

Abstract
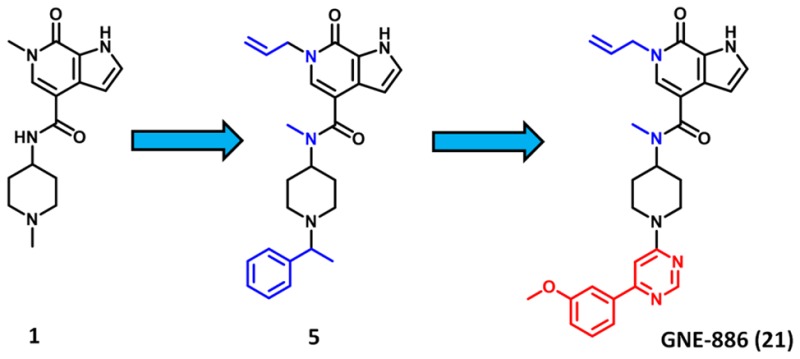
The biological function of bromodomains, epigenetic readers of acetylated lysine residues, remains largely unknown. Herein we report our efforts to discover a potent and selective inhibitor of the bromodomain of cat eye syndrome chromosome region candidate 2 (CECR2). Screening of our internal medicinal chemistry collection led to the identification of a pyrrolopyridone chemical lead, and subsequent structure-based drug design led to a potent and selective CECR2 bromodomain inhibitor (GNE-886) suitable for use as an in vitro tool compound.
Keywords: Epigenetics, Bromodomain, CECR2, Probe
Bromodomains are approximately 110 amino acid residue protein modules that function as epigenetic readers recognizing acylated lysine residues on histone tails, as well as other proteins.1−6 Bromodomains encompass 61 individual modules located on 46 proteins,7−9 but the biological function of individual bromodomain family members remains largely unclear. The most widely studied subclass of bromodomains is the bromodomain and extraterminal domain (BET) family, which is comprised of bromodomain-containing protein 2 (BRD2), bromodomain-containing protein 3 (BRD3), bromodomain-containing protein 4 (BRD4), and bromodomain testis-specific protein (BRDT), each of which contains dual bromodomains.8,9 However, interest in non-BET bromodomain inhibitors has increased significantly, as demonstrated by the number of selective bromodomain inhibitor tool compounds that have been disclosed recently.8−10
Cat eye syndrome chromosome region candidate 2 (CECR2) is a bromodomain-containing transcription factor that, with the ISWI-type catalytic subunit SMARCA1/SNF2L or SMARCA5/SNF2H, forms the heterodimeric chromatin remodeling complex CERF (CECR2-containing remodeling factor).11,12 CECR2 has been found to be important in DNA damage response (DDR), as CECR2 knockdown has been shown to result in decreased γ-H2AX foci formation.13Cecr2 deletion has also been associated with neural tube defect (NTD) exencephaly.14 The detailed biological function of the CECR2 bromodomain itself remains unknown, although overexpression of the bromodomain can inhibit formation of γ-H2AX and 53BP1 foci after irradiation of cells.13 This suggests that the CECR2 bromodomain has a binding function important in the response to DNA damage, and selective small-molecule inhibitors will be critical in better defining this function and any role it may have in disease states. Recently, the Structural Genomics Consortium (SGC) disclosed a CECR2 inhibitor identified in collaboration with Novartis.15 This compound, NVS-CECR2-1 has high-affinity activity (CECR2 AlphaScreen IC50: 47 nM) with strong cellular activity in a fluorescence recovery after photobleaching (FRAP) assay at 0.1 μM; however, selectivity data across the bromodomain family is not given. As part of our non-BET bromodomain probe discovery platform,16−18 we herein report our efforts to identify a potent and selective CECR2 bromodomain inhibitor. To identify CECR2 lead chemical matter, a collection of approximately 600 molecules from our internal bromodomain medicinal chemistry collection was screened in a single point CECR2 AlphaScreen (AS) assay. From this screen we identified a number of hits, including 6-methyl-N-(1-methylpiperidin-4-yl)-7-oxo-6,7-dihydro-1H-pyrrolo[2,3-c]pyridine-4-carboxamide (1). Subsequent titration in both AS and fluorescence resonance energy transfer (FRET) assays revealed 1 to have pronounced selectivity over the representative BET protein BRD4. Indeed, IC50 values were >20 μM for both BRD4 bromodomains. This was critically important, due to the strong phenotype that has been reported for BET inhibitors.8,9 While compound 1 was found to be more potent in TAF1(2) compared to CECR2, we observed that compound 1 had slight selectivity over BRD9, albeit only 2-fold (Figure 1A). This was the only compound from this screening set where we found a preference for CECR2 over BRD9, which coupled with the BRD4 selectivity made 1 an attractive starting point for subsequent lead optimization. We therefore endeavored to further improve the CECR2 potency while increasing our selectivity window against BRD4, BRD9, and TAF1(2) and maintaining high selectivity over other non-BET family members.
