

Abstract
An imbalance between oxidative stress and antioxidant activity plays an important role in the pathogenesis of chronic obstructive pulmonary disease (COPD). Cigarette smoke, a major risk factor of COPD, induces cellular oxidative stress, but levels of antioxidants such as heme oxygenase-1 (HO-1) are reduced in individuals with severe COPD. In this study, we evaluated the molecular mechanism of reduced HO-1 expression in human bronchial epithelial cells. We found that cigarette smoke extract (CSE) increases HO-1 levels via activation of NFE2-related factor 2 (Nrf2). However, pretreating cells with the protease neutrophil elastase (NE) suppressed the CSE-induced expression of HO-1 mRNA and protein. NE also decreased the sirtuin 1 (SIRT1) level, but did not inhibit CSE-induced nuclear translocation and DNA-binding activity of Nrf2. Transfection of cells with a Myc/His-tagged SIRT1 expression vector completely blocked the NE-mediated suppression of HO-1 expression. We further noted that the NE-induced down-regulation of SIRT1 was not due to decreased transcription or proteasomal/lysosomal degradation or loss of solubility. Immunofluorescence staining revealed that NE enters the cell cytoplasm, and we observed that NE directly cleaved SIRT1 in vitro, indicating that SIRT1 levels are decreased via direct degradation by internalized NE. Of note, we observed decreased SIRT1 levels in NE-treated primary human bronchial epithelial cells and in lung homogenates from both smokers and patients with COPD. In conclusion, NE suppresses CSE-induced HO-1 expression by cleaving SIRT1. This finding indicates the importance of cross-talk between oxidative stress and protease responses in the pathogenesis of COPD.
Keywords: antioxidant, chronic obstructive pulmonary disease (COPD), oxidative stress, pathogenesis, sirtuin 1 (SIRT1), cigarette smoke extract, heme oxygenase 1, neutrophil elastase
Introduction
Chronic obstructive pulmonary disease (COPD)2 is a type of obstructive lung disease characterized by chronic bronchitis and remodeling of the small airways and destruction of the lung parenchyma (1). Although the mechanism of COPD development and progression is not fully understood, various hypotheses have been proposed, including oxidative stress, inflammation, protease-antiprotease imbalance, autophagy, apoptosis, and aging (2). Among these, oxidative stress is a major factor in the pathogenesis of COPD (3). Oxidative stress arises from environmental exposure such as cigarette smoking and inflammatory responses to viral and bacterial infections in the lungs. Oxidative stress causes protein oxidation and lipid peroxidation, injuring the lung tissue (4). Therefore, targeting oxidative stress with antioxidants or increasing the endogenous levels of antioxidants is thought to be beneficial for COPD treatment.
Heme oxygenase (HO)-1 is a strongly inducible and highly regulated antioxidant (5). Ho-1-deleted cells were more sensitive to oxidative stress such as hydrogen peroxide and paraquat (6), and adenoviral overexpression of Ho-1 in the lungs protected mice from elastase-induced inflammation, pro-inflammatory cytokine production, and emphysema (7). In addition, polymorphisms in the HO-1 promoter that reduced HO-1 expression were reported to be associated with an increased susceptibility to emphysema in smokers (8). Pharmacological inhibition of Ho-1 or knockdown of the Ho-1 gene increased mucosal permeability and enhanced cigarette smoke (CS)-induced secretion of mucus (9, 10). Moreover, in lung epithelial cells, cigarette smoke extract (CSE)-induced apoptosis was suppressed by HO-1 induction (11). Similarly, HO-1 plays protective roles against destruction, mucus hypersecretion, and structural cell death in the lungs. These findings support the protective role of HO-1 in the development of COPD. When lung cells are exposed to oxidants, antioxidants such as HO-1 are typically up-regulated as a defense mechanism. However, patients with severe COPD show reduced levels of HO-1 (12); the mechanism for this has not been elucidated.
Nuclear factor (erythroid-derived 2) like-2 (Nrf2) is a major transcription factor that regulates the expression of antioxidant HO-1 (13). Under normal conditions, Nrf2 is maintained in the cytoplasm by Kelch-like ECH-associated protein 1 (KEAP1) and cullin3. Cullin3 ubiquitinates Nrf2 and mediates proteasomal degradation of Nrf2 (13). Oxidative stress such as CS and H2O2 increases the stability of Nrf2 by down-regulating the adaptor protein KEAP1, leading to increased Nrf2 nuclear accumulation/DNA-binding activity and the induction of antioxidant HO-1 (13, 14). Genetic ablation of Nrf2 as well as Ho-1 deficiency enhanced the susceptibility to CS-induced emphysema. In Nrf2-deficient mice, greater lung cell apoptosis and inflammation were observed compared with in control mice (15). In addition to Nrf2, several factors have been reported to regulate HO-1 gene expression. Signal transducer and activator of transcription 3 is associated with HO-1 gene activation (16, 17) and SIRT1 mediates CS-induced HO-1 expression via an Nrf2-independent and Foxo3-dependent mechanism (18).
In our previous study, we found that CSE induces the production of the neutrophilic chemokine interleukin-8 (IL-8) in lung epithelial cells and co-exposure to CSE and elastase (released from recruited neutrophils) enhances IL-8 secretion by accelerating extracellular signal-regulated kinase (ERK) activation (19). CSE up-regulates antioxidative and anti-inflammatory HO-1 protein as a defense mechanism. In this study, we evaluated the effect of neutrophil elastase (NE) on CSE-induced HO-1 expression and its regulatory mechanism in human bronchial epithelial cells. We found that NE suppresses the CSE-induced expression of HO-1 by cleaving SIRT1. This indicates the importance of cross-talk between oxidative stress (CSE) and protease (NE) in the pathogenesis of COPD.
Results
CSE increases HO-1 expression via Nrf2 activation in human bronchial epithelial cells
Oxidative stress is well known to activate Nrf2, which in turn binds cis-acting ARE enhancers and induces the expression of protective antioxidant genes (13). We first determined whether CSE activates Nrf2 and induces antioxidant in human bronchial epithelial cells (BEAS-2B) as a defense mechanism. BEAS-2B cells were treated with CSE (0, 1, 2, and 3%) for 4 h and DNA-binding activity of Nrf2 was measured using nuclear extracts. CSE increased the DNA-binding activity of Nrf2 in a dose-dependent manner (Fig. 1A). The levels of antioxidant HO-1 mRNA and protein were assessed by quantitative real-time PCR and Western blot analysis after treatment with CSE. CSE significantly up-regulated HO-1 mRNA and protein expression (Fig. 1, B and C). To evaluate whether Nrf2 activation mediates CSE-induced up-regulation of HO-1 protein, cells were infected with control (Mock), wild-type Nrf2 (Ad-WT-Nrf2), or dominant-negative Nrf2 (Ad-DN-Nrf2) adenovirus vector. Overexpression of DN-Nrf2, which lacks an N-terminal trans-activating domain and appears as a 29-kDa immunoreactive band, completely suppressed CSE-induced HO-1 expression (Fig. 1D). These results indicate that CSE up-regulates HO-1 via Nrf2 activation in human bronchial epithelial cells.
