

Abstract
Cardiac troponin C (cTnC) is the regulatory protein that initiates cardiac contraction in response to Ca2+. TnC binding Ca2+ initiates a cascade of protein–protein interactions that begins with the opening of the N-terminal domain of cTnC, followed by cTnC binding the troponin I switch peptide (TnISW). We have evaluated, through isothermal titration calorimetry and molecular-dynamics simulation, the effect of several clinically relevant mutations (A8V, L29Q, A31S, L48Q, Q50R, and C84Y) on the Ca2+ affinity, structural dynamics, and calculated interaction strengths between cTnC and each of Ca2+ and TnISW. Surprisingly the Ca2+ affinity measured by isothermal titration calorimetry was only significantly affected by half of these mutations including L48Q, which had a 10-fold higher affinity than WT, and the Q50R and C84Y mutants, each of which had affinities 3-fold higher than wild type. This suggests that Ca2+ affinity of the N-terminal domain of cTnC in isolation is insufficient to explain the pathogenicity of these mutations. Molecular-dynamics simulation was used to evaluate the effects of these mutations on Ca2+ binding, structural dynamics, and TnI interaction independently. Many of the mutations had a pronounced effect on the balance between the open and closed conformations of the TnC molecule, which provides an indirect mechanism for their pathogenic properties. Our data demonstrate that the structural dynamics of the cTnC molecule are key in determining myofilament Ca2+ sensitivity. Our data further suggest that modulation of the structural dynamics is the underlying molecular mechanism for many disease mutations that are far from the regulatory Ca2+-binding site of cTnC.
Keywords: calcium, cardiomyopathy, isothermal titration calorimetry (ITC), molecular dynamics, troponin
Introduction
Familial hypertrophic cardiomyopathy (FHC)4 is the inherited form of hypertrophic cardiomyopathy (HCM), the most common cause of sudden cardiac death in young athletes (1), with a prevalence of 1 in 200 individuals (2). There is a growing list of over 1000 mutations that have been associated with HCM, primarily in genes that code for sarcomeric proteins such as the cardiac troponin (cTn) complex (3) (4, 5). FHC is difficult to diagnose because it can be clinically asymptomatic prior to sudden cardiac death. The cTn complex is composed of three proteins: cTnC, the Ca2+ sensing component; cTnI, the inhibitory subunit, and cTnT, that tethers the cTn complex to the cardiac thin filament (6). Mutations in cTnC have a pronounced functional effect because the sequence of cTnC is highly conserved throughout vertebrates (7).
In cardiac contraction, the cytosolic Ca2+ concentration fluctuates between 100 nm in diastole and 400–1000 nm during systole (8, 9). When Ca2+ binds to the regulatory N-terminal domain of cTnC (N-cTnC), a conformational change exposes a hydrophobic region on the surface, which binds to the “switch” region of cTnI. The Ca2+ signal ultimately permits actomyosin cross-bridge formation and force production (6). Sequence substitutions in cTn components demonstrably affect the Ca2+ sensitivity of force production in myofibrils, skinned cardiomyocytes, and trabeculae (10–16). The N-cTnC–Ca2+ interaction has been measured with fluorescent probes such as anilino-napthalenesulfote iodoacetamide (17–19), in which a shift in the dynamic equilibrium between populations of open and closed cTnC is reported in response to the addition of Ca2+. These experiments produced different results for the isolated N-TnC compared with experiments that include the cTn complex and cardiac thin filament proteins actin and tropomyosin (19, 20). By understanding the thermodynamic basis of the function of N-cTnC, we can explain this variation and explore the specific effects of disease-associated mutations.
The function of cTnC and other Ca2+-sensing EF-hand proteins has been described as a balance between the opposing forces that push the cTnC molecule open and those that keep it closed (21). When Ca2+ binds N-cTnC, it creates a strain on the molecule, which is alleviated when N-cTnC changes conformation to better accommodate the presence of the ion; however, the energetic cost of the unfavorable exposure of a hydrophobic cleft provides a thermodynamic incentive to keep the N-cTnC molecule closed (21). The balance between these forces can be disrupted by sequence substitutions that alter the ability of N-cTnC to tolerate the conformational strain imposed by Ca2+ binding or substitutions that modify the hydrophobic cleft. The TnI switch peptide (TnISW) binds to TnC and stabilizes the open TnC conformation by occluding the hydrophobic cleft from the aqueous environment. The structural effects of Ca2+ binding have been examined through NMR and X-ray crystallographic data (22–25), including for the HCM-associated TnC mutant L29Q (24, 26). These structures and data from MD simulations have demonstrated minimal effects of sequence substitutions on the static structure but a greater effect on the dynamics of the protein (27–30).
In this study, we report the ITC-derived Ca2+-binding affinity of N-cTnC mutant constructs and their calculated effects on the dynamics, Ca2+ interaction, and TnISW interaction strengths. The N-cTnC mutations selected for analysis in this work are the FHC-associated mutations A8V (31–33), L29Q (12, 19, 35, 36), A31S (13), and C84Y (14); the engineered Ca2+-sensitizing mutation L48Q (11, 15, 16, 37, 38); and the dilated cardiomyopathy–associated mutation Q50R (39) (Fig. 1). The C84Y and Q50R mutations each conferred Ca2+ affinities 3-fold higher than WT, whereas the L48Q Ca2+ affinity was 10-fold higher than wild type. The combination of MD simulation techniques with ITC explains the molecular etiology of these mutations in terms of the energy landscape of the conformational change. The mutations that favor the open conformation of TnC indirectly increase the Ca2+ affinity of the isolated N-cTnC molecule (40–42). We propose that mutations that increase the Ca2+ sensitivity of the myofilament destabilize the closed conformation of N-cTnC, stabilize the open conformation of N-cTnC, and/or promote association with the TnISW peptide. The results presented in this work demonstrate that many N-cTnC mutations affect myofilament Ca2+ sensitivity by affecting the molecular motions that govern the regulation of cardiac contraction.
Figure 1.

A, structure of the core domain of the Tn complex (PDB code 1J1E) with each of the residues that were selected for this study highlighted. The troponin complex proteins are colored white (cTnC), gray (cTnT), and black (cTnI). B, the isolated N-cTnC domain bound to Ca2+ used in the PMF simulations. Helices and mutation sites are labeled. C, the N-cTnC domain bound to the TnISW and Ca2+ is the system used in the TnISW binding simulations. D, a schematic of the N-cTnC construct. The helices, Ca2+-binding loop, and the residues being examined in this study are labeled.
Results
ITC
The interaction between TnC and Ca2+ was endothermic for each of the TnC constructs except L48Q, which was exothermic. In each case the stoichiometric ratio (N) of Ca2+ binding to N-cTnC was 1:1, indicating that the regulatory site II was exclusively titrated during these experiments. Thermodynamic parameters are listed in Table 1, and ITC isotherms are shown in Fig. 2. At 25 °C each of the Kd values was within error from the WT, with the exceptions Q50R and C84Y, which each had Kd values approximately one-third of WT, and L48Q, in which the Kd was one-tenth of WT. The ΔS values were lower than WT for A31S, L48Q, Q50R, and C84Y. The ΔH values were less favorable than WT for A31S, Q50R, and C84Y but more favorable than WT for the L48Q construct.
Table 1.
