function in the liver (28, 36). Experiments using deletion and point mutants of SIK2 and Pin1 revealed the association to be mediated by the WW domain of Pin1 and two sites containing Ser/Thr-Pro motifs in and near the kinase domain of SIK2. The PPIase activity of Pin1 was shown to be necessary for the Pin1-induced enhancement of SIK2 activity. In fact, Pin1 knockdown raised the total p35 protein concentration in Min6 cells. In addition, treatment with olomoucine, a CDK5 inhibitor, reversed the decrease in insulin release resulting from Pin1 deficiency. Taken together, these findings allow us to conclude that Pin1 binds to and enhances SIK2 activity and thereby induces both the degradation of p35 and the down-regulation of CDK5 activity, eventually contributing to the enhanced insulin secretion response of β cells (Fig. 8).
Figure 8.
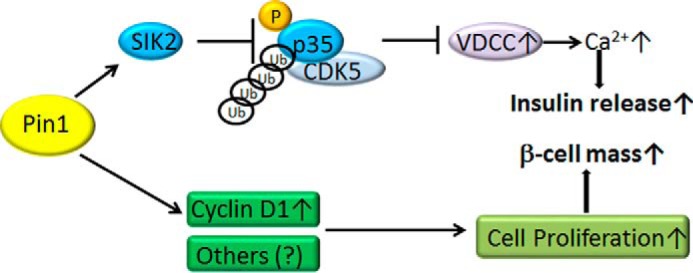
Regulation of pancreatic β-cell functions by Pin1. Pin1 associates with SIK2 and thereby decreases the expression of p35, a negative regulator of VDCC. The degradation of p35 protein promotes Ca2+ influx and subsequent insulin release. In addition, Pin1 controls pancreatic β-cell mass through, at least partially, the regulation of cyclin D1.
In summary, we have clearly demonstrated the roles of Pin1 in the regulation of both β-cell mass and insulin secretion. This mechanism presumably underlies the development of compensatory hyperinsulinemia accompanying β-cell hypertrophy, under conditions of obesity-related insulin resistance.
Experimental procedures
Materials
Antibodies were purchased from Santa Cruz Biotechnology (Pin1 mouse antibody sc-46660, lot D0115; tubulin mouse antibody sc-5286, lot B2715), Cell Signaling (Glucagon rabbit antibody 2760, lot 4; insulin rabbit antibody 3014, lot 4; cyclin D1 mouse antibody 2922, lot 7; and p35 rabbit antibody 2680, lot 4), Abcam (insulin mouse antibody ab6995, lot GR1753763; Ki67 rabbit antibody ab15580, lot GR278965-1; and S-tag rabbit antibody ab18588, lot GR124515-1), or Sigma (FLAG mouse antibody F3040, lot 035K6196). Phospho-p35 antibody was kindly provided by Dr. Hisanaga (Tokyo Metropolitan University) (24). HRP-linked mouse or rabbit IgG secondary antibody was purchased from GE Healthcare (NA931V and NA934V).
FLAG beads were purchased from Sigma. FITC- or Cy3-conjugated secondary antibodies were obtained from Jackson Immunoresearch, and BlueStatin was from Invitrogen.
Mice
Pin1 flox mice were produced, as described previously (17). Pin1 flox mice were back-crossed with C57BL/6J six times. To delete the Pin1 gene in pancreatic β cells, Pin1 flox mice were mated with MIP-Cre mice (RIKEN). Pin1 flox mice were mated with Pin1 flox MIP-Cre mice to achieve gain of both Pin1 flox and Pin1 flox MIP-Cre. These mice were maintained in a temperature- and light-controlled facility. In the analysis of βPin1 KO mice, Pin1 flox mice were used as a control (Figs. 2–5). The animals were handled in an accordance with the animal experimentation guidelines of Hiroshima University.
Plasmid
Human Pin1 cDNA was inserted into a pcDNA3.1 vector. S-tag was added to the N-terminal of Pin1 by the PCR method. Mouse SIK2 cDNA was inserted into the FLAG-pcDNA3.1 vector. To create a GST–p35 vector, mouse p35 cDNA was inserted into the pGEX4T-1 vector.