Figure 1.

CSE increases the level of HO-1 expression via Nrf2 activation. A, BEAS-2B cells were treated with CSE (0, 1, 2, and 3%) for 4 h. Nrf2 activity using nuclear proteins was measured using a DNA-binding ELISA kit for activated Nrf2 transcription factor. Data represent the mean ± S.D. of triplicate experiments. B, BEAS-2B cells were stimulated with CSE (0, 0.5, 1, 2, and 3%) for 8 or 12 h. Total RNA was isolated and quantitative real-time PCR for HO-1 and GAPDH was performed. Data represent the mean ± S.D. of triplicates. **, p < 0.05. C, BEAS-2B cells were treated with CSE (0, 0.5, 1, and 2%) for 24 h. D, BEAS-2B cells were infected with control (Mock), wild-type Nrf2 (Ad-WT-Nrf2), or dominant-negative Nrf2 (Ad-DN-Nrf2) adenovirus vector. Forty-eight hours after infection, the cells were treated with CSE (1%) for 24 h. Total cellular extracts were subjected to Western blot analysis for HO-1, Nrf2, and GAPDH (C and D). The results are representative of three independent experiments. S.E., short exposure; L.E., long exposure.
NE suppresses CSE-induced HO-1 expression
We next evaluated the effect of NE treatment on CSE-induced HO-1 expression. Cells were pretreated with NE (1 units/ml) and then stimulated with CSE. NE pretreatment alone slightly decreased the basal level of HO-1 mRNA and protein expression (Fig. 2, A and B). Moreover, up-regulation of HO-1 by CSE was significantly suppressed in NE-pretreated cells (Fig. 2, A and B).
Figure 2.
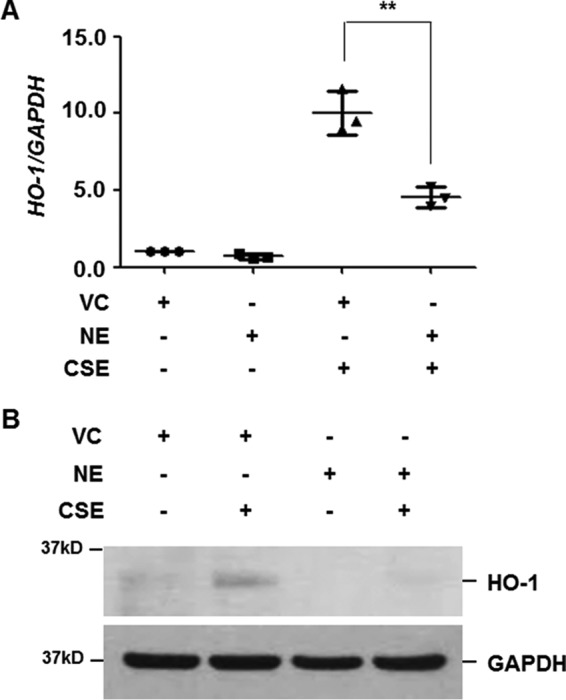
NE suppresses CSE-induced HO-1 expression. BEAS-2B cells were pretreated with vehicle control (VC) or NE (1 units/ml) for 4 h and then stimulated with CSE (1%) for 12 (A) or 24 h (B). A, total RNA was isolated and quantitative real-time PCR for HO-1 and GAPDH was performed. Data represent the mean ± S.D. of triplicate experiments. **, p < 0.05. B, total cellular extracts were subjected to Western blot analysis for HO-1 and GAPDH. The results are representative of three independent experiments.
NE does not inhibit CSE-induced nuclear translocation and DNA-binding activity of Nrf2
Under non-stress conditions, Nrf2 is anchored in the cytoplasm by binding to KEAP1, which, in turn, facilitates the ubiquitination and subsequent proteolysis of Nrf2 (13). CSE exposure leads to KEAP1 degradation, resulting in the stabilization and nuclear translocation of Nrf2 (20). To determine the molecular mechanism of NE-mediated suppression of HO-1, we first investigated whether NE suppresses CSE-induced KEAP1 degradation and nuclear translocation of Nrf2. BEAS-2B cells were pretreated with NE and then stimulated with the indicated concentrations of CSE for 4 h. Cytoplasmic and nuclear proteins were used to detect KEAP1 and Nrf2 expression, respectively. CSE decreased cytoplasmic KEAP1 expression and mediated nuclear translocation of Nrf2, which was not suppressed by NE pretreatment (Fig. 3A). We next evaluated the effect of NE on CSE-induced Nrf2 binding to ARE. Nuclear extracts from Nrf2-transfected COS-7 cells were used as a positive control (Fig. 3B). NE pretreatment did not inhibit the DNA-binding activity of Nrf2 induced by CSE. These results suggest that NE-induced suppression of HO-1 is Nrf2-independent.
Figure 3.
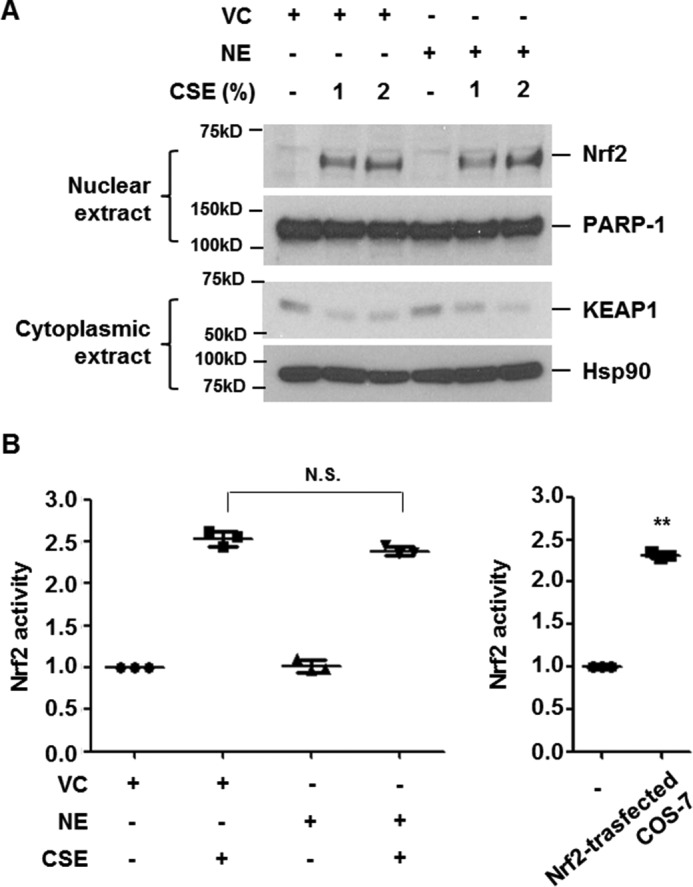
NE does not inhibit CSE-induced nuclear translocation and DNA-binding activity of Nrf2. A and B, BEAS-2B cells were pretreated with vehicle control (VC) or NE (1 units/ml) for 4 h and then stimulated with CSE (1 or 2%) for 4 h. Nuclear and cytoplasmic proteins were extracted and subjected to Western blot analysis for Nrf2, PARP-1, KEAP1, and Hsp90 (A). Nrf2 activity using nuclear extracts was determined. Nuclear proteins from COS-7 cells transfected with Nrf2 expressing vector were used as a positive control (B). Data represent the mean ± S.D. of triplicate experiments. **, p < 0.05. The results are representative of three independent experiments. N.S., not significant.