Thermodynamic parameters derived from ITC
For all mutants, n = 3.
| N-cTnC construct | N | Kd | ΔS | ΔH | ΔG |
|---|---|---|---|---|---|
| μm | J·mol−1·deg−1 | J·mol−1 | kJ·mol−1 | ||
| WT | 1.05 ± 1e-2 | 14.9 ± 0.7 | 140.5 ± 2 | 1.43e4 ± 6e2 | −2.76e4 ± 1e2 |
| A8V | 1.01 ± 2e-2 | 15.3 ± 0.9 | 141.6 ± 2 | 1.47e4 ± 4e2 | −2.75e4 ± 2e2 |
| L29Q | 0.99 ± 1e-2 | 14.2 ± 0.4 | 145.0 ± 1 | 1.56e4 ± 4e2 | −2.77e4 ± 5e1 |
| A31S | 0.88 ± 3e-2 | 11.8 ± 1.1 | 124.5 ± 2a | 8.99e3 ± 3e2a | −2.81e4 ± 2e2 |
| L48Q | 1.02 ± 3e-2 | 1.48 ± 0.1a | 50.1 ± 1a | −1.84e4 ± 2e2a | −3.33e4 ± 8e1a |
| Q50R | 1.05 ± 1e-2 | 5.85 ± 0.1a | 120.8 ± 1a | 6.15e3 ± 1e2a | −2.99e4 ± 8e1a |
| C84Y | 0.96 ± 2e-2 | 4.19 ± 0.9a | 113.9 ± 2a | 3.12e3 ± 1e2a | −3.09e4 ± 5e2a |
a Significant difference from WT (p < 0.05).
Figure 2.

Representative isotherms for each of the TnC constructs at 25 °C. The isotherms for WT and each mutant are endothermic, except for the exothermic L48Q isotherm.
Melting points
The melting points for all Apo TnC constructs were ∼65 °C, with the exceptions of A8V at 58.5 °C and L48Q at 42.5 °C (supplemental Table S2).
TnC + Ca2+ simulations
Each of the simulations diverged from the original coordinates (supplemental Fig. S1). The representative structures are very similar with a total backbone root mean square deviation of 1.9 Å. The local backbone dynamics are similar over 100 ns (supplemental Fig. S1). The mutations produce small backbone perturbations compared with WT in their respective local regions and have backbone root mean square deviation values that differ only by 1.2–2.4 Å. The mutations can; however, substantially disrupt the packing of interacting side chains for mutated residues that are not solvent-exposed: A8V, L48Q, Q50R, and C84Y. The L29Q and A31S substitutions affect fewer residues, but each introduces a hydrogen bond absent from the WT structure (Fig. 3). Compared with WT, the A8V model has a modified interaction between the N and D helices, specifically a hydrophobic interaction with that involves residues Tyr-5, Ala-8, Val-9, and Leu-12 on the N-helix and Leu-78, Val-79, and Val-82 on the D helix. In the L29Q model, Gln-29 is solvent-exposed, but the side chain amide is within hydrogen bonding distance of the Ile-26 backbone O. The A31S substitution is in the EF-hand site I loop, and Ser-31 is within interacting distance of the Cys-35 backbone O, which, in the WT model, interacts with the backbone O of Leu-29. The L48Q substitution disrupts a hydrophobic network in the core of the AB-helical interface that is composed of Phe-19, Ala-23, Cys-35, Val-44, Glu-76, and Met-80. The side chain amide of Gln-49 is within interacting distance of the backbone O of Met-47. The Q50R substitution, located between helices B and C, alters helix C of the molecule, and interaction between Gln-50 and the backbone O of Leu-48 is replaced by a series of hydrogen bonds between Arg-50 and Cys-84, in addition to both side chain O atoms of Glu-56. Finally, the C84Y substitution disrupts a hydrophobic interaction between Cys-84 with Met-60, Met-45, Pro-52 and Gln-50. Instead, Tyr-84 is within interacting distance with the backbone O of Met-60.
Figure 3.

Structural changes induced by each of the mutations. In each panel, the left side contains a representative structure of each mutation superimposed with the wild-type structure (white). To orient the reader, a red arrow indicates the location of the mutation on the structure. Changes to side chain packing in the immediate area of each mutation are demonstrated on the right for each of the mutations. Although the changes to the backbone are very subtle, there are side chain rearrangements in the local vicinity of the mutations, particularly for the mutations that occur at helix–helix interfaces such as A8V, L48Q, and Q50R.
The Ca2+ coordination geometry and distances are similar for all constructs with the exception of A8V, which has tighter Ca2+ coordination geometry (supplemental Table S3). This is likely correlated with the potential of mean force (PMF) results discussed in detail below.
The average number of H-bonds in each mutant structure is the same over the course of each simulation, as are the average hydrophobic solvent-accessible surface (h-sasa) in the closed and open forms (supplemental Table S4); however, monitoring the A/B interhelical angle (Fig. 4) and h-sasa (supplemental Fig. S3) over repeated 1-μs simulations demonstrates the frequency with which the TnC protein exposes the hydrophobic patch. The L29Q construct is least frequently open, followed in order by C84Y < A8V < WT < Q50R <A31S, and <L48Q. One of the replicated L29Q simulations had very high AB interhelical angles for a time; this was due to a transient loss of secondary structure, although it did not affect the exposed hydrophobic sasa (supplemental Fig. S2). The interhelical angle data can be expressed as the probability of observing the open conformation of the protein. An angle of 110° has been described as the maximum angle that can be considered open. The proportion of frames in which the N-cTnC molecule had an open AB interhelical angle was 0 for the A8V, L29Q, and C84Y constructs, 0.004% for the Q50R construct, 0.008% for WT, 0.016% for A31S, and 0.07% for the L48Q N-cTnC (Fig. 5). By defining cutoff angles for the transition between closed and open at 130° and 110°, we can determine the probability of observing the N-cTnC molecule in the closed state (AB interhelical angle >130°) and gain insight on the relative stability of the closed conformation (Fig. 5). The A8V construct has the lowest closed probability at 58%, followed by C84Y at 65%, WT and L48Q at 76%, Q50R at 86, A31S at 88%, and finally L29Q at 92%.
Figure 4.

The A/B interhelical angle is plotted as a function of time for five replicated simulations of each mutated model. Plots are a rolling average of 250 ps. An interhelical angle less than 110° is considered open, and above 130° is considered closed. There is little difference between WT and most of the constructs, with the exception of the L48Q model. The large angle values in one replicate of the L29Q simulations is an artifact caused by a transient loss and recovery of secondary structure in one of the replicated simulations; the hydrophobic solvent-accessible surface is not increased as a function of this change. The h-sasa as a function of time is reported in supplemental Fig. S4.
Figure 5.
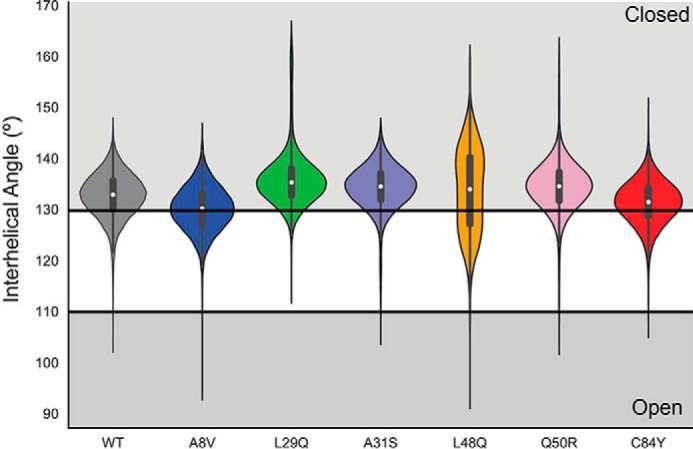
Violin plot demonstrating the distribution of open and closed N-cTnC structures observed over five replicated 1-μs simulations. The open conformation was defined by an AB interhelical angle less than 110°, whereas the closed conformation was defined by an AB interhelical angle greater than 130°. The proportion of open frames is not correlated with the proportion of closed frames. The L48Q construct has the most frames in the open conformation, whereas the A8V is the least closed. This suggests that the destabilization of the closed conformation does not necessarily imply that the open conformation is stabilized.
