

Abstract
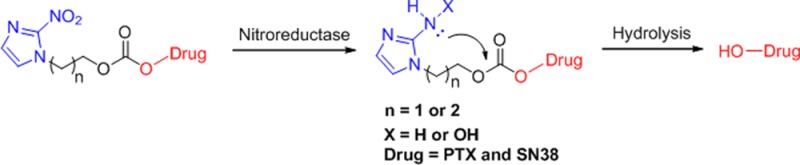
Due to the low esterase activity in human plasma, many ester and carbonate prodrugs tested in humans may be less effective than that in preclinical animals. In this letter, PTX and SN-38 were attached to the N-1 position of 2-nitroimidazole via a carbonate linker. Presumably, 2-aminoimidazole may help promote the intramolecular hydrolysis of the carbonate bond. The prodrugs exhibited a considerable stability in buffers at different pH values as well as in human plasma. Furthermore, a rapid reduction was exhibited in the presence of nitroreductase. An in vitro cytotoxicity assay demonstrated that hypoxic conditions could increase the toxicity of prodrugs. Potentially, the compound species may form a new class of promising antitumor agents.
Keywords: 2-Nitroimidazole, paclitaxel, SN-38, prodrug
Paclitaxel (PTX) and camptothecin (CPT) represent the most important anticancer drugs in clinical use to date and exhibit a high antitumor efficacy against a wide range of tumor species.1,2 Paclitaxel is a microtubule stabilizing agent isolated from Taxus brevifolia. Among others, paclitaxel and its analogue docetaxel are used for the treatment of solid tumors, including ovarian, breast, and lung cancesr.3−6 Camptothecin is a potent antitumor alkaloid isolated from Camptotheca acuminate and was found to target DNA topoisomerase I. Camptothecin derivatives such as 10-hydroxycamptothecin, topotecan, and irinotecan (active metabolite: SN-38) are used for the treatment of ovarian and colorectal cancers.7−10 Unfortunately, these compounds exhibit significant side effects on healthy tissues, poor solubility characteristics in aqueous media, and multidrug resistance. Ultimately, these drawbacks limit the applicability of the drugs in clinical treatments. In an effort to overcome these drawbacks, the development of prodrugs represents a common approach offering a site-specific release.11−14
A wide field of research studies has demonstrated that the lactone ring in camptothecin represents a critical feature for antitumor activity, corresponding to topoisomerase I inhibition. However, the instability of the lactone ring in the plasma decreases antitumor efficiency.15 Masking 20-OH by transforming it to the corresponding water-insoluble alkyl ester or carbonate has been shown to increase lactone stability in the plasma compared with their parent compounds.16 Some prodrugs conjugated with PEG in 20-OH position have been demonstrated to increase the solubility in aqueous media.24 However, one of the most common problems of such prodrugs is that they have been shown to be too stable in preclinical studies, ultimately not releasing the active compound at sufficiently high concentrations. Furthermore, insufficient prodrug conversion may reduce therapeutic efficacy due to the notion that the active compound may never reach a therapeutically relevant concentration. Overall, this issue may also occur in the case of ester paclitaxel prodrugs in 2′-OH position, representing a common strategy for paclitaxel prodrug design.17 A large body of research studies has shown that human plasma esterase activity is markedly different from the esterase activity in other animal species resulting in pharmacokinetic differences of these prodrugs. The efficiency of prodrugs tested in humans may be lower than that in preclinical animals, including mouse and rat, due to the lower esterase activity in human plasma.18,19 Therefore, we believe that it is necessary to introduce a trigger for bond cleavage in 20-OH position of camptothecin and in 2′-OH of paclitaxel in order to ensure active drug release at the target site.