NE decreases SIRT1 expression, which mediates the reduction of HO-1 expression in CSE-treated cells
Sirt1 deficiency has been reported to reduce the mRNA level of Ho-1 in mouse lungs exposed to CS. In contrast, Sirt1 transgenic overexpression or treatment with Sirt1 activator increased Ho-1 mRNA in both WT and Nrf2-deficient mouse lungs in response to CS exposure. This suggests that the protective effect of Sirt1 against CS-induced oxidative stress is caused by Ho-1 up-regulation independently of Nrf2 (18). Therefore, we evaluated the role of NE on SIRT1 expression. Interestingly, NE treatment significantly decreased SIRT1 expression. SIRT1 began decreasing at 2 h after NE treatment and its decrease was prolonged for up to 24 h (Fig. 4A). Moreover, when the cells were transiently transfected with Myc/His-tagged SIRT1 expression plasmids, CSE-induced HO-1 expression was enhanced and NE-mediated HO-1 suppression was completely blocked (Fig. 4B). These results demonstrate that NE negatively regulates HO-1 expression by down-regulating SIRT1.
Figure 4.
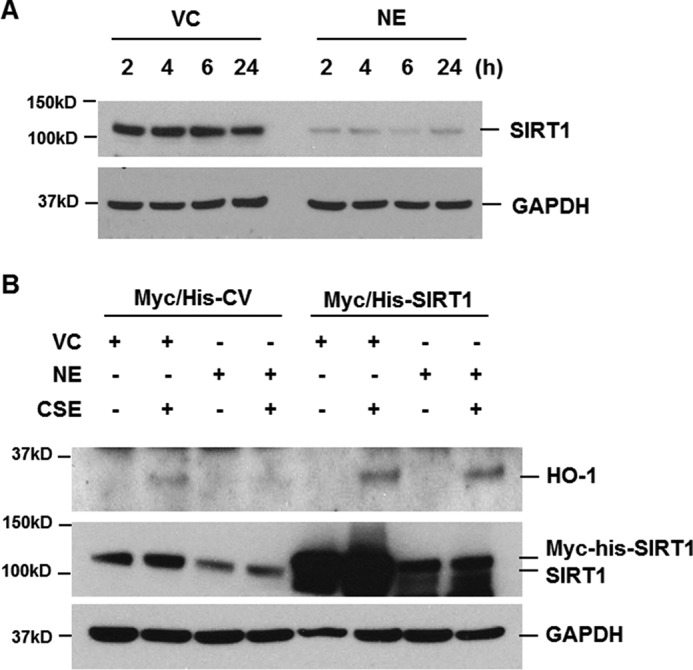
NE decreases SIRT1 expression, which mediates the reduction of HO-1 expression in CSE-treated cells. A, BEAS-2B cells were treated with vehicle control (VC) or NE (1 units/ml) for the indicated times. B, cells were transiently transfected with Myc/His-tagged SIRT1 (Myc/His-SIRT1) expression vector or control vector (Myc/His-CV). Forty-eight hours after transfection, the cells were pretreated with VC or NE (1 units/ml) for 4 h and then incubated with CSE (1%) for 24 h. Total cellular extracts were subjected to Western blot analysis for SIRT1, HO-1, and GAPDH. The results are representative of three independent experiments.
NE-mediated down-regulation of SIRT1 is not due to decreased transcription, proteasomal/lysosomal proteolysis, and loss of solubility
We next evaluated the mechanism of SIRT1 down-regulation by NE. To evaluate whether down-regulation of SIRT1 by NE is regulated at the transcriptional level, the mRNA levels of SIRT1 were assessed by RT-PCR after treatment with NE. NE treatment did not suppress the transcript levels of SIRT1 (Fig. 5A). We next examined whether NE-induced down-regulation of SIRT1 protein expression is due to intracellular degradation of SIRT1 protein. As the main sites of intracellular protein degradation are the proteasome and lysosome, we tested whether the down-regulation of SIRT1 protein by NE was caused by degradation via these sites. Proteasome or lysosome activity was blocked by proteasome inhibitors (PS-341, MG132) or lysosome inhibitors (ammonium chloride, chloroquine), respectively (supplemental Fig. S1). When both proteolysis sites were blocked, NE-induced down-regulation of SIRT1 was not affected (Fig. 5B). Because decreased solubility of protein contributes to reduced protein activity or expression (21), we tested this possibility for NE-induced down-regulation of SIRT1. To evaluate whether the decreased level of SIRT1 is mediated by the loss of solubility, the SIRT1 expression level in the soluble extracts and insoluble pellet were examined. Although SIRT1 expression in the soluble fraction decreased after NE treatment, SIRT1 in the insoluble pellet was not detected, indicating that NE-mediated SIRT1 down-regulation was not related to SIRT1 insolubilization (Fig. 5C).
Figure 5.

