

Graphical abstract
Keywords: Hyperuricemia, Gout, Pathogenesis, Clinical picture of gout, Imaging modalities, Management of gout
Abstract
Gout is a picturesque presentation of uric acid disturbance. It is the most well understood and described type of arthritis. Its epidemiology is studied. New insights into the pathophysiology of hyperuricemia and gouty arthritis; acute and chronic allow for an even better understanding of the disease. The role of genetic predisposition is becoming more evident. The clinical picture of gout is divided into asymptomatic hyperuricemia, acute gouty arthritis, intercritical period, and chronic tophaceous gout. Diagnosis is based on laboratory and radiological features. The gold standard of diagnosis is identification of characteristic MSU crystals in the synovial fluid using polarized light microscopy. Imaging modalities include conventional radiography, ultrasonography, conventional CT, Dual-Energy CT, Magnetic Resonance Imaging, nuclear scintigraphy, and positron emission tomography. There is remarkable progress in the application of ultrasonography and Dual-Energy CT which is bound to influence the diagnosis, staging, follow-up, and clinical research in the field. Management of gout includes management of flares, chronic gout and prevention of flares, as well as management of comorbidities. Newer drugs in the pharmacological armamentarium are proving successful and supplement older ones. Other important points in its management include patient education, diet and life style changes, as well as cessation of hyperuricemic drugs.
Introduction
Gout distinguished itself in the history of Homo sapiens since time immemorial. It appeared in medical records very early in the history of medical writing, and was also mentioned in the biographies of many famous names. It was depicted as the fate of a life of affluence as much as the challenge to a physician’s skill, and truly it was. Modern ages witnessed remarkable progress in managing gout. More recently, thanks to quantum leaps in molecular biology, diagnostic modalities, and pharmacotherapy, we enjoy deeper understanding of the disease and a more sophisticated armamentarium.
Gout is a systemic disease that results from the deposition of monosodium urate crystals (MSU) in tissues. Increased serum uric acid (SUA) above a specific threshold is a requirement for the formation of uric acid crystals. Despite the fact that hyperuricemia is the main pathogenic defect in gout, many people with hyperuricemia do not develop gout or even form UA crystals. In fact, only 5% of people with hyperuriceamia above 9 mg/dL develop gout. Accordingly, it is thought that other factors such as genetic predisposition share in the incidence of gout [1], [2].
MSU crystals can be deposited in all tissues mainly in and around the joints forming tophi. Gout is mainly diagnosed by identification of the pathognomonic MSU crystals by joint fluid aspiration or in tophi aspirate. Early presentation of gout is an acute joint inflammation that is quickly relieved by NSAIDs or colchicine. Renal stones and tophi are late presentations. Lowering SUA levels below deposition threshold either by dietary modification and using serum uric acid lowering drugs is the main goal in management of gout. This results in dissolution of MSU crystals preventing further attacks [3], [4].
Epidemiology
The general prevalence of gout is 1–4% of the general population. In western countries, it occurs in 3–6% in men and 1–2% in women. In some countries, prevalence may increase up to 10%. Prevalence rises up to 10% in men and 6% in women more than 80 years old. Annual incidence of gout is 2.68 per 1000 persons. It occurs in men 2–6 folds more than women. Worldwide incidence of gout increases gradually due to poor dietary habits such as fast foods, lack of exercises, increased incidence of obesity and metabolic syndrome [5].
Pathogenesis of hyperuricemia
Urate is the ionized form of uric acid present in the body. Uric acid is a weak acid with pH of 5.8. Urate crystals deposition in tissues starts to occur when serum uric acid level rises above the normal threshold. Pathological threshold of hyperuricemia is defined as 6.8 mg/dL [1], [6].
Some factors may affect the solubility of uric acid in the joint. These include synovial fluid pH, water concentration, electrolytes level, and other synovial components such as proteoglycans and collagen. SUA level in the body is determined by the balance between its production either from purine intake in diet or endogenous production by cellular turnover and its excretion by the kidneys and GIT. Increased production of UA is responsible for only 10% of cases of gout while the remaining 90% are caused by its renal under-excretion [7].
Factors affecting SUA levels include age and gender. SUA is low in children. After puberty, SUA levels start to increase to reach their normal levels. In men, levels are higher than in women. However, SUA levels in postmenopausal women increase to reach men’s levels. This explains why gout is usually a disease of middle aged and older men, and postmenopausal women. Rarely, it may happen in children and young adults in some rare inborn errors of purine metabolism. These enzymatic defects result in increased SUA with consequent production of UA crystals in kidneys and joints (Fig. 1) [8].
Fig. 1.

Pathogenesis of hyperuriceamia (perceived and designed by Dr. EL-Shahaly).
Overproduction of uric acid
Deficiency of enzymes involved in purine metabolism leads to overproduction of UA. For example, Lesch-Nyhan syndrome is an inborn error of metabolism resulting from deficiency of an enzyme involved in UA metabolism named hypoxanthine–guanine phosphoribosyltransferase. It is a genetic X-linked recessive disorder with varying degrees of severity according to the type of mutation. The clinical picture of this disease involves neurological abnormalities such as dystonia, chorea, cognitive dysfunction, compulsive injurious behavior, self-mutilation and articular manifestations (early onset gout) in addition to renal stones. If left untreated, it may lead to tophi formation and renal failure [9].
Another enzymatic abnormality that causes gout in the young is the superactivity of phosphoribosyl pyrophosphate synthetase. It is an X-linked dominant inherited disorder. The syndrome has two clinical forms, a severe early onset form in children and a mild late juvenile or early adult onset form. Clinical picture includes neurological abnormalities such as sensorineural hearing loss, hypotonia and ataxia in the severe form. The mild form manifests as uric acid renal stones and arthritis. However, these enzymatic disorders constitute only less than 10% of cases of overproduction of urates [10].
Diet
Ingestion of foods rich in purines such as cooked or processed food especially from animal and seafood origin is a key element of increasing uric acid precursors. While foods rich in purine of vegetable origin such as beans, lentils, mushrooms, peas, legumes, and dairy products do not carry any risk on hyperuriceamia and gout, thus, can be allowed in gout patients. Furthermore, foods rich in vitamin C, low fat dairy products, plant oils such as olive, sunflower and soy were associated with reduced risk for hyperuriceamia and gout. Vitamin C was found to increase renal excretion of uric acid so it can be used as a supplement during management of gout [11], [12].
Alcohol is a well-known risk factor for gout. Studies showed that alcohol consumption is related to the amount consumed. Additionally, the risk for gout and hyperuriceamia depends on the type of different alcoholic drinks. For instance, beer is the worst in increasing the risk for gout compared to liquor. While the lowest risk among alcoholic drinks was for wine [11].
Endogenous urate production
Increased endogenous production of uric acid occurs in accelerated cellular turnover such as in malignancies, heamatological and inflammatory diseases. Also, increased purine production may result from chemotherapy and tissue damage. Furthermore, increased body weight and obesity leads to enhanced production of uric acid aggravating the risk of hyperuriceamia. Leptin was found to increase serum levels of urate. So, weight loss and exercises are very useful in reducing SUA levels and gout risk [13], [14], [15], [16].
Decreased excretion of uric acid
Two thirds of urate excretion occurs in the kidneys while the rest is excreted through the gastrointestinal tract (GIT). Reduced secretory function of the transporter ABCG2 leads to decreased excretion of uric acid through the GIT resulting in rise of serum levels of uric acid and enhanced renal excretion[7], [17].
Uric acid crystals are not soluble so require specific membrane transporters in order to cross cell membranes. Of these transporters are the urate transporter/channel (URAT) mainly URAT1 and the organic anion transporters (OAT1 and OAT3) [7], [18].