Hypoxia, a deficiency in the amount of oxygen reaching the tissues, occurs in a variety of human disease states including solid tumors.20,21 Among others, the presence of hypoxia in solid tumors results in chemoresistance, radioresistance, angiogenesis, vasculogenesis, invasiveness, metastasis, and resistance to cell death.22,23 Considerable effort has been put forward to develop bioreductive prodrugs, which could be selectively activated under hypoxic microenvironments.24−27 Denny et al. have reported the use of prodrugs of cyclization-activated aromatic mustard species involving an ortho-nitrophenyl group.28 Ono et al. have reported a hypoxia-activated pro-oligo compounds based on 3-(2-nitrophenyl)propan-1-ol with a phosphoester bond activated via intramolecular cyclization of 2-aminophenol.29 For the latter study, 2-nitroimidazole was demonstrated to represent an improved bioreductive group compared to nitrobenzene.30 Therefore, we designed a new class of prodrugs in N-1 position of 2-nitroimidazole instead of the commonly used C-5 position. Here, PTX and SN-38 were conjugated to 2-(2-nitro-1H-imidazol-1-yl)ethanol and 3-(2-nitro-1H-imidazol-1-yl)propan-1-ol via a carbonate bond. We further found that 2-aminoimidazole derivatives have the ability to promote the hydrolysis of the carbonate bond and induce the release of the prodrugs.31 Moreover, we investigated two 2-nitroimidazole derivatives containing ethanol and propanol in N-1 position of 2-nitroimidazole. These prodrugs were designed with significantly increased activity at hypoxic tumor sites. One of the most important findings of this study was that the drug release mechanism via 2-aminoimidazole increased the drug release efficiency (Figure 1).
Figure 1.
Proposed mechanism of prodrug activated via bioreduction.
The prodrugs were synthesized according to the process outlined in Scheme 1. 2-(2-Nitro-1H-imidazol-1-yl)ethanol and 3-(2-nitro-1H-imidazol-1-yl)propan-1-ol were activated by addition of 4-nitrophenyl carbonochloridate and were then reacted with PTX to afford 2C-PTX and 3C-PTX. Conversely, 10-O-tert-butyldimethylsilyl-SN-38 was treated with triphosgene and then reacted with 2-(2-nitro-1H-imidazol-1-yl)ethanol and 3-(2-nitro-1H-imidazol-1-yl)propan-1-ol. The TBS group was deprotected by addition of TBAF to afford 2C-SN38 and 3C-SN38 (Scheme 1).
Scheme 1. Synthetic Route of Prodrugs.

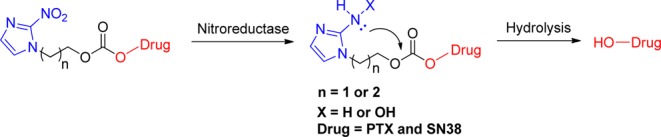
The stability of the compounds was tested first as this factor seems to be critical for biological evaluation. The compounds were evaluated in buffer at different pH conditions (pH 5, pH 6.5, and pH 7.4) for a period of 48 h using HPLC (Figure 2). The results indicated that the half-life (t1/2) value of 3C-SN38 and 2C-PTX in buffer at pH 5 was 40 and 48 h, respectively. The t1/2 values of 2C-SN38 and 3C-PTX were found to be beyond 48 h. In buffer at pH 6.5, the half-life value of 2C-SN38, 2C-PTX, and 3C-PTX was 41, 24, and 24 h, respectively, and the t1/2 of 3C-SN38 under these conditions was found to be beyond 48 h. In buffer at pH 7.4, the t1/2 value of 2C-PTX and 3C-PTX was 28 and 36 h, respectively, and the t1/2 values of 2C-SN38 and 3C-SN38 were beyond 48 h. We concluded that these compounds were stable in different buffers at different pH values. We then tested the compounds in vitro in mouse and human plasma. It was known that the much higher level of esterase in mouse plasma compared to human plasma may accelerate the hydrolysis of the carbonate bond. The results obtained indicate that the test compounds degrade rapidly in mouse plasma. The half-life of 2C-SN38, 3C-SN38, 2C-PTX, and 3C-PTX in mouse plasma was determined to be 47, 37, 20, and 55 min, respectively. However, the metabolic degradation rates of the compounds in human plasma were found to be much slower. The t1/2 of 2C-SN38, 3C-SN38, 2C-PTX, and 3C-PTX was 6, 2, 2, and 6 h. The compounds exhibited a considerable stability in buffers at different pH values and human plasma, a finding that is consistent with the other reports found in the literature.12,32 Our results indicate that the carbonate bond seems to be sufficiently stable in human plasma, a critical feature for the future development of bioreductive prodrug species.
Figure 2.
Stability of prodrugs in different buffer: (A) pH = 5.0 ABS; (B) pH = 6.5 PBS; (C) pH = 7.4 PBS; (D) mouse plasma; (E) human plasma.