Immunohistochemistry
Paraffin-embedded pancreatic sections were cut at 200-μm intervals. Immunohistochemistry was performed, as reported previously (18). Briefly, paraffin-embedded pancreatic sections were deparaffinized and then treated with 0.1% Triton. After washing with PBS, the sections were boiled in citrate buffer (pH 6.0) with a microwave to activate the antigens. The slides were then washed with PBS three times and incubated with primary antibody (insulin antibody 1:400, glucagon antibody 1:200, Ki67 antibody 1:400) at 4 °C overnight. Subsequently, the slides were reacted with FITC- or Cy3-conjugated secondary antibody in the presence or absence of BlueStatin for 1 h. After being washed with PBS three times, the sections were embedded in DAPI and then observed by fluorescent microscopy. To measure the areas of β cells, islets that had been stained with insulin antibody were randomly selected from at least 30 islets/mouse. The ratio of the area of β cells to that of entire pancreatic sections was calculated as an average of 3 sections/mouse. To calculate the ratios of cell proliferation and apoptotic cells, nuclei in the β-cell area were counted for at least 2000 cells/mouse.
Immunoblotting
Immunoblotting was performed, as reported previously (12, 16, 17). Briefly, proteins were separated by SDS-PAGE and transferred to PVDF membranes. After blocking with 3% bovine serum albumin for 1 h, the membranes were incubated with primary antibody (1:2000) for 1 h at room temperature. After being washed with PBS containing 0.1% Tween 20, the membranes were reacted with HRP-linked secondary antibody (1: 4000) for 1 h at room temperature. Membranes were washed with PBS three times, and proteins were detected by ECL. The results of several immunoblots were quantitatively analyzed using Chemi DocTMXRS Plus and Image LabTMSVP software (Bio-Rad).
Cell culture
293T cells were cultured in DMEM (Nissui) containing 10% FCS at 37 °C. Min6 cells were cultured in DMEM containing 15% FCS. For plasmid transfections, 293T cells were cultivated in collagen-coated 6-cm dishes, and plasmids were transferred, using X-treme (Roche Applied Science). For adenovirus infection, Min6 cells were cultured in 24-well plates and then infected with adenovirus coding LacZ shRNA or Pin1 shRNA for 2 h in serum-free DMEM. Then Min6 was incubated in DMEM containing FCS for 72 h.
Glucose/insulin tolerance test
Mice were fasted overnight, and glucose was intraperitoneally injected (2 g/kg). For insulin tolerance testing, mice were starved for 4 h, and insulin solution (0.75 IU/kg) was injected.
Real-time PCR
RNA extraction from isolated islets was performed using the NucleoSpin RNA XS kit (Takara). For reverse transcription, RNA was reacted with dNTP, random primers, RT enhancer, and reverse transcriptase (Thermo Fisher). Real-time PCR was performed, using SYBR Green (KAPA). Primer sets were as follows: mIns-1, aaacccacccaggcttttgt (forward) and atccacaatgccacgcttct (reverse); PDX1, cggacatctccccatacgaa (forward) and ctcgggttccgctgtgtaag (reverse); Glut2, tcatgtcggtgggacttgtg (forward) and ttgcagacccagttgctgaa (reverse); mNeuroD, ccccaagctggacagatgag (forward) and tttttgggaccccgtctctt (reverse).
Insulin release assay
Isolated islets were prepared from Pin1 flox or Pin1 MIP-Cre mice. After incubation in RPMI medium overnight, islets were incubated in HKR buffer containing 3 mm glucose for 30 min at 37 °C, followed by switching to HKR buffer containing 3 mm or 20 mm glucose for 1 h. Supernatants were collected, and insulin contents were measured with an ELISA kit (Morinaga). The rates of insulin release were adjusted by intracellular insulin contents.
Immunoprecipitation
Cells were lysed in buffer containing Tris-HCl (pH 7.4), EDTA, NaCl, Triton, orthovanadate, and phenylmethylsulfonyl fluoride. After centrifugation, the supernatants were transferred to new tubes, and FLAG beads were added. For p35 immunoprecipitation, both p35 antibody and Protein G beads were added. After the tubes had been rotated for 2 h at 4 °C, the beads were washed four times with lysis buffer.
In vitro kinase assay of SIK2 using GST–p35 as a substrate
Phosphorylation of p35 was measured as an indicator of SIK2 activity. The plasmid coding FLAG-SIK2 was transfected into 293T cells with or without the S-tag Pin1 plasmid. The cell lysates were incubated with anti-FLAG antibody-conjugated beads. After being washed 4 times with PBS, FLAG-tagged proteins, FLAG-SIK2, and bound Pin1 were eluted employing a FLAG peptide. GST–p35 was reacted with SIK2 or SIK2 binding with Pin1 in the presence of ATP for 15 min. The reactions were then stopped by adding DB buffer.