Figure 1.
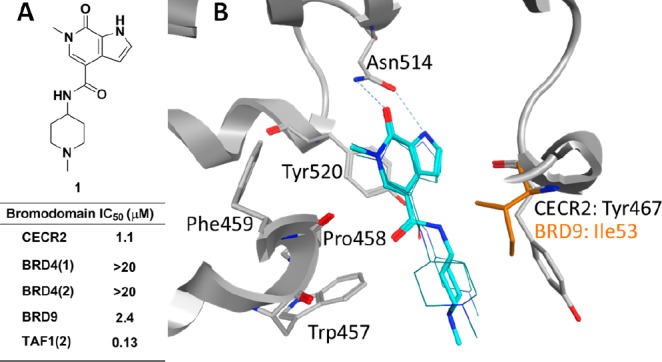
(A) Assay data for CECR2 screening hit 1. IC50 values are the average from at least two independent experiments. (B) Model of 1 (cyan) in CECR2 structure (PDB code 3NXB): thick sticks of 1 represent the conformation used to design the tertiary amide, while wire frames of 1 represent other possible conformations from the modeling. The CECR2 protein structure is shown as a gray ribbon with gray sticks for side chains containing the WPF shelf (Trp457, Pro458, and Phe459), the gatekeeper residue (Tyr520), and the conserved Asn514. Also shown are the side chain Tyr467 and its corresponding residue Ile53 in BRD9 (orange).
Routine crystallography proved to be challenging for our CECR2 inhibitors; however, we were confident in predicting the binding mode of 1 in CECR2 based on previous work with this class of chemical matter.17 Modeling of 1 bound to the CECR2 structure suggests that the pyridone carbonyl and N-methyl groups serve as the acetyl-lysine (KAc) mimetic, with the pyridone carbonyl and pyrrole forming a 2-point hydrogen-bonding interaction with the highly conserved Asn514 (Figure 1B). The gatekeeper Tyr520 forms a π-stacking interaction with the pyrrolopyridone. The N-methylpiperidine amide at the 4-position appeared capable of adopting multiple low-energy conformations, and three of the most probable conformations are shown in Figure 1B. In all three conformations, the piperidine extended into the ZA channel, forming hydrophobic interactions with Trp457 of the lipophilic shelf region (which conforms to the common “WPF” motif in CECR2)17 and Tyr467 from the αZ helix. From the model, we envisioned that the introduction of a tertiary amide would improve selectivity by creating unfavorable interactions with Ile53 in BRD9 and the WPF shelf in TAF1(2).
Gratifyingly, we observed that the incorporation of the N-methyl amide 2 produced a significant increase in selectivity over BRD9, as predicted by the model, as well as TAF1(2) while maintaining CECR2 affinity (Table 1). Extending further with an N-isopropyl piperidine 3 to the α-methylbenzyl piperidine 4 yielded a slight gain in potency although the nonspecific hydrophobic interactions yielded equivalent potency improvements across our internal bromodomain panel.
Table 1. SAR for N-Alkyl Substitutions Extending from the Piperidinea.

All bromodomain assays were run in TR-FRET format. IC50 data are an average of at least two independent experiments. Assay tables with standard deviations are located in the Supporting Information. A full table of data with standard deviations for these and other N-alkyl substituents can be found in the Supporting Information.
