

Abstract
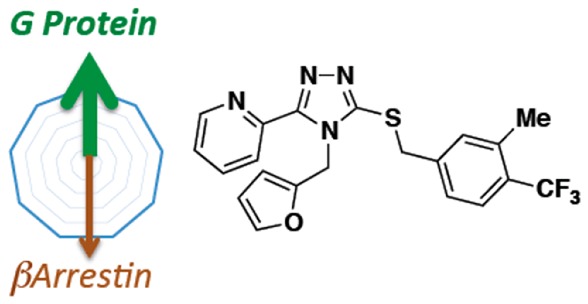
The discovery and characterization of two classes of kappa opioid receptor agonists that are biased for G protein over βarrestin signaling are described.
Keywords: βArrestin, biased ligands, functional selectivity, G protein-coupled receptors, kappa opioid receptor agonists
G protein-coupled receptors (GPCRs) are the single largest class of therapeutic targets, impinging on nearly every therapeutic area from cancer to neuroscience.1 This is in part due to their dominant role among cell-surface receptors in the body; their genes compose ca. 2% of the entire human genome. Moreover, many are eminently druggable, thanks to readily accessible binding pockets that evolved to accommodate small molecule ligands in the first place. As of now, more than half of all approved therapeutics function through a GPCR, with efforts to create new GPCR-targeted therapeutics continuing apace.2
The classical view of GPCR function as taught in introductory biochemistry courses begins with engagement of the receptor with an agonist and the subsequent loading of an attendant G protein with a molecule of GTP in exchange for GDP at its α subunit. Thus, activated, the Gα and combined Gβ/Gγ subunits separate and act upon downstream second messenger(s) associated with that GPCR.
As is often the case, reality turns out to be considerably more complicated than this elegant but incomplete picture. Originally identified as important mediators of receptor desensitization in response to sustained agonist exposure (along with G protein receptor kinases), a class of proteins known as βarrestins was also found to mediate alternative signaling pathways in response to receptor activation.3 βArrestins, so named because they often oppose the primary activation pathways associated with numerous GPCRs, are now considered to be a critical component of overall GPCR signaling.4
The existence of multiple pathways that can follow activation of a single GPCR, each of which will have unique pharmacological outcomes, leads to a fascinating proposition: what if it were possible to activate a given GPCR such that the “normal” G protein pathway and the βarrestin effects could be separated? And more importantly, what if different pathways are responsible for distinct physiological responses? An early demonstration that profound differences can arise from separating G protein and βarrestin function was the discovery that opioid antinociceptive tolerance and mu opioid receptor desensitization was significantly reduced in knockout mice lacking βarrestin.5
It is now appreciated that numerous GPCR targets may benefit from such separation.6−9 The concept has become known as “functional selectivity” or “ligand bias”, and compounds are being discovered that range from fully “balanced” (i.e., that activate each pathway with comparable efficiency) to highly “biased,” displaying preference for engaging one signaling pathway over another.
Our laboratories have been working toward the discovery of biased agonists of the kappa opioid receptor (KOR). A suitable compound would be able to function as a nociceptive agent or for the treatment of intractable itch (as of this writing, an unmet medical need) without the dysphoria, sedation and other side effects typically associated with this target (Figure 1).
Figure 1.
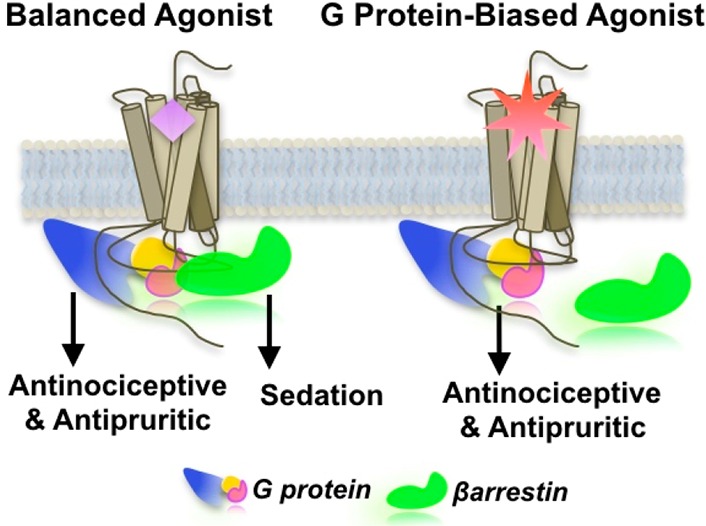
Model depicting functional selectivity of GPCR signaling. A balanced agonist would be predicted to activate multiple signaling cascades mediated by the effectors that associate with the receptor, while a biased agonist would preferentially engage with certain effectors over others to activate distinct signaling pathways.
At its outset, we were not specifically focused on finding biased compounds but rather concerned with developing chemically fresh KOR scaffolds for exploring general opioid pharmacology. As the project evolved, the focus rapidly shifted, however, and it is our hope that some of the lessons we have learned (and are still learning) will be of use to medicinal chemists broadly interested in GPCRs. As reflected by its title, this Innovations article is a follow-up to a 2010 prospective article coauthored by one of us (L.M.B.).10
Looking for Clues
A prerequisite for biased GPCR agonists is having high quality GPCR agonists in the first place.11 In our KOR program, we had identified appropriate chemical matter during the course of two separate projects. In one, an exploratory library of 72 isoquinolinones prepared as a part of the KU Chemical Methodology and Library Development program12 was screened in the UNC Psychoactive Drug Screening Program directed by Bryan Roth, which uses radioligand displacement assays to identify receptor binders. Remarkably, this screen directly afforded a highly potent and selective KOR agonist (Ki = 5 nM for KOR, 3550 nM for MOR, and >10 μM for DOR; see Figure 2 for an isoquinolinone hit).13−15
Figure 2.