Down-regulation of SIRT1 is due to direct cleavage after translocation of NE into the cells. A–C, down-regulation of SIRT1 is not due to decreased transcription, proteasomal/lysosomal proteolysis, and loss of solubility. A, BEAS-2B cells were treated with vehicle control (VC) or NE (1 unit/ml) for the indicated times. Total RNA was isolated and reverse transcription PCR (RT-PCR) for the SIRT1 and GAPDH genes was performed. B, cells were pretreated with proteasome inhibitors (200 nm PS-341, 10 μm MG132) or lysosomal inhibitors (50 mm ammonium chloride, 100 μm chloroquine) for 1 h and then stimulated with VC or NE (1 unit/ml) for 2 h. Total cellular extracts were subjected to Western blot analysis for SIRT1 and GAPDH. C, BEAS-2B cells were stimulated with VC or NE for 2 h. SIRT1 expression in soluble extracts or insoluble pellets were detected by Western blot analysis. D, cells were treated with VC or NE (20 units/ml) for 2 h. Immunofluorescence staining for internalized NE was performed. Cells were analyzed using an ECLIPSE TE300 (Nikon) fluorescence microscope. Scale bar: 20 μm. E, cell lysates were incubated with VC or NE (0.04, 0.2, and 1 unit/ml) in vehicle at 37 °C for 30 min. The reaction was stopped by adding 4× sample buffer. Samples were subjected to Western blot analysis for SIRT1 and NE under reducing conditions. F, BEAS-2B cells were treated with VC or NE (1 units/ml) in the presence or absence of elastase inhibitor (elaspol, 100 μg/ml) or serine protease inhibitor (PMSF, 1 mm). Cells were stimulated with heated forms of VC or NE (1 units/ml) for 4 h. Total cellular extracts were subjected to Western blot analysis for SIRT1 and GAPDH. The results are representative of three independent experiments.
SIRT1 is down-regulated because of direct cleavage after translocation of NE into cells
We previously reported that NE can enter into normal lung epithelial cells (19). We next investigated whether decreased expression of SIRT1 occurs by direct degradation by NE in the cytoplasm. Immunofluorescence staining for cytoplasmic/internalized NE was performed and analysis of NE-treated cells showed that NE was introduced into cells (Fig. 5D). To test whether the reduction of SIRT1 expression at the post-translational level occurs because of direct cleavage of SIRT1 by internalized NE, we assessed whether NE is capable of degrading SIRT1 in vitro. SIRT1 was highly sensitive to NE cleavage. Most SIRT1 was degraded after 30 min of incubation with NE (Fig. 5E). When cells were treated with NE in the presence of elastase inhibitor (elaspol) or serine protease inhibitor (phenylmethylsulfonyl fluoride), NE-mediated SIRT1 cleavage was completely blocked. Heated NE also failed to cleave SIRT1 (Fig. 5F). Taken together, these data suggest that NE enters into the cell and directly cleaves SIRT1 and activity of NE is required for SIRT1 cleavage.
To investigate cleavage sites of SIRT1 by NE, we used the program Peptidecutter to search for consensus cleavage sites. We identified 63 sites in the human SIRT1 amino acid sequence predicted to be recognition sites for the NE (supplemental Fig. S2). To confirm that NE cleaves SIRT1 to small fragments and the fragments are cleaved forms of SIRT1, cells were transiently transfected with control or SIRT1 siRNAs and then stimulated with NE. The expression of SIRT1 mRNA and protein was significantly reduced in SIRT1 siRNA-transfected cells (Fig. 6, A–C). Western blot analysis, using antibodies that recognize the carboxyl- or amino-terminal peptide of SIRT1, revealed the presence of faster migrating fragments in control siRNA/NE-treated cells. In SIRT1 knockdown cells, the expression level of migrating fragments was decreased or not detected, verifying that the fragments are cleaved SIRT1 (Fig. 6, B and C). These results demonstrate that NE cleaves SIRT1 at multiple sites.
Figure 6.

NE cleaves SIRT1 at multiple sites. BEAS-2B cells were transiently transfected with control or SIRT1 siRNA. Forty-eight hours after transfection, the cells were treated with NE (1 or 3 units/ml) for 4 h. Total RNA was isolated and quantitative real-time PCR for SIRT1 and GAPDH was performed (A). Data represent the mean ± S.D. of triplicates. **, p < 0.05. B and C, total cellular extracts were subjected to Western blot analysis for SIRT1 using two different SIRT1 antibodies that recognize the carboxyl-terminal region or the amino-terminal region of SIRT1, respectively. The results are representative of three independent experiments. n.s., nonspecific; S.E., short exposure; L.E., long exposure.
Decreased level of SIRT1 in NE-treated primary human bronchial epithelial cells (HBECs) and in lung homogenates from smokers and patients with COPD
We confirmed the effect of NE on SIRT1 expression in primary HBECs. Normal HBECs were collected using a bronchial brush. Airway brushings were obtained from the right lower lobe anterobasal segmental bronchus wall. The epithelial lineage was verified by immunocytochemical staining. Cultured HBECs stained intensely and exclusively for the epithelial specific markers (cytokeratin and E-cadherin). No expression was observed for macrophage and endothelial lineage markers (CD11b and CD31). Verified HBECs (passage No. 2) were used in the experiment. NE pretreatment significantly fragmented SIRT1 and CSE-induced HO-1 expression was suppressed in NE-pretreated primary HBECs (Fig. 7A). We compared the expression level of SIRT1 in protein extracts of lung tissues from normal (non-smoker), smoker, and COPD patients. SIRT1 expression was significantly lower in lung extracts of smokers and COPD patients (Fig. 7, B and C).
Figure 7.
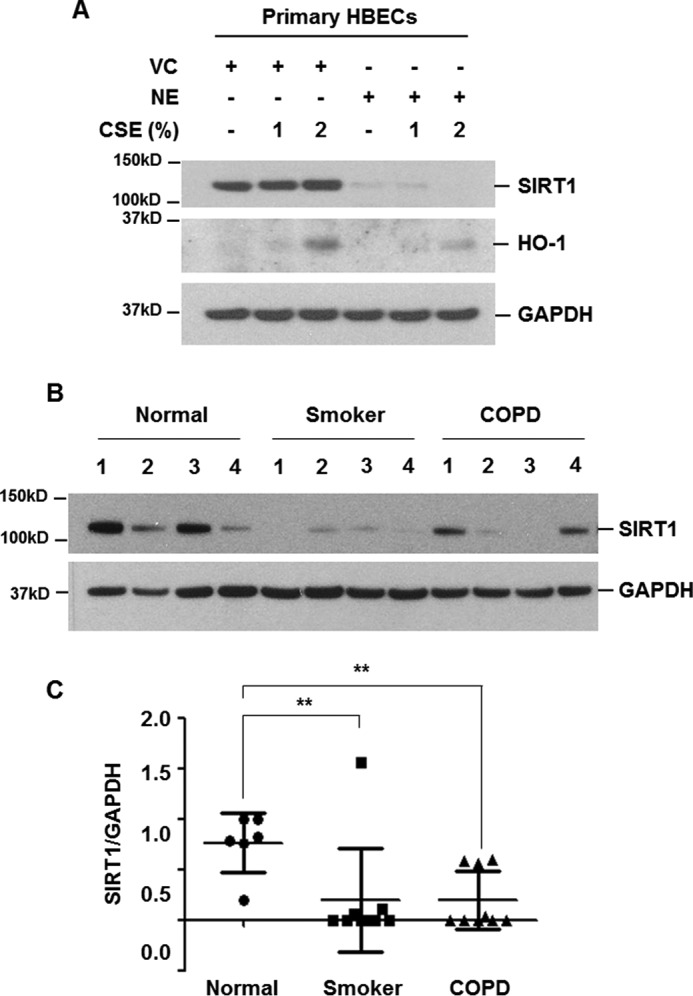
Decreased level of SIRT1 in NE-treated primary HBECs and in lung homogenates from smokers and COPD patients. A, primary human bronchial epithelial cells were pretreated with vehicle control (VC) or NE (1 units/ml) for 4 h and then stimulated with CSE (1 and 2%) for 24 h. Cell lysates were subjected to Western blot analysis for SIRT1, HO-1, and GAPDH. B, lung homogenates from normal (non-smoker) (n = 6), smoker (n = 9), and COPD (n = 9) patients were subjected to Western blot analysis for SIRT1 and GAPDH. C, gel data were quantified using Scion image densitometry. Data represent the mean ± S.D. normal versus smoker: **, p = 0.0299, normal versus COPD: **, p = 0.0027.
