

Abstract
Transactive response DNA-binding protein 43 (TDP-43) performs multiple tasks in mRNA processing, transport, and translational regulation, but it also forms aggregates implicated in amyotrophic lateral sclerosis. TDP-43's N-terminal domain (NTD) is important for these activities and dysfunctions; however, there is an open debate about whether or not it adopts a specifically folded, stable structure. Here, we studied NTD mutations designed to destabilize its structure utilizing NMR and fluorescence spectroscopies, analytical ultracentrifugation, splicing assays, and cell microscopy. The substitutions V31R and T32R abolished TDP-43 activity in splicing and aggregation processes, and even the rather mild L28A mutation severely destabilized the NTD, drastically reducing TDP-43's in vitro splicing activity and inducing aberrant localization and aggregation in cells. These findings strongly support the idea that a stably folded NTD is essential for correct TDP-43 function. The stably folded NTD also promotes dimerization, which is pertinent to the protein's activities and pathological aggregation, and we present an atomic-level structural model for the TDP-43 dimer based on NMR data. Leu-27 is evolutionarily well conserved even though it is exposed in the monomeric NTD. We found here that Leu-27 is buried in the dimer and that the L27A mutation promotes monomerization. In conclusion, our study sheds light on the structural and biological properties of the TDP-43 NTD, indicating that the NTD must be stably folded for TDP-43's physiological functions, and has implications for understanding the mechanisms promoting the pathological aggregation of this protein.
Keywords: amyotrophic lateral sclerosis (ALS) (Lou Gehrig disease), microscopic imaging, nuclear magnetic resonance (NMR), protein aggregation, site-directed mutagenesis, structure–function, subcellular fractionation
Introduction
Transactive DNA-binding protein 43 kDa (TDP-43) is an essential human protein that is vital to pre-mRNA (1) and microRNA processing (2). Key to these activities are TDP-43's two well-folded and stable RRM4 domains (spanning residues 106–177 and 192–259, respectively). TDP-43 contains a nuclear localization sequence (NLS, residues 80–102) and is mostly nuclear; however, its nuclear export sequence (residues 238–250) permits it to transport mRNAs to the cytoplasm and even to synapses as part of neuronal granules (3). TDP-43 also regulates translation by participating in stress granules (4).
Aberrant aggregate forms of TDP-43 are tightly linked to amyotrophic lateral sclerosis (ALS) as inclusions composed of polyubiquitinated, hyperphosphorylated, and truncated TDP-43 (5, 6) and have been reported in 95% of ALS patient motor neurons (7). TDP-43 aggregates are also observed in 60% of frontotemporal lobar degeneration (FTLD: a form of dementia whose symptoms overlap with ALS) patient neurons, and the observation of aggregates composed of TDP-43 plus Aβ or polyglutamine suggests that TDP-43 may contribute to other neurodegenerative diseases such as Alzheimer's or Huntington's (8, 9). Almost all the pathologically linked mutations are localized in the C-terminal region of TDP-43 (CTR, residues 270–414). In contrast to the RRM domains, the CTR is intrinsically disordered and consists of the following four segments: two of which (residues 267–320 and 367–414) are rich in G/S, aromatic, G/S motifs reminiscent of (G/S)Y(G/S) motifs in the RNA-binding protein Fused in Sarcoma/Translocated in Sarcoma, which drive the formation of a hydrogel or liquid phase. A third segment (residues 320–340) is hydrophobic and tends to adopt helical conformations (10–13) and also drives the formation of a distinct, non-aqueous liquid phase such as those present in stress granules and neuronal granules (13, 14).
The fourth CTR segment of TDP-43, composed of residues 341–367, is “Q/N-rich” as it contains a high proportion of Gln and Asn residues. Although variants lacking the Q/N-rich segment do not aggregate, variants containing 12 copies of it recapitulate in cells most of the pathological characteristics seen in ALS (15). In 2015, we advanced an amyloid-like conformer for this segment based on a variety of biochemical, spectroscopic, and computational data (16). In early 2016, the Q/N-rich segment, or part of it, was found to be intact in TDP-43 aggregates from ex vivo brain tissue, whereas all other segments of the CTR are heavily phosphorylated, deamidated, and oxidized (17). Because the exceptionally strong hydrogen-bonding networks of Q/N-rich amyloids (18) could impede such chemical modifications, we have recently interpreted these data as supporting our amyloid-like model of the Q/N-rich segment (19).
In 2012, the N-terminal domain of TDP-43 (NTD) was predicted to adopt a stable fold and was found to drive the formation of large oligomers (20). The NTD is required for TDP-43's physiological functions and pathological aggregation (21–23), but it has been less studied due to its unique sequence, which thwarts structural prediction based on homology modeling, and its strong tendency to aggregate. In 2014, an important advance in its structural characterization was reported by Song and co-workers (24), who discovered that NTD constructs with a C-terminal His tag are soluble in low pH, very low ionic strength solution conditions. This construct adopted a minor population of folded conformers; nevertheless, they were able to advance a medium resolution model for the tertiary fold of the NTD based on a small number of NMR NOE constraints and Rosetta ab initio structure prediction.
In 2016, we reported that the NTD is stably folded (Tm = 45–50 °C) in the context of short (residues 1–77, TDP-43(1–77)) or long (residues 1–102, TDP-43(1–102)) constructs with N-terminal His tags, which permitted the elucidation of the shorter construct's 3D structure to high resolution using NMR methods (25), PDB code 2N4P. In this study, we observed that the longer construct TDP-43(1–102), consisting of the NTD plus the NLS, which is rich in cationic residues, is more soluble. The NTD contains two highly conserved consecutive Leu residues at positions 27 and 28. We also found that the Leu-27's hydrophobic side chain is mostly solvent-exposed, which is unusual for a nonpolar residue, and that Leu-28 is mostly buried and makes important contacts linking different elements of secondary structure. Here, the roles of Leu-27 in promoting dimerization and of Leu-28 in stabilizing the tertiary structure are tested by substituting these residues by Ala. We also study another variant, V31R/T32R, designed to introduce two charges into the nonpolar core of the NTD. Because placing even one charge in the hydrophobic core strongly destabilizes proteins (26), this variant is expected to unfold the NTD.
Despite these advances, there is a continuing debate regarding whether the NTD is stably folded and whether this domain needs to be folded for TDP-43 to be active in cells (27–29). Previously, TDP-43 has been shown to exist as a monomer/dimer in vivo (30) and in vitro (31). The analysis of truncation mutants (30), size-exclusion chromatography, and a low resolution SAXS envelope (31) indicate the NTD is the domain chiefly responsible for this dimerization. In cells, TDP-43's concentration is exquisitely controlled (32, 33), which further suggests that the monomer/dimer equilibrium could have important functional consequences. However, the conformation of the dimer is currently unknown. Further studies in this area are therefore required, if we consider that TDP-43 affects the maturation and transport of thousands of mRNAs and that changes in TDP-43 concentration, due to aggregation or gene knockdown, strongly alter protein expression (34).
One objective of this study was to test whether stably folded NTD is required for TDP-43's activity by characterizing the biological functions of mutations, namely V31R/T32R and L28A, designed to disrupt the protein's tertiary structure. The latter variant was chosen for high resolution studies of its structure, stability, and dynamics. The second objective was to characterize the conformation of the NTD dimer on the basis of multidimensional heteronuclear NMR spectroscopy and corroborated by studies of a variant, L27A, that is designed to disrupt the dimer interface without perturbing the structure of the monomer.
Results
Characterization of the solution conformation of TDP-43(1–102)
Taking advantage of the superior solubility of the TDP-43(1–102) construct, we recorded a series of NOESY NMR spectra (2D NOESY and 3D HSQC-NOESY). These spectra yielded hundreds of new NOEs (over 1800 compared with the 1058 peaks used to calculate the structure of TDP-43(1–77) (25)) and allowed us to determine the NTD's structure to higher resolution (Fig. 1A and supplemental Table 1). The NMR assignments and final refined structures have been deposited in the Biological Magnetic Resonance Data Bank (BMRB access code 34081) and the RCSB (Protein Data Bank code 5MRG), respectively. Overall, the conformation is very similar to that of the TDP-43(1–77) construct studied previously, except some differences are observed for Asn-76 and Tyr-77 at the end of the last β-strand and for Cys-39 and Gly-40, which contact those residues. These minor differences could be due to the end effects, particularly the influence of Pro-78 on the conformation of Asn-76 and Tyr-77.
Figure 1.

Structure of TDP-43(1–102) confirms that the NTD is a stable, well folded domain. A, solution structure of TDP-43(1–102) calculated on the basis of 1628 restraints (see supplemental Table S1) shown in atomic (upper figure) and ribbon + atomic (lower figure) representations. Residues −11 to 0 (the His tag, sequence MRGSHHHHHHGS) is colored in dark brown, and residues 78–102 (the NLS) are in gray. The zoomed view on the right highlights the minor differences between the new structure (rose gold, PDB code 5MRG) and the previous one (green, PDB code 2N4P); i.e. the C-terminal residues Asn-76 and Tyr-77 now establish contacts with Cys-39 and Gly-40 (red labels), which allows elongation of the β-strands (red circles), and a half-turn of 3/10 helix spanning residues Pro-46–Ser-48 (purple labels and circle). This last element of structure is partially populated in the previous structure of the TDP-43(1–77) construct (PDB code 2N4P) and now appears in the lowest energy structure in the new calculation obtained with the longer construct, TDP-43(1–102). Gray lines indicate the close proximity in the tertiary fold of residues Pro-19, Leu-27, and Glu-58 (black labels), involved in the dimerization interface (see text). B, 2D 1H-15N HSQC spectrum of TDP-43(1–102) recorded at pH* 3.9, 25 °C in 1.0 mm deuterated acetic acid after transfer into 100% D2O. pH* is the pH reading of the pH meter without correction for the deuterium isotope effect. The blue, green, and red spectra were recorded after 1 h and 1.5 and 10 days, respectively, of exchange. For clarity, the green and red spectra are displaced 0.07 and 0.14 ppm, respectively, to the right along the x axis. Ser-20, Gln-34, and Tyr-73, whose slow exchanging HN groups are identified here, are circled.