Pulldown assay using GST–Pin1 protein
GST or GST–Pin1 protein was prepared from Escherichia coli harboring the transfected vector coding the gene of each protein. Wild-type SIK2 or SIK2 mutant was overexpressed in 293T cells, and cell lysates were prepared. Cell lysates were incubated with GST or GST–Pin1 binding beads for 2 h at 4 °C. The beads were then washed with buffer four times, and DB buffer was added.
Measurement of intracellular Ca2+ concentration
Min6 cells were cultivated in glass-bottom dishes. The cells were then loaded with 5 μm Fura-2 for 30 min and washed three times with PBS. The intracellular Ca2+ concentration changes were measured using the ARGAS-HiSCA system.
Measurement of intracellular ATP
Isolated islets were stimulated with 3 or 20 mm glucose for 15 min. After centrifugation, supernatant was removed, and intracellular ATP in the islets was measured employing an ATPlite kit (PerkinElmer Life Sciences). ATP levels were adjusted by the amount of protein.
Statistics
Results are shown as the means ± S.E. Student's t test was used to test the significant difference. Values of p < 0.05 were considered to be statistically significant.
Author contributions
Y. N., designed the study, performed experiments, and wrote the paper. K. M., Y. M., T. Y., K. U., Y. I., and K. M.-M. performed experiments. H. S., M. F., H. O., and A. K. contributed to experimental design. S. Y. and H. I. provided technical assistance and experimental design. T. A. designed experiments and wrote the paper.
This work was supported by a Grant-in-Aid for Scientific Research (C) (to Y. N.), a Grant-in-Aid for Scientific Research on Innovative Areas (area 3702) (to T. A.), and a Grant-in-Aid for Scientific Research (B) (to T. A.) from the Ministry of Education, Science, and Culture, Japan. This work was also supported by a Grant-in-Aid from The Salt Science Research Foundation. The authors declare that they have no conflicts of interest with the contents of this article.
- MIP-Cre
- mouse insulin promoter-Cre transgenic
- GTT
- glucose tolerance test
- HFHS
- high fat and high sucrose
- VDCC
- voltage-dependent calcium channel
- PPIase
- peptidyl prolyl isomerase.
References
- 1. DeFronzo R. A., Bonadonna R. C., and Ferrannini E. (1992) Pathogenesis of NIDDM: a balanced overview. Diabetes Care 15, 318–368 [DOI] [PubMed] [Google Scholar]
- 2. Kahn S. E., Hull R. L., and Utzschneider K. M. (2006) Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature 444, 840–846 [DOI] [PubMed] [Google Scholar]
- 3. Newgard C. B., and McGarry J. D. (1995) Metabolic coupling factors in pancreatic beta-cell signal transduction. Annu. Rev. Biochem. 64, 689–719 [DOI] [PubMed] [Google Scholar]
- 4. Rorsman P., and Renström E. (2003) Insulin granule dynamics in pancreatic beta cells. Diabetologia 46, 1029–1045 [DOI] [PubMed] [Google Scholar]
- 5. Okada T., Liew C. W., Hu J., Hinault C., Michael M. D., Krtzfeldt J., Yin C., Holzenberger M., Stoffel M., and Kulkarni R. N. (2007) Insulin receptors in beta-cells are critical for islet compensatory growth response to insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 104, 8977–8982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ebato C., Uchida T., Arakawa M., Komatsu M., Ueno T., Komiya K., Azuma K., Hirose T., Tanaka K., Kominami E., Kawamori R., Fujitani Y., and Watada H. (2008) Autophagy is important in islet homeostasis and compensatory increase of beta cell mass in response to high-fat diet. Cell Metab. 8, 325–332 [DOI] [PubMed] [Google Scholar]
- 7. Kulkarni R. N., Jhala U. S., Winnay J. N., Krajewski S., Montminy M., and Kahn C. R. (2004) PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J. Clin. Invest. 114, 828–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lu K. P., Hanes S. D., and Hunter T. A. (1996) A human peptidyl-prolyl isomerase essential for regulation of mitosis. Nature 380, 544–547 [DOI] [PubMed] [Google Scholar]
