

Abstract
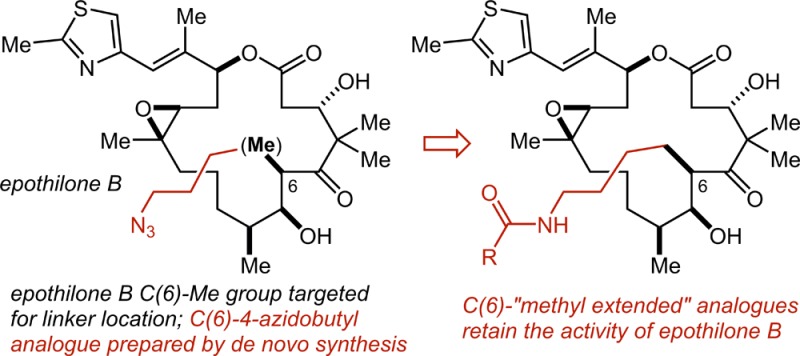
An approach to the validation of a linker strategy for the epothilone family of microtubule-stabilizing agents is reported. An analogue of epothilone B in which the C(6) methyl group has been replaced with a 4-azidobutyl group has been prepared by total chemical synthesis, and amides derived from the azido group have been shown to retain the activity of the parent compound. These results set the stage for an evaluation of the potential of the epothilones to serve as the drug component of antibody–drug conjugates and other selective tumor cell-targeting conjugates.
Keywords: Epothilones, microtubules, antibody−drug conjugates, linker, methyl extension
Among the large group of microtubule-stabilizing agents (MSAs) known to exert their antimitotic activity by binding to the luminal taxane binding site on the β-tubulin subunits of microtubules, the epothilone family of natural products1−3 constitute perhaps the most promising (Figure 1). Ixabepilone,4,5 the lactam analogue of epothilone B, was approved by the FDA in 2007 for use in the treatment of particularly aggressive and otherwise unresponsive forms of metastatic breast cancer, while several other fully synthetic analogues (e.g., sagopilone,6−8 iso-fludelone9) have recently undergone or are currently undergoing clinical evaluation. Because epothilone B, sagopilone, and iso-fludelone, among others, are possessed of subnanomolar potencies against a variety of cell lines including some taxol-resistant cell lines, we have become interested in establishing a validated linker strategy for this family of anticancer agents. Such a linker strategy would allow investigation of the potential of these compounds to serve as the drug component of antibody–drug conjugates (ADCs)10−12 and other selective tumor cell targeting conjugates.
Figure 1.

Potent MSAs epothilone A and B, ixabepilone, sagopilone, and iso-fludelone.
In order to prepare conjugates, the most basic requirement for the drug is that it must have a functional group (most straightforwardly an unhindered alcohol, a thiol, or an amine) that may be used selectively in synthetically mild and efficient conjugation reactions. With a traceless or self-immolative linker strategy this may be the only requirement, and two potent semisynthetic epothilone analogues equipped with a suitably reactive alcohol or amine (e.g., 1(13) and 2,14Figure 2a) have been reported and usefully employed in simple conjugation (acylation) reactions by the Bristol-Myers Squibb group in the course of their efforts to develop an epothilone–folic acid conjugate.14,15 An important additional design consideration for us, however, was to leave open the possibility of exploring conjugates wherein some portion of the linker may be retained (for example, to allow for the incorporation of additional functionality into the drug) in the active drug entity that is released from the conjugate following lysozomal degradation of the antibody. This adds the rather more stringent requirement that the conjugatable functionality must be located in a region of the drug that is solvent-exposed when the drug is bound in the receptor such that relatively large groups may be incorporated in the drug structure without any deleterious impact on its potency. So as to accomplish this while also perturbing the steric and electronic structure of the drug as little as possible, we decided to use the “methyl extension” strategy that we reported recently,16 which entails the identification of a solvent-exposed methyl group and the de novo synthesis of the analogue wherein the methyl group is replaced with a functional group-equipped linear alkyl group. To identify a suitable methyl group, we examined the recently reported high-resolution structure of epothilone A bound in the taxane binding site of an (αβ-tubulin)2-RB3-tubulin tyrosine ligase complex (Figure 2b).17 This structure reveals that the C(6) methyl group is solvent exposed and uninvolved in any interactions with the receptor, consistent with the observation that sagopilone, with an allyl group at C(6), is approximately equipotent with epothilone B.6 In this way, we decided to synthesize the C(6)-4-azidobutyl epothilone B analogue 3 (Figure 2c) and evaluate it and amides derived therefrom for potency.