To determine whether the prodrug fragments provide the desired active agent as intended, we carried out a chemical reduction over Pd/C in the presence of H2 in THF at 37 °C. The individual fragments were characterized by LC/MS. Compound 3C-SN38 afforded the best results and was found to be entirely reduced, resulting in the generation of two products, namely, SN-38 and the 2-aminoimidazole intermediate of 3C-SN38. The mass peak corresponding to the 2-aminoimidazole intermediate could be found in the MS-ESI spectrum (m/z = 561.02 [M + H]+) (Figure 3). Compound 2C-SN38 was determined to be mainly reduced to a byproduct, resulting in almost negligible release of SN-38. Presumably, the amino group is too close to the lactone ring ultimately causing a side reaction. However, the characterization results using the PTX derivatives were complex. Both the 2-aminoimidazole intermediate and PTX could be detected, while some unknown derivatives were found to be generated simultaneously. We believe that the prodrugs of PTX were not stable under the reductive conditions used here (Figure S1).
Figure 3.

LC/MS charts of 3C-SN38 after chemical reduction over Pd/C and H2 in THF.
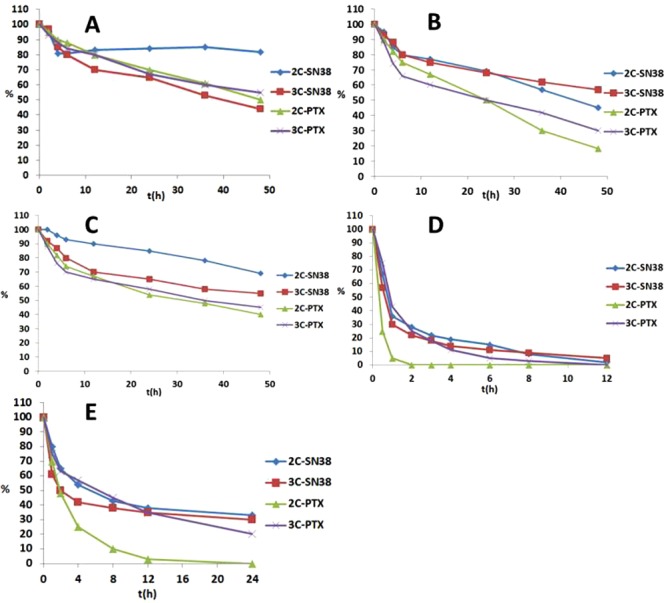
In hypoxic tumors, the reductase level has been reported to be much higher than in normal tissues. Presumably, this feature provides an opportunity to specifically target hypoxia. For the further investigation of the activate mechanism of the prodrugs, nitroreductase (NTR) extracted from Escherichia coli was used. The prodrugs (10 μM, 1% DMSO) dissolved in PBS (pH 7.4) were incubated with nitroreductase (50 μg/mL) and NADPH (1 μmol/mL) at 37 °C and the biological reduction process was monitored by HPLC (Figure 4). The prodrugs were found to be rapidly reduced by nitroreductase, except for 2C-SN38, which reacted slower. The t1/2 of 2C-SN38 was determined to be 30 min, and the t1/2 values of all other compound species were within 10 min. Approximately 30% of 2C-SN38 still remained after 1 h and only 30% of the active drug was found to be released. Presumably, 2C-SN38 was too stable under these bioreductive conditions described. Considering the results for 3C-SN38 and 3C-PTX, even though the prodrug disappeared rapidly, the generation rates of the active drug were not as fast as the compounds reduced. Therefore, we believe that the 2-aminointermediates, as well as some byproducts, may have been generated. However, 2C-PTX was determined to be the most suitable compound, with an overall PTX conversion rate of 85% within 1 h. Likely, the bioreductive process via nitroreductase was somewhat milder, resulting in a reduced formation of 2C-PTX byproducts compared to chemical reduction. The 2-aminoimidazole intermediate of 2C-PTX was not detected, which may indicate that the drug release process of the 2-aminoimidazole intermediate under physiological conditions was very fast.
Figure 4.
Percent of prodrug remaining (A) and active drug releasing (B) in 1 h of incubation with nitroreductase in the presence of NADPH.