TnC + Ca2+ + TnISW simulations
The effects on the structural interactions between the TnC molecule and TnISW are subtle when averaged across the simulations (Fig. 6), and as expected, the majority of the specific contacts are maintained when each of the N-cTnC mutants are compared with WT N-cTnC. The TnISW remains in contact with the TnC molecule for the duration of the simulations, but the specificity of that contact is somewhat different in response to certain mutations. For example: L29Q, A31S, and Q50R make more common contacts between the N-terminal region of TnISW and the N-helix of TnC than WT, whereas L48Q and C84Y make fewer contacts than WT in this region. These results suggest that alterations in this binding interface may be due to modified interactions between the TnISW and other regions of the protein near the mutation sites.
Figure 6.

Average distance between cTnC and cTnI residues. The mutated TnC residue in each plot is indicated by a gray bar. The structures to the right are representative structures of independent simulations and indicate the differences in the orientation and variability of the cTnISW peptide across replicates for each mutant. The TnISW is colored as a spectrum from blue at the N terminus to red at the C terminus. The calculated ΔG of interaction is maintained across mutations despite differences in the interaction distance profiles, which suggests a nonspecific interaction. The A31S mutant has a ΔG of interaction with the TnISW ∼25% lower than WT (Table 2), perhaps because of shorter interaction distances with the N-terminal region of N-cTnC but longer interaction distances in the vicinity of the A31S mutation and C-terminal portion of the TnISW.
Free energy calculations
The free energy change in Ca2+ binding, measured by PMF calculations (Fig. 7) indicates that the highest free energy change was observed in the A8V construct (−72 ± 5 kJ·mol−1), followed by Q50R (−41 ± 3 kJ·mol−1) and C84Y (−46 ± 3 kJ·mol−1), A31S (−46 ± 2 kJ·mol−1) and L29Q (−46 ± 2 kJ·mol−1), and finally L48Q (−32 ± 3 kJ·mol−1) and WT (−32 ± 4) kJ·mol−1. A representative structure from the five replicated 1-μs simulations of the A8V mutation yielded Ca2+ coordination distances that were similar to the other mutant constructs and a PMF-derived Ca2+ interaction ΔG of 46 ± 5 kJ·mol−1.
Figure 7.
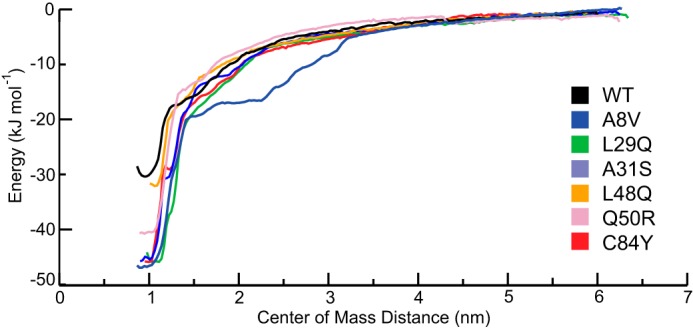
The potential of mean force profile of each of the mutated constructs as a function of center of mass distance between the TnC molecule and Ca2+ ion. Each of the mutated constructs has an increased ΔG of Ca2+ interaction. L48Q (−32 ± 3 kJ·mol−1) is the closest to WT (−32 ± 4 kJ·mol−1), followed by Q50R (−41.3 ± 3 kJ·mol−1), L29Q (−46 ± 2 kJ·mol−1), C84Y (−46 ± 3 kJ·mol−1) A31S (−46 ± 2 kJ·mol−1), and A8V (−46 ± 5 kJ·mol−1), which are similar to each other.
Interaction energies between TnC and the TnISW were estimated by molecular mechanics/Poisson Boltzmann surface area (MM/PBSA) calculations. The energies reported are much larger than could be reasonably expected, although this method has been used as a means to score the relative strength interactions with some success (43). The values are similar for each of the constructs, with the exception of the A31S mutant, which has a weaker calculated binding interaction (Table 2).
Table 2.
MM/PBSA results for the WT and mutant N-cTnC/TnISW interaction shown as averages ± S.D.
| Van der Waals | Electrostatic | Polar | sasa | Total | |
|---|---|---|---|---|---|
| A8V | −3.0E2 ± 3.5E1 | −1.7E3 ± 2.8E2 | 8.1E2 ± 2.3E2 | −3.9E1 ± 3.2E0 | −1.3E3 ± 1.2E2 |
| L29Q | −3.2E2 ± 4.1E1 | −2.0E3 ± 2.8E2 | 1.0E3 ± 2.7E2 | −4.1E1 ± 4.7E0 | −1.3E3 ± 6.3E1 |
| A31S | −3.0E2 ± 6.4E1 | −1.7E3 ± 2.9E2 | 1.1E3 ± 2.0E2 | −3.9E1 ± 7.7E0 | −9.6E2 ± 2.2E2 |
| L48Q | −3.1E2 ± 5.2E1 | −1.9E3 ± 2.6E2 | 1.0E3 ± 2.1E2 | −4.0E1 ± 4.5E0 | −1.3E3 ± 6.7E1 |
| Q50R | −3.1E2 ± 2.7E1 | −2.0E3 ± 1.8E2 | 1.1E3 ± 2.0E2 | −4.1E1 ± 3.0E0 | −1.2E3 ± 5.4E1 |
| C84Y | −3.2E2 ± 3.9E1 | −1.7E3 ± 2.2E2 | 8.4E2 ± 1.9E2 | −4.0E1 ± 4.3E0 | −1.2E3 ± 8.1E1 |
| WT | −3.2E2 ± 2.6E1 | −1.9E3 ± 2.8E2 | 1.0E3 ± 3.2E2 | −4.1E1 ± 3.1E0 | −1.2E3 ± 1.4E2 |
Discussion
The measurements described in this study have provided novel information about the molecular basis of N-cTnC function in the regulation of cardiac muscle contraction. Muscle contraction begins when N-cTnC binds to Ca2+, opens, and interacts with TnISW. The cTnC molecule also interacts with the cardiac-specific N-terminal extension of TnI (44), which responds to the phosphorylation of TnI residues 22 and 23 (19). In the presence of the Tn complex, the cTnC Ca2+ affinity increases by 10-fold over the isolated cTnC molecule. Improved affinity was attributed to the stabilized open conformation (5, 45). Our hypothesis was that the Ca2+ sensitizing mutations would directly increase Ca2+ affinity on the isolated N-cTnC, whereas desensitizing mutations reduce the N-cTnC Ca2+ affinity. Mutations were hypothesized to affect TnC function through directly modifying Ca2+ coordination or modifying the energetic cost of the N-cTnC conformational change. Mutations in the Tn complex have been shown to modify Ca2+ affinity indirectly through altering the interaction between N-cTnC and the TnISW (5, 45). We have explored this hypothesis through ITC measurements of Ca2+ affinity and MD simulations that 1) assess the strength of the Ca2+-binding interaction; 2) describe the properties of the N-cTnC conformational change; and 3) measure the strength of the interaction between N-cTnC and the TnISW peptide.