1H-15N HSQC spectra recorded on 15N-labeled TDP-43 (1–102), which had been transferred into 100% D2O buffer, revealed signals belonging to amide groups protected from H/D exchange. This experiment corroborated published results (25) and led to the identification of three new protected residues (Ser-20, Gln-34, and Tyr-73) (Fig. 1B). Ser-20 appears to donate an H-bond to Glu-3 in a minority of our family of 20 NMR structures, and Gln-34 and Tyr-73 are H-bonded in the α-helix and last β-strand, respectively. In our previous paper (25), H/D exchange was monitored by 1D 1H NMR spectra, and the protected peaks were identified on the basis of their 1HN chemical shift and based on a 2D 1H-1H NOESY spectrum that was recorded part way through the exchange. In this study, H/D exchange has been followed by 2D 1H-15N HSQC spectra. In the previous experiment, the closeness of the Ser-30 1HN signal to those of Thr-25, Ala-38, Val-72, Val-75, and Asn-76, the proximity of Gln-34's 1HN signal to aromatic side chain 1H resonances, and the closeness of Tyr-73's 1HN signal to those of Ile-5 and Val-7 made it difficult to identify these slow exchanging peaks. They could be unambiguously identified in the 2D 1H-15N HSQC spectra thanks to the superior separation of resonances in the 15N dimension.
Mutation L28A strongly destabilizes TDP-43
As a further test of the structural integrity of the NTD, the effect of a mutation, L28A, designed to disrupt NTD tertiary contacts and hydrophobic core packing, was studied. The 1H-15N HSQC NMR spectrum of this variant, called L28A, shown in Fig. 2A, revealed a large increase in the number and intensity of peaks clustered in the spectral region (15N, 114–126 ppm; 1H, 8.5–7.9 ppm) typical of denatured protein. For peaks outside this region, analysis of a 3D 1H-15N HSQC-NOESY spectrum allowed the corroboration of the assignment of peaks in the 1H-15N HSQC spectrum that had not shifted relative to their position in wild-type TDP-43(1–102) and the assignment of those that had. The largest chemical shift perturbations are localized at the mutated site, the second loop, the α-helix, the turn connecting it to the third β-strand, and the β-hairpin formed by β-strands 4 and 5 (Fig. 2B). This pattern of perturbations is consistent with the contacts formed by Leu-28's nonpolar side chain in the folded NTD (supplemental Fig. S1).
Figure 2.

L28A destabilizes and breaks tertiary contacts, whereas L27A disrupts the dimerization interface. A, 2D 1H-15N HSQC spectra of TDP-43(1–102) recorded at pH* 3.9, 25 °C in 1.0 mm deuterated acetic acid in 15% D2O: WT, blue; L27A, green; L28A, red. Selected residues are labeled. Black labels refer all three variants. Brown labels refer to very similar WT + L27A peaks. B and C, weighted average 1H and 15N chemical shift differences (Δδ) (calculated as the √((Δδ15N/5)2 + (Δδ1H)2) for WT-L28A (B) and WT-L27A (C). Asterisks mark residues, for example Thr-25, which moved or broadened so much in the variant so as to prevent assignment. The small “o” indicates residues 45, 47, 50, 51, 53, and 54 in the L27A variant where the chemical shift differences with WT are 0. Other positions lacking bars are either Pro (at sequence numbers 15, 19, 36, 46, 64, and 78) or do not have data due to ambiguous assignment in the variant. Blue arrows (β-strands), parallelogram (α-helix), and L (for loops) in B indicate the approximate position of secondary structural elements. D, thermal denaturation of WT TDP-43(1–77) = cyan, and TDP-43(1–102) WT = blue, and its mutants L27A = green, and L28A = red, followed by fluorescence spectroscopy as the ratio of the emission intensity at 330 nm (which is maximal for the folded protein) to 355 nm (where the emission of denatured protein peaks). The curves show the fit of a two-state denaturation model to the data points, from which the apparent temperature midpoint (Tm) values were calculated. These experiments were performed in 1 mm acetic acid, pH 3.9, at low protein concentrations (50–100 μm). E, sedimentation velocity of WT, blue; L27A, green; L28A, red. The darker solid lines correspond to the more concentrated samples, and the lighter dashes are the diluted samples. The concentrations studied were WT concentrated 230 μm, diluted 46 μm, L27A concentrated 200 μm, diluted 50 μm, and L28A concentrated 130 μm and diluted 32 μm. The minor shifts of the monomer peaks upon dilution are caused by nonideality effects due to low ionic strength. The experimental sensitivity permits detection of as little as 1% dimer, and none was detected for L27A or L28A. These experiments were performed 25 °C on the same samples utilized for the NMR experiments and contained 1.0 mm deuterated acetic acid, pH 3.9, and 15% D2O. For clarity, the profiles of the variants are displaced along the y axis.
Two 2D 1H-15N HSQC peaks, for the folded and denatured states, were observed for the side chain HNϵ of Trp-68 (Fig. 2A). Based on NMR peak integration, 19% of L28A TDP-43(1–102) is folded at pH 3.9, 25 °C. Upon lowering the temperature, the folded population, as gauged by this group, increases to 52% at 15 °C and 67% at 5 °C (supplemental Fig. S1). These values are in line with the results of fluorescence-monitored thermal denaturation described below. The lack of signal broadening and the observation of discrete native and denatured peaks for the side chain HNϵ group of Trp-68 are evidence that the folding/unfolding equilibrium of the L28A variant can be approximated as a two-state process, which is slow on the NMR time scale and that the population of folding intermediates is low.
To get more insight into the L28A variant's conformational stability, its thermal denaturation was followed by fluorescence spectroscopy. Even at low temperature, L28A is not completely folded as its wavelength of maximum emission is 333 nm compared with 329 nm for WT TDP-43(1–102). The apparent unfolding midpoint temperature is about 15 °C (Fig. 2D). Although this value is imprecise considering the inability to fit the pre-transition baseline, this apparent Tm is ∼30 °C lower than that of the WT TDP-43(1–102) construct, indicating that this variant is strongly destabilized. As will be shown below, this severe destabilization strongly affects TDP-43's subcellular localization, in cell aggregation and its ability to regulate mRNA splicing. By analytical ultracentrifugation, this variant sediments as a monomer; no dimer was observed, which is consistent with the lower concentration of folded protein (Fig. 2E).
NMR relaxation analysis shows increased dynamics in the L28A variant
The heteronuclear {1H} 15N NOE of WT TDP-43(1–77) and TDP-43(1–102) constructs and of the TDP-43(1–102) L27A and TDP-43(1–102) L28A variants provides insight into dynamics on the picosecond–nanosecond time scale (Fig. 3A). The low NOE ratios show that there are significant dynamics in both the N-terminal His tag and the C-terminal NLS; this is consistent with the lack of stable conformations in these segments. In contrast, the folded portion (residues 3–77) shows high NOE ratios that approach the values (0.85) expected for static behavior in the elements of secondary structure. Somewhat lower ratio values are observed in the loops, especially those connecting β-strands 3 and 4. Similar picosecond–nanosecond dynamic behavior was seen for the short (1–77) or long (1–102) WT constructs as well as the variant L27A, which was designed to disrupt dimer formation (see below). By contrast, the L28A variant's {1H} 15N NOE ratio values are considerably lower than WT NTD or the L27A variant, indicating higher dynamics on the fast picosecond–nanosecond time scale.
Figure 3.
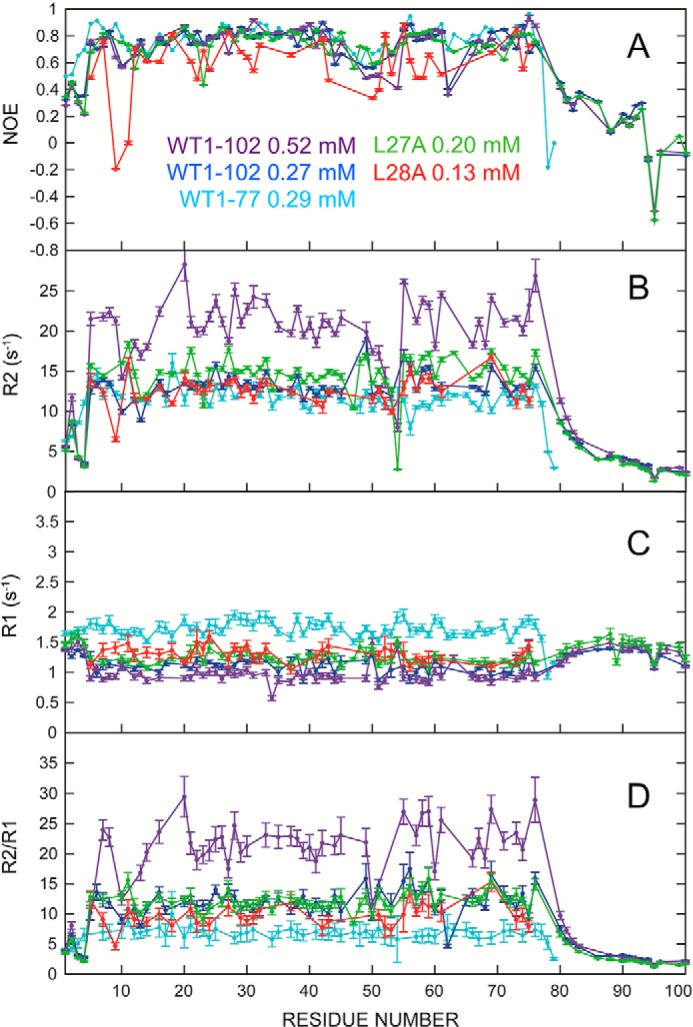
TDP-43 NTD dynamics and dimerization characterized by NMR relaxation measurements. All measurements were obtained at 800 MHz (1H), in 15% D2O, 85% H2O, in 1.0 mm acetic acid (d3), pH 3.9, 25 °C. WT TDP-43(1–102) 523 μm, purple; TDP-43(1–102) 270 μm, blue; TDP-43(1–77) 290 μm, turquoise; L27A TDP-43(1–102) 200 μm, green; L28A TDP-43(1–102) 130 μm, red. A, 15N{1H} NOE ratio; B, transversal rates, R2 (s−1); C, translational relaxation rates, R1 (s−1); D, R2/R1 ratio.