The compounds were evaluated for their cytotoxicity characteristics under normoxic and hypoxic (94% N2, 5% CO2, 1% O2) conditions in vitro using H460 human lung cancer cells and HT29 human colon cancer cells. These cell lines are known to express high levels of oxygen-insensitive reductase, potentially activating these prodrugs via enzymatic reduction and increasing the toxicity of the prodrugs under hypoxic conditions, and were therefore used for cytotoxicity tests. The cells were incubated in the presence of the test compounds at various concentrations for 72 h under normoxic or hypoxic conditions. The cell viability and proliferation behavior were assessed by MTT. The IC50 values for the proliferation inhibition of the tested compounds are shown listed in Table 1. The results obtained indicate that 3C-SN38 exhibited the highest selectivity ratio of 2.03 in the H460 cell line and 2C-PTX exhibited the highest selectivity ratio of 3.11 in the HT29 cell line. All tested compounds featured moderate selectivity toward hypoxic tumor cells. The hypoxic conditions could increase the toxicity of prodrugs.
Table 1. In Vitro Cytotoxicity Assay Data Summary.
| H460 |
HT29 |
|||||
|---|---|---|---|---|---|---|
| IC50 (μM) |
IC50 (μM) |
|||||
| AIR | N2 | IC50(AIR)/IC50(N2) | AIR | N2 | IC50(AIR)/IC50(N2) | |
| SN-38 | 0.050 ± 0.001 | 0.047 ± 0.007 | 1.06 | 0.096 ± 0.048 | 0.119 ± 0.018 | 0.82 |
| PTX | 0.006 ± 0.001 | 0.007 ± 0.005 | 0.86 | 0.014 ± 0.002 | 0.015 ± 0.001 | 0.93 |
| 2C-SN38 | 0.091 ± 0.022 | 0.063 ± 0.008 | 1.44 | >10 | 9.56 ± 0.66 | >1.05 |
| 3C-SN38 | 0.201 ± 0.022 | 0.099 ± 0.012 | 2.03 | >10 | 6.02 ± 0.52 | >1.67 |
| 2C-PTX | 0.009 ± 0.001 | 0.006 ± 0.002 | 1.5 | 0.140 ± 0.041 | 0.045 ± 0.035 | 3.11 |
| 3C-PTX | 0.021 ± 0.003 | 0.018 ± 0.003 | 1.17 | 0.290 ± 0.112 | 0.159 ± 0.038 | 1.82 |
In summary, a new class of carbonate prodrugs on the basis of 2-nitroimidazole was studied in order to release the active drug in an efficient manner. The prodrugs exhibited a considerable stability in different buffer and human plasma. The compounds were found to be reduced rapidly in the presence of nitroreductase. Among the studied compounds, compound 2C-PTX was determined to release 85% of the active drug, the highest conversion rate under the studied bioreduction conditions. Four prodrugs exhibited moderate hypoxia selectivity in H460 and HT29 cell lines. On the basis of the results obtained, we hypothesize that these prodrugs may find potential future applications as antitumor agents.
Glossary
ABBREVIATIONS
- SN-38
7-ethyl-10-hydroxy-camptothecin
- NADPH
nicotinamide adenine dinucleotide phosphate
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00189.