It is challenging to directly measure the N-cTnC–Ca2+ interaction. Fluorescence-based experiments employ reporters such as anilino-napthalenesulfote iodoacetamide (17–19) or an F27W mutation (46). Upon titration with Ca2+, the fluorophore reports the N-cTnC conformational change as a measure of N-cTnC Ca2+ affinity, whereas measurements made using stopped flow fluorospectroscopy report rates of Ca2+ dissociation (15). The Kd for WT TnC has been previously reported fluorometrically as 11.3 μm for WT N-cTnC, which was lowered to 8 μm with the L29Q mutation (12). Similarly, fluorescence titration of the N-cTnC–Ca2+ interaction yielded midpoint values of 12.3 μm for WT N-cTnC, 12.9 μm for A8V, and 37.2 μm for C84Y (5). Our ITC experiments, which consider the unmodified N-cTnC molecule, demonstrated no statistically significant differences in Kd between WT and the FHC-associated mutants, with the exception of C84Y (Table 1). Our measurements of the dilated cardiomyopathy-associated mutant Q50R and engineered Ca2+-sensitizing mutation L48Q have shown 3- and 10-fold increases in affinity for Ca2+, respectively. Our results agree with the previously reported Kd for full-length cTnC at 24 μm (16), which decreased to 1.9 μm in the L48Q construct (16). When compared with another ITC study of the WT N-cTnC–Ca2+ interaction, the ΔG values are ∼3 kJ·mol−1 lower (47). Each of A31S, L48Q, Q50R, and C84Y produced increased ΔH and decreased ΔS values relative to WT, although the molecular basis of these changes is not necessarily the same. The A31S mutation stabilizes the loop between helices A and B with a hydrogen bond, which accounts for changes in both ΔH and ΔS. The L48Q, Q50R, and C84Y mutations are along the interface between the NAD and BC helical bundles, which likely reduces the entropic cost of the closed/open transition by introducing a polar residue into a hydrophobic region. Each of these substitutions also creates at least one new hydrogen bond that affects ΔH (Fig. 3).
The increases in Ca2+ affinity of L48Q and Q50R N-cTnC were attributed to the reduced cost of exposing the hydrophobic patch. Our ITC experiments reported lower ΔS values for these mutants (Table 1), associated with the exposure of hydrophobic residues. The changes in ΔS are consistent with our measurement of the A/B interhelical angle, as the molecule transitions into the open form; hydrophobic residues in the interface between the NAD helical bundle and the BC helical bundle are exposed. This has been demonstrated in another MD-based study of the L48Q and V44Q N-cTnC mutations (38). In the A31S mutation, the ΔS value is lower than that of WT, perhaps because of the hydrogen bond formed by the serine, which reduces the mobility of the loop, a finding consistent with a previous exploration of A31S (13). The C84Y mutant disrupts the side-chain packing between Cys-84 and several residues between helices C and D and produces a more favorable open conformation (Fig. 3). Despite the overall structural similarity of the mutant constructs, there are changes in side-chain packing caused by each substitution compared with the WT model (Fig. 3). The melting points were lower for the A8V and L48Q mutants, consistent with the observation that they spend the least time in the closed conformation. That the hydrophobic interactions disrupted by these mutations have an important role in the stability of the N-TnC molecule (Fig. 5).
Small-angle X-ray scattering has been used to investigate tertiary protein contacts in the apo and Ca2+-bound states of the A8V and A31S mutants. Despite minimal structural changes in these mutants, this technique can be used to uncover potentially lower stability resulting from salient local changes in FHC mutants (48).
The strengths of the charge–charge interactions that govern the direct Ca2+–N-cTnC interaction are proportional to the distance between the Ca2+ ion and the coordinating oxygen atoms (supplemental Table S2). The PMF-derived ΔG of Ca2+ binding of the L48Q mutation was most similar to the WT protein, and the remaining mutations each yielded stronger Ca2+ interactions (Fig. 7). Notably the A8V interaction was the strongest, and the Ca2+ coordination was the tightest in 100-ns simulations; however, this was not found in the PMF calculation based on the 1-μs simulation (Fig. 7). The absolute values of the interaction energies are overestimated because of the parameterization of Ca2+ in the simulation; however, the results are useful as a relative measure of the change in free energy of binding (49).
In skeletal TnC, the affinity for Ca2+ is inversely related to protein stability. It is reasonable to assume that similar mechanisms govern the cardiac isoforms of this protein (50). The affinity of N-cTnC for Ca2+ is set by a balance between the conformational strain induced by Ca2+ binding that is acting to open the N-cTnC molecule and the energetic cost of exposing a hydrophobic cleft (21). Introduction of a polar amino acid into the hydrophobic cleft reduces the cost of opening N-cTnC by destabilizing the closed conformation or stabilizing the open conformation (41). Ca2+ binding causes a change in the dynamic equilibrium of the populations of open and closed TnC molecules. When Ca2+ is bound, 20–27% of the N-cTnC population is open (33, 51). Previous MD simulations have estimated the ΔG of the WT cTnC open/closed transition at 33.5 kJ·mol−1 (27). The N-cTnC conformational change necessarily precedes the TnISW interaction, and therefore changes in the favorability of the conformational change are related to the probability of N-cTnC binding to the TnISW (27).
Monitoring the A/B interhelical angle through 1-μs simulations has revealed that mutations affect the conformational dynamics of N-cTnC (Fig. 5). A previous study had defined the N-cTnC molecule as “open” with an AB interhelical angle <110° (27). We define a closed structure as any with an AB interhelical angle >130°, similar to the NMR structure of N-cTnC (22). This allows for the quantification of the relative stability of the closed state (Fig. 5). The open conformation is found most frequently in simulations of the L48Q construct, followed by the A31S mutation. Work by Marques et al. (48) shows that A31S may cause greater exposure in the primed state of the full-length TnC (i.e. when Ca2+ is bound to sites III/IV). A similar degree of openness was observed for the remaining constructs, a finding corroborated the h-sasa (supplemental Fig. S3). The L29Q mutant was the least open; interhelical angles below 115° were not observed and had the highest closed probability. The closed state was least frequently observed in the A8V construct, which is consistent with paramagnetic NMR data showing that A8V cTnC opened more readily than WT N-cTnC (33) and contribute to the increased affinity between A8V TnC and TnISW (31). These data suggest that the molecular etiology is different for each mutation, despite producing a similar disease phenotype.
The ΔG of N-cTnC and TnISW interaction is similar to WT for each of the N-cTnC mutants with the exception of A31S, which is ∼75% of the WT ΔG (Table 2). As with the PMF calculations, the ΔG values derived from MM/PBSA calculations provide insight into the relative strengths, but the absolute values are not expected to correspond to more computationally expensive calculations or experimentally derived measurements (43). The A31S N-cTnC has a modified interface with TnISW, which favors closer interactions with the N-terminal region of TnC and longer distance interactions with remainder of N-cTnC, particularly in the vicinity of the mutation (Fig. 6). Relative to WT, each mutant has a slightly different interaction with TnISW but did not produce a change in interaction energy. We have previously observed a similar effect in the zebrafish TnC–TnISW interaction in which TnI substitution had a greater effect on the interaction than TnC substitution (52). There is fluorescence-based evidence that both the L48Q (53) and A8V (31) N-cTnC mutations increase the affinity of N-cTnC for the TnISW; our results suggest that this affinity change may be due, in part, to the ability of these mutants to open more readily than WT, which generates more opportunity for TnC–TnI interaction.