WT TDP-43(1–102) dimerizes in vitro
As a first step, we studied the oligomerization behavior of WT TDP-43(1–102) by analytical centrifugation, and we found evidence for dimer formation at higher concentrations (Fig. 2E), namely a peak with a sedimentation constant of 1.9 S corresponding to dimer was observed at higher concentrations along with a larger peak at 0.8–1.1 S, which corresponds to monomer. Upon dilution, the peak corresponding to dimer disappears. Similar results were obtained on an independent sample prepared in 100% D2O (see below). Based on these data, an estimated KD of 1 mm for the dimer dissociation could be calculated under these experimental conditions: pH 4.0, 25 °C. Additional thermal denaturation experiments on WT TDP-43(1–102) monitored by fluorescence spectroscopy showed a mean increase in Tm of 2.2 °C at 500 μm relative to 77 μm (data not shown). This increased Tm is consistent with a dimer dissociation constant of 0.94 mm.
Previous studies of TDP-43 in vitro and in cells at near neutral pH and physiological salt concentrations reported that Cys residues can form disulfide bonds under oxidizing conditions that promote TDP-43 oligomerization (35, 36). Cys residues in the RRM domains were found to be mainly responsible, but the participation of the two Cys of the NTD was not ruled out. Here, MALDI-TOF mass spectra revealed a major peak, which is consistent with the sequence of 15N-labeled TDP-43(1–102) and a generally high incorporation (>90%) of 15N (supplemental Fig. S2). Only trace peaks whose mass corresponds to a dimer were detected (supplemental Fig. S2). The MALDI-TOF procedure dissociates non-covalently linked dimers but does not separate dimers linked by disulfide bonds. This point is relevant considering that the NTD contains two rather exposed Cys residues (25) (supplemental Fig. S3C). Because a dimer peak was not detected, these results, as well as 13Cβ chemical shift values characteristic of reduced Cys, rule out dimerization through the formation of intermolecular disulfide bonds under the conditions studied, namely pH 4.0, 25 °C. MALDI-TOF mass spectra recorded on the L27A and L28A variants also revealed mass peaks expected for these variants, a high level of 15N incorporation and no evidence for dimers linked by covalent bonds (data not shown).
Next, NOESY spectra on a freshly prepared sample dissolved in buffer containing 1 mm deuterated acetic acid and 100% D2O were recorded on concentrated (340 μm) and diluted (100 μm) samples. By these spectra, we observed that some signals, for example those of Leu-27, were considerably perturbed (supplemental Fig. S3A). This strongly suggests that this residue is directly involved in the dimerization interface. The relatively small number of changes is likely due to the fact that these spectra were recorded in D2O, and most surface HN have exchanged and become invisible. Analytical centrifugation was performed on this sample and revealed a significant population of dimers (data not shown). The small number of changes also suggests the interface involves a relatively low number of residues, which is in agreement with the weak association constant and the relatively low population of dimers.
The NTD surface, shown supplemental Fig. S3B, features several exposed hydrophobic residues. Of them, Met-1, Val-57, Ile-60, Met-63, and Val-74 are discontinuous and are not conserved by evolution, whereas Ile-16, Pro-19, Leu-27, Ala-33, and Pro-36 are clustered near each other and are invariable from Homo sapiens to Drosophila (37) and are strictly conserved in Hydra, save a conservative substitution of Leu-27 by Met (38) (supplemental Fig. S3C). Exposed and evolutionarily conserved hydrophobic residues commonly form binding surfaces (39, 40).
As an additional test for dimer formation, the translational and transversal relaxation rates for the following 15N-labeled samples, 523 μm WT TDP-43(1–102), 145 μm WT TDP-43(1–102), 200 μm L27A TDP-43(1–102), 132 μm L28A TDP-43(1–102), at 25 °C in 1 mm acetic acid, pH 3.9, were compared with values previously determined for the TDP-43(1–77) construct at 290 μm (Fig. 3) (25). These results show that the transversal relaxation rates, R2, of well-folded residues of the concentrated WT TDP-43(1–102) sample are significantly higher than their counterparts in the more dilute TDP-43(1–102) sample, the shorter TDP-43(1–77) construct, and the two variants TDP-43(1–102) L27A and L28A. The translational relaxation rate, R1, values of the 523 μm WT TDP-43(1–102) sample are also slightly lower. These results, which can also be represented as the R2/R1 ratio, are clear evidence for dimer formation in the 523 μm TDP-43(1–102) sample. From the mean R1 and R2 rates, values of the correlation time, Tc, which measures the tumbling time in solution and is related to the size, were calculated (supplemental Table 2) These Tc values are not directly comparable because of the influence of disordered segments on the experimental Tc values but not the calculated Tc values. Nevertheless, all the samples' Tc values are in reasonable agreement with the values calculated based on the monomer structure (supplemental Table S2) and with Tc values for proteins of this size, except the 523 μm WT TDP-43(1–102) sample. The latter's Tc value is consistent with a mixed sample content of monomer and dimer.
Atomistic model for the solution structure of the NTD dimer
A sample containing 50% unlabeled TDP-43(1–102) and 50% 13C,15N-TDP-43(1–102) labeled (total concentration = 330 μm) was studied to obtain atomic-level information on the TDP-43 dimer's structure. In this sample, half the dimers will contain one labeled subunit and one unlabeled subunit, which affords the exclusive detection of pure intermolecular proton–proton contacts using a 13C-edited/12C-filtered 2D NOESY experiment, in which only cross-peaks between the unlabeled and labeled monomers within the same dimer are visible. Following this strategy, we could detect 17 intermolecular cross-peaks (supplemental Table S3 and supplemental Fig. S3D).
Whereas this limited number of unambiguous NOEs is insufficient to enable the determination of a high-resolution dimer structure, it does reveal many structural features of the interface. Several of these NOEs arise from inter-monomer contacts between Leu-27 side chains, as well as between Pro-19 (at the end of β-strand 2) with Glu-58 (in the loop that connects β-strands 4 and 5). By utilizing a limited number of conformational restraints based just on these NOEs (supplemental Table S3 and supplemental Fig. S3), the structure of a minimal dimer interface could be calculated (Fig. 4A). In this structure, the burial of the hydrophobic side chain of Leu-27 (Fig. 4A), which is exposed in the monomer (see above and Fig. 1A and supplemental Tables S1 and S3) would provide a favorable free energy change to drive dimerization.
Figure 4.
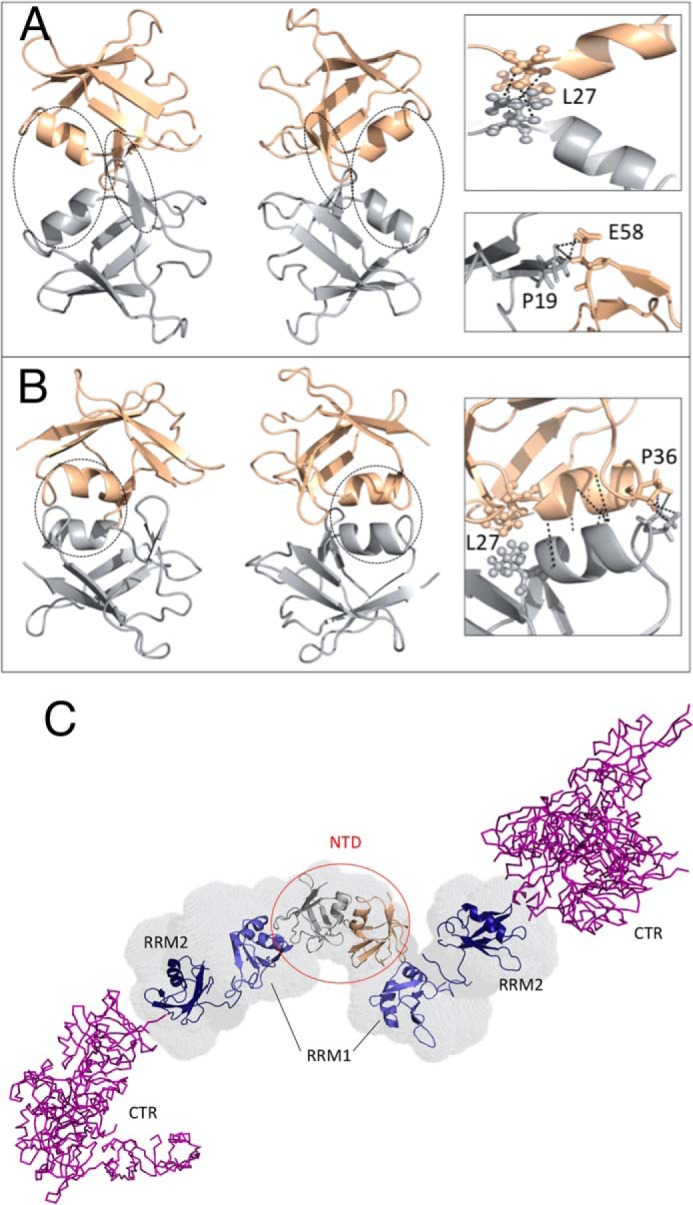
Atomistic structural model of TDP-43 dimerization via its NTD. A, minimal dimerization interface as defined by the intersubunit NOEs involving Pro-19, Leu-27, and Glu-58. These residues are circled on the left and shown in a zoomed view on the right. The two subunits are colored silver and rose gold. B, additional NOEs detected between Pro-36 residues in the two monomers and certain residues located in the α-helix suggest a larger dimerization interface, which includes dynamic, transient coiled-coil-like packing of the two helices. These regions are circled on the left and are represented in a zoomed view on the right. C, structural model of the complete TDP-43 dimer structure, based on the NTD dimer model and high resolution monomer structure of the NTD reported here (silver/rose gold), the structures of the RRM domains (blue) (41), and a representative disordered conformer of the C-terminal region (purple). The N-terminal domains are labeled and circled in red. The RRM domains and the CTR are labeled black.