Experimental procedures, characterization data, and NMR spectra (PDF)
Author Contributions
‡ These authors contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Harvey A. L. Natural products in drug discovery. Drug Discovery Today 2008, 13, 894–901. 10.1016/j.drudis.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Newman D. J.; Cragg G. M. Natural products as sources of new drugs over the last 25 years. J. Nat. Prod. 2007, 70, 461–477. 10.1021/np068054v. [DOI] [PubMed] [Google Scholar]
- Wani M. C.; Taylor H. L.; Wall M.; Coggon E. P.; McPhail A. T. Plant antitumor agents. VI. Isolation and structure of taxol, a novel antileukemic and antitumor agent from Taxus brevifolia. J. Am. Chem. Soc. 1971, 93, 2325–2327. 10.1021/ja00738a045. [DOI] [PubMed] [Google Scholar]
- Mekhail T. M.; Markman M. Paclitaxel in cancer therapy. Expert Opin. Pharmacother. 2002, 3, 755–766. 10.1517/14656566.3.6.755. [DOI] [PubMed] [Google Scholar]
- Ferlini C.; Ojima I.; Distefano M.; Gallo D.; Riva A.; Morazzoni P.; Bombardelli E.; Mancuso S.; Scambia G. Second generation taxanes: from the natural framework to the challenge of drug resistance. Curr. Med. Chem.: Anti-Cancer Agents 2003, 3, 133–138. 10.2174/1568011033353489. [DOI] [PubMed] [Google Scholar]
- Nabholtz J. M.; Vannetzel J. M.; Llory J. F.; Bouffette P. Advances in the use of taxanes in the adjuvant therapy of breast cancer. Clin. Breast Cancer 2003, 4, 187–192. 10.1016/S1526-8209(11)70624-3. [DOI] [PubMed] [Google Scholar]
- Wall M. E.; Wani M. C.; Cook C. E.; Palmer K. H.; McPhail A. T.; Sim G. A. Plant Antitumor Agents. I. The Isolation and Structure of Camptothecin, a Novel Alkaloidal Leukemia and Tumor Inhibitor from Camptotheca acuminate. J. Am. Chem. Soc. 1966, 88, 3888–3890. 10.1021/ja00968a057. [DOI] [Google Scholar]
- Hsiang Y. H.; Hertzberg R.; Hecht S.; Liu L. F. Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem. 1985, 260, 14873–14878. [PubMed] [Google Scholar]
- Wang J. C. DNA Topoisomerases - Annual Review of Biochemistry. Annu. Rev. Biochem. 1996, 65, 635–692. 10.1146/annurev.bi.65.070196.003223. [DOI] [PubMed] [Google Scholar]
- Ulukan H.; Swaan P. W. Camptothecins: a review of their chemotherapeutic potential. Drugs 2002, 62, 2039–2057. 10.2165/00003495-200262140-00004. [DOI] [PubMed] [Google Scholar]
- Lis L. G.; Smart M. A.; Luchniak A.; Gupta M. L.; Gurvich V. J. Synthesis and biologicalevaluation of a biotinylatedpaclitaxel with an extra-long chain spacer arm. ACS Med. Chem. Lett. 2012, 3, 745–748. 10.1021/ml300149z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-berna G.; Cabañas M. J. D.; Mangas-Sanjuán V.; Gonzalezalvarez M.; Gonzalezalvarez I.; Abasolo I.; Schwartz S.; Bermejo M.; Corma A. Semisynthesis, cytotoxic activity, and oral availability of new lipophilic 9-substituted camptothecin derivatives. ACS Med. Chem. Lett. 2013, 4, 651–655. 10.1021/ml400125z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.; Xie S.; Ma L. J.; Chen Y.; Lu W. 10-Boronic acid substituted camptothecin as prodrug of SN-38. Eur. J. Med. Chem. 2016, 116, 84–89. 10.1016/j.ejmech.2016.03.063. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Tang K.; Wang H.; Liu Y.; Bao B.; Fang Y.; Zhang X.; Lu W. Design, Synthesis, and Biological Evaluation of New Cathepsin B-Sensitive Camptothecin Nanoparticles Equipped with a Novel Multifuctional Linker. Bioconjugate Chem. 2016, 27, 1267–1275. 10.1021/acs.bioconjchem.6b00099. [DOI] [PubMed] [Google Scholar]
- Hertzberg R. P.; Caranfa M. J.; Holden K. G.; Jakas D. R.; Gallagher M. R.; Mong S. M.; Bartus J.; Johnson R. K.; Kingsbury W. D. Modification of the hydroxylactone ring of camptothecin: inhibition of mammalian topoisomerase I and biological activity. J. Med. Chem. 1989, 32, 715–720. 10.1021/jm00123a038. [DOI] [PubMed] [Google Scholar]