Renal excretion of uric acid is the end result of 4 phases. The first phase is the passage of UA across the Bowman’s capsule (glomerular filtration); followed by reabsorption of almost all urates passing in the proximal tubules. The third phase involves secretion of part of the reabsorbed UA ending with another reabsorption phase in the proximal tubules. The excreted UA is almost 10% of the filtered urate through Bowman’s capsule and the rest is reabsorbed in the body (Fig. 2) [19].
Fig. 2.

Renal excretion of uric acid (perceived and designed by Dr. EL-Shahaly).
Reduced renal excretion of urate is associated with some autosomal dominant disorders. Uromodulin is a gene that is expressed in the thick ascending limb of the loop of henle. It is responsible for regulating water permeability. Mutations of uromodulin gene result in decreased fractional excretion of UA, which in turn increases SUA [20].
URAT1 transports UA in the filtered fluid passing through the proximal tubules into the tubulules by an active transport process. Uricosuric drugs such as probenecid, benzbromarone and sulfinpyrazone decrease URAT1 activity, and consequently UA reabsorption in proximal tubules. On the other hand, drugs such as pyrazinamide, nicotinate and lactate increase urate reabsorption by acting on URAT1, moving UA from the lumen into the tubular cells. They both increase glomerular filtration and tubular reabsorption of UA preventing its loss in urine and increasing UA levels in serum [21].
Substances that affect URAT1 activity can both potentiate or inhibit its activity according to their dose. For example, low doses of aspirin have an anti-uricosuric effect while high doses have a uricosuric effect. High dose aspirin inhibits URAT1, hence its uricosuric effect. This process is called cis-inhibition of URAT1. The anti-uricosuric effect is caused by trans-stimulation of URAT1 by aspirin [22].
Genes responsible for uric acid regulation
SLC22A12 gene encodes for the transporter URAT1 present on the apical membrane of renal tubules. SLC2A9 is another gene involved in regulation of UA excretion. It encodes for a transporter protein in the membrane of renal tubules. Polymorphism of both genes results in decreased fractional excretion of UA leading to increased SUA levels. ABCG2 is a gene transporter for UA in the proximal tubular cells of the kidney as well as in the GIT. SLC17A1, SLC17A3 genes are important determinants of SUA levels acting as membrane transporters in the kidenys. Other genes involved in determination of SUA levels include SLC22A11, the glucokinase regulatory protein (GCKR), Carmil (LRRC16A), and near PDZ domain containing 1 (PDZK1) genes [23], [24].
Pathogenesis of acute gouty arthritis
Deposition of UA crystals in the joint cavity is the triggering cause of gout. These crystals initiate the inflammatory process by being engulfed by synovial phagocytic cells leading to release of lysosomal enzymes and production of inflammatory chemokines. Another mechanism is that UA crystals change the stability of cell membrane of phagocytic cells by direct crosslinkage with membrane lipids and glycoproteins. This involves the triggering of G protein, phospholipase A2, C and D, tyrosine kinase and other kinases such as mitogen-activated kinases (ERK1/ERK2, p38) and c-Jun N-terminal kinase. This interaction leads to increased IL-8 in phagocytes resulting in activation of neutrophils [25], [26].
The pathogenesis of gouty arthritis involves initial activation of monocytes and mast cells followed by neutrophils. Before the first attack of gout and in the inter-critical period, macrophages engulf UA crystals. Well-differentiated macrophages have the capability to contain these crystals without inducing an inflammatory response. While less-differentiated monocytes produce abundant amounts of TNF, IL-1, IL-6 and IL-8 along with endothelial activation following phagocytosis of urate crystals. Also, mast cells are key players in inducing the acute gouty attack by producing histamine and IL-1. This results in increasing vascular permeability and vasodilatation. Interestingly, it is thought that may even end the inflammatory phase by engulfing the crystals and the inflammatory debris [26], [27].
The chemotactic factors produced by monocytes and mast cells and the local vasodilatation stimulates neutrophilic chemotaxis. Also, endothelial cells activation further aggravates the inflammatory response and migration of neutrophils. This leads to an influx of neutrophils locally. Colchicine is thought to act by stopping the acute attack through changing the affinity of selectins on endothelial cells and neutrophils to inflammatory mediators and also by blocking the neutrophilic stimulation induced by endothelial cells [28], [29].
Inside the synovium, the abundance of chemotactic factors such as leukotrienes, platelet activating factor and interleukins mainly IL-8 is responsible for 90% of neutrophils activation and exacerbation of acute inflammation. Accordingly, targeting IL-8 can be promising for stopping the acute attack of gout [26].
The acute attack of gout is usually self-limited. It resolves within hours to few days of its beginning. This occurs by removal and phagocytosis of crystals by macrophages, hence suppressing cellular and chemokine activation. Also, macrophages clear the cellular apoptotic remnants to help stop the inflammatory cascade. Additionally, macrophages secrete TGF-β that eliminates IL-1, another key player in enhancing the inflammatory process [30].
Anti-inflammatory cytokines play an important role in inhibiting the inflammatory process. Other mechanisms involved in terminating the acute attack include proteolysis of pro-inflammatory cytokines, decreasing expression of receptors for TNFα and interleukins on the surface of leukocytes. Vasodilatation and increased vascular permeability is also important to allow extravasation of macrophages into the synovial fluid to clear the inflammatory area (Fig. 3) [30].
Fig. 3.

Pathogenesis of acute gouty inflammation (perceived and designed by Dr. EL-Shahaly).
Pathogenesis of chronic gout
Chronicity is a feature of gout. It results from chronic inflammation that follows recurrent attacks of gout. Chronic gout manifests by chronic synovitis, bony erosions, cartilage damage and tophi formation. This can be explained by different mechanisms. Presence of urate crystals in the synovium leads to stimulation of chondrocytes to produce inflammatory cytokines, nitric oxide and matrix metalloproteases resulting in cartilage damage [31], [32].
On the bone level, IL-1β and activation of receptor for nuclear factor κ B (RANK) and RANK ligand (RANK-RANKL) pathway are key players in osteoclastogensis and the formation of bone erosions. Gouty erosions are characterized by having overhanging edges and partial preservation of joint space. Furthermore, osteoblasts release pro-inflammatory cytokines leading to erosions and bone destruction in addition to compromising their own bone formation function. In the intercritical phase, there is persistent low-grade inflammation in affected joints. The same cytokines responsible for the acute flare up can be found at lower concentrations inbetween attacks. Although chronicity may result even with the use of uric acid lowering drugs and appropriate management of acute flare ups, yet its incidence is lower compared to patients with recurrent inappropriately treated attacks. Chronicity can be decreased by long-term use of low dose anti-inflammatory agents such as colchicine and lowering SUA to safe levels (<6 mg/dL) [32], [33].
Increased uric acid excretion in urine is usually calculated by the fractional excretion of urate compared to creatinine clearance. Both urine and blood samples are taken at the same time. The formula to calculate this parameter is [urine UA × serum Cr/serum UA x urine Cr]. The normal fractional excretion of uric acid is 7–10%. When it decreases, this reflects a reduction of uric acid excretion resulting in increased serum urate level [34].
Interestingly, it appears that levels of SUA actually decrease during the acute attack of gout. Furthermore, precipitation of an attack is common following the introduction of allopurinol or febuxostat without the prophylactic use of NSAID or colchicine. Also, states with increased excretion of SUA such as during surgery can trigger an acute gouty attack. Accordingly, it is assumed that sudden reduction of SUA precipitates acute gout [35].
Although hyperuriceamia is the main cause of gout, uric acid itself is an anti-oxidant that has a protective role on vascular endothelium. So, the presence of uric acid is essential for vascular integrity and homeostasis of human body’s functions. On the other hand, some studies found that allopurinol, a xanthine oxidase inhibitor used for treatment of hyperuriceamia and gout, has protective effects on vascular endothelial cells reducing cardiovascular risk. What determines whether presence of uric acid is beneficial or not is the type of tissue affected, whether it is intracellular or extracellular and its concentration. [36], [37].