Structures and G protein/βarrestin data for selected compounds. (a) Standard balanced agonists. (b) Initial probe structures and advanced analogues generated in these studies.
Several years later, as part of the Molecular Libraries Initiative (MLI), we became aware of a screening project wherein the NIH Small Molecule Repository was to be examined for novel KOR ligands. This project was originally set up between the Conrad Prebys Center for Chemical Genomics at the Sanford–Burnham Medical Research Institute and a team of pharmacologists comprising Lawrence S. Barak and Marc G. Caron (both at Duke) and one of the current coauthors (L.M.B.). Upon hearing about this project, for which the screening was done and was about to enter the chemistry phase, the other coauthor of the present article (J.A., then at the University of Kansas (KU)) lobbied the Sanford–Burnham scientists to allow him and his colleagues to serve as the chemistry team on the project. Generously, the Sanford–Burnham chemistry team of Gregory Roth and Nicholas Cosford blessed the swap and the project was transferred to J.A. and the Specialized Chemistry Center team at KU (see dedication).
The screening used the now-ubiquitous DiscoveRx PathHunter assay expressing human KOR in U2OS (human osteosarcoma) cells.10 This enzyme fragment complementation (EFC) assay produces a chemiluminescence readout resulting from recombination of two portions of galactosidase (one on the receptor, one on the βarrestin) in the presence of the substrate. It provides a direct measure of βarrestin recruitment and can be used to identify receptor agonists or antagonists. Of a number of hits obtained in this screen, four were subjected to initial medicinal chemistry optimization and follow-up pharmacological characterization. These were presented to the scientific community as MLI probes, two agonists16 and two antagonists.17 These initial communications were followed up with a 2012 full paper that detailed the overall project and an amount of postprobe potency and selectivity enhancement.18
As the MLI KOR project wound to a close in about 2010, we decided to shift our interest to the discovery of biased KOR agonists. At the time, most investigators concentrated on a relatively concise set of accepted KOR agonists as tool compounds or for drug development: naturally occurring or synthetic morphinoids, fentanyl-type compounds, peptide analogues of dynorphin,19 the valuable heterocyclic compounds discovered at Upjohn (U69,593 (U69) or U50,488H (U50)),20 and the non-nitrogenous natural product salvinorin A.21,22 Although exceptions would soon emerge, compounds in these classes were balanced for comparable activation of the G protein cascade or βarrestin recruitment. Given this context, we chose to focus on our trove of new kappa chemotypes for signs of bias.
We began by measuring the relative potencies of a given agent to activate two proximal outcomes of receptor activation: coupling to G proteins and recruitment of βarrestin2. The βarrestin2 data was already in hand for many examples thanks to the screening efforts, although most were repeated to obtain more accurate data with freshly synthesized compounds. The classical way of measuring G protein signaling is through monitoring the extent of GDP–GTP exchange by using radiolabeled, hydrolysis-resistant [35S]-GTPγS for binding to agonist-stimulated membranes. This was readily adapted to 96-well plate format using Chinese hamster ovary (CHO) cells stably expressing the human KOR. To a first approximation, bias was optimized by seeking compounds with the highest ratios of [35S]-GTPγS recruitment/βarrestin recruitment. Although a more rigorous quantification of bias is required for validation of advanced compounds (see below), this straightforward analysis worked well for the initial stages of compound optimization.
We came to the project with numerous analogues already in hand. The original KOR structure–activity relationship (SAR) studies had focused mainly on the easily modified phenyl groups in both series. Figure 2b show data from the first probes published in each series and the compounds that were ultimately selected for in-depth investigation. An essential feature was the selection of an unbiased standard as a comparator; here, we used U69 or U50 for this purpose (Figure 2a).
One does not often have cause to celebrate irony in research, but here is a clear opportunity to do so: in at least one series, we were able to identify a G protein-biased agent through a screen based on βarrestin recruitment!
One Way or Another
Although a simple comparison of IC50 values allows for a general sense of potential bias, it can be misleading in assays that are contextually hard to compare. For example, a compound may have a potency (EC50) of 10 nM in one assay and a potency of 100 nM in another assay. At first glance, one might conclude: the agonist is biased for assay 1 over assay 2. However, this fails to consider the overall efficiency of the assay systems being considered. In a cell based signaling system that has a highly amplified response, an agonist may appear to be more potent than in a system that has a much more reserved window of response. Therefore, one can conclude little about biased agonism by simply comparing its performance between two assays. In order to account for the efficiency of the system, the performance of the agonist must be compared to the performance of a “known” agonist that can reveal the full potential of the assay system to generate a response. The “known” agonist is called the “reference agonist” and must produce the maximum response the system is capable of producing. Then the performance of the test compound can be compared to the reference, allowing for a normalization of its performance and accounting for the limitations of the system context.
A useful operational model that allows one to make valid comparisons across series is based on the models proposed by Black and Leff.23−26 A simplified version entails the calculation of normalized transduction coefficients to compare the action of a given test compound with the balanced standard (here, U69) in each assay (eq 1, where τ is agonist efficacy and KA is the equilibrium affinity constant). This being done, one can now calculate a bias factor for a given agonist across any two assays of choice (eq 2); the balanced standard by definition has a bias factor of 1.