We recently reported that small hydrophobic channels can be induced in BRD9 and CECR2 bromodomains between the tyrosine gatekeeper and the lipophilic shelf (GPP in BRD9,WPF in CECR2) by replacing the pyrrolopyridone methyl with small aliphatic unsaturated chains.17 In that study we observed that, in the case of CECR2, an allyl group extending from the pyridone nitrogen was the optimal substituent, yielding CECR2 potency similar to the N-methyl analogue and significantly enhanced selectivity over the majority of our internal bromodomain panel. Incorporation of the N-allyl substituent onto our 4-carboxamide scaffold to make compound 5 yielded a similar improvement in selectivity to that we had previously reported. We were also pleased to observe significant selectivity over BRD4(2), as well as CBP, BRPF1, and TAF1(1) (TR-FRET IC50’s > 20 μM, data not shown); however, only small improvements in selectivity were observed over BRD9. This is not surprising, as BRD9 is able to form the same hydrophobic pocket as CECR2 to accommodate the hydrophobic allyl chain. TAF1(2) potency only decreased 4-fold, which was expected, as we have found that TAF1(2) is able to tolerate such hydrophobic substituents through rearrangement of the conserved binding-pocket water network.17
To improve selectivity against TAF1(2), we next focused on substitution at the pyrrolopyridone 2-position leading into the BC loop region, which we hypothesized might not be tolerated by TAF1(2) but might introduce further favorable van der Waals interactions with CECR2. Introduction of a methyl at the 2-position (compounds 6, 7) did, in fact, result in improved selectivity over TAF1(2) (Table 1). However, the BRD9 affinity was increased at least 5-fold. Interestingly, we observed very little preference between enantiomers 6 and 7 in binding to any of the bromodomains assayed.
A cocrystal structure of compound 6 bound to CECR2 (2.7 Å, PDB 5V84) confirmed the predicted overall binding mode of the core (Figure 2). The allyl folds into the hydrophobic channel with the backstop residue Phe459 from the WPF shelf shifting slightly toward Met506 in order to accommodate binding. The allyl forms hydrophobic contacts with Pro458 and Phe459 as well as with Tyr520, which also serve to constrain the channel and prevent the deeper penetration that we have observed with crotonyl ligands binding to BRD9. The amide was found to be nearly orthogonal to the pyrrolopyridone core and occupied space that became accessible due to the induced fit (Supporting Information Figure S1). The carbonyl forms a direct hydrogen bond with the ZA loop Asp464, while the N-methyl rests in the cavity, where it makes hydrophobic interactions with the WPF shelf and the gatekeeper. The piperidine makes additional hydrophobic contacts extending through the ZA channel with a water mediated interaction between the protonated piperidine and Asp464. The benzylic group was not observed to make specific interactions and indeed had significant rotational freedom as it exited the ZA channel into solvent, which would explain that lack of potency difference between the separated enantiomers.
Figure 2.
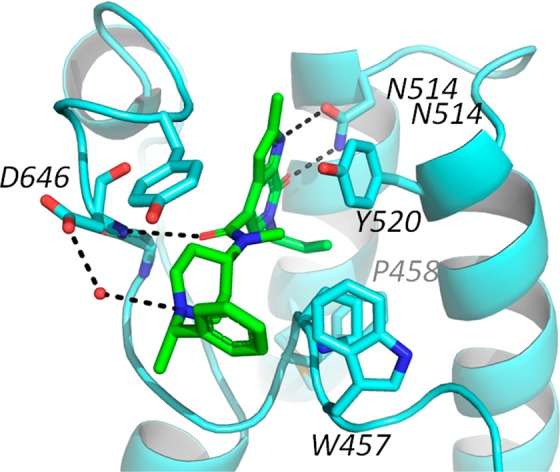
Compound 6 bound to CECR2 [PDB 5V84]. Compound 6 is shown as green sticks and H-bond interactions are represented as dashes.
Additional substituted benzylic analogs yielded little to no improvements in potency (data not shown). We therefore examined more constrained substitutions on the piperidine to reduce the entropic penalty resulting from the freely rotating benzylic substituent, which led to the identification of the 4-pyrimidine 8. Compound 8 has potency comparable to that of 5, although selectivity versus BRD9 and TAF1(2) is decreased (Table 2). We hypothesized that the pyrimidine heteroatoms may be able to form water-mediated interactions to residues in the ZA channel, as docking exercises did not indicate a potential for a direct hydrogen bonding interaction. Additional substitution was found to be beneficial at the 4-position of the pyrimidine ring. Starting from trifluoromethyl 9, we observed that increasing steric bulk to the methoxy 10, cyclopropyl 11, and t-butyl 12 yielded small potency increases for CECR2, with IC50 values now below 100 nM.