Discussion
COPD is a complex disease process. Oxidative stress, inflammation, and protease-antiprotease imbalance have been suggested as a pathogenic triad in COPD and many studies have demonstrated that these processes closely interact with each other. We recently reported that CSE induces IL-8, a well known neutrophil chemotactic factor. CSE-induced IL-8 mediates neutrophil recruitment, and recruited neutrophils release several proteinases including elastase. When lung epithelial cells were co-stimulated with CSE and NE, IL-8 production was enhanced. Consequently, the interactive action of oxidative stress (CSE) and protease (NE) lead to continued neutrophil accumulation and inflammation (19). Both CS and CSE exert strong oxidative stress and this action is limited by the simultaneous activation of antioxidative pathways. Although NE enhanced CSE-induced inflammation synergistically, the effect of NE on CSE-induced activation of the antioxidative pathway has not been elucidated. Therefore, in this study, we evaluated the interactive action of CSE and NE on the antioxidative pathway and its molecular mechanism in bronchial epithelial cells.
In this study, we demonstrated that CSE-induced up-regulation of HO-1 depends on the activation of its transcriptional factor Nrf2, which agrees with the results of previous studies (22). In contrast, NE markedly suppressed the level of HO-1 mRNA and protein expression induced by CSE. Considering the well known protective effects of HO-1 in lung diseases (23), these results suggest that NE plays a role in maintaining oxidative stress by CS or CSE. We next examined the molecular mechanism of down-regulation of HO-1 by NE.
Because Nrf2 is the representative transcriptional factor regulating the expression of antioxidant genes (13), we first evaluated the role of Nrf2 in the down-regulation of HO-1 by NE. NE not only did not affect the basal level of Nrf2 mRNA and protein (data not shown), but also did not suppress CSE-induced KEAP1 down-regulation in the cytoplasm and nuclear translocation/DNA-binding activity of Nrf2. These results indicate that NE-mediated suppression of HO-1 is independent of Nrf2 activity.
SIRT1 is a member of the sirtuin family (class III histone/protein deacetylases), which play important roles in a variety of cellular processes including aging, inflammation, and oxidative stress resistance (24–26). A recent report showed that Sirt1 protected mice against CS-induced lung oxidative stress via a Foxo3-dependent mechanism (18). Although Sirt1 deficiency reduced the level of CS-induced Ho-1 mRNA, transgenic overexpression of Sirt1 or treatment with Sirt1 activator augmented Ho-1 in response to CS exposure (18). This study is the first to show that NE treatment rapidly and effectively decreased the SIRT1 protein level, which was not due to decreased transcription. NE-mediated reduction of HO-1 was restored by SIRT1 overexpression, suggesting that abrogation of CSE-induced HO-1 up-regulation by NE occurred because of down-regulation of SIRT1. In accordance with this result, the level of SIRT1 was reported to be significantly lower in the nuclear extracts of peripheral lung tissue of smokers and patients with COPD than in the lungs of nonsmokers (27). We also observed decreased levels of SIRT1 protein expression in lung tissues from smokers and COPD patients.
We next evaluated how SIRT1 is down-regulated by NE treatment. SIRT1 was reported to be cleaved in adipose tissue by high-fat diets. SIRT1 cleavage was mediated by inflammation-activated caspase-1 (28). However, NE did not activate caspase-1 (supplemental Fig. S3). The concentration of CSE or NE used in this study did not change the cell viability, and CSE treatment up to 24 h did not decrease the level of SIRT1 protein expression (data not shown). CSE or H2O2 decreased SIRT1 protein levels in both a human monocyte-macrophage cell line (MonoMac6) and primary and transformed epithelial cell lines (27, 29) because of proteasomal degradation (29). In this study, however, NE-mediated SIRT1 degradation was not blocked by proteasome inhibition. Although NE has been shown to activate intracellular signaling mediators through cell membrane receptors including PARs (30, 31), we did not find an association between NE-mediated down-regulation of SIRT1 and cellular reactive oxygen species generation or Akt/ERK activation. Moreover, we found that NE activated intracellular signaling mediators via receptor, PAR2, in human bronchial epithelial cells and knockdown of PAR2 did not affect NE-mediated down-regulation of SIRT1 (supplemental Fig. S4). Finally, as previous studies, including ours, reported that NE was internalized into cells (19, 32), we assessed the direct cleavage of SIRT1. We found that NE can fragment SIRT1 in vitro. On the other hand, NE did not cleave several cellular proteins including Akt, ERK, PARP, Hsp90, KEAP1 (supplemental Fig. S5, Fig. 3), and IL-8 (19), which indicates that NE selectively fragments SIRT1 protein. NE pre-mixed with elaspol (NE inhibitor) or PMSF (serine protease inhibitor) failed to degrade SIRT1 and heated NE had no effect on SIRT1 expression, suggesting that NE activity is necessary for SIRT1 degradation.
NE activity in patients with COPD is known to be increased (33), which may be associated with a decreased level of SIRT1. This indicates the importance of NE-induced SIRT1 down-regulation in the pathogenesis of COPD. Although many studies have demonstrated the anti-inflammatory and antioxidative effects of SIRT1 activator (18, 25, 34), pretreatment with resveratrol, a well known SIRT1 activator, did not suppress NE-mediated reduction of SIRT1 (data not shown).
In conclusion, our study demonstrates the importance of the cross-talk between oxidative stress (CSE) and protease (NE) in the pathogenesis of COPD. NE suppresses CSE-induced activation of the antioxidant pathway by down-regulating SIRT1 (Fig. 8).
Figure 8.