While it is attractive to assign a number to a compound, it is important to keep in mind that bias is a comparison that depends on the reference ligand and circumstances that include the cell line, the species of the receptor, the coexpressed proteins, the modifications to the receptor, and the assay conditions. While it is attractive to assign a number to a compound, it should be kept in mind that bias is dependent upon a comparison. However, the assignment of bias can still provide useful in determining SAR; particularly when a desirable vs an undesirable signaling pathway has been determined.
 |
1 |
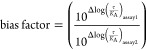 |
2 |
Using the operational model, isoquinolinone 2.1 and triazole 1.1 have bias factors of 31.4 and 61.2 for the [35S]GTPγS vs βarrestin2 recruitment (using the EFC method), respectively. They are clearly biased by this measure.
Contemporarily, other laboratories have published biased KOR ligands based on previously known structural classes. In separate work, the Javitz27 and Bohn28 laboratories reported that 6′-guanidinonaltrindole (6′-GNTI), a synthetic derivative of morphine first reported in 2001,29 has a complex pharmacology that includes “extreme bias” as partial agonist toward G protein and against the βarrestin pathway. One calculation of the bias factor for 6′-GNTI was 9.8.28 Another previously known morphine derivative, nalfurafine, was first synthesized in 199830 and approved in Japan for the treatment of pruritis in 2009.31 In 2017, Chavkin and co-workers reported that nalfurafine is highly biased toward the G protein pathway (measuring p38 phosphorylation vs extracellular signal-regulated kinases 1/2 (ERK1/2) phosphorylation, using U50 as the standard ligand).32 In 2013, the Roth group reported that the salvinorin A derivative 22-thiocyanatosalvinorin A, RB-64, was biased toward the G protein pathway (bias factor 25 at hKOR, using salvinorin A as the reference ligand33 or 96 at the mouse KOR34). The fact that RB-64 differs structurally at a single position from the nonbiased salvinorin A supports the view that ligand bias is a property susceptible to traditional SAR optimization.
Another aspect of SAR examined in our laboratories was the effect of the triazole ligand class on the downstream phosphorylation of ERK1/2 kinase (Figure 3).35 At the outset, we had no expectations how biased agents would affect this MAP kinase because ERK1/2 is involved in both G protein36−38 and βarrestin pathways.39,40 When additional structural modification was carried out in the triazole series, we learned that the nature of the aromatic ring attached to the N-4 position of the triazole centroid dramatically affected the degree of bias toward G protein activation over ERK1/2 in this series.
Figure 3.

Effect of N-4 substitution on G protein/ERK1/2 bias in a series of triazoles.35
Additional verification of bias was sought through the examination of other cellular measurements.41 A vivid demonstration of βarrestin recruitment is obtained from confocal imaging of βarrestin tagged with green fluorescent protein (GFP; Figure 4a). Upon βarrestin recruitment, the βarrestin–GFP construct, which is normally distributed throughout the cytosol, localizes at the nucleus (cf. the effect of 10 μM U50 at a high concentration to that from the biased isoquinolinone 2.1 at the same concentration). In addition, independent measurements of ligand binding in the presence of the cell permeant saponin and cellular impedance provided additional verification of strong bias for both isoquinolinone 2.1 and triazole 1.1. This is evident from the spider graph profiles for U69 versus our biased exemplars in Figure 4b.
Figure 4.
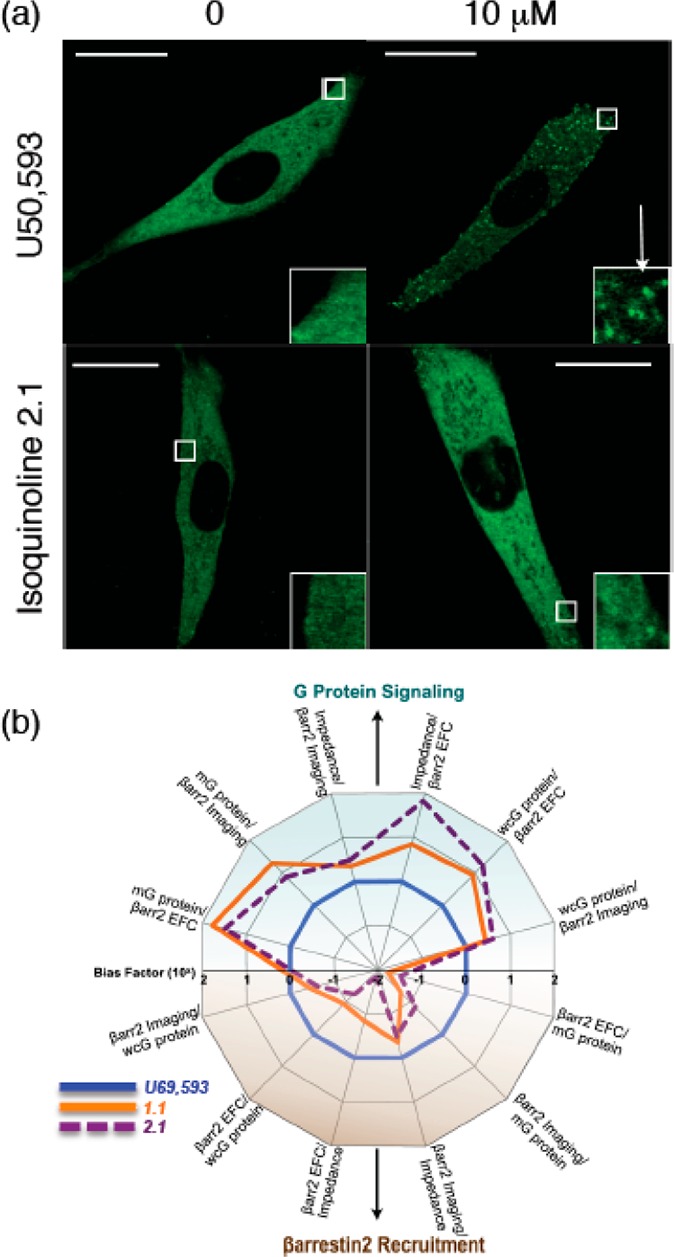
Additional assay and bias data for triazole 1.1 and isoquinolinone 2.1. (a) Confocal imaging of the effect of U69 or isoquinolinone 2.1 on βarrestin recruitment. (b) Bias trend across various measurement pairs for U69, triazole 1.1, and isoquinolinone 2.1. Adapted with permission from ref (41)
The primary cellular pathway of KOR signaling in the brain is the striatum, which is also a major regulator of dopamine activity. Accordingly, an important step toward verifying the action of KOR ligands is to examine their pharmacology in striatal membrane preparations from wild-type mice as well as from mice with knockouts of the KOR and, as a control, MOR. Such studies were carried out using endogenous agonistic dynorphin peptides, the literature antagonists norBNI and 5′-GNTI, and an example of the sulfonamide class discovered in the course of our MLI work.18,42 Besides showing that the activity of these agents could be reproduced in this closer-to-realistic cellular environment (as opposed to transfected CHO cells), the use of parallel knockout models enabled the insight that some ligands generally considered to be selective in fact operate though both the KOR and MOR pathways.