This work provides insight into how the dynamics of N-cTnC can govern the interactions with each of Ca2+ and cTnI, which in turn influence sarcomeric Ca2+ sensitivity. The engineered L48Q mutation has the most salient effect on the N-cTnC ITC-derived Ca2+ binding and on the dynamics of the N-cTnC molecule. A finding that has been corroborated, experimentally (16, 53), in silico (38) and in vivo (11, 37). The L48Q mutation has a large disruptive effect on the hydrophobic interactions that maintain N-cTnC in the closed conformation, which creates increased Ca2+ affinity through a modification of the thermodynamic landscape of the conformational change and allows the TnISW to bind to N-cTnC more readily (53). Through these molecular changes, the L48Q mutation can produce a positive inotropic effect or, with higher sarcomere incorporation and β-blocking drugs, can produce hypertrophy in murine models (11, 37). The L29Q mutation was similar to WT in each of our measurements; However, L29Q has been shown to affect length-dependent activation and modify the response to phosphorylation of serine 22/23 of cTnI (19, 26, 54, 55). The A8V and C84Y mutations are near the interface with the N-helix of TnI (Fig. 1) and may have a more pronounced effect on the orientation of N-cTnC in the Tn complex (56).
Our results support the model for the molecular mechanism of Ca2+ binding that is dictated by the favorability of the conformational change and the stability of the TnI–TnC interaction (42). Mutations modify the structural dynamics of TnC rather than the regulatory Ca2+-binding site. The changes are observed in the relative favorability of the protein conformations that transduce the contraction signal (Fig. 8). This can increase Ca2+ sensitivity of contraction by destabilizing the closed conformation of N-cTnC, stabilizing the open conformation, or stabilizing the TnISW interaction. These changes lead to an increase in the Ca2+ buffering capacity of the myofilament that may increase the duration of the Ca2+ transient and is consistent with observations of the greater capacity of the Tn complex to bind Ca2+ than the isolated N-cTnC molecule (16, 20, 45). The complimentary use of MD, ITC, and fluorometric techniques provides detailed information about the molecular etiology of cTnC mutations. A complete understanding of the molecular and thermodynamic basis for myofilament Ca2+ sensitivity will inform the risk stratification of existing and novel FHC-associated mutations and the selection of appropriate therapeutics and enable rational drug development that specifically target or compensate for the unique deleterious effects of these disease mutations.
Figure 8.
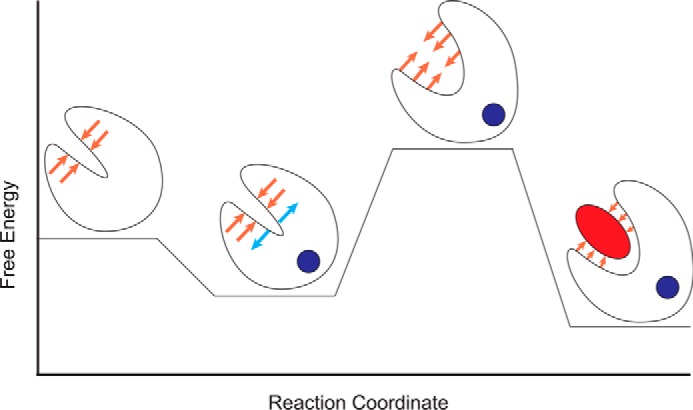
Schematic of the energetic landscape of N-cTnC activation. N-cTnC is shown as a cartoon. Ca2+ is a blue circle, and the TnI switch peptide is represented as a red ellipse. Lower energy states are more favorable. The orange arrows represent the resistance to the conformational change caused by the hydrophobic cleft. The blue arrows indicate conformational strain introduced by Ca2+ binding. The Ca2+-bound, open conformation relieves the conformational strain while occluding the hydrophobic cleft and is therefore the most favorable conformation. Mutations that affect the relative stabilities of these states will modify the probability of transitions between them and increase or decrease the Ca2+ sensitivity of the myofilament.
Experimental procedures
The codon-optimized gene sequence for human TNNC1 was cloned into the pET21a(+) vector (Novagen). A stop codon was introduced at position 90 by following the Phusion site-directed mutagenesis protocol (Thermo). Protein expression, purification, melting point determination, and ITC were carried out using protocols modified from previous work (30). The N-cTnC construct corresponds to the human TNNC1 gene (Uniprot ID P63316), which was cloned into the pET-21a+ expression vector (Novagen). The codon corresponding to residue 90 was mutated to a stop codon, and individual mutations were introduced with the Phusion site-directed mutagenesis kit (Thermo). Mutated constructs were sequenced and transformed into the BL21(DE3) host strain. Overnight cultures were grown in lysogeny broth supplemented with 50 μg/ml ampicillin at 37 °C overnight with shaking at 250 RPM, 1% subcultures were grown for 3 h followed by induction with 1 mm isopropyl β-d-1-thiogalactopyranoside and a further 3 h of growth. The cells were harvested by centrifugation and resuspended in 50 mm Tris-HCl, pH 8.0, 1 mm PMSF, 5 mm EDTA, and a cOmpleteTM protease inhibitor tablet (Roche). The cells were sonicated on ice at 80% amplitude with 30-s pulses separated by 30 s. The lysate was centrifuged at 30,000 × g for 30 mins, and the supernatant was decanted. The protein was purified with a fast-flow DEAE column (GE Healthcare). The column was equilibrated with 50 mm Tris-HCl, pH 8.0, 1 mm DTT, and 5 mm EDTA, and the protein was eluted with a 180-ml NaCl gradient up to 0.55 m. Fractions containing the TnC protein were retained and concentrated to 5 ml with an Amicon centrifugal concentrator with a molecular mass cutoff of 3 kDa (Millipore). The protein was further purified with a HiPrep 26/60 Sephacryl S-100 column (GE Healthcare) equilibrated with 50 mm Tris-HCl, pH 8.0, 100 mm NaCl, and 1 mm DTT. The fractions that contain the pure N-cTnC protein were pooled, concentrated, and stored at −80 °C.
ITC buffer contained 50 mm HEPES, pH 7.2, and 150 mm KCl, protein samples were dialyzed three times against 2 liters of ITC buffer and diluted to 200 μm. In successive dialysis steps, 15 mm β-mercaptoethanol (BME) and 2 mm DTT, 15 mm of BME, and 2 mm of BME were added to the buffer. An extinction coefficient of 1490 m−1 cm−1 and a molecular mass of 10.1 kDa were used to measure protein concentration. The Ca2+ solution was diluted from a 1 m Ca2+ solution (Sigma) into the buffer from the final dialysis step to a final concentration of 4 mm. Ca2+ was titrated into the protein solution by a single 0.4-μl injection, followed by a series of eighteen 2-μl injections at 2-min intervals while stirring at 1000 rpm. The experiments were carried out at 25 °C. The heat of dilution of Ca2+ was accounted for by subtracting the average of the final three data points from the titration curve. Analysis was performed with Origin 8.0 (OriginLab, Northampton, MA).
To measure the melting point of the proteins, the samples were first dialyzed four times against 2 liters of MT buffer (10 mm HEPES, pH 7.5, 150 mm KCl, 3 mm MgCl2, 2 mm EGTA) and combined with 2.5 μl of 100-fold diluted SYPRO orange (Thermo) to a final concentration of 3 mg/ml. The temperature was increased from 4 to 95 °C at 5-s intervals using a CFX96 Touch real-time PCR system (Bio-Rad). The melting point was determined at the midpoint of the unfolding transition, which is indicated by the peak of the first derivative curve.
Equilibrium MD simulations of WT and mutant TnC were performed as previously described (30, 52) with the exception that all calculations in this study were performed at 300 K. Structural models of the mutant constructs were generated with the Swiss model workspace (57), The N-cTnC + Ca2+ models used the NMR-derived structure of human N-TnC as a template (PDB code 1AP4) (22), and the models of the human N-TnC in complex with TnISW were based on the WT NMR derived structure of N-cTnC in complex with Ca2+ and cTnI residues 147–163 (PDB code 1MXL) (23).