- 9. Ryo A., Suizu F., Yoshida Y., Perrem K., Liou Y. C., Wulf G., Rottapel R., Yamaoka S., and Lu K. P. (2003) Regulation of NF-κB signaling by Pin1-dependent prolyl isomerization and ubiquitin-mediated proteolysis of p65/RelA. Mol. Cell. 12, 1413–1426 [DOI] [PubMed] [Google Scholar]
- 10. Wulf G. M., Ryo A., Wulf G. G., Lee S. W., Niu T., Petkova V., and Lu K. P. (2001) Pin1 is overexpressed in breast cancer and cooperates with Ras signaling in increasing the transcriptional activity of c-Jun towards cyclin D1. EMBO J. 20, 3459–3472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lu K. P., and Zhou X. Z. (2007) The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell Biol. 8, 904–916 [DOI] [PubMed] [Google Scholar]
- 12. Nakatsu Y., Sakoda H., Kushiyama A., Zhang J., Ono H., Fujishiro M., Kikuchi T., Fukushima T., Yoneda M., Ohno H., Horike N., Kanna M., Tsuchiya Y., Kamata H., Nishimura F., et al. (2011) Peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 associates with insulin receptor substrate-1 and enhances insulin actions and adipogenesis. J. Biol. Chem. 286, 20812–20822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lv L., Zhang J., Zhang L., Xue G., Wang P., Meng Q., and Liang W. (2013) Essential role of Pin1 via STAT3 signalling and mitochondria-dependent pathways in restenosis in type 2 diabetes. J. Cell Mol. Med. 17, 989–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liu X., Liang E., Song X., Du Z., Zhang Y., and Zhao Y. (2016) Inhibition of Pin1 alleviates myocardial fibrosis and dysfunction in STZ-induced diabetic mice. Biochem. Biophys. Res. Commun. 479, 109–115 [DOI] [PubMed] [Google Scholar]
- 15. Nakatsu Y., Matsunaga Y., Yamamotoya T., Ueda K., Inoue Y., Mori K., Sakoda H., Fujishiro M., Ono H., Kushiyama A., and Asano T. (2016) Physiological and pathogenic roles of prolyl isomerase Pin1 in metabolic regulations via multiple signal transduction pathway modulations. Int. J. Mol. Sci. 10.3390/ijms17091495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nakatsu Y., Sakoda H., Kushiyama A., Ono H., Fujishiro M., Horike N., Yoneda M., Ohno H., Tsuchiya Y., Kamata H., Tahara H., Isobe T., Nishimura F., Katagiri H., Oka Y., et al. (2010) Pin1 associates with and induces translocation of CRTC2 to the cytosol, thereby suppressing cAMP-responsive element transcriptional activity. J. Biol. Chem. 285, 33018–33027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Nakatsu Y., Iwashita M., Sakoda H., Ono H., Nagata K., Matsunaga Y., Fukushima T., Fujishiro M., Kushiyama A., Kamata H., Takahashi S., Katagiri H., Honda H., Kiyonari H., Uchida T., and Asano T. (2015) Prolyl isomerase Pin1 negatively regulates AMP-activated protein kinase (AMPK) by associating with the CBS domain in the γ subunit. J. Biol. Chem. 290, 24255–24266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nakatsu Y., Otani Y., Sakoda H., Zhang J., Guo Y., Okubo H., Kushiyama A., Fujishiro M., Kikuch T., Fukushima T., Ohno H., Tsuchiya Y., Kamata H., Nagamachi A., Inaba T., et al. (2012) Role of Pin1 protein in the pathogenesis of nonalcoholic steatohepatitis in a rodent model. J. Biol. Chem. 287, 44526–44535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen Z. J., Hu J., Ali A., Pastor J., Shiizaki K., Blank R. D., Kuro-o M, Malter J. S. (2013) Pin1 null mice exhibit low bone mass and attenuation of BMP signaling. PLoS One 8, e63565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoon W. J., Islam R., Cho Y. D., Woo K. M., Baek J. H., Uchida T., Komori T., van Wijnen A., Stein J. L., Lian J. B., Stein G. S., Choi J. Y., Bae S. C., and Ryoo H. M. (2013) Pin1-mediated Runx2 modification is critical for skeletal development. J. Cell. Physiol. 228, 2377–2385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cengiz M., Ozenirler S., Yücel A. A., and Yılmaz G. (2014) Can serum pin1 level be regarded as an indicative marker of nonalcoholic steatohepatitis and fibrotic stages? Digestion 90, 35–41 [DOI] [PubMed] [Google Scholar]