Figure 2.
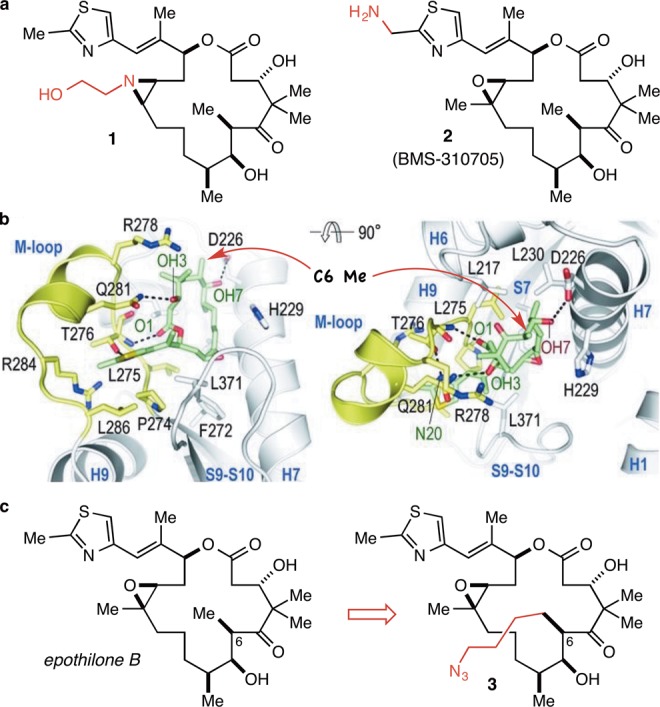
(a) Two previously described analogues of the epothilones that may be used in conjugation reactions with a traceless or self-immolative linker strategy. (b) The crystal structure of epothilone A bound in the taxane binding site reveals that the C(6) methyl group is solvent exposed, consistent with available SAR data. Reprinted with permission from ref (17). Copyright 2013 AAAS. (c) C(6)-methyl extended analogue 3 is predicted to be approximately equipotent with the parent natural product.
We have developed two conceptually related syntheses of the C(1)–C(9) fragment of the epothilone family of natural products18,19 based on our silylformylation/crotylsilylation/Tamao oxidation methodology20 we developed for the step-economical and efficient syntheses of complex polyketide fragments. The requisite starting material for the synthesis of the C(6)-4-azidobutyl analogue, homopropargyl alcohol 4, was prepared using our recently reported synthesis19 (see the Supporting Information file for details), and we began by examining our second-generation approach in which the three parts of the sequence were carried out in a stepwise fashion (Figure 3a).19 As shown, this sequence was successful in delivering the desired C(1)–C(9) fragment 10 with a 4-chlorobutyl group installed at C(6). Unfortunately, however, the aldehyde crotylation step (7 + 8 → 9),21 which had worked well in the C(6)-methyl series, proceeded with low (3.5:1) diastereoselectivity and was inefficient, presumably due to the extreme steric hindrance of aldehyde 7. As a result, we were able to isolate the desired product (9) in only 37% yield. Given that one of the primary intended benefits of the second-generation stepwise sequence was to allow for high levels of diastereoselectivity in the crotylation event, we decided to evaluate the first-generation tandem and one-pot version of the sequence (Figure 3b). This was carried out according to our previous report18 and produced 10 as the major product of a 7:1.3:1 mixture of diastereomers in 44% combined yield from 4. Though due to the added steric hindrance of the C(6)-4-chlorobutyl group this sequence, too, proceeded with reduced efficiency relative to the version with the C(6)-methyl substrate, it also proceeds in just two pots, and because of this extraordinary step-economy, we were able to rapidly produce multigram quantities of 10 using this route.
Figure 3.