The structural models were simulated using GROMACS 4.6.5 (58), and the AMBER99sb-ILDN force field (59); the models were placed in a periodic, cubic simulation system and solvated with the TIP3P water model (60); and the charges were neutralized with the addition of K+ or Cl− ions. The composition of each system is listed in supplemental Table S1. The systems were energy minimized using the steepest descent algorithm to a tolerance of 10 kJ·mol−1·nm−1 followed by conjugate gradient minimization for 10,000 steps. 100 kJ·mol−1·nm−1 restraints were placed on every protein and Ca2+ atom, and the system was simulated for 1 ns to allow the water to equilibrate around the protein.
The WT and mutant N-cTnC+Ca2+ systems were then simulated for either 100 ns or 1 μs total time, and the WT and mutant N-cTnC+cTnISW+Ca2+ constructs were simulated for 100 ns total (Fig. 1, B and C). All of the simulations were performed with Berendsen pressure coupling (61) with a τT of 0.1. V-rescale temperature coupling (62) with a τP of 4.0, PME (particle mesh Ewald) electrostatics (63) with a grid spacing of 0.12 nm and interpolation order of 6, and the Verlet cutoff scheme was used with a 1.0-nm cutoff (64). Bond lengths were constrained with the LINCS algorithm (65).
Clustering was carried out over the backbone and Cβ atoms of each construct using the Daura algorithm (66). The degree of the open/closed N-cTnC conformational change and protein stability were assessed through measurements of the solvent-accessible surface area, the interhelical angles, and the number of hydrogen bonds, which were calculated using g_sas (67), Interhlx (68), and g_hbond (58), respectively. The g_hbond program used a cutoff radius of 3.5 Å and a 30° angle to define a given hydrogen bond.
Umbrella sampling and PMF calculations were performed as described previously (30). The Ca2+ was extracted from the N-cTnC molecule by restraining the α-helical Cα atoms with a force constant of 1000 kJ·mol−1·nm−1 and restraining the Ca2+ ion in the Y and Z dimensions with a force constant of 1000 kJ·mol−1·nm−1. A constraint pulling force in the X direction as applied at 0.01 Å per second until the Ca2+ ion was 5 nm from the N-cTnC molecule. The conformations for umbrella sampling were extracted from the resulting trajectory at distance intervals of 0.5 Å between 0 and 1 nm, every 1 Å between 1 and 2 nm, and every 2 Å between 2 and 5 nm. Umbrella simulations were run with the same parameters as the pull simulations, with the “pull rate” parameter set to 0, and were unrestrained aside from a single restraining potential between the center of mass of the N-cTnC molecule and the center of mass of the Ca2+ ion. These simulations were run for 30 ns each, and a potential of mean force was calculated with the weighted histogram analysis method through the use of g_wham (69), and errors were estimated with 5000 bootstraps of the weighted histogram analysis method calculation. To further explore the outlier A8V mutation, the process was repeated using the center structure of the most populous cluster from the five replicated 1-μs simulations.
Interaction energies between the TnC models and TnISW were calculated with g_mmpbsa (34) 100 ps apart over the last 10 ns of each equilibrium simulation of the TnISW–N-cTnC complex. The MM/PBSA calculations used the non-linear Poisson-Boltzmann equation, and calculations were performed at 300 K, with a solvent dielectric constant of 80, and a probe radius of 1.4 Å. Contact maps of the interacting surfaces between N-cTnC and the cTnISW were calculated over the final 50 ns of each simulation and were based on measurements made with g_mindist (58).
Author contributions
C. M. S. and B. L. performed preliminary PMF simulations; K. R. introduced mutations, purified protein, and collected ITC and Tm data; C. M. S. and G. S. developed MD simulation and analysis protocols; C. M. S. built homology models and performed and analyzed MD simulations, PMF calculations, MM/PBSA calculations, long-time scale simulations; K. R. and C. M. S. analyzed ITC and Tm data; C. M. S. and K. R. wrote the manuscript; and C. M. S., K. R., D. P. T., and G. F. T. reviewed the manuscript
Supplementary Material
Acknowledgments
We are grateful for GROMACS tutorials by Justin Lemkul and and to Christine Genge and Laura Dewar for careful reading of the manuscript. The molecular dynamics simulations were carried out on the Westgrid and Cacul Quebec Complexes, which are under the aegis of Compute Canada.
This work was supported by grants from the Natural Sciences and Engineering Research Council of Canada (to G. F. T. and to D. P. T.) and Canadian Institute of Health Research (to G. F. T.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1–S4 and Figs. S1–S4.
- FHC
- familial hypertrophic cardiomyopathy
- HCM
- hypertrophic cardiomyopathy
- cTn
- cardiac troponin
- cTnC
- cardiac troponin C
- N-TnC
- N-terminal domain of cTnC
- ITC
- isothermal titration calorimetry
- MD
- molecular dynamics
- PMF
- potential of mean force
- MM
- molecular mechanics
- PBSA
- Poisson Boltzmann surface area
- BME
- β-mercaptoethanol
- PDB
- Protein Data Bank
- h-sasa
- hydrophobic solvent-accessible surface.
References
- 1. Maron B. J., Shirani J., Poliac L. C., Mathenge R., Roberts W. C., and Mueller F. O. (1996) Sudden death in young competitive athletes: clinical, demographic, and pathological profiles. JAMA 276, 199–204 [PubMed] [Google Scholar]
- 2. Semsarian C., Ingles J., Maron M. S., and Maron B. J. (2015) New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 65, 1249–1254 [DOI] [PubMed] [Google Scholar]
- 3. Harada K., and Morimoto S. (2004) Inherited cardiomyopathies as a troponin disease. Jpn. J. Physiol. 54, 307–318 [DOI] [PubMed] [Google Scholar]
- 4. Seidman C. E., and Seidman J. G. (2011) Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ. Res. 108, 743–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pinto J. R., Parvatiyar M. S., Jones M. A., Liang J., Ackerman M. J., and Potter J. D. (2009) A functional and structural study of troponin C mutations related to hypertrophic cardiomyopathy. J. Biol. Chem. 284, 19090–19100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Parmacek M. S., and Solaro R. J. (2004) Biology of the troponin complex in cardiac myocytes. Prog. Cardiovasc. Dis. 47, 159–176 [DOI] [PubMed] [Google Scholar]
- 7. Gillis T. E., Marshall C. R., and Tibbits G. F. (2007) Functional and evolutionary relationships of troponin C. Physiol. Genomics 32, 16–27 [DOI] [PubMed] [Google Scholar]
- 8. Bers D. M. (2000) Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 87, 275–281 [DOI] [PubMed] [Google Scholar]
- 9. Kirschenlohr H. L., Grace A. A., Vandenberg J. I., Metcalfe J. C., and Smith G. A. (2000) Estimation of systolic and diastolic free intracellular Ca2+ by titration of Ca2+ buffering in the ferret heart. Biochem. J. 346, 385–391 [PMC free article] [PubMed] [Google Scholar]
- 10. Feest E. R., Steven Korte F., Tu A. Y., Dai J., Razumova M. V., Murry C. E., and Regnier M. (2014) Thin filament incorporation of an engineered cardiac troponin C variant (L48Q) enhances contractility in intact cardiomyocytes from healthy and infarcted hearts. J. Mol. Cell Cardiol. 72, 219–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shettigar V., Zhang B., Little S. C., Salhi H. E., Hansen B. J., Li N., Zhang J., Roof S. R., Ho H. T., Brunello L., Lerch J. K., Weisleder N., Fedorov V. V., Accornero F., Rafael-Fortney J. A., et al. (2016) Rationally engineered troponin C modulates in vivo cardiac function and performance in health and disease. Nat. Commun. 7, 10794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liang B., Chung F., Qu Y., Pavlov D., Gillis T. E., Tikunova S. B., Davis J. P., and Tibbits G. F. (2008) Familial hypertrophic cardiomyopathy-related cardiac troponin C mutation L29Q affects Ca2+ binding and myofilament contractility. Physiol. Genomics 33, 257–266 [DOI] [PubMed] [Google Scholar]