Table 2. SAR for N-Aryl Substitutions on the Piperidine Corea.

All bromodomain assays were run in TR-FRET format. IC50 data are an average of at least two independent experiments.
Introducing a methyl group at the pyrimidine 2-position to make disubstituted analogs 13, 14, and 15 maintained the improved CECR2 potency, although BRD9 potency was also maintained. As the tert-butyl group of 12 may serve as a spatial isostere to a phenyl, we introduced increased steric bulk with aromatic substituents. Five-membered aromatic rings such as pyrazole 16 and furan 17 did not significantly improve either selectivity or potency, nor did ortho-substituted phenyl groups 18–20. Meta-substitution quickly proved superior, possibly due to a decreased torsion-angle strain relative to ortho-substituted analogs, as the 3-methoxy phenyl 21 yielded significantly improved potency (16 nM).
Cellular permeability of compound 21 was assessed by measuring the displacement of a CECR2-ZsGreen fusion protein from chromatin.19 Protein displaced from the chromatin forms large aggregates, which can be visualized as fluorescent puncta by microscopy (Supporting Information). These features (dots) can be quantified as a function of compound dose. Compound 21 was found to have a CECR2 "dot" EC50 of 370 nM, and was therefore subjected to further analysis. The selectivity of compound 21 across the broader bromodomain family was assessed by profiling in a BROMOscan panel (Supporting Information for full data table).20
Compound 21 had a wide selectivity over the 40 bromodomains tested, with only BRD9 (KD = 2000 μM) and its homologue BRD7 (KD = 1100 μM), as well as TAF1(2) (KD = 0.62 μM) and its homologue TAF1L(2) (KD = 2400 μM), showing significant binding. While the selectivity window was reduced in comparison to the TR-FRET data, compound 21 still maintained a 2 log window over BET family members, therefore minimizing the risk of observing BET inhibition phenotypes. Compound 21 was also screened against a panel of 35 diverse kinases, with no kinase inhibited over 20% at 1 μM (Supporting Information). Finally, compound 21 (GNE-886) had a highly favorable measured kinetic solubility of 122 μM and was therefore considered an appropriate in vitro tool compound for the CECR2 bromodomain (Figure 3).
Figure 3.

Profile of GNE-886. All bromodomain assays were run in FRET format. IC50 data are an average of at least two independent experiments. bLE = −RT ln(IC50)/N heavy atoms. LLE = pIC50 – cLogP.
In summary, we identified pyrrolopyridone-4-carboxamide compound 1 through screening of our internal medicinal chemistry collection as a promising chemical lead for identification of a small-molecule inhibitor of the CECR2 bromodomain. Structure-based lead optimization efforts involved the inducement of a hydrophobic pocket with an allyl substituent, occupying a small cavity in the binding pocket with an N-methyl amide and introducing rigidified biaryl substituents into the ZA channel toward solvent-exposed space. This led to the identification of compound 21, suitable for use in in vitro phenotypic screening.
Acknowledgments
We thank the staff at beamline SER-CAT 22BM of the Advanced Photon Source. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We also thank Phil Bergeron and Tom Pillow for assistance during manuscript preparation, and Mengling Wong, Michael Hayes, and Amber Guillen for compound purification. Baiwei Lin, Yanzhou Liu, Deven Wang, and Yutao Jiang are acknowledged for analytical support. Grady Howes, Jan Seerveld, Hao Zheng, Ted Peters, Gigi Yuen, and Jeff Blaney are also recognized for help with compound management and logistics.
Glossary
Abbreviations
- BET
bromodomain and extra terminal domain
- BRD2
bromodomain-containing protein 2
- BRD3
bromodomain-containing protein 3
- BRD4(1)
the first bromodomain of bromodomain-containing protein 4
- BRD4(2)
the second bromodomain of bromodomain-containing protein 4
- BRD9
bromodomain-containing protein 9
- BRDT
bromodomain testis-specific protein
- BRPF1
bromodomain and PHD finger containing, 1
- CBP
CREB binding protein bromodomain
- CECR2
cat eye syndrome critical region protein 2
- TAF1(1)
the first bromodomain of human transcription initiation factor TFIID subunit 1
- TAF1(2)
the second bromodomain of human transcription initiation factor TFIID subunit 2
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.7b00132.