(a) Synthesis of the C(1)–C(9) fragment of the epothilones with a 4-chlorobutyl group installed at C(6) using our second generation approach (ref (19)). (b) Synthesis of the C(1)–C(9) fragment of the epothilones with a 4-chlorobutyl group installed at C(6) using our first generation approach (ref (18)).
To complete the synthesis of the target compound 3, we adapted the chemistry22 pioneered by the Danishefsky team in the course of their development of the fludelone family of epothilone analogues.23 As shown in Figure 4, coupling of acid 12 and alcohol 13 proceeded smoothly and was followed by ring-closing metathesis with the second generation Grubbs catalyst24 and Wittig reaction to install the thiazole side chain. Removal of the triethylsilyl (TES) protecting groups was followed by selective reduction of the C(9)–C(10) alkene and displacement of the chloride with azide. Finally, epoxidation of the macrocyclic alkene with dimethyldioxirane according to Danishefsky’s procedure25 gave the C(6)-4-azidobutyl epothilone B analogue 3.
Figure 4.
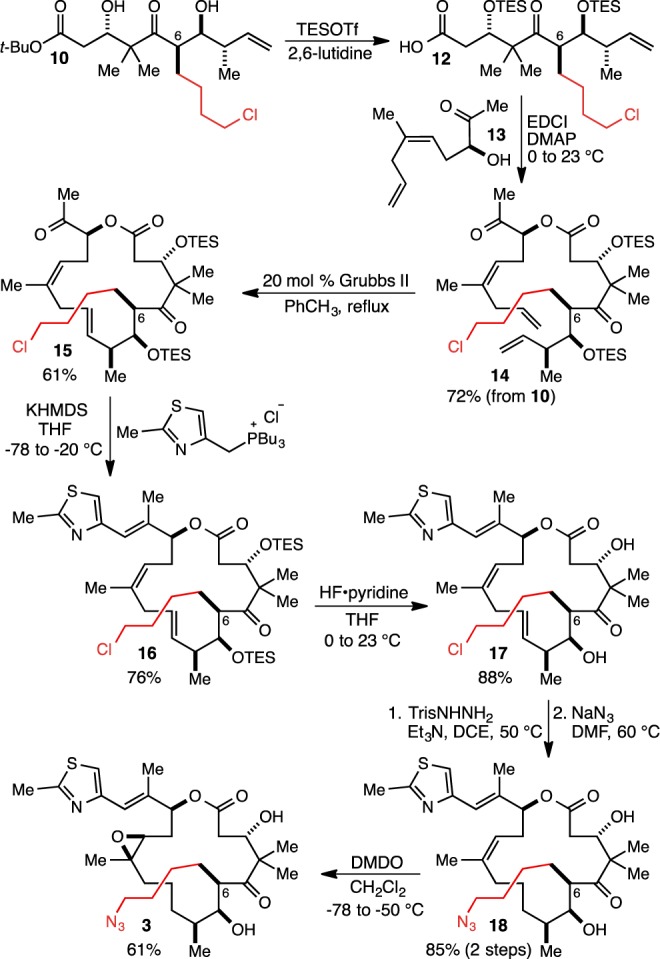
Completion of the synthesis of the C(6)-4-azidobutyl epothilone B analogue 3 according to the Danishefsky strategy.
With access to 3 secured, we explored various linking/conjugation strategies. While strain-accelerated azide–alkyne cycloaddition reactions with cyclooctynes26,27 proved straightforward and efficient, we also found that the azide could be reduced using the classical Staudinger reaction,28 and the derived amine (19) smoothly acylated with N-hydroxysuccinate esters (Figure 5). This was judged to be the more convenient and straightforward approach, and in short order, amides 20 and 21 were produced as models for linker equipped analogues of epothilone B. Amides 20 and 21 were assayed for cell growth inhibition against the PC3 (prostate) and A549 (lung) cell lines along with paclitaxel and epothilone B as positive controls, and gratifyingly, they were found to be approximately equipotent with epothilone B (Figure 5b).
Figure 5.