- 13. Parvatiyar M. S., Landstrom A. P., Figueiredo-Freitas C., Potter J. D., Ackerman M. J., and Pinto J. R. (2012) A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 287, 31845–31855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Landstrom A. P., Parvatiyar M. S., Pinto J. R., Marquardt M. L., Bos J. M., Tester D. J., Ommen S. R., Potter J. D., and Ackerman M. J. (2008) Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell Cardiol. 45, 281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tikunova S. B., Liu B., Swindle N., Little S. C., Gomes A. V., Swartz D. R., and Davis J. P. (2010) Effect of calcium-sensitizing mutations on calcium binding and exchange with troponin C in increasingly complex biochemical systems. Biochemistry 49, 1975–1984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tikunova S. B., and Davis J. P. (2004) Designing calcium-sensitizing mutations in the regulatory domain of cardiac troponin C. J. Biol. Chem. 279, 35341–35352 [DOI] [PubMed] [Google Scholar]
- 17. Dong W. J., Wang C. K., Gordon A. M., and Cheung H. C. (1997) Disparate fluorescence properties of 2-[4′-(iodoacetamido)anilino]-naphthalene-6-sulfonic acid attached to Cys-84 and Cys-35 of troponin C in cardiac muscle troponin. Biophys. J. 72, 850–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hazard A. L., Kohout S. C., Stricker N. L., Putkey J. A., and Falke J. J. (1998) The kinetic cycle of cardiac troponin C: calcium binding and dissociation at site II trigger slow conformational rearrangements. Protein Sci. 7, 2451–2459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li A. Y., Stevens C. M., Liang B., Rayani K., Little S., Davis J., and Tibbits G. F. (2013) Familial hypertrophic cardiomyopathy related cardiac troponin C L29Q mutation alters length-dependent activation and functional effects of phosphomimetic troponin I*. PLoS One 8, e79363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Davis J. P., Norman C., Kobayashi T., Solaro R. J., Swartz D. R., and Tikunova S. B. (2007) Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys. J. 92, 3195–3206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gifford J. L., Walsh M. P., and Vogel H. J. (2007) Structures and metal-ion-binding properties of the Ca2+-binding helix-loop-helix EF-hand motifs. Biochem. J. 405, 199–221 [DOI] [PubMed] [Google Scholar]
- 22. Spyracopoulos L., Li M. X., Sia S. K., Gagné S. M., Chandra M., Solaro R. J., and Sykes B. D. (1997) Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry 36, 12138–12146 [DOI] [PubMed] [Google Scholar]
- 23. Li M. X., Spyracopoulos L., and Sykes B. D. (1999) Binding of cardiac troponin-I147–163 induces a structural opening in human cardiac troponin-C. Biochemistry 38, 8289–8298 [DOI] [PubMed] [Google Scholar]
- 24. Zhang X. L., Tibbits G. F., and Paetzel M. (2013) The structure of cardiac troponin C regulatory domain with bound Cd2+ reveals a closed conformation and unique ion coordination. Acta Crystallogr. D Biol. Crystallogr. 69, 722–734 [DOI] [PubMed] [Google Scholar]
- 25. Takeda S., Yamashita A., Maeda K., and Maéda Y. (2003) Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41 [DOI] [PubMed] [Google Scholar]
- 26. Robertson I. M., Sevrieva I., Li M. X., Irving M., Sun Y. B., and Sykes B. D. (2015) The structural and functional effects of the familial hypertrophic cardiomyopathy-linked cardiac troponin C mutation, L29Q. J. Mol. Cell Cardiol. 87, 257–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lindert S., Kekenes-Huskey P. M., and McCammon J. A. (2012) Long-timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in troponin C. Biophys. J. 103, 1784–1789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dewan S., McCabe K. J., Regnier M., McCulloch A. D., and Lindert S. (2016) Molecular effects of cTnC DCM mutations on calcium sensitivity and myofilament activation: an integrated multiscale modeling study. J. Phys. Chem. B 120, 8264–8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lindert S., Kekenes-Huskey P. M., Huber G., Pierce L., and McCammon J. A. (2012) Dynamics and calcium association to the N-terminal regulatory domain of human cardiac troponin C: a multiscale computational study. J. Phys. Chem. B 116, 8449–8459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Stevens C. M., Rayani K., Genge C. E., Singh G., Liang B., Roller J. M., Li C., Li A. Y., Tieleman D. P., van Petegem F., and Tibbits G. F. (2016) Characterization of zebrafish cardiac and slow skeletal troponin C paralogs by MD simulation and ITC. Biophys. J. 111, 38–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zot H. G., Hasbun J. E., Michell C. A., Landim-Vieira M., and Pinto J. R. (2016) Enhanced troponin I binding explains the functional changes produced by the hypertrophic cardiomyopathy mutation A8V of cardiac troponin C. Arch. Biochem. Biophys. 601, 97–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Martins A. S., Parvatiyar M. S., Feng H. Z., Bos J. M., Gonzalez-Martinez D., Vukmirovic M., Turna R. S., Sanchez-Gonzalez M. A., Badger C. D., Zorio D. A., Singh R. K., Wang Y., Jin J. P., Ackerman M. J., and Pinto J. R. (2015) In vivo analysis of troponin C knock-In (A8V) mice: evidence that TNNC1 is a hypertrophic cardiomyopathy susceptibility gene. Circ. Cardiovasc. Genet. 8, 653–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cordina N. M., Liew C. K., Gell D. A., Fajer P. G., Mackay J. P., and Brown L. J. (2013) Effects of calcium binding and the hypertrophic cardiomyopathy A8V mutation on the dynamic equilibrium between closed and open conformations of the regulatory N-domain of isolated cardiac troponin C. Biochemistry 52, 1950–1962 [DOI] [PubMed] [Google Scholar]
- 34. Kumari R., Kumar R., Lynn A., and Open Source Drug Discovery Consortium (2014) g_mmpbsa: a GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 54, 1951–1962 [DOI] [PubMed] [Google Scholar]
- 35. Schmidtmann A., Lindow C., Villard S., Heuser A., Mügge A., Gessner R., Granier C., and Jaquet K. (2005) Cardiac troponin C-L29Q, related to hypertrophic cardiomyopathy, hinders the transduction of the protein kinase A dependent phosphorylation signal from cardiac troponin I to C. FEBS J. 272, 6087–6097 [DOI] [PubMed] [Google Scholar]
- 36. Hoffmann B., Schmidt-Traub H., Perrot A., Osterziel K. J., and Gessner R. (2001) First mutation in cardiac troponin C, L29Q, in a patient with hypertrophic cardiomyopathy. Hum. Mutat. 17, 524. [DOI] [PubMed] [Google Scholar]
- 37. Davis J., Davis L. C., Correll R. N., Makarewich C. A., Schwanekamp J. A., Moussavi-Harami F., Wang D., York A. J., Wu H., Houser S. R., Seidman C. E., Seidman J. G., Regnier M., Metzger J. M., Wu J. C., et al. (2016) A tension-based model distinguishes hypertrophic versus dilated cardiomyopathy. Cell 165, 1147–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kekenes-Huskey P. M., Lindert S., and McCammon J. A. (2012) Molecular basis of calcium-sensitizing and desensitizing mutations of the human cardiac troponin C regulatory domain: a multi-scale simulation study. PLoS Comput. Biol. 8, e1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. van Spaendonck-Zwarts K. Y., van Tintelen J. P., van Veldhuisen D. J., van der Werf R., Jongbloed J. D., Paulus W. J., Dooijes D., and van den Berg M. P. (2010) Peripartum cardiomyopathy as a part of familial dilated cardiomyopathy. Circulation 121, 2169–2175 [DOI] [PubMed] [Google Scholar]