Impact of systemic diseases on uric acid
Gout seems to affect osteoarthritic joints more often. This observation shows that cartilage damage resulting from OA induces formation of MSU crystals. Interestingly, UA crystals seem to affect the cartilage from its outer surface. Oppositely, pseudogout crystals appear inside the cartilage. Accumulation of UA crystals in the joint results from decreased vascularity and susceptibility of the synovial membrane to pass the crystals. Thus, gout tends to affect peripheral joints such as the big toe [38].
Hypertension is known as a risk factor for hyperuricemia and gout. Increased systemic blood pressure results in reduced glomerular filtration rate leading to decreased glomerular blood flow and decreased excretion of UA [34]. However, recent data suggest that hyperuricemia leads to increased blood pressure and that uric acid is a true modifiable risk factor for development of essential hypertension [39].
Diabetes mellitus (DM) is also a significant risk factor for hyperuriceamia and gout. Failure of oxidative phosphorylation increases adenosine levels resulting in increased production of uric acid and reduction of its renal excretion. Insulin treatment increases SUA by increasing its renal reabsorption from renal tubules. Metabolic syndrome is also associated with hyperuriceamia and gout [40].
Diagnosis
Clinical diagnosis
Asymptomatic hyperuriceamia
Gout undergoes 4 stages during its course starting with asymptomatic hyperuriceamia. In this stage, patients have no symptoms or signs and are usually accidentally discovered when measuring SUA (serum level greater than 7 mg/dL). However, some patients with hyperuriceamia may develop an acute gouty attack.
Acute gouty attack
Acute gouty attack is usually monoarthritic that peaks within hours to severely inflamed joint with cardinal signs of inflammation including redness, hotness, tenderness, swelling and loss of function. In large joints such as knees and ankles, skin signs are infrequent, but swelling and pain can be intense.
Gout has a predilection for lower extremities such as the first MTP, which is the commonest site for acute gout known as podagra [41]. Other joints that can be affected are the tarsal and metatarsal joints, ankles, knees, wrists, MCPs as well as interphalangeal joints of the hands. Rarely, hip and shoulder joints can be involved. Vertebral column involvement is extremely rare. Soft tissue inflammation may also occur including olecranon bursitis and Achilles tendonitis [42].
Arthritis of more than one joint at the same time is not very rare. It is more common in long-term untreated gout or in postmenopausal women. Constitutional symptoms such as fever, headache, and malaise can be present. In such case, the joint has to be managed as septic arthritis until proven otherwise. Extreme caution should be taken when dealing with such cases, as septic arthritis may happen in a gouty joint with the presence of MSU crystals. On the other hand, gouty attack can be mild with low-grade inflammation [43].
Intercritical period
When the acute attack settles down within hours to days following the introduction of colchicine or NSAIDs, patients enter into a remission phase. This period is characterized by the absence of symptoms. It may be interrupted suddenly by newer attacks if proper treatment for hyperuriceamia has not been introduced. This quiescent stage can be prolonged after the first attack. Without proper treatment, however, attacks become more frequent and more severe [44].
Chronic tophaceous gout
Untreated disease progresses into destruction of joints with formation of palpable tophi. A tophus is a mass formed of large amounts of accumulated crystals. It happens in chronic untreated gout. It can be present around the joints in the ears, the subcutaneous tissue or the skin. It is a manifestation of chronicity and uncontrolled disease. Macroscopically, tophi contain a white chalky material. Tophi may lead to joint destruction and deformity. Bony erosions may also occur as growing tophi extend to the bone. Differentiation of tophi from other nodules such as rheumatoid nodules, osteoarthritic Heberden’s and Bouchard’s nodules, lipomas or is essential for further management. This can be easily done by taking a simple needle biopsy that will show MSU crystals characteristic of gout [45].
Clinical diagnosis of gout is widely used allover the world especially in developing countries where resources are limited. However, when clinical diagnosis has been compared to microscopic diagnosis of crystals, it appeared to have low sensitivity and specificity [46].
In certain circumstances with atypical presentation of gout such as in multiple joint affection or atypical joint distribution, identification of MSU is mandatory to differentiate gout from other diagnoses. Elevated levels of SUA associated with typical joint involvement such, as podagra is usually a straightforward diagnosis. However, according to the EULAR recommendations, synovial fluid analysis is still advised to exclude other causes mainly septic arthritis [47].
Formation of tophi is a late clinical manifestation of gout (Fig. 4), though it may develop early in the disease course. When present, it can be a good indicator of gout. But still differentiation from other arthritides associated with nodules needs to be excluded before jumping to a definite diagnosis of gout [48].
Fig. 4.
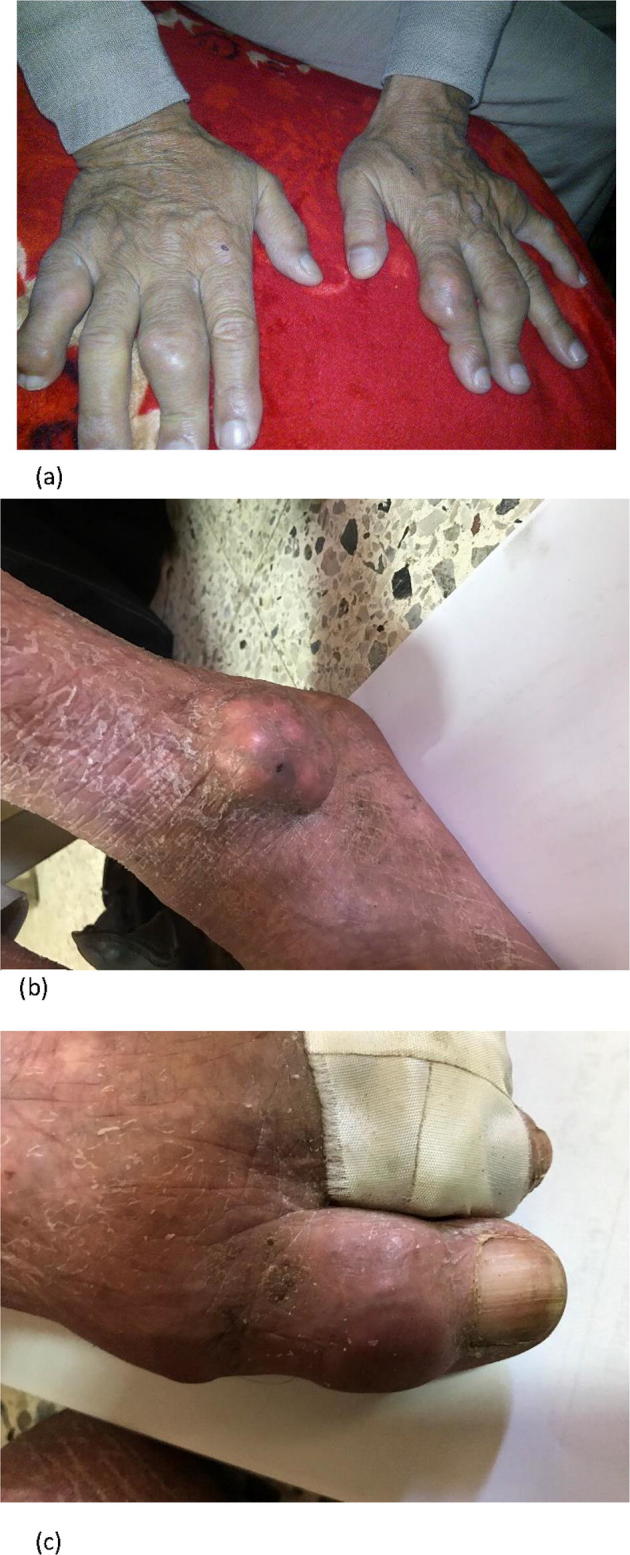
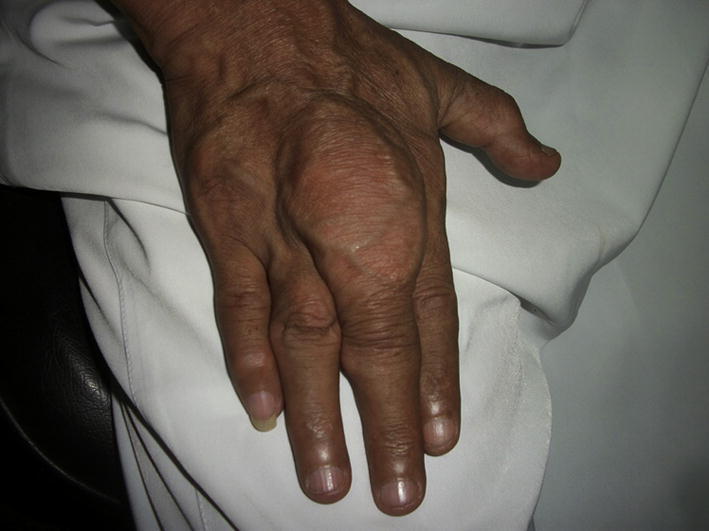
Chronic tophaceous gout: (a) hands, (b) ankle, (c) left greater toe (from the private collection of the authors).