The remaining NOE signals arise from residues 30 to 32 in the α-helix and Pro-36, which lies right at the end of this element of secondary structure. Employing all possible NOE-derived conformational restraints, a larger dimer interface, which features helix-helix “knobs into grooves” packing could be determined (Fig. 4B). Whereas this structure represents our proposal for the TDP-43 NTD dimer in solution, it is important to point out that on the basis of this structure, one may expect to see additional NOE cross-peaks between 1H in the α-helices; nevertheless, these signals were unobserved. Dynamic behavior on the micro- to millisecond time scale, which might arise from interconversion between the conformers comprising the minimal and large dimer interfaces, or low signal intensity due to the low population of the dimer could account for why these signals were not detected.
Leu-27 is a key residue for TDP-43 NTD dimerization
To further validate the structural model for the NTD dimer, a variant containing the Leu-27 to Ala mutation was prepared and studied. The 2D 1H-15N HSQC of the TDP-43(1–102) L27A variant shows features of the natively folded domain (Fig. 2A). A 3D 15N-NOESY-HSQC spectrum was also recorded, and its analysis permitted the verification of assignments in the 2D 1H-15N HSQC spectrum, the confirmation of essential side chain assignments, and the corroboration that crucial NOEs defining the tertiary structure were still present in this variant with respect to the WT (supplemental Fig. S4). A plot of the differences in the 1H-15N backbone chemical shifts in the L27A mutant versus the WT construct reveals minor variations except for the residues neighboring the mutation site and residues 21, 22, and 58 (Fig. 2C). The latter differences may reflect the lack of a populated dimer. In addition, we monitored the thermal denaturation of this variant using fluorescence spectroscopy, and we found that the WT TDP-43(1–102) L27A variant (Tm = 45 °C) is almost as stable as the WT TDP-43(1–102) WT (Tm = 49 °C) at pH 3.9, as illustrated in Fig. 2D. Analytical centrifugation revealed that the TDP-43(1–102) L27A variant is completely monomeric at concentrations where WT TDP-43(1–102) exists partly as a dimer (Fig. 2E).
We also analyzed the effect of substituting both Leu-27 and Leu-28 by alanine residues, in the context of the L27A/L28A double mutant. This variant appears to be even less stable than L28A as its 2D 1H-15N HSQC spectrum is typical of a completely unfolded protein (supplemental Fig. S5). The destabilizing effect caused by the mutations increases from L27A to L28A to L27A/L28A can be appreciated in supplemental Fig. S6 where the upfield region of the 1H NMR spectrum near 0 ppm shows intense peaks for methyls retaining the native fold in the hydrophobic core of L27A, weaker and shifted signals in L28A, and the absence of these peaks in the double mutant.
Conformational model of the TDP-43(1–414) dimer
A model of the complete TDP-43 protein, built on the basis of the NTD monomer structure and dimer interface reported here, and the SAXS envelope of Ref. 31, shows that the NTD and RRM domains are arranged like beads on a string or rosary and that the disordered C-terminal region extends out away from them (Fig. 4C). However, considering that the segment connecting the NTD to RRM1 is flexible and that the RRM1 to RRM2 linker is also flexible in the absence of RNA (41), it is quite possible that the folded domains and C-terminal regions can flex and bend to interact with each other, when they are buffeted by other macromolecules or due to attractive interactions with other molecules such as RNA. This structure, and the contacts defining the dimer interface in the N-terminal domain, may well be affected by changes in pH and ionic strength. This is in agreement with this consideration, and evidence for interdomain contacts in TDP-43 has been recently reported (28).
Functional experiments on NTD structural mutants in the Leu-28–Phe-35 α-helix
To study the biological implications of NTD variants in a cellular context, we engineered a mutation, V31R/T32R, that was predicted to completely destroy the α-helix structure and unfold the NTD, because of the well known severely destabilizing effect of introducing charges into the hydrophobic core (26). This mutant was then assayed in a variety of functional assays to test its ability to affect the splicing and aggregation functionality of TDP-43.
First of all, we tested this mutation in an add-back assay (Fig. 5). This assay, which is described in detail by Ref. 42, is based on a minigene system carrying CFTR exon 9 that includes a mutation in a splicing enhancer element within its sequence (C155T) to obtain a 50:50 ratio of exon inclusion/skipping in normal conditions (Fig. 5, 1st lane). This is the optimal condition to see whether a change in TDP-43 structure can result in either a loss-of-function (less exon 9 skipping) or a gain-of-function effect (more exon 9 skipping). In this system, when endogenous TDP-43 is removed from the cells by siRNA treatment, the levels of CFTR exon recognition substantially increase to more than 80% (Fig. 5, 2nd lane). As expected, splicing inhibition was fully rescued following expression of an si-resistant TDP-43 wild-type protein (Fig. 5, 3rd lane) but not when an siRNA-resistant F4L TDP-43 mutant that cannot bind RNA due to mutations in the RRM1 and RRM2 domains was expressed at similar levels (Fig. 5, 4th lane). The same result was also observed for the V31R/T32R mutant (Fig. 5, 5th lane).
Figure 5.
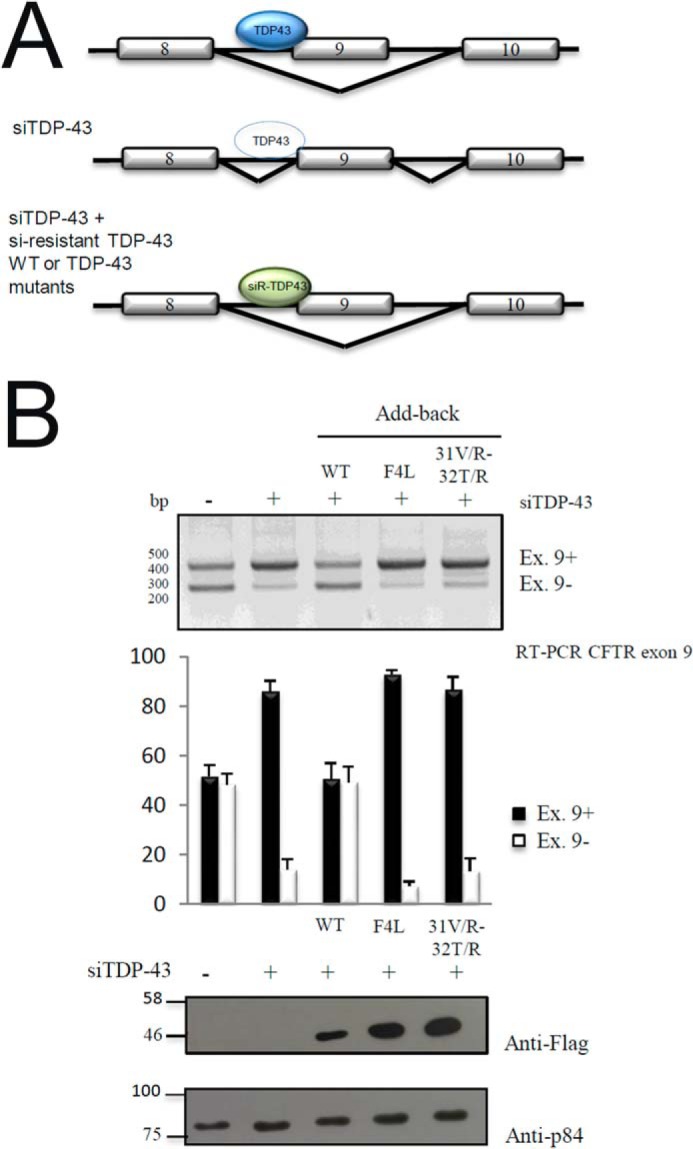
Functional add-back of TDP-43 V31R/T32R mutant. A, schematic drawing showing the experimental design. WT TDP-43 (blue, top) reduces splicing; this action is minimal when TDP-43 is knocked down (middle). In the context of knockdown endogenous WT TDP-43, the ability of TDP-43 mutant proteins to modulate splicing can be tested (bottom). B, RT-PCR of CFTR exon 9 isoforms upon transient transfection of HeLa cell line added back with si-resistant TDP-43 WT, F4L, and V31R/T32R mutants. Quantification of three independent experiments is also shown. Western blotting of transfected plasmids using an anti-FLAG antibody and loading control using anti-tubulin antibody is also shown.
To determine whether this change could also be associated with loss-of-function, we took advantage of an aggregation system that we previously set up by adding 12 repetitions of the Q/N prion-like domain of TDP-43 (15). The induction of this protein, when stably transfected in cells, caused the accumulation of aggregates capable of sequestering the endogenous TDP-43 protein and inducing a very well defined loss-of-function phenotype (22, 34, 43). Therefore, we also inserted the V31R/T32R mutation in these stable cell lines (FLAG-TDP-12X-V31R/T32R) to see their effect on aggregation and endogenous TDP-43 sequestration (supplemental Figs. S7A and S8A). As shown in supplemental Figs. S7B and S8B, compared with the expression of a wild-type TDP-12X-Q/N, the expression of this protein was not capable of inducing loss-of-function effects in the pre-mRNA splicing of POLDIP3. The reason for this loss is due to the fact that, as shown in co-immunoprecipitation experiments, the FLAG-TDP-12X-V31R/T32R protein has a very reduced ability to interact with endogenous TDP-43 (supplemental Figs. S7C and S8B). Additional experiments also confirmed that endogenous TDP-43 remains soluble in the nucleus even in the presence of aggregated FLAG-TDP-12X-V31R/T32R (data not shown).