- 40. Schlecht W., Li K. L., Hu D., and Dong W. (2016) Fluorescence based characterization of calcium sensitizer action on the troponin complex. Chem. Biol. Drug Des. 87, 171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tikunova S. B., Rall J. A., and Davis J. P. (2002) Effect of hydrophobic residue substitutions with glutamine on Ca2+ binding and exchange with the N-domain of troponin C. Biochemistry 41, 6697–6705 [DOI] [PubMed] [Google Scholar]
- 42. Li M. X., and Hwang P. M. (2015) Structure and function of cardiac troponin C (TNNC1): implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene. 571, 153–166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lindert S., Cheng Y., Kekenes-Huskey P., Regnier M., and McCammon J. A. (2015) Effects of HCM cTnI Mutation R145G on troponin structure and modulation by PKA phosphorylation elucidated by molecular dynamics simulations. Biophys. J. 108, 395–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hwang P. M., Cai F., Pineda-Sanabria S. E., Corson D. C., and Sykes B. D. (2014) The cardiac-specific N-terminal region of troponin I positions the regulatory domain of troponin C. Proc. Natl. Acad. Sci. U.S.A. 111, 14412–14417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Johnson J. D., Collins J. H., Robertson S. P., and Potter J. D. (1980) A fluorescent probe study of Ca2+ binding to the Ca2+-specific sites of cardiac troponin and troponin C. J. Biol. Chem. 255, 9635–9640 [PubMed] [Google Scholar]
- 46. Gillis T. E., Blumenschein T. M., Sykes B. D., and Tibbits G. F. (2003) Effect of temperature and the F27W mutation on the Ca2+ activated structural transition of trout cardiac troponin C. Biochemistry 42, 6418–6426 [DOI] [PubMed] [Google Scholar]
- 47. Skowronsky R. A., Schroeter M., Baxley T., Li Y., Chalovich J. M., and Spuches A. M. (2013) Thermodynamics and molecular dynamics simulations of calcium binding to the regulatory site of human cardiac troponin C: evidence for communication with the structural calcium binding sites. J. Biol. Inorg. Chem. 18, 49–58 [DOI] [PubMed] [Google Scholar]
- 48. Marques M. A., Pinto J. R., Moraes A. H., Iqbal A., de Magalhães M. T., Monteiro J., Pedrote M. M., Sorenson M. M., Silva J. L., and de Oliveira G. A. (2017) Allosteric transmission along a loosely structured backbone allows a cardiac troponin C mutant to function with only one Ca2+ ion. J. Biol. Chem. 292, 2379–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li H., Ngo V., Da Silva M. C., Salahub D. R., Callahan K., Roux B., and Noskov S. Y. (2015) Representation of ion-protein interactions using the drude polarizable force-field. J. Phys. Chem. B 119, 9401–9416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suarez M. C., Machado C. J., Lima L. M., Smillie L. B., Pearlstone J. R., Silva J. L., Sorenson M. M., and Foguel D. (2003) Role of hydration in the closed-to-open transition involved in Ca2+ binding by troponin C. Biochemistry 42, 5522–5530 [DOI] [PubMed] [Google Scholar]
- 51. McKay R. T., Saltibus L. F., Li M. X., and Sykes B. D. (2000) Energetics of the induced structural change in a Ca2+ regulatory protein: Ca2+ and troponin I peptide binding to the E41A mutant of the N-domain of skeletal troponin C. Biochemistry 39, 12731–12738 [DOI] [PubMed] [Google Scholar]
- 52. Genge C. E., Stevens C. M., Davidson W. S., Singh G., Tieleman D. P., and Tibbits G. F. (2016) Functional divergence in teleost cardiac troponin paralogs guides variation in the interaction of TnI switch region with TnC. Genome Biol. Evol. 8, 994–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang D., Robertson I. M., Li M. X., McCully M. E., Crane M. L., Luo Z., Tu A. Y., Daggett V., Sykes B. D., and Regnier M. (2012) Structural and functional consequences of the cardiac troponin C L48Q Ca2+-sensitizing mutation. Biochemistry 51, 4473–4487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Robertson I. M., Sevrieva I., Li M. X., Irving M., Sun Y.-B., and Sykes B. D. (2014) In vitro and in situ structure and function of the cardiac troponin C familial hypertrophic cardiomyopathy-linked mutation, L29Q. Biophys. J. 106, 723a–724a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Messer A. E., and Marston S. B. (2014) Investigating the role of uncoupling of troponin I phosphorylation from changes in myofibrillar Ca2+-sensitivity in the pathogenesis of cardiomyopathy. Front. Physiol. 5, 315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sevrieva I., Knowles A. C., Kampourakis T., and Sun Y. B. (2014) Regulatory domain of troponin moves dynamically during activation of cardiac muscle. J. Mol. Cell. Cardiol. 75, 181–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Bordoli L., and Schwede T. (2012) Automated protein structure modeling with SWISS-MODEL Workspace and the Protein Model Portal. Methods Mol. Biol. 857, 107–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., and Shaw D. E. (2010) Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mahoney M. W., and Jorgensen W. L. (2000) A five-site model for liquid water and the reproduction of the density anomaly by rigid, nonpolarizable potential functions. J. Chem. Phys. 112, 8910–8922 [Google Scholar]
- 61. Berendsen H. J., Postma J. P., Vangunsteren W. F., Dinola A., and Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 [Google Scholar]
- 62. Bussi G., Donadio D., and Parrinello M. (2007) Canonical sampling through velocity rescaling. J. Chem. Phys. 126:014101, [DOI] [PubMed] [Google Scholar]
- 63. Cerutti D. S., Duke R. E., Darden T. A., and Lybrand T. P. (2009) Staggered mesh Ewald: an extension of the smooth particle-mesh Ewald method adding great versatility. J. Chem. Theory Comput. 5, 2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pall S., and Hess B. (2013) A flexible algorithm for calculating pair interactions on SIMD architectures. Comp. Phys. Commun. 184, 2641–2650 [Google Scholar]
- 65. Hess B., Bekker H., Berendsen H. J., and Fraaije J. G‘. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [Google Scholar]
- 66. Daura X., Gademann K., Jaun B., Seebach D., Van Gunsteren W. F., and Mark A. E. (1999) Peptide folding: when simulation meets experiment. Angew. Chem. Int. Ed. 38, 236–240 [Google Scholar]
- 67. Eisenhaber F., Lijnzaad P., Argos P., Sander C., and Scharf M. (1995) The double cubic lattice method: efficient approaches to numerical-integration of surface-area and volume and to dot surface contouring of molecular assemblies. J. Comput. Chem. 16, 273–284 [Google Scholar]
- 68. Yap K. L., Ames J. B., Swindells M. B., and Ikura M. (2002) Vector geometry mapping: a method to characterize the conformation of helix-loop-helix calcium-binding proteins. Methods Mol. Biol. 173, 317–324 [DOI] [PubMed] [Google Scholar]
- 69. Hub J. S., de Groot B. L., and van der Spoel D. (2010) g_wham: a free weighted histogram analysis implementation including robust error and autocorrelation estimates. J. Chem. Theory Comput. 6, 3713–3720 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.