- Penco S.; Merlini L.; Zunino F.; Dallavalle S. Perspectives in camptothecin development. Expert Opin. Ther. Pat. 2002, 12, 837–844. 10.1517/13543776.12.6.837. [DOI] [Google Scholar]
- Zhu Q.; Guo Z.; Huang N.; Wang M.; Chu F. Comparative molecular field analysis of a series of paclitaxel analogues. J. Med. Chem. 1997, 40, 4319–4328. 10.1021/jm970442u. [DOI] [PubMed] [Google Scholar]
- Berry L. M.; Wollenberg L.; Zhao Z. Esterase Activities in the Blood, Liver and Intestine of Several Preclinical Species and Humans. Drug Metab. Lett. 2009, 3, 70–77. 10.2174/187231209788654081. [DOI] [PubMed] [Google Scholar]
- Bahar F. G.; Ohura K.; Ogihara T.; Imai T. Species Difference of Esterase Expression and Hydrolase Activity in Plasma. J. Pharm. Sci. 2012, 101, 3979–3988. 10.1002/jps.23258. [DOI] [PubMed] [Google Scholar]
- Wilson W. R.; Hay M. P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- Brown J. M. Tumor Microenvironment and the Response to Anticancer Therapy. Cancer Biol. Ther. 2002, 1, 453–458. 10.4161/cbt.1.5.157. [DOI] [PubMed] [Google Scholar]
- Cairns R. A.; Harris I. S.; Mak T. W. Regulation of cancer cell metabolism. Nat. Rev. Cancer 2011, 11, 85–95. 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Ohh M. Oxygen-mediated endocytosis in cancer. J. Cell. Mol. Med. 2010, 14, 496–503. 10.1111/j.1582-4934.2010.01016.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baran N.; Konopleva M. Molecular Pathways: Hypoxia-activated prodrugs in cancer therapy. Clin. Cancer Res. 2017, 23, 2382. 10.1158/1078-0432.CCR-16-0895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thambi T.; Park J. H.; Lee D. S. Hypoxia-responsive nanocarriers for cancer imaging and therapy: recent approaches and future perspectives. Chem. Commun. 2016, 52, 8492–8500. 10.1039/C6CC02972H. [DOI] [PubMed] [Google Scholar]
- Wigerup C.; Påhlman S.; Bexell D. Therapeutic targeting of hypoxia and hypoxia-inducible factors in cancer. Pharmacol. Ther. 2016, 164, 152–169. 10.1016/j.pharmthera.2016.04.009. [DOI] [PubMed] [Google Scholar]
- Damen E. W. P; Nevalainen T. J.; van den Bergh T. J. M.; de Groot F. M. H.; Scheeren H. W. Cheminform abstract: synthesis of novel paclitaxel prodrugs designed for bioreductive activation in hypoxic tumor tissue. Bioorg. Med. Chem. 2002, 10, 71–77. 10.1016/S0968-0896(01)00235-8. [DOI] [PubMed] [Google Scholar]
- Atwell G. J.; Sykes B. M.; O’Connor C. J.; Denny W. A. Relationships between structure and kinetics of cyclization of 2-aminoaryl amides: potential prodrugs of cyclization-activated aromatic mustards. J. Med. Chem. 1994, 37, 371–380. 10.1021/jm00029a009. [DOI] [PubMed] [Google Scholar]
- Saneyoshi H.; Iketani K.; Kondo K.; Saneyoshi T.; Okamoto I.; Ono A. Synthesis and Characterization of Cell-Permeable Oligonucleotides Bearing Reduction-Activated Protecting Groups on the Internucleotide Linkages. Bioconjugate Chem. 2016, 27, 2149–2156. 10.1021/acs.bioconjchem.6b00368. [DOI] [PubMed] [Google Scholar]
- O’Connor L. J.; Cazares-Körner C.; Saha J.; Evans C. N. G.; Stratford M. R. L.; Hammond E. M.; Conway S. J. Design, synthesis and evaluation of molecularly targeted hypoxia-activated prodrugs. Nat. Protoc. 2016, 11, 781–794. 10.1038/nprot.2016.034. [DOI] [PubMed] [Google Scholar]
- Hanaya K.; Yoshioka S.; Ariyasu S.; Aoki S.; Shoji M.; Sugai T. Development of a novel sulfonate ester-based prodrug strategy. Bioorg. Med. Chem. Lett. 2016, 26, 545–550. 10.1016/j.bmcl.2015.11.074. [DOI] [PubMed] [Google Scholar]
- He X.; Lu W.; Jiang X.; Cai J.; Zhang X.; Ding J. Synthesis and biological evaluation of bis and monocarbonate prodrugs of 10-hydroxycamptothecins. Bioorg. Med. Chem. 2004, 12, 4003–4008. 10.1016/j.bmc.2004.06.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.