Laboratory diagnosis
Diagnosis of gout based on hyperuricemia is a common misconcept among non-rheumatologists. Hyperuriceamia is usually asymptomatic and does not necessitate the diagnosis of gout. Among patients with SUA levels between 7 and 7.9 mg/dL only 0.09% will develop gout every year. As for patients with SUA between 8 and 8.9 mg/dl, 0.4% out of them may develop gout. With hyperuriceamia above 9 mg/dl, only 0.5% of patients may get gout [49].
Although hyperuriceamia is a characteristic feature of gout; it should be noted that during gouty attacks, SUA might drop to normal levels. Hyperuricemia is a weak marker for gout diagnosis and the disease might still be diagnosed even with normal serum levels [50].
The gold standard of diagnosis is the identification of MSU crystals in synovial fluid aspirate using polarized light microscopy. Better diagnostic yields can be obtained when using compensator. However, a regular light microscope can also be used for identification of crystals and differentiating MSU from other crystals such as calcium pyrophosphate dehydrate (CPPD) crystals. MSU crystals are found in the synovial fluid in all stages of the disease; during attacks, in the intercritical period or in chronic tophaceous gout [51].
Samples should be examined as soon as possible; better within 6 h. Though, they can be examined within 24 h if kept refrigerated at 4 °C. This is to avoid cellular dissolution and disappearance of crystals. In order to examine a specimen, a small drop is placed on a glass slide and covered with another, then placed under the microscope [52].
Using simple light microscopy, UA crystals are needle-like in shape, with different sizes. They can be seen clearly with 600× magnification. More magnification allows identification of further details. These can be easily distinguished from pseudogout (CPPD) crystal, which are usually rhomboidal in shape. Size can be similar to MSU crystals. Similar magnification to UA crystals ranging from 600× to 1000× can easily differentiate both crystals from each other [52].
Using a polarized filter helps better detection of crystals and birefringence. MSU crystals appear as shiny strong negatively birefringent crystals against dark background. They appear yellow when aligned parallel to the axis of red plate compensator. CPPD crystals, on the other hand, show positive birefringence and appear blue in color under the same circumstances [53].
Further analysis of synovial fluid should include leukocytic count, chemistry, culture and sensitivity. In acute gout, synovial fluid leukocytic count may exceed 50.000 cells/µL in some cases mostly polymorphs. Chemistry reveals normal glucose levels contrary to septic arthritis, in which bacteria consume glucose leading to low levels. Care should be taken to exclude septic arthritis in gouty cases, as both may be present in the same joint. So, culture and sensitivity along with gram stain is crucial to confirm the diagnosis [54].
Analysis of amount of uric acid in urine over 24 h is useful in assessing the etiology of hyperuriceamia in gout patients. Urinary uric acid of more than 800 mg/24 h indicates that such patients have increased production of uric acid, thus they excrete a large amount of uric acid. They require a drug that prevents uric acid production such as xanthine oxidase inhibitors rather than a uricosuric agent. Renal function tests should be done regularly for such patients due to the high risk for stone formation [55].
Radiological diagnosis
The significance of imaging in gouty arthritis cannot be overemphasized. It is extremely important for diagnosis and follow-up in clinical practice. Also, its potential as an outcome measure in clinical trials is growing. Recently, developments in the field of technology are influencing the staging, and even the type of gout nomenclature.
Conventional Radiography (CR)
It is the most widely used method in clinical practice, however in early stages of the disease it is not very helpful [56]. Radiographic changes may be missed for a minimum of 10 years after the first gouty attack [57]. During the early stages of gout, radiographic images are usually normal or may show asymmetric soft tissue swelling near the affected joints, but subtle early lesions such as small erosions and tophi are difficult to detect [58].
In chronic tophaceous gout, the main radiographic features are:
-
(a)
Tophi which are articular or periarticular soft tissue dense nodules [59], [60]
-
(b)
MSU deposits in the cartilaginous part
-
(c)
Joint space narrowing in advanced disease [61]
-
(d)
Bone erosions are characteristic. They are well circumscribed intraarticular or juxtarticular lesions with overhanging margins [62]. They result from the growth of tophi into the bone, hence are usually seen near tophi [63].
-
(e)
Periarticular osteopenia is usually absent and proliferating bone can be seen mostly as irregular spicules [64]
-
(f)
Calcified MSU deposits can penetrate in the bone; in severe cases, they should not be confused with bone infarcts or enchondromas. Radiography has low sensitivity (31%), however, its specificity is high (93%) [65].
CR is widely available, inexpensive, quick, and acceptable to patients. Radiation hazard is small [66]. The CR Sharp-van de Hejde scoring system for gout (SvdH-G), has been adapted from its RA counterpart and modified. The gout version, includes scoring for bone erosions as well as joint space narrowing with the distal interphalangeal joints (DIPjs) added [67].
Ultrasound (US)
Recently, progress in US technology (machines, transducers, techniques), encouraged its use by rheumatologists for the diagnosis and management of gout. In their excellent review, Nestrova and Foder [68], listed the main indications for using US in crystal-induced arthritis. These include detection of joint effusion and synovitis, differentiating between active and inactive synovitis, studying cartilage, describing bone contour for erosions and osteophytes, evaluation of tendons, evaluation of crystal deposition, execution of US-guided procedures (diagnostic and/or therapeutic), monitoring disease evolution as well as being helpful for the differential diagnosis with other arthritides (Fig. 5).
Fig. 5.

Three examples of Ultrasonography in gout. (a) Intraarticular tophus, metatarsophalangeal joint; (b) Double contour sign; (c) Longitudinal image of extensor digitorum longus (EDL) tendon showing markedly distended sheath with synovial effusion, synovial hypertrophy and crystal aggregates (arrows) (Courtesy of Dr. Adham Aboul-Fotouh, Kasr Alainy Teaching Hospital, Cairo University).
In gout US features can be either nonspecific or specific. Nonspecific features include:
-
(1)
Synovial fluid
Synovial fluid varies from being totally anechoic to containing aggregates of variable echogenicity. Aggregates of MSU microcrystals can be detected as hyperechoic spots or bright stippled foci. They tend to float in the joint space sometimes giving a snow-storm appearance when applying gentle pressure on the skin surface [69], [70].