Mice
We approached the critical phase of testing our compounds in animal models with a combination of expectation and curiosity. The expectation was that our agonists, pending appropriate vetting for suitable pharmacokinetic (PK) properties, would be active nociceptive agents based on their in vitro potencies. Although slightly less certain, if only because there is less guidance from previous literature, it seemed reasonable to expect that we might see good anti-itch activity as well. Thus, the critical question was how functionally selective KOR agonists would differ physiologically and behaviorally compared to classical agents. A related question, for which we had no clue at the time, was how much bias would be needed to translate into any meaningful differences in biological outcomes.
The pharmacokinetic properties of KOR ligands have been a subject of particular concern. Specifically, the antagonists norBNI and JDTic have been reported to have an extended duration of action (over 2 weeks), a pragmatic concern with therapeutic usage (and leading to some controversy as to the origin of the effect).43−45 Cognizant of this history, we performed preliminary PK studies at an early stage of the present project, finding that parenterally delivered isoquinolinones and triazoles did indeed penetrate the blood–brain barrier and were cleared with reasonable (ca. 2 h) half-lives.41
Moving forward, our efforts mainly used triazole 1.1. Since many of the in vivo studies used U50 as the standard molecule, we repeated the bias measurements for 1.1 against U50 using the EFC assay to measure βarrestin recruitment and the [35S]-GTPγS binding assay for G protein pathway engagement. It remains a highly biased molecule under these circumstances, with a calculated bias factor of 28.46
Triazole 1.1 was found to have excellent antinociceptive activity in the mouse tail-flick model and suppressed chloroquine phosphate-induced scratching, with activity close to that of U50 in each assay. A series of experiments showed that the effects were due to on-target activity. Thus, the above effects were blocked by the KOR antagonist norBNI, and the antinociceptive effects were absent in KOR knockout mice. Moreover, the penetrance of the compounds to the striatum was confirmed by HPLC of homogenized brain extracts and pretreatment with either triazole 1.1 or U50 in vivo prevented subsequent binding of [3H]U69 in dissected striata.
But just as it was evident that the in vitro pharmacology translated nicely to the in vivo setting, it quickly became clear that these were not traditional KOR agonists. In general, KOR activation leads to down-regulation of dopamine release and therefore sedation. This can be observed in mice through opioid-induced changes in locomotion, a readily measurable parameter. Indeed, triazole 1.1 was found to result in essentially no change in ambulatory behavior in test mice under doses and conditions when U50 would lead to dramatically lowered movement.
At this stage, we teamed up with Professors Sara Jones and Thomas J. Martin at Wake Forest University for advanced brain physiology and behavioral studies. In the former, we wished to address the central question of whether there are differences in dopamine tone arising from treatment of a biased KOR agonist relative to a classical one. Remarkable differences were observed when voltammetry analysis of ex vivo slices from the nucleus accumbens cores or shells was carried out using U50 vs triazole 1.1.46 At most, only a modest dip in dopamine levels were observed in the latter case at the very highest doses for the triazole 1.1, which stands in strong contrast to the continuous dose-dependent changes typical of a classical KOR agonist (and seen here for U50; Figure 5).
Figure 5.
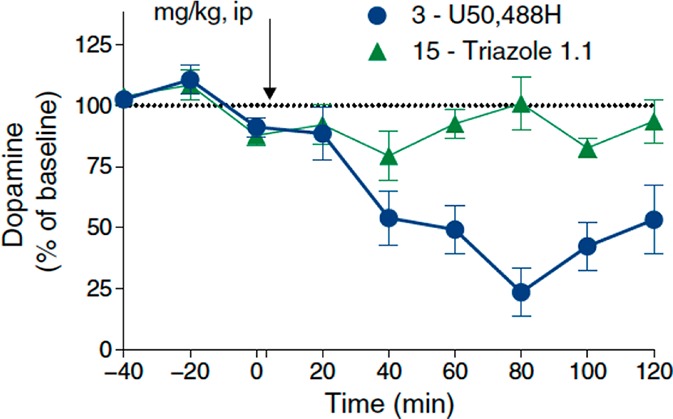
Effect of dopamine levels in vivo following administration of U50 v. triazole 1.1. Reproduced by permission from ref (46). Copyright 2016 AAAS.