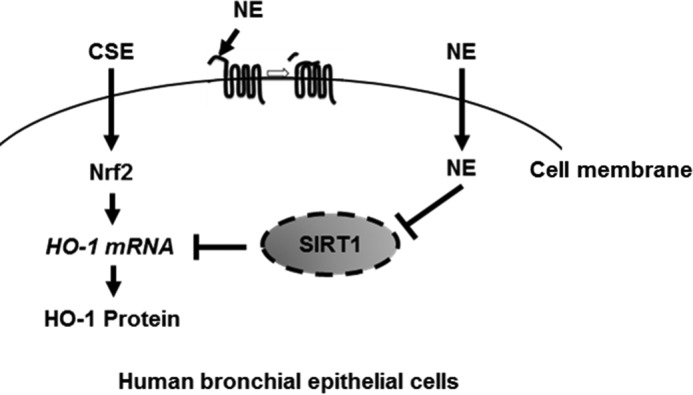
Schematic diagram of the effect of NE on CSE-induced HO-1 expression. CSE up-regulates HO-1 through Nrf2 activation as a defense mechanism in human bronchial epithelial cells. NE suppresses CSE-induced HO-1 by cleaving SIRT1 protein.
Experimental procedures
Cells
Normal human bronchial epithelial cells, BEAS-2B, were maintained in defined keratinocyte-SFM (GIBCO by Life Technologies, Grand Island, NY) at 37 °C under 5% CO2. Normal primary HBECs were obtained after review and approval by the Seoul National University Hospital Institutional Review Board (SNUH IRB number: H-1602-108-742). Primary HBECs were isolated from bronchial brushing samples during bronchoscopy. The bronchial brushing samples were obtained from the right lower lobe anterobasal segmental bronchus wall. The brush was immediately immersed in a tube containing 10 ml of ice-cold RPMI containing 20% fetal bovine serum. Within a few minutes, the cells were centrifuged and resuspended in defined keratinocyte-SFM. Submerged cells were grown as monolayers to 80–100% confluence and then used for experiments.
Cigarette smoke extract
CSE was prepared as previously described (35). Commercial cigarettes (THIS; 84 mm long, diameter of 8 mm; purchased from KT&G, Republic of Korea) were smoked continuously by a bottle system connected to a vacuum machine. The smoke from 20 cigarettes was bubbled in 60 ml of phosphate-buffered saline (GIBCO). The large, insoluble particles in the resulting suspension were removed by filtering the solution through a 0.22-μm filter (35).
Reagents
Rabbit monoclonal anti-SIRT1 (C-term) and anti-KEAP1 antibodies were from Cell Signaling (Danvers, MA). Rabbit polyclonal anti-SIRT1 (N-term) antibody was from Millipore (Darmstadt, Germany). Rabbit polyclonal anti-Nrf2 (to detect C terminus of Nrf2), anti-poly(ADP-ribose) polymerase-1 (PARP-1), goat polyclonal anti-heme oxygenase 1 (HO-1), anti-GAPDH, mouse monoclonal anti-heat shock protein 90 (Hsp90) antibodies, and secondary antibodies conjugated to horseradish peroxidase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Human sputum leukocyte elastase (NE) was purchased from Elastin Product Company (Owensville, MO). Elastase was dissolved in 50% glycerol and 50% 0.02 m NaOAc (pH 5). PS-341 was obtained from Selleckchem (Houston, TX), and MG132 was from Calbiochem (Darmstadt, Germany). Chloroquine, ammonium chloride, and PMSF were from Sigma. Elaspol was from Ono Pharmaceutical Co., Ltd. (Osaka, Japan).
Nrf2 activity assay
Nrf2 activity in nuclear extracts was determined using the TransAM® Nrf2 activity assay kit (Active Motif, Carlsbad, CA). Briefly, nuclear extracts were incubated in a 96-well plate, to which oligonucleotides containing the antioxidant response element (ARE) consensus binding site (5′-GTCACAGTGACTCAGCAGAATCTG-3′) had been immobilized. The primary antibody recognized an epitope on Nrf2 protein upon DNA binding. A horseradish peroxidase-conjugated secondary antibody was added to each well and a colorimetric reaction was performed. Absorbance was read at 450 nm with a reference wavelength of 655 nm.
Real-time PCR and reverse transcription-PCR (RT-PCR)
Total RNA was isolated using the RNeasy kit (Qiagen, Hilden, Germany). cDNA was synthesized from 1 μg of total RNA, using the Reverse Transcription system (Promega, Madison, WI). PCR amplification was performed with 2× TaqMan gene expression master mix (Applied Biosystems, Foster City, CA) or SYBR Green (Applied Biosystems). HO-1 probe (Hs00157965_m1) and GAPDH probe (Hs99999905_m1) were obtained from Applied Biosystems. Human SIRT1 primers (fwd, 5′-TCGCAACTATACCCAGAACATAGACA-3′, rev, 5′-CTGTTGCAAAGGAACCATGACA-3′) and GAPDH (fwd, 5′-GAAGGTGAAGGTCGGAG TC-3′, rev, 5′-GAAGATGGTGATGGGATTTC-3′) were used. RT-PCR was performed using the following primers: human SIRT1 (fwd, 5′-TCAGTGGCTGGAACAGTGAG-3′, rev, 5′-TCTCCATCAGTCCCAAATCC-3′) and GAPDH (fwd, 5′-CTTTTGCGTCGCCAGCCGAG-3′, rev, 5′-CAGCCTTGGCAGCGCCAGTA-3′).
Protein extraction
Cells were allowed to equilibrate in ice-cold cytoplasmic extraction buffer (CEB) consisting of 10 mm Tris-HCl (pH 7.8), 10 mm KCl, 1.5 mm EDTA, and 0.5 mm DTT for 5 min. The cells were lysed on ice for 10 min in 0.4% Nonidet P-40/CEB/protease inhibitor mixture (Roche Diagnostics Corporation, Basel, Switzerland). Following centrifugation at 3500 rpm for 5 min, the supernatants (cytoplasmic extracts) were collected. The nuclear pellets were washed with CEB and then suspended in nuclear extraction buffer consisting of 20 mm Tris-HCl (pH 7.8), 150 mm NaCl, 50 mm KCl, 1.5 mm EDTA, 5 mm DTT, and 0.4% Nonidet P-40/protease inhibitor mixture. Following centrifugation at 13,000 rpm for 15 min, the supernatant (nuclear extract) was collected. Total cellular extracts were prepared in 1× cell lysis buffer (Cell Signaling). Triton X-100-insoluble proteins remaining in the pellets were dissolved in SDS sample buffer consisting of 10 mm Tris-Cl (pH 7.4), 1 mm sodium orthovanadate, and 2% SDS. Frozen lung tissues (SNUH IRB Number: H-1309-073-521) were homogenized in tissue extraction buffer (Life Technologies) containing protease inhibitor mixture (Sigma) and phosphatase inhibitor mixture (Sigma). Protein concentrations were determined by the Bradford method (Bio-Rad, Hercules, CA).