IC50 values for compounds 1-21 with smile strings (XLS)
Experimental procedures and compound characterization for compounds 1–21, protein and assay methods, CECR2 cellular assay methods, dot images, curve, and replicate data for 21, crystallography data and refinement for 6, modeling methods, kinase and bromodomain selectivity data for 21 (PDF)
The authors declare the following competing financial interest(s): All authors were employed by Constellation, Genentech, and Wuxi, and experiments described were performed in Constellation, Genentech, or Wuxi laboratories.
Supplementary Material
References
- Gershey E. L.; Vidali G.; Allfrey V. G. Chemical studies of histone acetylation. J. Biol. Chem. 1968, 243, 5018–5022. [PubMed] [Google Scholar]
- Dhalluin C.; Carlson J. E.; Zeng L.; He C.; Aggarwal A. K.; Zhou M.-M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. 10.1038/20974. [DOI] [PubMed] [Google Scholar]
- Winston F.; Allis C. D. The bromodomain: a chromatin-targeting module?. Nat. Struct. Biol. 1999, 6, 601–604. 10.1038/10640. [DOI] [PubMed] [Google Scholar]
- Choudhary C.; Kumar C.; Gnad F.; Nielsen M. L.; Rehman M.; Walther T. C.; Olsen J. V.; Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Sanchez R.; Zhou M. M. The role of human bromodomains in chromatin biology and gene transcription. Curr. Opin. Drug Discovery Dev. 2009, 12, 659–665. [PMC free article] [PubMed] [Google Scholar]
- Haynes S. R.; Dollar C.; Winston F.; Beck S.; Trowsdale J.; Dawid I. B. The bromodomain: a conserved sequence found in human, Drosophila and yeast proteins. Nucleic Acids Res. 1992, 20, 2603. 10.1093/nar/20.10.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J.-P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A.-C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural Analysis of the human bromodomain family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero F. A.; Taylor A. M.; Crawford T. D.; Tsui V.; Côté A.; Magnuson S. Disrupting acetyl-lysine recognition: Progress in the development of bromodomain inhibitors. J. Med. Chem. 2016, 59, 1271–1298. 10.1021/acs.jmedchem.5b01514. [DOI] [PubMed] [Google Scholar]
- Brand M.; Measures A. M.; Wilson B. G.; Cortopassi W. A.; Alexander R.; Höss M.; Hewings D.; Rooney T.; Paton R.; Conway S. Small molecule inhibitors of bromodomain-acetyl-lysine interactions. ACS Chem. Biol. 2015, 10, 22–39. 10.1021/cb500996u. [DOI] [PubMed] [Google Scholar]
- Theodoulou N. H.; Tomkinson N. C. O.; Prinjha R. K.; Humphreys P. G. Progress in the development of non-BET bromodomain chemical probes. ChemMedChem 2016, 11, 477–487. 10.1002/cmdc.201500540. [DOI] [PubMed] [Google Scholar]
- Banting G. S.; Barak O.; Ames T. M.; Burnham A. C.; Kardel M. D.; Cooch N. S.; Davidson C. E.; Godbout R.; McDermid H. E.; Shiekhattar R. CECR2, a protein involved in neurulation, forms a novel chromatin remodeling complex with SNF2L. Hum. Mol. Genet. 2005, 14, 513–524. 10.1093/hmg/ddi048. [DOI] [PubMed] [Google Scholar]
- Thompson P. J.; Norton K.; Niri F. H.; Dawe C. E.; McDermid H. E. CECR2 is involved in spermatogenesis and forms a complex with SNF2H in the testis. J. Mol. Biol. 2012, 415, 793–806. 10.1016/j.jmb.2011.11.041. [DOI] [PubMed] [Google Scholar]
- Lee S.-K.; Park E.-J.; Lee H.-S.; Kwon J. Genome-wide screen of human bromodomain-containing proteins identifies Cecr2 as a novel DNA damage response protein. Mol. Cells 2012, 34, 85–91. 10.1007/s10059-012-0112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawe C. E.; Kooistra M. K.; Fairbridge N. A.; Pisio A. C.; McDermid H. E. Role of chromatin remodeling gene Cecr2 in neurulation and inner ear development. Dev. Dyn. 2011, 240, 372–383. 10.1002/dvdy.22547. [DOI] [PubMed] [Google Scholar]
- NVS-CECR2-1: A Chemical Probe for CECR2; SGC, 2015; http://www.thesgc.org/chemical-probes/NVS-1 (accessed August 2, 2016).