The dysphoria associated with KOR activation and hypothesized to result from βarrestin involvement is notoriously hard to measure. It would be nice if mice were able to fill out questionnaires pertaining to mood on tiny clipboards, but this ability is currently lacking, despite our best efforts. Moreover, “aversion” is a complex phenomenon that may reflect anything from changes in fundamental brain chemistry, which is what we hope to measure here, to GI distress caused by a drug molecule. Of the various indirect ways of assessing aversion available, we looked for changes in intracranial self-stimulation (ICSS) behavior in rats with ventral tegmental area (VTA)-implanted brain electrodes that are trained to press a lever for self-stimulation in response to a light cue. The suppression of a VTA ICSS response is interpreted to mean that the animal is less “interested” in pleasure due to decreased dopamine levels.
Once again, significant differences were observed following treatment of the rats with triazole 1.1 under conditions when substantial changes could be observed with U50.46 While U50 decreased ICSS as expected consistent with its known dysphoric and sedating properties, 1.1 had no effect in this assay consistent with its lack of effects on forebrain DA. More interesting, these same doses of 1.1 were able to inhibit the ability of abdominal inflammation to decrease ICSS, an effect shared by clinically useful analgesics such as ketoprofen and morphine. These data indicate that the biased signaling of 1.1 found in vitro translated to the desired effects in vivo, namely, a preservation of the analgesic properties with no signs of the sedating or dysphoric effects of typical balanced KOR agonists that have limited their development as therapeutics. A similar separation of desired/undesired effects was observed by the Roth group using the biased salvinorin A-derivative RB-64.34
Where Do We Go From Here?
To date, the results are consistent with the primary premise that KOR G protein pathway activation over βarrestin recruitment will enhance therapeutic activity and reduce unfavorable side effects. However, as always, it is advisable to modulate one’s expectations for advancement of any translational candidate, and we are still in the early going here.
We expect that future efforts toward GPCR targeted therapeutics will increasingly take bias into account. From the perspective of a working medicinal chemist, this pragmatically means adding just one more optimization parameter to the already-daunting list facing drug discovery aspirants (albeit a parameter that can morph dependent on the experimental context). For GRCRs with information about the relative roles of different intracellular pathways, this represents an appealing hypothesis driver for research. Conversely, new tools able to differentiate between pathways may enable new understanding of less-explored GPCR targets.
Of course, any journey toward a new drug starts with a single step or, more literally, a single molecule. Although determining “where to begin” is always challenging, there is no reason to think that there is anything particularly difficult about finding a suitable starting point for developing a biased agonist for GPCR drug discovery. In our case, we were seeking new ligands for an extremely well established drug target for which essentially all chemotypes led to balanced activation of the receptor. By deliberately setting out to find structurally novel ligands, and with the benefit of a little luck (for which we think no apology is necessary), we found multiple biased classes that we could optimize using standard medicinal chemistry. Similarly, successful efforts to create new chemotypes for the KOR47 vs the MOR48 from de novo in silico design have been reported.
Such stories will only become more common, particularly as the scientific community learns more about the molecular origins of GPCR bias through structural biology49,50 and with the aid of novel chemical tools. We hope to continue to contribute to this renaissance of GPCR biology through the development of our biased agents for translational work and further discovery and functional elucidation in other settings.
Acknowledgments
We gratefully acknowledge the dedication and efforts of our many collaborators and co-workers named in the text and references. We particularly thank Kevin Frankowski, who has made extensive contributions to this project from its outset to the present. We also thank Thomas J. Martin for his contributions to this manuscript.
Glossary
ABBREVIATIONS
- EFC
enzyme fragment complementation
- GFP
green fluorescent protein
- 6′-GNTI
6′-guanidinonaltrindole
- GPCR
G protein-coupled receptor
- GTP
guanosine triphosphate
- ICSS
intracranial self-stimulation
- KOR
kappa opioid receptor
- MOR
mu opioid receptor
- norBNI
nor-binaltorphimine
- PK
pharmacokinetics
- SAR
structure–activity relationship
- U50
U50,488H
- U69
U69,683
- VTA
ventral tegmental area
Author Contributions
The manuscript was written by both authors.
Two NIH center programs enabled our discovery of isoquinolinones (the KU Chemical Methodology and Library Development center, funded by NIGMS 5P50GM069663, to J.A., PI) and the triazoles (the MLI, which was a collaboration between the KU Specialized Chemistry Center (5U54HG005031, to J.A., PI), the Conrad Prebys Center for Chemical Genomics at the Sanford-Burnham Medical Research Institute (5U54HG005033 (John Reed, PI), and screening grants awarded by the National Institute on Drug Abuse (NIDA) to Lawrence S. Barak (1X01DA026208) and Marc G. Caron (5U01DA022950)). Initial screening and ongoing characterization were also carried out by the Psychoactive Drug Screening Program at the University of North Carolina, Chapel Hill (National Institute of Mental Health contract # HHSN-271-2008-00025-C, Bryan Roth, PI). We gratefully acknowledge NIDA for continuing support through 5R01DA031927 (to L.M.B. and J.A., co-PIs).
The authors declare the following competing financial interest(s): The coauthors are co-inventors on several patents that are related to the studies described in this article.
Dedication
We dedicate this paper to the memory of Greg Roth.