Having established the importance of folded NTD and its α-helix in TDP-43 splicing and aggregation properties, it was therefore of interest to see the effects of the L27A and L28A mutants for which we had obtained the structural data described above.
Functional experiments on NTD structural mutants designed to disrupt quaternary (L27A) and tertiary (L28A) contacts
First of all, we performed add-back experiments on all the three mutants (L27A/L28A, L27A, and L28A) in HeLa cells (Fig. 6A and B). These results show that the double mutant L27A/L28A cannot recover CFTR exon 9 skipping following its add-back in TDP43-depleted cells (Fig. 6A, 5th lane) just like the V31R/T32R. Furthermore, it almost all localizes in the cytoplasm as observed with immunofluorescence experiments (Fig. 6C). This is expected, as we have shown that this mutant lacks any native structure in solution, as gauged from the NMR studies described above. More interesting is the L28A variant, which is also unable to recover the exon skipping activity (Fig. 6A, 7th lane) and is also predominantly localized in the cytoplasm. This similar behavior of L28A and the L27A/L28A double mutant is evidence that TDP-43 needs to be completely folded, and not partially folded, to perform its biological functions and maintain its predominantly nuclear localization.
Figure 6.

Functional add-back and immunolocalization assay of TDP-43 Leu-27 and Leu-28 mutants. A, RT-PCR of CFTR exon 9 isoforms upon transient transfection of HeLa cell line added back with si-resistant TDP-43 WT and F4L, L27A, L28A, and L27A/L28A mutants. B, Quantification of three independent experiments is also shown. C, immunofluorescence experiment of HeLa cells transiently transfected with TDP-43 WT and L27A, L28A, and L27A/L28A. All FLAG-TDP-43 proteins are shown in red, and nuclei are stained with TO-PRO3 in blue, and the merge is shown.
In fact, the L27A variant that is shown above to retain the native fold can restore exon skipping just like WT does (Fig. 6A, 6th lane). Interestingly, although this L27A is well localized in the nucleus, it is also quite present in the cytoplasm (Fig. 6C). We attribute this behavior to this variant's ability to retain the native fold, while being less able than the WT to dimerize.
These results are also reflected when these mutants are transfected in cell lines stably expressing a GFP-TDP-43–12X-Q/N-F4L protein (Fig. 7). First of all, it can be observed that the add-back of L27A can substantially recover the CFTR exon 9 skipping following the formation of the aggregates (Fig. 7A, 6th lane), although this was not observed for the L28A and L27A/L28A mutants (Fig. 7A, 7th and 9th lanes). Moreover, the immunohistochemical localization of L27A in the presence of the aggregates still remains diffuse in the nucleus (Fig. 7B) as opposed to the L28A and L27A/L28A, which colocalize extensively with the aggregates.
Figure 7.

Functional assay of TDP-43 Leu-27 and Leu-28 mutants in the presence of TDP-43 aggregates. A, RT-PCR of CFTR exon 9 isoforms upon transient transfection of GFP-TDP-12X-F4L stable cell line with TDP-43 WT, TDP-43-F4L, TDP-43-L27A, TDP-43-L28A, and TDP-43-L27A/L28A mutants. Quantification of three independent experiments is also shown. B, immunofluorescence experiment of GFP-TDP-12X-F4L stable cell line transiently transfected with TDP-43 WT, TDP-43-F4L, TDP-43-L27A, TDP-43-L28A, and TDP-43-L27A/L28A mutants. A merge of GFP (green)/anti-FLAG (red)/TO-PRO3 (blue) is shown.
Discussion
Thanks to the superior solubility of the TDP-43(1–102) construct, we have been able to extend the structure of the NTD to higher resolution, corroborate the identity of well protected amide groups in secondary structure, and identify three new protected residues. These results are additional evidence that the NTD is well folded and stable (25) and not poorly folded and marginally stable (24, 28).
Our findings are pertinent to the open debate on whether the NTD is stably folded and functionally relevant. In particular, the strong destabilization and structural disruption of the NTD in the context of the Leu-28, L27A/L28A, and V31R/T32R mutations, these variants' dramatically reduced activity in modulating RNA splicing, subcellular mislocalization, and strong tendency to aggregate in cells all emphasize the importance of folded NTD for TDP-43's native activity. Based on these results, we conclude that the NTD must be stably folded for TDP-43 to be able to carry out its physiological functions. This conclusion is likely to hold at the level of whole organisms, because the deletion of a short segment of the NTD corresponding to the first β-strand, which we expect would provoke the domain's denaturation, has severe physiological effects in mice (23).
TDP-43 is known to exist as a dimer/monomer equilibrium in vivo (30). Under the conditions studied here (25 °C, pH 3.9), the TDP-43 NTD dimerizes through interactions mediated by conserved exposed hydrophobic residues such as Leu-27, Pro-19, and Pro-36 as well as Glu-58. Residues in the α-helix could contribute additional interactions. The dimerization interface is relatively small, and the association is weak under the conditions examined here. However, it could be relevant for TDP-43's physiological activities, particularly in “crowded” conditions, for example when concentrated in subcellular compartments such as stress granules or as part of microdroplets (as discussed below).
Considering that the KD value of the TDP-43 NTD dimer is estimated to be in the range of 1 mm, which is well above the in vivo concentration of TDP-43, there is a doubt regarding how much dimer is present in cells. TDP-43 levels in cells are subjected to very fine regulation as the protein binds and regulates the concentration of its own mRNA (32) and also acts to retain its own mRNA in the nucleus to prevent translation (33). Nevertheless, crowding effects from the high in vivo concentrations of macromolecules and metabolites act to promote protein oligomerization (44), including TDP-43 dimerization. In addition, in vivo post-translational modifications, such as the acetylation of Lys-145 (45) or phosphorylation (46), are known to increase or decrease, respectively, TDP-43 self-association. It is also possible that the monomer association is strengthened at neutral pH and higher ionic strength conditions (20). In addition, gel filtration and SDS-PAGE experiments have also shown that TDP-43 truncation constructs consisting of both RRM domains either linked together or as separate entities also dimerize or tetramerize, respectively, in vitro (47). Therefore, TDP-43 dimerization is likely to be aided by interactions among the RRM domains and could be further strengthened when distinct TDP-43 molecules are bound to the same RNA.
It is fascinating that the L27A variant, which is unable to dimerize but can stably fold, is active in modulating splicing. This is logical considering that no TDP-43 dimers were previously detected in the nucleus where splicing occurs (30). In addition to regulating splicing, TDP-43 performs several other functions, and dimerization may well be required for some of them. TDP-43 dimers were previously observed in the cytoplasm (30), despite the fact that TDP-43 is more concentrated in the nucleus. This suggests that some mechanism concentrates TDP-43 in the cytoplasm so as to promote its dimerization. Over the last several years, the idea that cells contain several types of discrete liquid phases known as “microdroplets” such as nucleoli (48), stress granules (49), or neuronal granules that serve to concentrate and organize certain proteins and RNAs has gained acceptance (50–52). TDP-43 has been recently shown to bind to stress granules through the hydrophobic subsegment in the CTR (13, 14), and as a working hypothesis, we speculatively propose that the concentration of TDP-43 molecules within these microdroplets could well promote its dimerization and eventually lead to pathological aggregation or altered functionality. Future experiments will be necessary to test this proposal as well as to ascertain the physiological and eventually pathological function(s) of the TDP-43 dimer. Nonetheless, our results provide a better understanding of the relationship between the TDP-43 NTD and its functionality. Most importantly, the level of structural detail achieved by our analyses to characterize the dimerization interface at a single amino acid level may eventually open the way for the development of small molecules/peptides capable of interfering with this process. These effectors could then eventually be used to specifically inhibit TDP-43 aggregation tendencies and enhance its solubility within cells.
Experimental procedures
Protein expression and purification
Protein expression was essentially performed as described previously in Mompean et al. (25). Briefly, the sequences coding for TDP-43 N terminus residues 1–102 and the various mutants were cloned in the BamHI-HindIII of pQE306xHis plasmid. The plasmids were then used to transform M15 bacteria grown on kanamycin/ampicillin-resistant plates. The bacteria were then grown in M9 media supplemented with d-glucose (U-13C, 99%, Cambridge Isotope Laboratories, Inc.) at 0.3% v/v and 15NH4Cl (Sigma) at 0.1% v/v, and protein expression was induced overnight with 1 mm isopropyl 1-thio-β-d-galactopyranoside at 30 °C. All constructs contained an N-terminal His tag (MRGSHHHHHHGS), which facilitated the proteins' purification following the manufacturer's instructions using nickel-nitrilotriacetic acid-agarose Qiagen resin in the presence of Complete protease inhibitor cocktail (Roche Applied Science). Eluted fractions were then analyzed by 15% SDS-PAGE before staining with Colloidal Coomassie Blue to check for purity. For spectroscopic studies, samples were transferred to buffer containing 85% milliQ water, 15% D2O (Cambridge Isotope Laboratories, Inc.), and 1.0 mm acetic acid-d3 (Aldrich) using PD-10 desalting columns (GE Healthcare) and concentrated utilizing Vivaspin centrifugal concentrators (Sartorius). We used deuterated acetic acid to avoid the 1H signal from the methyl group of acetic acid.