-
(2)
Synovial proliferation and hypervascularization
The doppler mode can differentiate active from inactive synovial tissue by assessing its vascularity. This is essential for diagnosis and in monitoring the disease and its response to therapy [68].
-
(3)
Bone erosions
These are defined in gout as “intra- and/or extra-articular discontinuity of the bone surface (visible in two perpendicular planes) [71]. They are more likely found in patients who experience frequent attacks, or who have long disease duration, and tophi [71]. US has a threefold sensitivity than CR in detecting erosions < 2 mm (P < 0.001) [68]. There is, however, standardized US scoring system for erosions in gout [68].
Specific US features in gout
Articular cartilage “double contour Sign” (DCS)
DCS is very specific for gout. It is defined as abnormal hyperechoic band over the superficial margin of the articular hyaline cartilage, independent of the angle of insonation and which may be either irregular or regular, continuous or intermittent and can be distinguished from the cartilage interface sign [71].
DCS is reported in acute flare-up in clinically uninvolved joints, and in patients with asymptomatic hyperuricemia [68]. False-positive results have also been reported [72], [73]. Threle and Schlesinger demonstrated that DCS can disappear when SUA levels were lowered to 6 mg/dl for 7 months or more [74].
MSU deposits (Tophi and Aggregates)
-
i-
A tophus is a circumscribed, inhomogeneous, hyperechoic, and/or hypoechoic aggregation (that may or may not generate posterior acoustic shadow), which may be surrounded by a small anechoic rim. Aggregates are heterogeneous hyperechoic foci that maintain their high degree of reflectivity even when the gain setting is minimized or the insonation angle is changed and which occasionally may generate posterior acoustic shadow.
-
ii-
Tophi have been also described by US as “wet sugar clumps” with an oval or irregular shape [75].
-
iii-
Intra-articular and intrabursal tophi have been defined as heterogeneous hyperechoic (relative to subdermal fat) aggregates with poorly defined margins with or without areas with acoustic shadowing within the synovial recesses or bursae, respectively [76].
Doppler US can distinguish between active/hot tophi and inactive/cold ones based on their doppler signal [68]. Tophi can directly be measured by US using special calipers. There is good sensitivity to change associated with ULT [77]. US is feasible as it can be performed in the clinic and there are no radiation hazards involved. The time required for scanning, however may be significant and training costs may be considerable [66].
Conventional CT (CCT)
CT is characterized by excellent resolution and high contrast, hence it is the best technique for the assessment and characterization of crystal arthropathies [61].
CCT is not helpful in the diagnosis of acute gout as it can’t detect inflammation, synovitis, tenosynovitis and osteitis. This handicap is however, more than counterbalanced by its role in chronic gout. It is able to detect erosions better than Magnetic Resonance Imaging (MRI) or CR [78]. These are described as well defined, punched out lytic bone lesions, with sclerotic overhanging edges [79].
The specificity of CCT for the assessment of tophi exceeds that of US or MRI [80]. CT of tophi has been confirmed microscopically by identifying MSU crystals [66]. Its measurement of tophi has also been compared to physical exam using Vernier calipers [81], [82]. Tophi, soft tissue, intra-articular as well as intra-osseous ones appear as soft tissue masses with well described attenuation, making it easier to distinguish them from other soft tissue lesions [79], [80], [81], [82], [83].
CCT can help to monitor disease burden and response to therapy [81], but has the disadvantage of radiation exposure [84], [85].
Dual-Energy CT (DECT)
The introduction of this new imaging technique opened a new horizon. It allows the differentiation of deposits based on their different X-ray spectra. It applies the concept that the attenuation of tissues depends on their density, atomic number as well as the photon beam energy [86]. Like CCT, it can detect damage but does not help in inflammation. It is superior to all other available imaging technologies in its ability to identify all urate deposition in the area imaged [66] (Fig. 6).
Fig. 6.
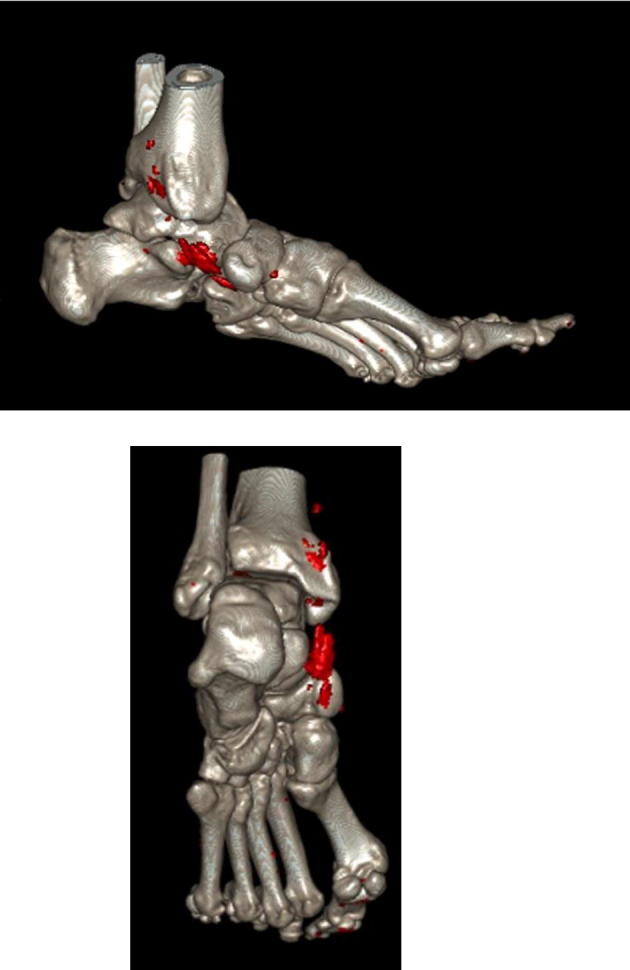
DECT of a gouty patient showing two views of MSU deposits (in red) in the tibialis posterior tendon (from the private collection of prof. Bardin.
DECT can offer a quick, non-invasive method to visualize MSU crystals, soft tissue changes, and early erosions at high-resolution, even before CR. This, particularly, helps in the differential diagnosis from pigmented villo-nodular synovitis, psoriasis, and septic arthritis which can share clinical features with gout [87]. DECT is highly accurate in detecting MSU crystals in joints, tendons, ligaments and soft tissues and can be used to identify subclinical gout with high specificity [79]. It, however, misses crystal deposition on the surface of cartilage, a feature US can detect as the DCS [88].
There are many causes of false negatives; lower density of tophi due to lower crystal concentrations, small size of tophi or crystals (less than 2 mm.) or technical parameters [89], [90], [91]. On the other hand, false-positive results were described around nail beds, in the skin, in regions of metal artifacts and in severe OA [89], [90], [91], [92].
DECT is not widely available, which limits its application for clinical and research purposes. Its costs are equivalent or higher than CCT and it entails radiation exposure [66].
MRI
MRI features of arthritis are those of nonspecific inflammation, synovial thickening, effusion, erosion, and bone marrow edema. Tophi show homogenous T1 signal intensity (low to intermediate) and heterogeneous T2 signal intensity (variable low to intermediate), depending on the degree of its hydration and classification [93].
MRI role is limited because of expense and limited availability. It is, however, useful for evaluation of gout at unusual sites. The literature abounds with case reports in the axial skeleton [94], [95], [96], [97], [98], [99], or presentation as spondyloarthritis [100], [101], carpal tunnel syndrome [102], [103], crown dens syndrome [104], paraspinal abscess [105], or intra-abdominal mass [106]. The diagnosis in these reports was made by MRI, which was occasionally combined with other modalities.