References
- Rosenbaum D. M.; Rasmussen S. G. F.; Kobilka B. K. The Structure and Function of G-Protein-Coupled Receptors. Nature 2009, 459, 356–363. 10.1038/nature08144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overington J. P.; Al-Lazikani B.; Hopkins A. L. How Many Drug Targets Are There?. Nat. Rev. Drug Discovery 2006, 5, 993–996. 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Lefkowitz R. J.; Shenoy S. K. Transduction of Receptor Signals by β-Arrestins. Science 2005, 308, 512–517. 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- Smith J. S.; Rajagopal S. The β-Arrestins: Multifunctional Regulators of G Protein-coupled Receptors. J. Biol. Chem. 2016, 291, 8969–8977. 10.1074/jbc.R115.713313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn L. M.; Lefkowitz R. J.; Gainetdinov R. R.; Peppel K.; Caron M. G.; Lin F.-T. Enhanced Morphine Analgesia in Mice Lacking β-Arrestin 2. Science 1999, 286, 2495–2498. 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Jacobson K. A. New Paradigms in GPCR Drug Discovery. Biochem. Pharmacol. (Amsterdam, Neth.) 2015, 98, 541–555. 10.1016/j.bcp.2015.08.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luttrell L. M.; Maudsley S.; Bohn L. M. Fulfilling the Promise of ″Biased″ G Protein-Coupled Receptor Agonism. Mol. Pharmacol. 2015, 88, 579–588. 10.1124/mol.115.099630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rankovic Z.; Brust T. F.; Bohn L. M. Biased Agonism: An Emerging Paradigm in GPCR Drug Discovery. Bioorg. Med. Chem. Lett. 2016, 26, 241–250. 10.1016/j.bmcl.2015.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wootten D.; Miller L. J.; Koole C.; Christopoulos A.; Sexton P. M. Allostery and Biased Agonism at Class B G Protein-Coupled Receptors. Chem. Rev. (Washington, DC, U. S.) 2017, 117, 111–138. 10.1021/acs.chemrev.6b00049. [DOI] [PubMed] [Google Scholar]
- Bohn L. M.; McDonald P. H. Seeking Ligand Bias: Assessing GPCR Coupling to β-Arrestins For Drug Discovery. Drug Discovery Today: Technol. 2010, 7, e37–e42. 10.1016/j.ddtec.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye S. V. The Art of the Chemical Probe. Nat. Chem. Biol. 2010, 6, 159–161. 10.1038/nchembio.296. [DOI] [PubMed] [Google Scholar]
- Frankowski K. J.; Hirt E. E.; Zeng Y.; Neuenswander B.; Fowler D.; Schoenen F.; Aubé J. Synthesis of N-Alkyl-octahydroisoquinolin-1-one-8-carboxamide Libraries Using a Tandem Diels–Alder/Acylation Sequence. J. Comb. Chem. 2007, 9, 1188–1192. 10.1021/cc700127f. [DOI] [PubMed] [Google Scholar]
- Frankowski K. J.; Ghosh P.; Setola V.; Tran T. B.; Roth B. L.; Aubé J. N-Alkyl-octahydroisoquinolin-1-one-8-carboxamides: Selective and Nonbasic kappa-Opioid Receptor Ligands. ACS Med. Chem. Lett. 2010, 1, 189–193. 10.1021/ml100040t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh P.; Aubé J. Resolution of Carboxylic Acids Using Copper(I)-Promoted Removal of Propargylic Esters under Neutral Conditions. J. Org. Chem. 2011, 76, 4168–4172. 10.1021/jo200433w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slauson S. R.; Pemberton R.; Ghosh P.; Tantillo D. J.; Aubé J. Domino Acylation/Diels–Alder Synthesis of N-Alkyl-octahydroisoquinolin-1-one-8-carboxylic Acids under Low-Solvent Conditions. J. Org. Chem. 2015, 80, 5260–5271. 10.1021/acs.joc.5b00804. [DOI] [PubMed] [Google Scholar]
- Hedrick M. P.; Gosalia P.; Frankowski K.; Shi S.; Prisinzano T. E.; Schoenen F.; Aubé J.; Su Y.; Vasile S.; Sergienko E.; Gray W.; Hariharan S.; Ghosh P.; Milan L.; Heynen-Genel S.; Chung T. D. Y.; Dad S.; Caron M.; Bohn L. M.; Barak L. S.. Selective KOP Receptor Agonists: Probe 1 & Probe 2. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, 2010. [PubMed] [Google Scholar]
- Hedrick M. P.; Gosalia P.; Frankowski K.; Whipple D. A.; Shi S.; Prisinzano T. E.; Schoenen F.; Aubé J.; Su Y.; Vasile S.; Sergienko E.; Gray W.; Hariharan S.; Milan L.; Heynen-Genel S.; Chung T. D. Y.; Dad S.; Caron M.; Bohn L. M.; Barak L. S.. Selective KOP Receptor Antagonists: Probe 1. In Probe Reports from the NIH Molecular Libraries Program; National Center for Biotechnology Information (US): Bethesda, MD, 2010. [PubMed] [Google Scholar]
- Frankowski K. J.; Hedrick M. P.; Gosalia P.; Li K.; Shi S.; Whipple D.; Ghosh P.; Prisinzano T. E.; Schoenen F. J.; Su Y.; Vasile S.; Sergienko E.; Gray W.; Hariharan S.; Milan L.; Heynen-Genel S.; Mangravita-Novo A.; Vicchiarelli M.; Smith L. H.; Streicher J. M.; Caron M. G.; Barak L. S.; Bohn L. M.; Chung T. D.; Aubé J. Discovery of Small Molecule Kappa Opioid Receptor Agonist and Antagonist Chemotypes through a HTS and Hit Refinement Strategy. ACS Chem. Neurosci. 2012, 3, 221–236. 10.1021/cn200128x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udenfriend S.; Meienhofer J.. Opioid Peptides: Biology, Chemistry, and Genetics: The Peptides: Analysis, Synthesis, Biology; Elsevier: 2014; Vol. 6. [Google Scholar]