Fluorescence spectroscopy
The thermal denaturation of WT TDP-43(1–102) and that of the L27A, L28A, and L27A/L28A variants was monitored using a Fluoromax4 spectrofluorimeter (Jobin-Yvon Inc., Edison, NJ). The xenon emission (467 nm) line and the water Raman signal (which is 397 nm when exciting at 350 nm) were used to calibrate the excitation and emission wavelengths, respectively. The experiments were made using fractions from gel filtration or NMR samples (concentrations 50–100 μm, save one experiment at 500 μm) and contained the same buffer (1 mm acetic acid, pH 3.9) as the NMR experiments. Emission spectra were recorded over the emission range of 270–380 nm, utilizing an excitation wavelength of 280 nm, 2–2.5 nm excitation, 2 nm emission slit widths, and a Peltier module to control the temperature. Data were collected using a scan speed of 2 nm·s−1 over intervals of 1–2 °C with 1- or 2-min equilibration times, respectively, at the new temperature before recording the spectra.
Analytical ultracentrifugation
The samples used for the NMR experiments were subjected to sedimentation velocity experiments at 25 °C, using an Optima XL-A analytical ultracentrifuge with a UV-visible variable wavelength detection system. The sedimentation assays were performed on NMR samples at their initial concentrations or diluted 3–5-fold by centrifugation at 48,000 rpm and detected using a wavelength of 280 nm. The concentrations of TDP-43(1–102) studied were as follows: WT 0.23 and 0.046 mm; L27A 0.20 and 0.050 mm; L28A 0.13 and 0.032 mm. A second independent WT sample was studied and gave similar results. For the experiments shown in Fig. 2E, thin cells (pathlength = 3 mm) were used for the concentrated L27A and L28A samples, and normal cells (pathlength = 12 mm) were utilized for the dilute samples. This accounts for why the peak absorbance does not decrease despite the substantial dilution. The program SEDFIT (version 15.01b) (53) was used to analyze the sedimentation profiles and obtain the sedimentation coefficients.
NMR experiments
All NMR experiments were recorded at 25 °C and pH 3.9, using a Bruker Avance 800 MHz spectrometer equipped with triple axis resonance cryo-probe with Z-gradients. Based on our reported chemical shift assignments for WT TDP-43(1–77) (25) (BMRB access code 25675), we performed additional standard 3D experiments, including 1H-1H-15N HSQC-NOESY, CBCA(CO)NH, HNCO, and HNCA spectra, to ensure that no structural changes occurred, to assign the segment 78–102, and to confirm that it is unfolded and does not interact with the folded domain comprising residues 1–77. This assignment process was carried out manually using SPARKY (54), and the 1H chemical shifts were referenced to sodium 4,4-dimethyl-4-silapentane-1-sulfonate. The 13C and 15N chemical shifts were referenced indirectly to 1H based on the nuclei's gyromagnetic ratios as recommended (55). Interproton NOE distance restraints were obtained by 2D 1H–1H and 13C-edited/12C-filtered NOESY spectra (120-ms mixing time). The NMR assignments have been deposited in the Biological Magnetic Resonance Data Bank (BMRB access code 34081).
NMR 15N relaxation measurements
The 15N relaxation data for WT TDP-43(1–102), (523 and 270 μm), L27A (200 μm), and L28A (130 μm) variants were recorded at 298 K on an 800 MHz spectrometer, with 1024 and 64 complex points along the direct and indirect dimensions and 32 scans. Steady-state 15N-1H heteronuclear NOE measurements (56) were carried out with an overall recycling delay of 10 s to ensure the maximal development of NOEs before acquisition and to allow solvent relaxation (57). Heteronuclear NOEs were calculated from the ratio of cross-peak intensities in spectra collected with and without amide proton saturation during the recycle delay. Uncertainties in peak heights were determined from the standard deviation (σ) of the distribution of intensities in the region of the spectra where no signal and only noise were observed.
The characterization of the dynamics and dimer formation by measuring the translational (R1), off-resonance rotating frame (R1ρ), and transversal (R2) relaxation rates using the parameters and procedures was previously described for the TDP-43(1–77) construct (25). For the 523 μm WT TDP-43(1–102) sample, four additional spectra were recorded with longer relaxation delays (2.0, 2.2, 2.5, and 3.0 s) to accurately fit the R1 rates, which are somewhat shorter than those of the other samples. The overall correlation time, Tc, was calculated from the ratio of the mean values of R1 and R2 excluding disordered residues. Values of NOE ratios < 0.65, R2 < the mean R2 − 1σ and R1 > the mean R1 + 1σ were used to define disordered residues (58). In addition to the experimental measurements, the correlation times were also calculated for the well-folded regions of the monomer and the dimer structures using the HYDRO NMR program (59).
NMR structure calculations
The dimeric structure of TDP-43(1–102) was determined in a two-step procedure using CYANA-3.97 (60). In the first step, we resolved the monomeric structure using dihedral angle restraints as determined by TALOS+ based on the chemical shift information, in addition to a large number NOE cross-peaks obtained from a 1H–1H 2D NOESY spectrum. These data were used as the input for interactive structure calculation, to elaborate, seven cycles of simultaneous automatic assignment of the cross-peaks and torsion angle dynamics. Next, a final simulated annealing calculation was carried out using the distance restraints derived from the consensus assignment achieved in the seventh cycle. The resulting structure is slightly more refined than that of monomeric TDP-43(1–77) that we recently reported (25), essentially because the samples used in this work yielded superior spectra, as explained under “Results.” The final refined structures have been deposited in the RCSB Protein Data Bank (code 5MRG), respectively.
In the second step, the 13C,15N-edited/12C,14N-filtered 2D NOESY experiment, which is specifically designed to detect intermolecular contacts, was recorded on a hemi-13C,15N-labeled 330 μm TDP-43(1–102) sample. The cross-peaks obtained from this experiment were converted to upper distance limits of 5.5 Å. These data were used as distance restraints to complement the previously obtained dihedral and distance restraints of the monomer. As we noted in our previous study, there is a unique H-bonding pattern that can account for residues showing protection against H/D exchange and used as constraints along with the experimental cross-peaks resulting in a bundle of conformers without violations. Thus, we used all this information together and submitted two copies of TDP-43(1–102) to a second simulated annealing calculation to obtain the final dimeric structure. Finally, the structural model of the NTD dimer and the tandem RRM domain structure solved in 2013 (41) were manually fitted into the previously published SAXS envelope to produce the model for the complete TDP-43 dimer shown in Fig. 4C.
Add-back splicing assay and in cell aggregation and subcellular localization with WT and the mutants
Using specific sets of primers the V31R/T32R, L27A, L28A, and L27A/L28A were inserted in the previously described si-resistant plasmid expressing wild-type TDP-43 (42). Briefly, to maximize TDP-43 silencing efficiency, HeLa cells were plated at 30% of confluence (day 0), and two rounds of TDP-43 siRNA transfections were carried out on days 1 and 2, according to the procedure already described (61). On the afternoon of day 2, cotransfection was performed with 1 μg of pFLAG-fusion protein expression vector and 0.5 μg of the CFTR C155T reporter minigene. On day 3, cells were harvested, and total RNA was extracted with EuroGold TRifast (Euroclone, Milan, Italy). Reverse transcription was performed using murine leukemia virus reverse transcriptase (Invitrogen), according to the manufacturer's protocol. PCR with DNA polymerase (New England Biolabs, Ipswich, MA) using minigene-specific primers was carried out for 35 amplification cycles (95 °C for 45 s, 54 °C for 45 s, and 72 °C for 45s). The expression levels of the added-back TDP-43 proteins were monitored through Western blotting, using a commercially available antibody against TDP-43 (Protein Tech, 10782-2-AP). Endogenous tubulin (in-house made mouse monoclonal antibody) and p84 (Abcam, Ab487) were used as loading controls. Inclusion levels of exons were quantified using the Qiaexcel platform used to run the PCR products or through ImageJ quantification.
Stable cell line generation
HEK293 flip-in cell line (Invitrogen) was grown in DMEM/Glutamax-I (Gibco) supplemented with 10% fetal bovine serum (Gibco) and antibiotic/antimycotic-stabilized suspension (Sigma). The plasmid transfections were carried out using Effectene transfection reagent (Qiagen) according to the manufacturer's instructions. To generate the stable clone, 0.5 g of the FLAG-TDP-12X-V31R/T32R expressing plasmid were cotransfected together with 0.5 g of pOG44 vector that expresses the Flp-recombinase (Invitrogen).
Cells were grown in DMEM/Glutamax-I supplemented with 10% fetal bovine serum, antibiotic/antimycotic-stabilized suspension (Sigma) and gradually selected using 100 g/ml hygromycin B and 10 g/ml blasticidin (both purchased from Invivogen). The induction of FLAG-TDP-43–12X-Q/N, FLAG-TDP-12X-V31R/T32R, and GFP-TDP-43–12X Q/N-F4L proteins was achieved by adding 1 g/ml tetracycline (Sigma) to the culture medium.
Protein expression was analyzed through Western blotting experiments using anti-FLAG antibody (Sigma, F1804), anti-TDP-43 (Protein Tech, 10782-2-AP), and anti-GAPDH (Santa Cruz Biotechnology, sc-25778) as loading control.
Coimmunoprecipitation assays
For coimmunoprecipitation assays, HEK293 flip-in stable cell lines expressing FLAG-TDP-43–12X-Q/N and FLAG-TDP-12X-V31R/T32R were induced for 24 h with 1 μg/ml tetracycline. Cells were collected in RIPA lysis buffer (50 mm Tris/HCl, pH 7.4, 150 mm NaCl, 1% Nonidet P-40, 0,1% SDS, 1 mm EDTA, pH 8, 1 mm PMSF, 0.5% SDC, H2O up to the final volume) supplemented with protease inhibitors (Roche Applied Science, catalog no. 11836145001) and incubated for 30 min at 4 °C. After spin down at 500 × g at 4 °C, cells were lysed by sonication. The lysates were then incubated with 40 μl of A/G plus agarose beads (Santa Cruz Biotechnology) to perform a pre-clearing for 1 h at 4 °C. In the meantime, an incubation of 40 μl of A/G plus agarose beads with 3 μg of anti-FLAG antibody (Sigma, F1804) in RIPA buffer for 2 h at 4 °C was performed. After both incubations, the pre-cleared lysate was incubated with A/G plus agarose beads/anti-FLAG overnight at 4 °C. The day after, the beads were precipitated and washed with PBS once for 10 min at 4 °C. The beads were finally resuspended in 50 μl of Resuspension Buffer (50 mm Tris/HCl, pH 7.4, 5 mm EDTA, 10 mm DTT, 1% SDS, H2O up to the final volume), and 20 μl of SDS 5× loading buffer were added.