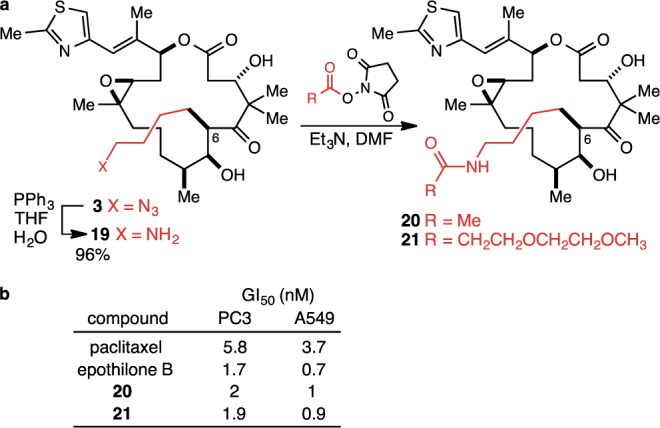
(a) Synthesis of C(6)-4-aminobutyl epothilone B (19) and conjugation reactions to produce model linker-epothilone B constructs 20 and 21. (b) Cell growth inhibition GI50 values for 20 and 21 (values are the average over two–four experiments; see the Supporting Information for details).
Guided by the high-resolution structure of epothilone A bound in the taxane binding site (Figure 2b), we applied the methyl extension approach16 to design the C(6)-4-azidobutyl epothilone B analogue 3 and synthesized it using our previously developed routes to the C(1)–C(9) fragment of the epothilones18,19 and Danishefsky’s fragment coupling and end-game strategies.22,23,25 Amides 20 and 21 were then produced and evaluated in an in vitro cell growth inhibition assay and found to be approximately equipotent with epothilone B. These results thus constitute a proof-of-concept result toward a validated linker strategy for the epothilone family of MSAs, and set the stage for a full exploration of linker structure–activity relationships and ultimately an evaluation of the potential of these clinically validated compounds to serve as the drug component of ADCs and other selective tumor cell-targeting conjugates. Current efforts are focused on those objectives, as well as on a streamlined and more efficient and selective method for the synthesis of the C(1)–C(9) fragment of the epothilones with a 4-chlorobutyl group installed at C(6).
Acknowledgments
This work was supported by NIGMS/NIH (GM058133) and was supported in part by the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development, NIH.
Biographies
James Leighton received his Ph.D. from Harvard University in 1994 working under the direction of Professor David Evans. After a National Science Foundation Postdoctoral Fellowship with Professor Eric Jacobsen at Harvard University, he joined the faculty at Columbia University as an Assistant Professor in 1996. He was promoted to the rank of Associate Professor in 1999 and to the rank of Professor in 2004. His research interests are in organic chemistry, with a particular current focus on the synthesis and development of bioactive polyketide natural products.
Dan Sackett received his Ph.D. in Molecular and Cellular Biology from Brown University. His work since has continued his focus on cell division and microtubule biochemistry and biophysics, beginning with Post Doctoral research at the NIADDKD, NIH. His work continued in the National Cancer Institute until, in 1999, he joined the Program in Physical Biology at the NICHD. He is currently Senior Researcher in the Division of Basic and Translational Biophysics at the NICHD, NIH, where his research focuses on biochemistry and pharmacology of tubulin and biophysics of microtubules and microtubule arrays.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00131.