- Szmuszkovicz J., U-50,488 and the κ Receptor: A Personalized Account Covering the Period 1973 to 1990. In Progress in Drug Research; Springer: 1999; pp 167–195. [DOI] [PubMed] [Google Scholar]
- Roth B. L.; Baner K.; Westkaemper R.; Siebert D.; Rice K. C.; Steinberg S.; Ernsberger P.; Rothman R. B. Salvinorin A: A Potent Naturally Occurring Nonnitrogenous Kappa Opioid Selective Agonist. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 11934–11939. 10.1073/pnas.182234399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding W. W.; Tidgewell K.; Byrd N.; Cobb H.; Dersch C. M.; Butelman E. R.; Rothman R. B.; Prisinzano T. E. Neoclerodane Diterpenes as a Novel Scaffold for Opioid Receptor Ligands. J. Med. Chem. 2005, 48, 4765–4771. 10.1021/jm048963m. [DOI] [PubMed] [Google Scholar]
- Black J. W.; Leff P. Operational Models of Pharmacological Agonism. Proc. R. Soc. London, Ser. B 1983, 220, 141. 10.1098/rspb.1983.0093. [DOI] [PubMed] [Google Scholar]
- Kenakin T.; Watson C.; Muniz-Medina V.; Christopoulos A.; Novick S. A Simple Method For Quantifying Functional Selectivity and Agonist Bias. ACS Chem. Neurosci. 2012, 3, 193–203. 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T.; Christopoulos A. Signalling Bias in New Drug Discovery: Detection, Quantification and Therapeutic Impact. Nat. Rev. Drug Discovery 2013, 12, 205–216. 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Stahl E. L.; Zhou L.; Ehlert F. J.; Bohn L. M. A Novel Method for Analyzing Extremely Biased Agonism at G Protein-Coupled Receptors. Mol. Pharmacol. 2015, 87, 866–877. 10.1124/mol.114.096503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rives M.-L.; Rossillo M.; Liu-Chen L.-Y.; Javitch J. A. 6′-Guanidinonaltrindole (6′-GNTI) Is A G Protein-Biased κ-Opioid Receptor Agonist That Inhibits Arrestin Recruitment. J. Biol. Chem. 2012, 287, 27050–27054. 10.1074/jbc.C112.387332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmid C. L.; Streicher J. M.; Groer C. E.; Munro T. A.; Zhou L.; Bohn L. M. Functional Selectivity Of 6′-Guanidinonaltrindole (6′-GNTI) at κ-Opioid Receptors in Striatal Neurons. J. Biol. Chem. 2013, 288, 22387–22398. 10.1074/jbc.M113.476234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S. K.; Jones R. M.; Metzger T. G.; Ferguson D. M.; Portoghese P. S. Transformation of a κ-Opioid Receptor Antagonist to a κ-Agonist by Transfer of a Guanidinium Group from the 5‘- to 6‘-Position of Naltrindole. J. Med. Chem. 2001, 44, 2073–2079. 10.1021/jm010095v. [DOI] [PubMed] [Google Scholar]
- Nagase H.; Hayakawa J.; Kawamura K.; Kawai K.; Takezawa Y.; Matsuura H.; Tajima C.; Endo T. Discovery of a Structurally Novel Opioid Kappa-Agonist Derived from 4,5-Epoxymorphinan. Chem. Pharm. Bull. 1998, 46, 366–369. 10.1248/cpb.46.366. [DOI] [PubMed] [Google Scholar]
- Inui S. Nalfurafine Hydrochloride to Treat Pruritus: A Review. Clin., Cosmet. Invest. Dermatol. 2015, 8, 249–255. 10.2147/CCID.S55942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schattauer S. S.; Kuhar J. R.; Song A.; Chavkin C. Nalfurafine is a G-Protein Biased Agonist Having Significantly Greater Bias at the Human than Rodent Form of the Kappa Opioid Receptor. Cell. Signalling 2017, 32, 59–65. 10.1016/j.cellsig.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K. L.; Scopton A. P.; Rives M. L.; Bikbulatov R. V.; Polepally P. R.; Brown P. J.; Kenakin T.; Javitch J. A.; Zjawiony J. K.; Roth B. L. Identification of Novel Functionally Selective Kappa-Opioid Receptor Scaffolds. Mol. Pharmacol. 2014, 85, 83–90. 10.1124/mol.113.089649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White K. L.; Robinson J. E.; Zhu H.; DiBerto J. F.; Polepally P. R.; Zjawiony J. K.; Nichols D. E.; Malanga C. J.; Roth B. L. The G Protein-Biased Kappa-Opioid Receptor Agonist RB-64 Is Analgesic with a Unique Spectrum of Activities In Vivo. J. Pharmacol. Exp. Ther. 2015, 352, 98–109. 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovell K. M.; Frankowski K. J.; Stahl E. L.; Slauson S. R.; Yoo E.; Prisinzano T. E.; Aubé J.; Bohn L. M. Structure–Activity Relationship Studies of Functionally Selective Kappa Opioid Receptor Agonists that Modulate ERK 1/2 Phosphorylation While Preserving G Protein Over βArrestin2 Signaling Bias. ACS Chem. Neurosci. 2015, 6, 1411–1419. 10.1021/acschemneuro.5b00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn L. M.; Belcheva M. M.; Coscia C. J. Mitogenic Signaling via Endogenous κ-Opioid Receptors in C6 Glioma Cells. J. Neurochem. 