Immunoprecipitates were then analyzed by Western blotting using anti-FLAG (Sigma, F1804), anti-TDP-43 (Protein Tech, 10782-2-AP), or anti-tubulin (Calbiochem, CP-06) antibodies. 10% of input from total lysate was loaded on SDS-PAGE and analyzed as control.
Immunofluorescence microscopy
For indirect immunofluorescence HEK-FLAG-TDP-12X-Q/N, HEK-FLAG-TDP-12X-V31R/T32R and HEK-GFP-TDP-12X-F4L stable cell lines were induced and processed as described previously (61). The primary antibodies used were anti-FLAG (Sigma, F1804) and anti-TDP-43 (Protein Tech, 10782-2-AP). The secondary antibodies anti-mouse AlexaFluor 594 (catalog no. A21203), anti-rabbit AlexaFluor 488 (catalog no. A21200), and TO-PRO3 (catalog no. T3605) were all purchased from Life Technologies, Inc. Cells were analyzed on a Zeiss LSM 510 Meta confocal microscope.
Cell lysate fractionation
To perform cell lysate fractionation in soluble and pellet fractions, 2 × 106 cells (HEK-FLAG-TDP-12X-Q/N, HEK-FLAG-TDP-12X-V31R/T32R) were seeded and induced with 1 μg/ml tetracycline for 24 h. Then, cells were collected and lysed with 1 ml of RIPA lysis buffer + protease inhibitor for 30 min at 4 °C. After centrifugation at 4000 rpm for 20 min, the whole supernatant was further sonicated for 5 min to allow a better lysis. Two hundred μg of cell lysate were ultracentrifuged in a clean Beckman polycarbonate thick wall centrifuge tube (rotor type 70.1Ti) for 1 h at 25 °C at 33,000 rpm.
The supernatant was collected, and the pellet was washed twice with 100 μl of RIPA buffer. Pellet was finally dissolved in urea buffer (7 m urea, 4% CHAPS, 30 mm Tris, pH 8.5). To analyze each fraction by Western blotting, 10% of input, 10% of soluble fraction, and 30% of pellet volume were loaded in a 10% SDS-polyacrylamide gel.
Author contributions
V. R., C. S., F. E. B., and E. B. produced TDP-43 N-terminal domain protein and site-directed mutant constructs. M. M., D. P. U., and D. V. L. designed, performed, and analyzed the experiments shown in Figs. 1–4. V. R., C. S., F. E. B., and E. B. designed, performed, and analyzed the experiments shown in Figs. 5–7. M. M., F. E. B., E. B., and D. V. L. wrote the manuscript with help from V. R., D. P. U., and C. S. All authors reviewed the results and approved the final version of the manuscript.
Supplementary Material
Acknowledgments
We thank Dr. Hanna Yuan, from the Institute of Molecular Biology (Sinica Academy, Taiwan), for sharing the SAXS envelope of the TDP-43 dimer. We are grateful to Prof. Marta Bruix for critically reading the manuscript and to Dr. Vivian de los Rios and Dr. Juan Roman Luque Ortega (Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas) for expert mass spectrometry and analytical ultracentrifugation, respectively. We thank Dr. Marco Baralle and Dr. Sanja Skaro for preparing the V31R, T32R sample and Francesca Paron for the immunofluorescence photography shown in Fig. 6C.
This work was supported by Grants SAF2013-49179-C2–2-R and SAF2016-76678-C2-2-R, European Union Grant JPND AC14/00037 (to D. V. L.), European Union Grant JPND RiMOdFTD, Italy, Ministero della Sanitá/MIUR (to E. B.), and the Thierry Latran Foundation REHNPALS (to E. B.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains supplemental Tables S1–S3 and Figs. S1–S8.
The atomic coordinates and structure factors (code 5MRG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- RRM
- RNA recognition motif
- NLS
- nuclear localization sequence
- ALS
- amyotrophic lateral sclerosis
- NTD
- N-terminal domain
- CTR
- C-terminal region
- PDB
- Protein Data Bank
- SAXS
- small angle x-ray scattering
- CFTR
- cystic fibrosis transmembrane conductance regulator
- H/D
- hydrogen/deuterium.
References
- 1. Buratti E., and Baralle F. E. (2012) TDP-43: Gumming up neurons through protein-protein and protein-RNA interactions. Trends Biochem. Sci. 37, 237–247 [DOI] [PubMed] [Google Scholar]
- 2. Kawahara Y., and Mieda-Sato A. (2012) TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. U.S.A. 109, 3347–3352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu-Yesucevitz L., Lin A. Y., Ebata A., Boon J. Y., Reid W., Xu Y. F., Kobrin K., Murphy G. J., Petrucelli L., and Wolozin B. (2014) ALS-linked mutations englard TDP-43 enriched neuronal RNA granules in the dendritic arbor. J. Neurosci. 34, 4167–4174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Colombrita C., Zennaro E., Fallini C., Weber M., Sommacal A., Buratti E., Silani V., and Ratti A. (2009) TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 111, 1051–1061 [DOI] [PubMed] [Google Scholar]
- 5. Neumann M., Sampathu D. M., Kwong L. K., Truax A. C., Micsenyi M. C., Chou T. T., Bruce J., Schuck T., Grossman M., Clark C. M., McCluskey L. F., Miller B. L., Masliah E., Mackenzie I. R., Feldman H., et al. (2006) Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science 314, 130–133 [DOI] [PubMed] [Google Scholar]
- 6. Arai T., Hasegawa M., Akiyama H., Ikeda K., Nonaka T., Mori H., Mann D., Tsuchiya K., Yoshida M., Hashizume Y., and Oda T. (2006) TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem. Biophys. Res. Commun. 351, 602–611 [DOI] [PubMed] [Google Scholar]
- 7. Ling S.-C., Polymenidou M., and Cleveland D. W. (2013) Converging mechanisms in ALS and FTLD: disrupted RNA and protein homeostasis. Neuron 79, 416–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin W. L., and Dickson D. W. (2008) Ultrastructural localization of TDP-43 in filamentous neuronal inclusions in various neurodegenerative diseases. Acta Neuropathol. 116, 205–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fuentealba R. A., Udan M., Bell S., Wegorzewska I., Shao J., Diamond M. I., Weihl C. C., and Baloh R. H. (2010) Interaction with polyglutamine aggregates reveals a Q/N-rich domain in TDP-43. J. Biol. Chem. 285, 26304–26314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jiang L. L., Che M. X., Zhao J., Zhou C. J., Xie M. Y., Li H. Y., He J. H., and Hu H. Y. (2013) Structural transformation of the amyloidogenic core region of TDP-43 protein initiates its aggregation and cytoplasmic inclusion. J. Biol. Chem. 288, 19614–19624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang L. L., Zhao J., Yin X. F., He W. T., Yang H., Che M. X., and Hu H. Y. (2016) Two mutations G335D and Q343R within the amyloidogenic core region of TDP-43 influence its aggregation and inclusion formation. Sci. Rep. 6, 23928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lim L., Wei Y., Lu Y., and Song J. (2016) ALS-causing mutations significantly perturb the self-assembly and interactions with nucleic acid of the intrinsically disordered prion-like domain of TDP-43. PLoS Biol. 14, e1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Conicella A. E., Zerze G. H., Mittal J., and Fawzi N. L. (2016) ALS mutations disrupt phase separation mediated by α-helical structure in the TDP-43 low complexity C-terminal domain. Structure 24, 1537–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schmidt H. B., and Rohatgi R. (2016) In vivo formation of vacuolated multiphase compartments lacking membranes. Cell Rep. 16, 1228–1236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Budini M., Buratti E., Stuani C., Guarnaccia C., Romano V., De Conti L., and Baralle F. E. (2012) Cellular model of TAR DNA-binding protein 43 (TDP-43) aggregation based on its C-terminal Gln/Asn-rich region. J. Biol. Chem. 287, 7512–7525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mompeán M., Hervás R., Xu Y., Tran T. H., Guarnaccia C., Buratti E., Baralle F., Tong L., Carrión-Vázquez M., McDermott A. E., and Laurents D. V. (2015) Structural evidence of amyloid fibril formation in the putative aggregation domain of TDP-43. J. Phys. Chem. Lett. 6, 2608–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kametani F., Obi T., Shishido T., Akatsu H., Murayama S., Saito Y., Yoshida M., and Hasegawa M. (2016) Mass spectrometric analysis of accumulated TDP-43 in amyotrophic lateral sclerosis brains. Sci. Rep. 6, 23281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mompeán M., Nogales A., Ezquerra T. A., and Laurents D. V. (2016) Complex system assembly underlies a two-tiered model of highly delocalized electrons. J. Phys. Chem. Lett. 7, 1859–1864 [DOI] [PubMed] [Google Scholar]
- 19. Mompeán M., Baralle M., Buratti E., and Laurents D. V. (2016) An amyloid-like pathological conformation of TDP-43 is stabilized by hyper-cooperative hydrogen bonds. Front. Mol. Neurosci. 9, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chang C. K., Wu T. H., Wu C. Y., Chiang M. H., Toh E. K., Hsu Y. C., Lin K. F., Liao Y. H., Huang T. H., and Huang J. J. (2012) The N-terminus of TDP-43 promotes its oligomerization and enhances its DNA binding. Biochem. Biophys. Res. Commun. 425, 219–224 [DOI] [PubMed] [Google Scholar]