Western blot analysis
Equal amounts of protein were resolved by gradient SDS-polyacrylamide gel electrophoresis (Invitrogen) and transferred to nitrocellulose membranes (GE Healthcare, Little Chalfont, UK). The membranes were blocked with 5% skim milk blocking buffer for 1 h before being incubated overnight at 4 °C with primary antibodies. The membranes were then washed three times and incubated with horseradish peroxidase-conjugated secondary antibodies in blocking buffer for 1 h. After successive washes, the membranes were developed using SuperSignal West Pico Chemiluminescent kit (Thermo Scientific, Waltham, MA).
Transfection of plasmid vectors or siRNA
Transfection of control vector, Myc/His-tagged SIRT1 expression vector, control siRNA, or SIRT1 siRNA was carried out using the Neon Transfection System (Thermo Fisher Scientific) according to the manufacturer's specifications. After 48 h, the cells were used in experiments.
Transduction of adenoviruses vectors
Cells were plated in a 60-mm tissue culture plate. After overnight incubation, the cells were transduced at multiplicities of infection of 50 by adenovirus vectors expressing wild-type Nrf2 (WT-Nrf2) or dominant-negative Nrf2 (DN-Nrf2), which lacks an N-terminal transactivating domain, in media for 2 h with gentle shaking and then incubated at 37 °C and 5% CO2. After 48 h, the cells were used in the experiments as indicated.
Immunofluorescent staining for NE
BEAS-2B cells grown in 35-mm dishes in the presence or absence of NE for 2 h were fixed in methanol and incubated with rabbit polyclonal anti-NE antibody diluted 1:100 in 3% bovine serum albumin for 24 h. The cells were subsequently incubated with Alexa Fluor 555 goat anti-rabbit antibody diluted 1:100 in 3% bovine serum albumin for 30 min. After successive washes, the cells were analyzed with a fluorescence microscope (ECLIPSE TE300, Nikon, Tokyo, Japan).
Fragment generation of SIRT1
Cell lysates were incubated in vehicle control (50% glycerol 50% 0.02 m NaOAc, pH 5) or NE (0.04, 0.2, 1 units/ml) at 37 °C for 30 min. The reaction was stopped by adding Western blotting sample buffer and samples were subjected to Western blotting analysis for SIRT1 and NE.
Statistical analysis
Data were subjected to Student's t test to analyze statistical significance, and a p value of <0.05 was considered significant.
Author contributions
K. H. L. and C. G. Y. designed the study; K. H. L., J. J., Y. J. K., A. H. J., and C. H. L. conducted the experiments; K. H. L., C. H. L., and C. G. Y. contributed to the data analysis; K. H. L. and C. G. Y. wrote the manuscript.
Supplementary Material
The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Figs. S1–S5.
- COPD
- chronic obstructive pulmonary disease
- NE
- neutrophil elastase
- HO-1
- heme-oxygenase 1
- CS
- cigarette smoke
- CSE
- cigarette smoke extract
- Nrf2
- nuclear factor (erythroid-derived 2) like-2
- KEAP1
- kelch like-ECH-associated protein1
- ERK
- extracellular signal-regulated kinase
- HBEC
- human bronchial epithelial cell
- PARP-1
- poly(ADP-ribose) polymerase-1
- ARE
- antioxidant response element
- CEB
- cytoplasmic extraction buffer
- PAR
- protease-activated receptor.
References
- 1. Vestbo J., Hurd S. S., Agustí A. G., Jones P. W., Vogelmeier C., Anzueto A., Barnes P. J., Fabbri L. M., Martinez F. J., Nishimura M., Stockley R. A., Sin D. D., and Rodriguez-Roisin R. (2013) Global strategy for the diagnosis, management, and prevention of chronic obstructive pulmonary disease: GOLD executive summary. Am. J. Respir. Crit. Care Med. 187, 347–365 [DOI] [PubMed] [Google Scholar]
- 2. Fischer B. M., Pavlisko E., and Voynow J. A. (2011) Pathogenic triad in COPD: oxidative stress, protease-antiprotease imbalance, and inflammation. Int. J. Chron. Obstruct. Pulmon. Dis. 6, 413–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mak J. C. (2008) Pathogenesis of COPD: part ii. oxidative-antioxidative imbalance. Int. J. Tuberc. Lung Dis. 12, 368–374 [PubMed] [Google Scholar]
- 4. Rahman I., and Adcock I. M. (2006) Oxidative stress and redox regulation of lung inflammation in COPD. Eur. Respir. J. 28, 219–242 [DOI] [PubMed] [Google Scholar]
- 5. Choi A. M., and Alam J. (1996) Heme oxygenase-1: function, regulation, and implication of a novel stress-inducible protein in oxidant-induced lung injury. Am. J. Respir. Cell Mol. Biol. 15, 9–19 [DOI] [PubMed] [Google Scholar]
- 6. Poss K. D., and Tonegawa S. (1997) Reduced stress defense in heme oxygenase 1-deficient cells. Proc. Natl. Acad. Sci. U.S.A. 94, 10925–10930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shinohara T., Kaneko T., Nagashima Y., Ueda A., Tagawa A., and Ishigatsubo Y. (2005) Adenovirus-mediated transfer and overexpression of heme oxygenase 1 cDNA in lungs attenuates elastase-induced pulmonary emphysema in mice. Hum. Gene Ther. 16, 318–327 [DOI] [PubMed] [Google Scholar]
- 8. Yamada N., Yamaya M., Okinaga S., Nakayama K., Sekizawa K., Shibahara S., and Sasaki H. (2000) Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility toemphysema. Am. J. Hum. Genet. 66, 187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tagawa A., Kaneko T., Shinohara T., Ueda A., Sato T., and Ishigatsubo Y. (2005) Heme oxygenase-1 inhibits cigarette smoke-induced increase in the tracheal mucosal permeability in guineapigs in vivo. Inflamm. Res. 54, 229–234 [DOI] [PubMed] [Google Scholar]
- 10. Almolki A., Guenegou A., Golda S., Boyer L., Benallaoua M., Amara N., Bachoual R., Martin C., Rannou F., Lanone S., Dulak J., Burgel P. R., El-Benna J., Leynaert B., Leynaert A. B., Aubier M., and Boczkowski J. (2008) Heme oxygenase-1 prevents airway mucus hypersecretion induced by cigarette smoke in rodents and humans. Am. J. Pathol. 173, 981–992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Slebos D. J., Ryter S. W., van der Toorn M., Liu F., Guo F., Baty C. J., Karlsson J. M., Watkins S. C., Kim H. P., Wang X., Lee J. S., Postma D. S., Kauffman H. F., and Choi A. M. (2007) Mitochondrial localization and function of heme oxygenase-1 in cigarette smoke-induced cell death. Am. J. Respir. Cell Mol. Biol. 36, 409–417 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 12. Maestrelli P., Páska C., Saetta M., Turato G., Nowicki Y., Monti S., Formichi B., Miniati M., and Fabbri L. M. (2003) Decreased haem oxygenase-1 and increased inducible nitric oxide synthase in the lung of severe COPD patients. Eur. Respir. J. 21, 971–976 [DOI] [PubMed] [Google Scholar]