2000, 74, 564–573. 10.1046/j.1471-4159.2000.740564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belcheva M. M.; Clark A. L.; Haas P. D.; Serna J. S.; Hahn J. W.; Kiss A.; Coscia C. J. μ and κ Opioid Receptors Activate ERK/MAPK Via Different Protein Kinase C Isoforms and Secondary Messengers in Astrocytes. J. Biol. Chem. 2005, 280, 27662–27669. 10.1074/jbc.M502593200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLennan G. P.; Kiss A.; Miyatake M.; Belcheva M. M.; Chambers K. T.; Pozek J. J.; Mohabbat Y.; Moyer R. A.; Bohn L. M.; Coscia C. J. Kappa Opioids Promote The Proliferation Of Astrocytes Via Gβγ and β-Arrestin 2-Dependent MAPK-Mediated Pathways. J. Neurochem. 2008, 107, 1753–1765. 10.1111/j.1471-4159.2008.05745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas M. R.; Macey T. A.; Lowe J. D.; Chavkin C. Kappa Opioid Receptor Activation of p38 MAPK is GRK3-and Arrestin-Dependent in Neurons and Astrocytes. J. Biol. Chem. 2006, 281, 18081–18089. 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas M. R.; Land B. B.; Aita M.; Xu M.; Barot S. K.; Li S.; Chavkin C. Stress-Induced p38 Mitogen-Activated Protein Kinase Activation Mediates κ-Opioid-Dependent Dysphoria. J. Neurosci. 2007, 27, 11614–11623. 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou L.; Lovell K. M.; Frankowski K. J.; Slauson S. R.; Phillips A. M.; Streicher J. M.; Stahl E.; Schmid C. L.; Hodder P.; Madoux F.; Cameron M. D.; Prisinzano T. E.; Aubé J.; Bohn L. M. Development of Functionally Selective, Small Molecule Agonists at Kappa Opioid Receptors. J. Biol. Chem. 2013, 288, 36703–36716. 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankowski K. J.; Slauson S. R.; Lovell K. M.; Phillips A. M.; Streicher J. M.; Zhou L.; Whipple D. A.; Schoenen F. J.; Prisinzano T. E.; Bohn L. M.; Aubé J. Potency Enhancement of the Kappa Opioid Receptor Antagonist Probe ML140 Through Sulfonamide Constraint Utilizing a Tetrahydroisoquinoline Motif. Bioorg. Med. Chem. 2015, 23, 3948–3956. 10.1016/j.bmc.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas M. R.; Yang T.; Schreiber S.; DeFino M.; Kwan S. C.; Li S.; Chavkin C. Long-Acting κ Opioid Antagonists Disrupt Receptor Signaling and Produce Noncompetitive Effects By Activating C-Jun N-Terminal Kinase. J. Biol. Chem. 2007, 282, 29803–29811. 10.1074/jbc.M705540200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishioka S.; Kiguchi N.; Kobayashi Y.; Yamamoto C.; Saika F.; Wakida N.; Ko M.-C.; Woods J. H. Pharmacokinetic Evidence for the Long-Lasting Effect of nor-Binaltorphimine, a Potent Kappa Opioid Receptor Antagonist, In Mice. Neurosci. Lett. 2013, 552, 98–102. 10.1016/j.neulet.2013.07.040. [DOI] [PubMed] [Google Scholar]
- Patkar K. A.; Wu J.; Ganno M. L.; Singh H. D.; Ross N. C.; Rasakham K.; Toll L.; McLaughlin J. P. Physical Presence of Nor-Binaltorphimine in Mouse Brain over 21 Days after a Single Administration Corresponds to Its Long-Lasting Antagonistic Effect on κ-Opioid Receptors. J. Pharmacol. Exp. Ther. 2013, 346, 545. 10.1124/jpet.113.206086. [DOI] [PubMed] [Google Scholar]
- Brust T. F.; Morgenweck J.; Kim S. A.; Rose J. H.; Locke J. L.; Schmid C. L.; Zhou L.; Stahl E. L.; Cameron M. D.; Scarry S. M.; Aubé J.; Jones S. R.; Martin T. J.; Bohn L. M. Biased Agonists of the Kappa Opioid Receptor Suppress Pain and Itch Without Causing Sedation or Dysphoria. Sci. Signaling 2016, 9, ra117. 10.1126/scisignal.aai8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Z.; Huang X.-P.; Mangano T. J.; Zou R.; Chen X.; Zaidi S. A.; Roth B. L.; Stevens R. C.; Katritch V. Structure-Based Discovery of New Antagonist and Biased Agonist Chemotypes for the Kappa Opioid Receptor. J. Med. Chem. 2017, 60, 3070–3081. 10.1021/acs.jmedchem.7b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manglik A.; Lin H.; Aryal D. K.; McCorvy J. D.; Dengler D.; Corder G.; Levit A.; Kling R. C.; Bernat V.; Hübner H.; Huang X.-P.; Sassano M. F.; Giguère P. M.; Löber S.; Da D.; Scherrer G.; Kobilka B. K.; Gmeiner P.; Roth B. L.; Shoichet B. K. Structure-Based Discovery of Opioid Analgesics with Reduced Side Effects. Nature 2016, 537, 185–190. 10.1038/nature19112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J. Y.; Lee S. Y.; Kim H. R.; Seo M.-D.; Chung K. Y. Structural mechanism of GPCR-Arrestin Interaction: Recent Breakthroughs. Arch. Pharmacal Res. 2016, 39, 293–301. 10.1007/s12272-016-0712-1. [DOI] [PubMed] [Google Scholar]
- Latorraca N. R.; Venkatakrishnan A. J.; Dror R. O. GPCR Dynamics: Structures in Motion. Chem. Rev. (Washington, DC, U. S.) 2017, 117, 139–155. 10.1021/acs.chemrev.6b00177. [DOI] [PubMed] [Google Scholar]