Nuclear scintigraphy
Nuclear Scintigraphy is rarely used for evaluation. Positive results are often found incidentally when a study is performed for other indications.
Positron emission tomography (PET)
Case reports of (PET/CT) in gout showed articular and periarticular FDG (18 F-fluoro-2-deoxy-D-glucose) uptake. Soft tissue FDG uptake identifying tophi has also been reported. This can be helpful when gout presents at unusual locations [93].
Major advances in the imaging of gout took place in the last decade. Despite this, it is currently unknown which, if any, imaging techniques can provide valid outcome measures for clinical studies in gout [66]. It has been advocated that multiple imaging modalities need to be further developed for use as outcome measures in chronic gout as different modalities have relevance and potential for different domains [107].
Dalbeth and Choi [108] proposed a roadmap to improve upon the current generation of global outcomes and their associated outcomes in gout. To refine the disease stages they suggested prospective studies of individuals with hyperuricemia and gout, using advanced techniques such as US and DECT. They also proposed the development of novel prognostic markers and gout-specific disease activity indices beyond SUA levels including new applications of advanced imaging (US, DECT and potentially MRI) [108].
Management
Gout appears as the best-understood and most manageable rheumatic disease. Lifelong lowering of uricemia under specific targets allows dissolving the pathogenic crystals and suppressing disease manifestations. However, therapeutic failure is frequent [109] and has led to the production of recommendations [110], [111], [112]. Failure is often due to poor adherence to urate-lowering drugs ULD [113], underlining the need for patient and physician education.
Management of flares
Gout flare medications include colchicine, Non-Steroidal anti-inflammatory Drugs (NSAIDs) and steroids, which can be taken together in severe cases and are most efficient when taken early after the flare onset (Fig. 7). This has led the European League against Rheumatism (EULAR) panel to recommend that patients be educated to auto medicate [112].
Fig. 7.

EULAR recommendation for the management of flares in patients with gout [112].
Colchicine
When taken within 12 h after flare onset, 1.8 mg (1.2 mg then 0.6 mg one hour later) of colchicine has been shown to be as effective as the traditional higher doses [114]. In clinical practice this drug appears as much less efficient when given long after the flare onset. The EULAR and American College of Rheumatology (ACR) have restricted the use of colchicine to patients presenting within 12 and 24 h of flare onset respectively.
Practitioners should keep in mind that colchicine has a narrow therapeutic toxicity window and can be very toxic when used inappropriately. Gastrointestinal intolerance (diarrhea, nausea, or vomiting) is common. It is usually the first feature of colchicine toxicity and should lead to dose reduction or interruption. Further toxicity includes neutropenia and multi-organ failure, which can be lethal. The maximum daily dose has been recently reduced to 2 mg (in divided doses) in France.
Renal failure decreases colchicine excretion. Doses should be limited to 0.5–0.6 mg/d in patients with moderate renal insufficiency (eGFR from 30 to 60 mL/min) and to 0.5–0.6 mg every 2 or 3 days in those with eGFR from 15 to 30 mL/min. Colchicine is contra-indicated in CKD stage 5 patients (eGFR < 15 mL/min or dialysis).
Doses should also be reduced in patients with hepatic failure, as the drug is predominantly eliminated through the hepato-biliary system. Inhibitors of cytochrome P450 3A4 or P glycoprotein increase plasma concentration and toxicity of colchicine. The doses of colchicine should be reduced to 0.3 mg every 3 days when cyclosporine, ketokonazole, erythromycin, retronavir are co-prescribed and to 1.2 mg every 3 days when diltiazem or verapramil are used [115]. The French regulatory agency contraindicates co-prescription of macrolide antibiotics and colchicine, even though azythromycin has been found to have no pharmacokinetic interaction with colchicine. Muscle toxicity, including rhabdomyolysis has been reported with the concomitant use of colchicine and statins, especially in renal failure patients [116]. Nerve and muscle toxicity can be observed in long term low dose colchicine users who have kidney transplant or chronic kidney disease CKD, usually with 30 mL/min of eGFR or less [117]. This toxicity is usually slowly reversible after drug cessation and requires CK monitoring.
NSAIDS
NSAIDs or COXIBs are used at the maximum authorized dose, with proton inhibitors when indicated. Their efficacy is largely accepted, even though no placebo controlled trial has been performed. Early prescription allows reducing administered doses.
Steroids
Oral prednisone, at a daily dose of 30 mg/d for 7 days has been shown to be effective [118], [119], [120] and is recommended by the ACR and EULAR panels as potential first line therapy in the management of gout flares [111], [112]. However steroids can worsen hypertension and diabetes [121] and are, in our view, best indicated in patients contra indicated for NSAIDs or colchicine (i.e. CKD patients). Co-prescription of a small dose (0.5–1 mg/d) of colchicine, when not contraindicated, may avoid rare inflammation relapses after steroid cessation. Open studies also suggest that ACTH can relieve gouty inflammation [122].
Intra-articular steroid injections appear as very efficient and are recommended by both the ACR and the EULAR in the management of mono or pauci-articular flares, despite the lack of randomized clinical trials (RCT).
IL-1 blockers
Open studies of the IL-1 receptor antagonist anakinra [123], [124] support its off-label use in patients resistant or contraindicated to NSAIDs, colchicine and steroids. Canakinumab, a long lasting antibody to IL-1 beta, has been approved by the European Medical Agency, following 2 RCT trials against intramuscular triamcinolone acetonide [125]. The EULAR recommends considering IL-1 blockers for the management of gout flares in patients with frequent flares contraindicated to NSAIDs, colchicine and steroids (oral or injectable). Current infection is a contra indication [112].
Management of chronic gout and prevention of flares
Uricemia targets
To obtain MSU crystal dissolution, SUA should be lowered to values which are under the MSU saturation point. Both the ACR and the EULAR indicate that the SUA target is below 6 mg/dL in all gouty patients and below 5 mg/dL in severe gout patients, to allow more rapid dissolution of the crystal load. Hyperuricemia must be routinely checked by measuring SUA levels [110], [111], [112]. This approach has been recently challenged by the American College of Physicians (ACP) who recommended to treat gout to control symptoms rather than to target an uricemia level [126]. The main reason for this GP guideline is the present lack of rigorous treat to target trial. Such a trial is underway in New-Zealand and should definitively settle the issue. However, numerous clinical and pathophysiological data already tell us that lowering uricemia under the saturation point is the best and most reliable way to control gout symptoms in the long run, and that prescribing ULTs without checking that uricemia is lowered enough is a frequent cause of gout treatment failure. This ACP guideline therefore appears, in our view, as detrimental and should not be followed.
Patient education
As already emphasized above, patient education is key to gout management success, as shown by a preliminary study that explored the effect of a predominantly nurse-delivered education program. Following this program, 98 of 103 included patients had their uricemia at target after one year of allopurinol treatment, in sharp contrast with what is usually observed [127]. Information should be given on the pathophysiology of the disease, its relationship with uricemia, its curable nature, uricemic targets to be reached, the life-long nature of urate-lowering treatment, the importance to treat flares early, the mechanisms of ULD-induced flares and ways to prevent them. Patient education takes time and must frequently be repeated, but it is a mandatory tool to achieve success in long-term gout management.
Diet and life style changes
Following epidemiologic demonstration of the influence of these lifestyle factors on the risk of gout, EULAR and ACR recommended weight loss in obese patients; avoidance of beer (including non-alcoholic), spirit, and sugar sodas; restriction of meat and seafood intake; and increased intake of skimmed-milk products, together with enhanced physical activity [110], [111], [112]. Very scarce evidence however supports the efficacy of these changes. Small and short term controlled studies showed that milk decreased uricemia and that weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fats were found to have a beneficial effect on serum urate and lipoprotein levels [16], [128].