- 22. Paneni F., Costantino S., Castello L., Battista R., Capretti G., Chiandotto S., D'Amario D., Scavone G., Villano A., Rustighi A., Crea F., Pitocco D., Lanza G., Volpe M., Del Sal G., Lüscher T. F., and Cosentino F. (2015) Targeting prolyl-isomerase Pin1 prevents mitochondrial oxidative stress and vascular dysfunction: insights in patients with diabetes. Eur. Heart J. 36, 817–828 [DOI] [PubMed] [Google Scholar]
- 23. Tattikota S. G., Rathjen T., McAnulty S. J., Wessels H. H., Akerman I., van de Bunt M., Hausser J., Esguerra J. L., Musahl A., Pandey A. K., You X., Chen W., Herrera P. L., Johnson P. R., O'Carroll D., et al. (2014) Argonaute2 mediates compensatory expansion of the pancreatic β cell. Cell Metab. 19, 122–134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hosokawa T., Saito T., Asada A., Fukunaga K., and Hisanaga S. (2010) Quantitative measurement of in vivo phosphorylation states of Cdk5 activator p35 by Phos-tag SDS-PAGE. Mol. Cell. Proteomics 9, 1133–1143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brouwers B., de Faudeur G., Osipovich A. B., Goyvaerts L., Lemaire K., Boesmans L., Cauwelier E. J., Granvik M., Pruniau V. P., Van Lommel L., Van Schoors J., Stancill J. S., Smolders I., Goffin V., Binart N., et al. (2014) Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 20, 979–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Carboneau B. A., Le T. D., Dunn J. C., and Gannon M. (2016) Unexpected effects of the MIP-CreER transgene and tamoxifen on β-cell growth in C57Bl6/J male mice. Physiol. Rep. 10.14814/phy2.12863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liou Y. C., Ryo A., Huang H. K., Lu P. J., Bronson R., Fujimori F., Uchida T., Hunter T., and Lu K. P. (2002) Loss of Pin1 function in the mouse causes phenotypes resembling cyclin D1-null phenotypes. Proc. Natl. Acad. Sci. U.S.A. 99, 1335–1340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sakamaki J., Fu A., Reeks C., Baird S., Depatie C., Al Azzabi M., Bardeesy N., Gingras A. C., Yee S. P., and Screaton R. A. (2014) Role of the SIK2-p35-PJA2 complex in pancreatic β-cell functional compensation. Nat. Cell Biol. 16, 234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brüning J. C., Winnay J., Bonner-Weir S., Taylor S. I., Accili D., and Kahn C. R. (1997) Development of a novel polygenic model of NIDDM in mice heterozygous for IR and IRS-1 null alleles. Cell. 88, 561–572 [DOI] [PubMed] [Google Scholar]
- 30. Hirose H., Lee Y. H., Inman L. R., Nagasawa Y., Johnson J. H., and Unger R. H. (1996) Defective fatty acid-mediated beta-cell compensation in Zucker diabetic fatty rats: pathogenic implications for obesity-dependent diabetes. J. Biol. Chem. 271, 5633–5637 [DOI] [PubMed] [Google Scholar]
- 31. Ryo A., Nakamura M., Wulf G., Liou Y. C., and Lu K. P. (2001) Pin1 regulates turnover and subcellular localization of beta-catenin by inhibiting its interaction with APC. Nat. Cell Biol. 3, 793–801 [DOI] [PubMed] [Google Scholar]
- 32. Min S. H., Zhou X. Z., and Lu K. P. (2016) The role of Pin1 in the development and treatment of cancer. Arch. Pharm. Res. 39, 1609–1620 [DOI] [PubMed] [Google Scholar]
- 33. Liou Y. C., Zhou X. Z., and Lu K. P. (2011) Prolyl isomerase Pin1 as a molecular switch to determine the fate of phosphoproteins. Trends Biochem. Sci. 36, 501–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X., Gaspard J. P., Mizukami Y., Li J., Graeme-Cook F., and Chung D. C. (2005) Overexpression of cyclin D1 in pancreatic beta-cells in vivo results in islet hyperplasia without hypoglycemia. Diabetes 54, 712–719 [DOI] [PubMed] [Google Scholar]
- 35. Kushner J. A., Ciemerych M. A., Sicinska E., Wartschow L. M., Teta M., Long S. Y., Sicinski P., and White M. F. (2005) Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol. Cell. Biol. 25, 3752–3762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dentin R., Liu Y., Koo S. H., Hedrick S., Vargas T., Heredia J., Yates J. 3rd, and Montminy M. (2007) Insulin modulates gluconeogenesis by inhibition of the coactivator TORC2. Nature 449, 366–369 [DOI] [PubMed] [Google Scholar]