- 21. Zhang Y. J., Caulfield T., Xu Y. F., Gendron T. F., Hubbard J., Stetler C., Sasaguri H., Whitelaw E. C., Cai S., Lee W. C., and Petrucelli L. (2013) The dual functions of the extreme N terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 22, 3112–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Budini M., Romano V., Quadri Z., Buratti E., and Baralle F. E. (2015) TDP-43 loss of cellular function through aggregation requires additional structural determinants beyond its C-terminal Q/N prion-like domain. Hum. Mol. Genet. 24, 9–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Sasaguri H., Chew J., Xu Y. F., Gendron T. F., Garrett A., Lee C. W., Jansen-West K., Bauer P. O., Perkerson E. A., Tong J., Stetler C., and Zhang Y. J. (2016) The extreme N terminus of TDP-43 mediates the cytoplasmic aggregation of TDP-43 and associated toxicity in vivo. Brain Res. 1647, 57–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qin H., Lim L. Z., Wei Y., and Song J. (2014) TDP-43 N terminus encodes a novel ubiquitin-like fold and its unfolded form in equilibrium that can be shifted by binding to ssDNA. Proc. Natl. Acad. Sci. U.S.A. 111, 18619–18624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mompeán M., Romano V., Pantoja-Uceda D., Stuani C., Baralle F. E., Buratti E., and Laurents D. V. (2016) The TDP-43 N-terminal domain structure at high resolution. FEBS J. 283, 1242–1260 [DOI] [PubMed] [Google Scholar]
- 26. Karp D. A., Gittis A. G., Stahley M. R., Fitch C. A., Stites W. E., and García-Moreno E B (2007) High dielectic constant inside a protein reflects structural reorganization coupled to the ionization of an internal Asp. Biophys. J. 92, 2041–2053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang C. K., and Huang T. H. (2016) Untangling the structure of the TDP-43 N-terminal domain. FEBS J. 283, 1239–1241 [DOI] [PubMed] [Google Scholar]
- 28. Wei Y., Lim L., Wang L., and Song J. (2016) Inter-domain interactions of TDP-43 as decoded by NMR. Biochem. Biophys. Res. Commun. 473, 614–619 [DOI] [PubMed] [Google Scholar]
- 29. Sun Y., and Chakrabartty A. (2017) Phase to phase with TDP-43. Biochemistry 56, 809–823 [DOI] [PubMed] [Google Scholar]
- 30. Shiina Y., Arima K., Tabunoki H., and Satoh J. (2010) TDP-43 dimerizes in human cells in culture. Cell. Mol. Neurobiol. 30, 641–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang Y. T., Kuo P. H., Chiang C. H., Liang J. R., Chen Y. R., Wang S., Shen J. C., and Yuan H. S. (2013) The truncated C-terminal RNA recognition motif of TDP-43 protein plays a key role in forming proteinaceous aggregates. J. Biol. Chem. 288, 9049–9057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ayala Y. M., De Conti L., Avendaño-Vázquez S. E., Dhir A., Romano M., D'Ambrogio A., Tollervey J., Ule J., Baralle M., Buratti E., and Baralle F. E. (2011) TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 30, 277–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Avendaño-Vázquez S. E., Dhir A., Bembich S., Buratti E., Proudfoot N., and Baralle F. E. (2012) Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 26, 1679–1684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Prpar Mihevc S., Baralle M., Buratti E., and Rogelj B. (2016) TDP-43 aggregation mirrors TDP-43 knockdown, affecting the expression levels of a common set of proteins. Sci. Rep. 6, 33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Cohen T. J., Hwang A. W., Unger T., Trojanowski J. Q., and Lee V. M. (2012) Redox signalling directly regulates TDP-43 via cysteine oxidation and disulfide bond cross-linking. EMBO J. 31, 1241–1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chang C. K., Chiang M. H., Toh E. K., Chang C. F., and Huang T. H. (2013) Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 587, 575–582 [DOI] [PubMed] [Google Scholar]
- 37. Ayala Y. M., Pantano S., D'Ambrogio A., Buratti E., Brindisi A., Marchetti C., Romano M., and Baralle F. E. (2005) Human, Drosophila and C. elegans TDP-43: nucleic acid binding properties and splicing regulatory function. J. Mol. Biol. 348, 575–588 [DOI] [PubMed] [Google Scholar]
- 38. Chapman J. A., Kirkness E. F., Simakov O., Hampson S. E., Mitros T., Weinmaier T., Rattei T., Balasubramanian P. G., Borman J., Busam D., Disbennett K., Pfannkoch C., Sumin N., Sutton G. G., Viswanathan L. D., et al. (2010) The dynamic genome of Hydra. Nature 464, 592–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ma B., Elkayam T., Wolfson H., and Nussinov R. (2003) Protein-protein interactions: structurally conserved residues distinguish between binding sites and exposed proteins surfaces. Proc. Natl. Acad. Sci. U.S.A. 100, 5772–5777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Guharoy M., and Chakrabarti P. (2010) Conserved residue clusters at protein-protein interfaces and their use in binding site identification. BMC Bioinformatics 11, 286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lukavsky P. J., Daujotyte D., Tollervey J. R., Ule J., Stuani C., Buratti E., Baralle F. E., Damberger F. F., and Allain F. H. (2013) Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 20, 1443–1449 [DOI] [PubMed] [Google Scholar]
- 42. D'Ambrogio A., Buratti E., Stuani C., Guarnaccia C., Romano M., Ayala Y. M., and Baralle F. E. (2009) Functional mapping of the interaction between TDP-43 and hnRNP A2 in vivo. Nucleic Acids Res. 37, 4116–4126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Romano V., Quadri Z., Baralle F. E., and Buratti E. (2015) The structural integrity of TDP-43 N terminus is required for efficient aggregate entrapment and consequent loss of protein function. Prion 9, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rivas G., Fernandez J. A., and Minton A. P. (1999) Direct observation of the self-association of dilute proteins in the presence of inert macromolecules at high concentration via tracer sedimentation equilibrium: theory, experiment, and biological significance. Biochemistry 38, 9379–9388 [DOI] [PubMed] [Google Scholar]
- 45. Cohen T. J., Hwang A. W., Restrepo C. R., Yuan C. X., Trojanowski J. Q., and Lee V. M. (2015) An acetylation switch controls TDP-43 function and aggregation propensity. Nat. Commun. 6, 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Li H. Y., Yeh P. A., Chiu H. C., Tang C. Y., and Tu B. P. (2011) Hyperphosphorylation as a defense mechanism to reduce TDP-43 aggregation. PLoS ONE 6, e23075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Kuo P. H., Doudeva L. G., Wang Y. T., Shen C. K., and Yuan H. S. (2009) Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 37, 1799–1808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Feric M., Vaidya N., Harmon T. S., Mitrea D. M., Zhu L., Richardson T. M., Kriwacki R. W., Pappu R. V., and Brangwynne C. P. (2016) Coexisting liquid phases underlie nucleolar subcompartments. Cell 165, 1686–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Patel A., Lee H. O., Jawerth L., Maharana S., Jahnel M., Hein M. Y., Stoynov S., Mahamid J., Saha S., Franzmann T. M., Pozniakovski A., Poser I., Maghelli N., Royer L. A., Weigert M., et al. (2015) A liquid-to-solid phase transition of the ALS protein FUS accelerated by disease mutation. Cell 162, 1066–1077 [DOI] [PubMed] [Google Scholar]
- 50. Brangwynne C. P., Eckmann C. R., Courson D. S., Rybarska A., Hoege C., Gharakhani J., Jülicher F., and Hyman A. A. (2009) Germline P granules are liquid droplets that localize by controlled dissolution/condensation. Science 324, 1729–1732 [DOI] [PubMed] [Google Scholar]
- 51. Chong P. A., and Forman-Kay J. D. (2016) Liquid-liquid phase separation in cellular signaling systems. Curr. Opin. Struct. Biol. 41, 180–186 [DOI] [PubMed] [Google Scholar]
- 52. Uversky V. N. (2016) Intrinsically disordered proteins in overcrowded milieu: membrane-less organelles, phase separation and intrinsic disorder. Curr. Opin. Struct. Biol. 44, 18–30 [DOI] [PubMed] [Google Scholar]
- 53. Schuck P. (2000) Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 78, 1606–1619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Goddard T. D., and Kneller D. G. (2008) Sparky 3, University of California, San Francisco [Google Scholar]
- 55. Markley J. L., Bax A., Arata Y., Hilbers C. W., Kaptein R., Sykes B. D., Wright P. E., and Wüthrich K. (1988) Recommendations for the presentation of NMR structures of proteins and nucleic acids. Pure Appl. Chem. 70, 117–142 [DOI] [PubMed] [Google Scholar]
- 56. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., and Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 [DOI] [PubMed] [Google Scholar]
- 57. Renner C., Schleicher M., Moroder L., and Holak T. A. (2002) Practical aspects of the 2D 15N-[1H]-NOE experiment. J. Biomol. NMR 23, 23–33 [DOI] [PubMed] [Google Scholar]
- 58. Pawley N. H., Wang C., Koide S., and Nicholson L. K. (2001) An improved method for distinguishing between anisotropic tumbling and chemical exchange in analysis of 15N relaxation parameters. J. Biomol. NMR 20, 149–165 [DOI] [PubMed] [Google Scholar]
- 59. Ortega A., and García de la Torre J. (2005) Efficient, accurate calculation of rotational diffusion and NMR relaxation of globular proteins from atomic-level structures and appropriate hydrodynamic calculations. J. Am. Chem. Soc. 127, 12764–12765 [DOI] [PubMed] [Google Scholar]
- 60. Güntert P. (2004) Automated NMR structure calculation with CYANA. Methods Mol. Biol. 278, 353–378 [DOI] [PubMed] [Google Scholar]
- 61. Ayala Y. M., Zago P., D'Ambrogio A., Xu Y. F., Petrucelli L., Buratti E., and Baralle F. E. (2008) Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 121, 3778–3785 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.