Diet modification appears to be less effective than ULD to control hyperuricemia. However, combining both is very successful in management of chronic gout. Furthermore, to allow moderate SUA reduction, lifestyle changes, exercises and most importantly loss of weight are important tools to control the metabolic syndrome and cardiovascular diseases associated with gout [129], [130]. Diet modification aiming to correct hypertension or metabolic syndrome have been shown to lower uricemia [131], [132].
Targeting SUA is a key component of gout treatment, which allows, when properly done in the long run, disappearance of disease features [133].
Cessation of hyperuricemic drugs
Attempts should be made to stop drugs that increase uricemia. This is mainly the case with antihypertensive drugs. Thiazide and loop diuretics increase uricemia by an average of 0.65 and 0.96 mg/dL respectively (134). Beta-blockers, non-losartan ARBs and ACE inhibitors have also been associated with an increased risk of gout (135) and increased uricemia. Calcium channel inhibitors and losartan should be privileged. In cardiac failure, spironolactone, which has no effect on uric acid (136) can be advised when possible. Cardio-protective aspirin modestly increases uricemia and replacement by clopidogrel can be considered.
Urate lowering drugs (ULDs)
Indications
Indications for ULD have increased over the years following better awareness of potential adverse effects of hyperuricemia on the cardio-vascular system and that long standing gout associates with comorbidities and large MSU deposits which will make crystal dissolution more difficult. According to the recent EULAR recommendations, ULDs are indicated in severe gout or when associated with uric acid lithiasis as traditionally, but also in patients with cardio-vascular or renal comorbidities, or with high (>8 mg/dL) uricemia or young age (<40 years), as these are likely to have frequent attacks. In patients with a definite diagnosis of gout, the EULAR advises to discuss with the patient the indication of ULD as soon as after the first flare [112].
General principles
The initiation of ULDs is associated with an increased risk of gout flares due to crystal mobilization: schematically, when they start to dissolve, deposits become more fragile and crystals can shed into the joint space and trigger inflammation. This should be explained to the patient and the risk should be reduced by progressive titration of ULDs and prescription of low dose (0.5–1 mg/d) of colchicine or NSAID (e.g. naproxen 250 mg/d) as a prophylaxis against ULD-induced gout flares in patients with no contraindications to these drugs [110], [112], [134]. Prophylaxis is usually indicated during the first 6 months of ULD prescription. It may be prolonged in tophaceous gout in which complete dissolution of crystal deposits takes a longer time. Interestingly long term low dose colchicine has been shown to be cardio-protective [135], an additional benefit for gouty patients who are known to be at increased cardiovascular risk [136].
Even after obtaining whole crystal dissolution, uricemia should be kept lifelong under 6 mg/dL, to avoid recurrence of crystal formation and flares. Uricemia should be checked every 6 months in the long run [110], [137], to encourage patient’s adherence to ULD and avoid increases of uricemia above the target, due to new medication or weight gain. Gout must therefore be considered as a chronic disease and the frequent misconception of considering gout as the archetype of an acute disease should be combated through education of patients and doctors.
Lack of adherence to ULD is the main cause of gout management failure. Gout is the worst chronic disease in term of treatment adherence [138]. This is probably due to the lack of explanation and prevention of the ULD-induced flares, the perception of gout as an acute disease, which would only require flare management, and the lack of proper patients’ information. Patient education is a key toll to increase management success.
Allopurinol
Allopurinol is an oral xanthine oxidase inhibitor, first introduced to the clinic in the sixties. Allopurinol is a purine, which is rapidly converted into its active metabolite, oxypurinol, by the xanthine oxidase enzyme. Xanthine accumulation has been seldom reported to cause urinary xanthine stone [139] which can be fully prevented by sufficient fluid intake. In addition to inhibiting xanthine oxidase, oxypurinol inhibits the synthesis of purines, a mechanism that requires the intervention of the enzymes hypoxanthine guanine phosphoribosyl transferase and phosphoribosyl pyrophosphate synthetase and is not observed when these enzymes are deficient. Excretion of oxypurinol is mainly through the kidney and is decreased by renal failure and increased by uricosurics. Dose requirements to reach uricemia targets are increased by body weight increase and diuretic use [140]. ABCG2 loss of function polymorphism decreases its urate-lowering effect [141].
Because oxypurinol has a long half-life, allopurinol can be prescribed once a day. The urate-lowering effect is dose dependent. Over the world, allopurinol is prescribed at the dose of 300 mg/d or less in more than 90–95% of gouty patients [142]. At the daily dose of 300 mg, allopurinol used to bring uricemia to less than 6 mg/dL in nearly every gouty patient when the drug was initially launched [143]. But this has now largely changed, possibly because gouty patients’ uricemia and weight have substantially increased since the sixties: recent studies have shown that only a minority of patients receiving 300 mg/d of allopurinol reached the desirable uricemia target (<6 mg/dL) [144], [145], [146].
The maximal approved daily dose of allopurinol is 800 or 900 mg/d according to countries and in, patients with normal renal function, increasing the dose above 300 mg/d is often necessary to attain the uricemia target [147]. Allopurinol and other xanthine oxidase inhibitors should not be prescribed with azathioprine and 6-mercaptopurine, as xanthine oxidase is involved in the metabolism of these drugs.
Allopurinol is usually well tolerated. Abdominal discomfort, nausea and diarrhea, liver [148] or bone marrow toxicity [149], acute interstitial nephritis [150] are very rare early side effects which can be part of the allopurinol hypersentivity syndrome; gynecomastia and peripheral neuropathy [151] have been observed, very rarely, during long-term allopurinol treatment. Patients should be told that cutaneous side effects may develop during the 2 or 3 first months of treatment and should lead to immediate, life-long, allopurinol cessation. They include benign maculo-papular rash, reported in 2–4% of allopurinol initiators, and life- threatening severe skin reactions which can take the form of an acute generalized exanthematous pustulosis [152], toxic epidermolysis/Steven Johnson syndrome [153] or Drug Related Eosinophilia with Systemic Symptoms (DRESS) syndrome [154]. The incidence of these severe skin reactions has been estimated at 0.7 [95%CI0;5–9.9] per 1000 allopurinol initiators-years in the USA [155], but they are more frequent in Asia, due to more common genetic predisposition [156].
Risk factors include recent (<3 months) allopurinol initiation, use of allopurinol for asymptomatic hyperuricemia, female gender [156], a history of skin reaction to allopurinol, HLA∗B-5801 carriage [157], [158], high initial dose [159] and renal failure [160], [161].
In Taiwan, where both the frequency of the allele and related relative risk are high, HLA∗B-5801 typing allows preventing major skin reactions by avoiding allopurinol prescription in carriers of the allele [162] and the ACR recommends to genotype patients from Han, Korean and Thai ancestries before prescribing allopurinol [110].
In Caucasians and Japanese, the association exists but the risk allele is very rare and most of the patients who developed serious skin reactions did not carry the allele so that genotyping is seldom used. It is important to start allopurinol at a low dose (50-100 mg/d) to be progressively increased every 2–4 weeks until the targeted uricemia is obtained, as recommended by the EULAR [112]. Lack of titration has been associated with severe skin reaction [159].
The impact of renal function on allopurinol dosage is debated. The ACR does not follow the traditional guideline, still implemented by most regulatory agencies, that is to reduce the maximum allopurinol dose according to the creatinine clearance, but recommends increasing allopurinol until the target is reached, with no limitation in CKD patients [110]. Reasons for this are that adjustment of allopurinol according to creatinine clearance seldom allows proper control of uricemia in patients with renal failure [163] and small series have not shown an increased incidence of severe reactions in patients with allopurinol progressively titrated above the authorized dose [164].