- Taylor A. M.; Côte A.; Hewitt M. C.; Pastor R.; Leblanc Ý.; Nasveschuk C. G.; Romero F. A.; Crawford T. D.; Cantone N.; Jayaram H.; Setser J.; Murray J.; Beresini M. H.; de Leon Boenig G.; Chen Z.; Conery A. R.; Cummings R. T.; Dakin L. A.; Flynn E. M.; Huang O. W.; Kaufman S.; Keller P. J.; Kiefer J. R.; Lai T.; Li Y.; Liao J.; Liu W.; Lu H.; Pardo E.; Tsui V.; Wang J.; Wang Y.; Xu Z.; Yan F.; Yu D.; Zawadzke L.; Zhu X.; Zhu X.; Sims R. J. III; Cochran A. G.; Bellon S.; Audia J. E.; Magnuson S.; Albrecht B. K. Fragment-based discovery of a selective and cell-active benzodiazepinone CBP/EP300 bromodomain inhibitor (CPI-637). ACS Med. Chem. Lett. 2016, 7, 531–536. 10.1021/acsmedchemlett.6b00075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford T. D.; Tsui V.; Flynn E. M.; Wang S.; Taylor A. M.; Côté A.; Audia J. E.; Beresini M. H.; Burdick D. J.; Cummings R. T.; Dakin L. A.; Duplessis M.; Good A. C.; Hewitt M. C.; Huang H.-R.; Jayaram H.; Kiefer J. R.; Jiang Y.; Murray J. M.; Nasveschuk C. G.; Pardo E.; Poy F.; Romero F. A.; Tang Y.; Wang J.; Xu Z.; Zawadzke L. E.; Zhu X.; Albrecht B. K.; Magnuson S. R.; Bellon S. F.; Cochran A. G. Diving Into the water: Inducible binding conformations for BRD4, TAF1(2), BRD9, and CECR2 bromodomains. J. Med. Chem. 2016, 59, 5391–5402. 10.1021/acs.jmedchem.6b00264. [DOI] [PubMed] [Google Scholar]
- Crawford T. D.; Romero F. A.; Lai K. W.; Tsui V.; Taylor A. M.; de Leon Boenig G.; Noland C. L.; Murray J. M.; Ly J.; Choo E. F.; Hunsaker T. L.; Chan E. W.; Merchant M.; Kharbanda S.; Gascoigne K. E.; Kaufman S.; Beresini M. H.; Liao J.; Liu W.; Chen K. X.; Chen Z.; Conery A. R.; Côté A.; Jayaram H.; Jiang Y.; Kiefer J. R.; Kleinheinz T.; Li Y.; Maher J.; Pardo E.; Poy F.; Spillane K. L.; Wang F.; Wang J.; Wei X.; Xu Z.; Bellon S. F.; Cummings R. T.; Cochran A. G.; Albrecht B. K.; Magnuson S. R. Discovery of a potent and selective in vivo probe (GNE-272) for the bromodomains of CBP/EP300. J. Med. Chem. 2016, 59, 10549–10563. 10.1021/acs.jmedchem.6b01022. [DOI] [PubMed] [Google Scholar]
- Ghosh S.; Taylor A.; Chin M.; Huang H.-R.; Conery A. R.; Mertz J. A.; Salmeron A.; Dakle P. J.; Mele D.; Cote A.; Jayaram H.; Setser J. W.; Poy F.; Hatzivassiliou G.; DeAlmeida-Nagata D.; Sandy P.; Hatton C.; Romero F. A.; Chiang E.; Reimer T.; Crawford T.; Pardo E.; Watson V. G.; Tsui V.; Cochran A. G.; Zawadzke L.; Harmange J.-C.; Auida J. E.; Bryant B. M.; Cummings R. T.; Magnuson S. R.; Grogan J. L.; Bellon S. F.; Albrecht B. K.; Simms R. J. III; Lora J. M. Regulatory T Cell modulation by CBP/EP300 bromodomain inhibition. J. Biol. Chem. 2016, 291, 13014–13027. 10.1074/jbc.M115.708560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BROMOscan bromoKdMAX recombinant protein binding assays performed at DiscoveRx; https://www.discoverx.com.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.