Experimental procedures, characterization data, and NMR spectra (PDF)
Author Contributions
‡ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Bollag D.; McQueney P.; Zhu J.; Hensens O.; Koupal L.; Liesch J.; Goetz M.; Lazarides E.; Woods C. M. Epothilones, a New Class of Microtubule-stabilizing Agents with a Taxol-like Mechanism of Action. Cancer Res. 1995, 55, 2325–2333. [PubMed] [Google Scholar]
- Gerth K.; Bedorf N.; Höfle G.; Irschik H.; Reichenbach H. Epothilons A and B: Antifungal and Cytotoxic Compounds from Sorangium cellulosum (Myxobacteria) Production, Physico-chemical and Biological Properties. J. Antibiot. 1996, 49, 560–563. 10.7164/antibiotics.49.560. [DOI] [PubMed] [Google Scholar]
- Höfle G.; Bedorf N.; Steinmetz H.; Schomburg D.; Gerth K.; Reichenbach H. Epothilone A and B—Novel 16-Membered Macrolides with Cytotoxic Activity: Isolation, Crystal Structure, and Conformation in Solution. Angew. Chem., Int. Ed. Engl. 1996, 35, 1567–1569. 10.1002/anie.199615671. [DOI] [Google Scholar]
- Lee F. Y. F.; Borzilleri R.; Fairchild C. R.; Kim S.-H.; Long B. H.; Reventos-Suarez C.; Vite G. D.; Rose W. C.; Kramer R. A. BMS-247550: A Novel Epothilone Analog with a Mode of Action Similar to Paclitaxel but Possessing Superior Antitumor Efficacy. Clin. Cancer Res. 2001, 7, 1429–1437. [PubMed] [Google Scholar]
- Lee F. Y. F.; Borzilleri R.; Fairchild C. R.; Kamath A.; Smykla R.; Kramer R.; Vite G. Preclinical discovery of ixabepilone, a highly active antineoplastic agent. Cancer Chemother. Pharmacol. 2008, 63, 157–166. 10.1007/s00280-008-0724-8. [DOI] [PubMed] [Google Scholar]
- Klar U.; Buchmann B.; Schwede W.; Skuballa W.; Hoffmann J.; Lichtner R. B. Total Synthesis and Antitumor Activity of ZK-EPO: The First Fully Synthetic Epothilone in Clinical Development. Angew. Chem., Int. Ed. 2006, 45, 7942–7948. 10.1002/anie.200602785. [DOI] [PubMed] [Google Scholar]
- Krause W.; Klar U. Differences and Similarities of Epothilones. Curr. Cancer Ther. Rev. 2011, 7, 10–36. 10.2174/157339411794474155. [DOI] [Google Scholar]
- Klar U.; Platzek J. Asymmetric Total Synthesis of the Epothilone Sagopilone – From Research to Development. Synlett 2012, 23, 1291–1299. 10.1055/s-0031-1290163. [DOI] [Google Scholar]
- Chou T.-C.; Zhang X.; Zhong Z.-Y.; Li Y.; Feng L.; Eng S.; Myles D. R.; Johnson R.; Wu N.; Yin Y. I.; Wilson R. M.; Danishefsky S. J. Therapeutic effect against human xenograft tumors in nude mice by the third generation microtubule stabilizing epothilones. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 13157–13162. 10.1073/pnas.0804773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber H.-P.; Koehn F. E.; Abraham R. T. The antibody-drug conjugate: an enabling modality for natural product-based cancer therapeutics. Nat. Prod. Rep. 2013, 30, 625–639. 10.1039/c3np20113a. [DOI] [PubMed] [Google Scholar]
- Perez H. L.; Cardarelli P. M.; Deshpande S.; Gangwar S.; Schroeder G. M.; Vite G. D.; Borzilleri R. M. Antibody–drug conjugates: current status and future directions. Drug Discovery Today 2014, 19, 869–881. 10.1016/j.drudis.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Chari R. V. J.; Miller M. L.; Widdison W. C. Antibody–Drug Conjugates: An Emerging Concept in Cancer Therapy. Angew. Chem., Int. Ed. 2014, 53, 3796–3827. 10.1002/anie.201307628. [DOI] [PubMed] [Google Scholar]
- Vlahov I. R.; Vite G. D.; Kleindl P. J.; Wang Y.; Santhapuram H. K. R.; You F.; Howard S. J.; Kim S.-H.; Lee F. F. Y.; Leamon C. P. Regioselective synthesis of folate receptor-targeted agents derived from epothilone analogs and folic acid. Bioorg. Med. Chem. Lett. 2010, 20, 4578–4581. 10.1016/j.bmcl.2010.06.016. [DOI] [PubMed] [Google Scholar]