- 13. Boutten A., Goven D., Artaud-Macari E., Boczkowski J., and Bonay M. (2011) NRF2 targeting: a promising therapeutic strategy in chronic obstructive pulmonary disease. Trends Mol. Med. 17, 363–371 [DOI] [PubMed] [Google Scholar]
- 14. Mercado N., Thimmulappa R., Thomas C. M., Fenwick P. S., Chana K. K., Donnelly L. E., Biswal S., Ito K., and Barnes P. J. (2011) Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem. Biophys. Res. Commun. 406, 292–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rangasamy T., Cho C. Y., Thimmulappa R. K., Zhen L., Srisuma S. S., Kensler T. W., Yamamoto M., Petrache I., Tuder R. M., and Biswal S. (2004) Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J. Clin. Invest. 114, 1248–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee P. J., Camhi S. L., Chin B. Y., Alam J., and Choi A. M. (2000) AP-1 and STAT mediate hyperoxia-induced gene transcription of heme oxygenase-1. Am. J. Physiol. Lung Cell Mol. Physiol. 279, L175–L182 [DOI] [PubMed] [Google Scholar]
- 17. Zhang X., Shan P., Jiang G., Zhang S. S., Otterbein L. E., Fu X. Y., and Lee P. J. (2006) Endothelial STAT3 is essential for the protective effects of HO-1 in oxidant-induced lung injury. FASEB J. 20, 2156–2158 [DOI] [PubMed] [Google Scholar]
- 18. Yao H., Sundar I. K., Ahmad T., Lerner C., Gerloff J., Friedman A. E., Phipps R. P., Sime P. J., McBurney M. W., Guarente L., and Rahman I. (2014) SIRT1 protects against cigarette smoke-induced lung oxidative stress via a FOXO3-dependent mechanism. Am. J. Physiol. Lung Cell. Mol. Physiol. 306, L816–L828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lee K. H., Lee C. H., Jeong J., Jang A. H., and Yoo C. G. (2015) Neutrophil elastase differentially regulates interleukin 8 (IL-8) and vascular endothelial growth factor (VEGF) production by cigarette smoke extract. J. Biol. Chem. 290, 28438–28445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Boutten A., Goven D., Boczkowski J., and Bonay M. (2010) Oxidative stress targets in pulmonary emphysema: focus on the Nrf2 pathway. Expert Opin. Ther. Targets 14, 329–346 [DOI] [PubMed] [Google Scholar]
- 21. Lee K. H., Hwang Y. H., Lee C. T., Kim Y. W., Han S. K., Shim Y. S., and Yoo C. G. (2004) The heat-shock-induced suppression of the IκB/NF-κB cascade is due to inactivation of upstream regulators of IκBα through insolubilization. Exp. Cell Res. 299, 49–56 [DOI] [PubMed] [Google Scholar]
- 22. Knörr-Wittmann C., Hengstermann A., Gebel S., Alam J., and Müller T. (2005) Characterization of Nrf2 activation and heme oxygenase-1 expression in NIH3T3 cells exposed to aqueous extracts of cigarette smoke. Free Radic. Biol. Med. 39, 1438–1448 [DOI] [PubMed] [Google Scholar]
- 23. Raval C. M., and Lee P. J. (2010) Heme oxygenase-1 in lung disease. Curr. Drug Targets 11, 1532–1540 [DOI] [PubMed] [Google Scholar]
- 24. Hwang J. W., Yao H., Caito S., Sundar I. K., and Rahman I. (2013) Redox regulation of SIRT1 in inflammation and cellular senescence. Free Radic. Biol. Med. 61, 95–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rahman I., Kinnula V. L., Gorbunova V., and Yao H. (2012) SIRT1 as a therapeutic target in inflammaging of the pulmonary disease. Prev. Med. 54, S20–S28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yao H., and Rahman I. (2012) Perspectives on translational and therapeutic aspects of SIRT1 in inflammaging and senescence. Biochem. Pharmacol. 84, 1332–1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rajendrasozhan S., Yang S. R., Kinnula V. L., and Rahman I. (2008) SIRT1, an antiinflammatory and antiaging protein, is decreased in lungs of patients with chronic obstructivepulmonary disease. Am. J. Respir. Crit. Care Med. 177, 861–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chalkiadaki A., and Guarente L. (2012) High-fat diet triggers inflammation-induced cleavage of SIRT1 in adipose tissue to promote metabolic dysfunction. Cell Metab. 16, 180–188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Caito S., Rajendrasozhan S., Cook S., Chung S., Yao H., Friedman A. E., Brookes P. S., and Rahman I. (2010) SIRT1 is a redox-sensitive deacetylase that is post-translationally modified by oxidants and carbonyl stress. FASEB J. 24, 3145–3159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Suzuki T., Yamashita C., Zemans R. L., Briones N., Van Linden A., and Downey G. P. (2009) Leukocyte elastase induces lung epithelial apoptosis via a PAR-1-, NF-κB-, and p53-dependent pathway. Am. J. Respir. Cell Mol. Biol. 41, 742–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mihara K., Ramachandran R., Renaux B., Saifeddine M., and Hollenberg M. D. (2013) Neutrophil elastase and proteinase-3 trigger G protein-biased signaling through proteinase-activated receptor-1 (PAR1). J. Biol. Chem. 288, 32979–32990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Gregory A. D., Hale P., Perlmutter D. H., and Houghton A. M. (2012) Clathrin pit-mediated endocytosis of neutrophil elastase and cathepsin G by cancer cells. J. Biol. Chem. 287, 35341–35350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rodriguez J. R., Seals J. E., Radin A., Lin J. S., Mandl I., and Turino G. M. (1979) Neutrophil lysosomal elastase activity in normal subjects and in patients with chronic obstructive pulmonary disease. Am. Rev. Respir. Dis. 119, 409–417 [DOI] [PubMed] [Google Scholar]
- 34. Zhu X., Liu Q., Wang M., Liang M., Yang X., Xu X., Zou H., and Qiu J. (2011) Activation of Sirt1 by resveratrol inhibits TNF-α induced inflammation in fibroblasts. PLoS ONE 6, e27081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ishii T., Matsuse T., Igarashi H., Masuda M., Teramoto S., and Ouchi Y. (2001) Tobacco smoke reduces viability in human lung fibroblasts: protective effect of glutathione S-transferase P1. Am. J. Physiol. Lung Cell. Mol. Physiol. 280, L1189–L1195 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.