- Kamath A. V.; Chang M.; Lee F. Y.; Zhang Y.; Marathe P. H. Preclinical pharmacokinetics and oral bioavailability of BMS-310705, a novel epothilone B analog. Cancer Chemother. Pharmacol. 2005, 56, 145–153. 10.1007/s00280-004-0928-5. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; de Mas N.; Parlanti L.; Lyngberg O. K.; Ströhlein G.; Guo Z.; Dambalas K.; Rosso V. W.; Yang B. S.; Girard K. P.; Manaloto Z. A.; D’Arasmo G.; Frigerio R. E.; Wang W.; Lu X.; Bolgar M. S.; Gokhale M.; Thakur A. B. Synthesis, Chromatographic Purification, and Isolation of Epothilone–Folic Acid Conjugate BMS-753493. Org. Process Res. Dev. 2011, 15, 797–809. 10.1021/op200023g. [DOI] [Google Scholar]
- Ho S.; Sackett D. L.; Leighton J. L. A “Methyl Extension” Strategy for Polyketide Natural Product Linker Site Validation and Its Application to Dictyostatin. J. Am. Chem. Soc. 2015, 137, 14047–14050. 10.1021/jacs.5b09869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prota A. E.; Bargsten K.; Zurwerra D.; Field J. J.; Díaz J. F.; Altmann K.-H.; Steinmetz M. O. Molecular Mechanism of Action of Microtubule-Stabilizing Anticancer Agents. Science 2013, 339, 587–590. 10.1126/science.1230582. [DOI] [PubMed] [Google Scholar]
- Harrison T. J.; Rabbat P. M. A.; Leighton J. L. An “Aprotic” Tamao Oxidation/Syn-Selective Tautomerization Reaction for the Efficient Synthesis of the C(1)–C(9) Fragment of Fludelone. Org. Lett. 2012, 14, 4890–4893. 10.1021/ol302221s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foley C. N.; Leighton J. L. A Highly Stereoselective, Efficient, and Scalable Synthesis of the C(1)–C(9) Fragment of the Epothilones. Org. Lett. 2015, 17, 5858–5861. 10.1021/acs.orglett.5b03034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spletstoser J. T.; Zacuto M. J.; Leighton J. L. Tandem Silylformylation–Crotylsilylation/Tamao Oxidation of Internal Alkynes: A Remarkable Example of Generating Complexity from Simplicity. Org. Lett. 2008, 10, 5593–5596. 10.1021/ol802489w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H.; Ho S.; Leighton J. L. A More Comprehensive and Highly Practical Solution to Enantioselective Aldehyde Crotylation. J. Am. Chem. Soc. 2011, 133, 6517–6520. 10.1021/ja200712f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivkin A.; Yoshimura F.; Gabarda A. E.; Cho Y. S.; Chou T.-C.; Dong H.; Danishefsky S. J. Discovery of (E)-9,10-Dehydroepothilones through Chemical Synthesis: On the Emergence of 26-Trifluoro-(E)-9,10-dehydro-12,13-desoxyepothilone B as a Promising Anticancer Drug Candidate. J. Am. Chem. Soc. 2004, 126, 10913–10922. 10.1021/ja046992g. [DOI] [PubMed] [Google Scholar]
- Rivkin A.; Chou T.-C.; Danishefsky S. J. On the Remarkable Antitumor Properties of Fludelone: How We Got There. Angew. Chem., Int. Ed. 2005, 44, 2838–2850. 10.1002/anie.200461751. [DOI] [PubMed] [Google Scholar]
- Scholl M.; Ding S.; Lee C. W.; Grubbs R. H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999, 1, 953–956. 10.1021/ol990909q. [DOI] [PubMed] [Google Scholar]
- Stachel S. J.; Danishefsky S. J. Chemo- and stereoselective epoxidation of 12,13-desoxyepothilone B using 2,2′-dimethyldioxirane. Tetrahedron Lett. 2001, 42, 6785–6787. 10.1016/S0040-4039(01)01209-6. [DOI] [Google Scholar]
- Agard N. J.; Prescher J. A.; Bertozzi C. R. A Strain-Promoted [3 + 2] Azide–Alkyne Cycloaddition for Covalent Modification of Biomolecules in Living Systems. J. Am. Chem. Soc. 2004, 126, 15046–15047. 10.1021/ja044996f. [DOI] [PubMed] [Google Scholar]
- Lutz J.-F. Copper-Free Azide–Alkyne Cycloadditions: New Insights and Perspectives. Angew. Chem., Int. Ed. 2008, 47, 2182–2184. 10.1002/anie.200705365. [DOI] [PubMed] [Google Scholar]
- Staudinger H.; Meyer J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta 1919, 2, 635–646. 10.1002/hlca.19190020164. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.