The EULAR recommends to keep limiting allopurinol dose according to the creatinine clearance and to use alternative drugs when this fails to reach uricemia targets [112].
Febuxostat
Febuxostat is an oral, once a day, non-purine xanthine oxidase inhibitor, which is available as 40 and 80 mg tablets in the USA and 80 and 120 mg tablets in Europe. At the doses of 80 and 120 mg/d, the maximum doses approved in the USA and Europe respectively, febuxostat is a more potent ULD than allopurinol 300 mg/d [144], [145]. Because of its mixed renal and hepatic metabolism, the drug can be prescribed with no dose reduction in patients with moderate renal failure. Recent small studies have suggested that efficacy and safety is maintained in patients with creatinine clearance below 30 mL/min [165]. Because the drug inhibits xanthine oxidase, febuxostat should not be co-prescribed with azathioprine or 6-mercaptopurine.
Side effects to febuxostat include rare and early liver or kidney hypersensitivity reactions, and benign skin rashes which have been reported in about 5% of patients during phase 3 trials. Serious cutaneous reactions have been very rarely reported [166]. Skin reactions to febuxostat appear as slightly more frequent in patients with previous cutaneous intolerance to allopurinol; the increased risk does not result from cross-reactivity but from the increased likelihood for any known allergic patient to develop skin reaction to another drug [167]. In phase 3 studies, cardiovascular side effects and mortality were numerically increased in the febuxostat-treated as compared to the allopurinol-treated patients. This has led the European agency to recommend caution in prescribing febuxostat in patients with a history of heart disease and to ask for a postlicensing cardiovascular safety trial comparing febuxostat to allopurinol, the results of which are still pending [168].
ACR considers febuxostat as a first line ULD [110], whereas for EULAR, the drug is indicated in patients intolerant or refractory to allopurinol. Dose titration is recommended to reduce ULD-induced flares, even though there is no evidence that this improves febuxostat tolerance [112].
Uricosurics
Uricosurics lower uricemia by increasing uric acid output in the urine. Therefore they expose the patients to the risk of uric acid stone, which is worse at the onset of treatment. When uricemia has decreased, uricuria and the risk become lower, as uricuria also decreases. They should not be administered as a monotherapy in patients with a history of uric acid stone or hyperuricuria and should be taken with abundant water intake; the urinary pH should also be checked and kept above 6 to decrease the concentration of uric acid in urine, which governs the risk of lithiasis. Except for lesinurad, uricosurics can be used alone; they are now most often used in combination with xanthine oxidase inhibitors, when these fail to obtain the uricemia targets.
Probenecid has been the first commercialized ULD [169] and was at first a very popular drug. When allopurinol became available, probenecid was much less used because it had to be given in divided doses and required high fluid intakes and adjustment of the urine pH. In addition, probenecid, which was first developed to decrease the renal excretion of penicillin, could interfere with the excretion of other organic acid drugs and gastro-intestinal or cutaneous intolerance were fairly common. It has been recently confirmed to be a decent ULD, including those patients with moderate kidney involvement and remains one of the therapeutic options in patients intolerant or refractory to allopurinol [170]. The initial dose is 250 mg twice daily, which can be weekly increased up to 1 g twice daily. Larger doses expose to major central nervous system toxicity.
Sulfinpyrazone is not universally available. This uricosuric drug is usually given twice daily at the total daily doses of 200–400 mg, progressively attained. Side effects include gastrointestinal intolerance, skin rashes, platelet aggregation impairment and rare bone marrow toxicity.
Benzbromarone is a powerful uricosuric drug, which is used once a day at a dose of 100–200 mg/d. [147], [171], [172]. Following reports of severe liver toxicity, the drug has been retrieved from Europe, where it can still be prescribed on a named patient basis, but is still largely used in Asia. The uricosuric property is largely retained in renal failure patients [172].
Lesinurad is selective URAT1 inhibitor which has been recently approved at the dose of 200 mg/d in the USA and Europe, as an add-on therapy to xanthine oxidase inhibitors when these failed to lower uricemia down to the suitable target [173], [174]. Serum creatinine elevations have been observed, which although most often transient, require renal function monitoring.
Fenofibrate, atorvastatin and losartan are non-licensed uricosurics which can be used to treat gout comorbidities or in association with xanthine oxidase inhibitors [175], [176].
Urate oxidases
Rasburicase is a short-life IV uricase, which is approved for the management of tumor lysis syndrome. Its non-licensed use has been reported in tophaceous gout [177]. Pegloticase is a PEGylated uricase which has been approved, in the USA and Europe, for the management of severe gout, refractory to oral ULDs, and is commercially available in the USA. The drug is administered by IV infusions of 8 mg every 2 weeks and has been shown to be very effective [178]. Antibodies develop at high titers in about half of the patients, leading to loss of uricemia response and to an increased risk of serious infusion reactions. It is therefore recommended to measure uricemia in the 24 h preceding every planned reinfusion and to stop the drug if uricemia is not decreased. No other ULD should be prescribed concomitantly to keep this warning signal (Fig. 8).
Fig. 8.

EULAR recommendation for the management of hyperuricemia in patients with gout [112].
Management of comorbidities
Cardiovascular and renal diseases, the metabolic syndrome should be screened and properly treated in gouty patients, and smoking cessation should be advised to decrease mortality [112]. In order to help lowering SUA levels, hypertension treatment should favor losartan and calcium channel inhibitors, statins or fenofibrate should be used in dyslipidemic patients, insulin lowering drugs should be privileged in type 2 diabetic patients. The recently introduced SGLT2 inhibitors also have interesting urate lowering effects [179].
Conclusions and future perspectives
To understand gout, and consequently to manage it, has been a challenge to the skill of physicians along the history of medicine. Advances in this field that took the shape of continuous progress, have recently witnessed quantum leaps. We enjoy a deeper insight into the disease pathogenesis. We can rely on more sophisticated diagnostic modalities, and most importantly, we have at our disposal, a wider array of therapeutic options to deal with it.
Gout has been considered the nemesis of longevity. That gloomy outlook is exemplified in Lord Byron’s [180] (1788–1824) reflections on old age: “They kindly leave us, though not quite alone; but in good company- the gout or the stone”
We are now entitled to a more optimistic view that we owe to modern science.
The future agenda in the field is likely to address the following issues:
-
1-
Intensive studies in genomics and proteomics. These further our understanding of disease predisposition and susceptibility to drug adverse effects. They also offer potential therapeutic breakthroughs.
-
2-
Investigating the possible role of the microbiome in gout parallel to its metabolic counterparts.
-
3-
Attempts to standardize international medical approaches regarding gout; outcome measures, staging and management.
-
4-
Emphasis on patient and physician education. This is a highly cost-effective approach.
Conflict of Interest
The authors have declared no conflict of interest.
Compliance with Ethics Requirements
This article does not contain any studies with human or animal subjects.
Biographies

Prof. Gaafar Ragab graduated from the Faculty of Medicine, Cairo University, 1976, where he got his degrees in Internal Medicine: MSc, 1980, and PhD, 1985. He served his faculty as a Resident, Assistant Lecturer (1980), Lecturer (1985), Assistant Professor (1990), and Professor (1995) till now. He spent a sabbatical year 1989 at the UAB, USA in the department of Clinical Immunology and Rheumatology. He headed the Clinical Immunology & Rheumatology Unit affiliated to the Internal Medicine Department 2010–2015. He was Chief of the Internal Medicine Department’s Research Committee 1995–2013. He is Fellow of the American College of Rheumatology (ACR) since 1989. He is a Co-founder of the Egyptian Society of Immunology and Rheumatology (EGYSIR) which he headed as its President 2010–2013. He was chosen as President of the Egyptian League Against Rheumatism (ELAR) annual meeting in Alexandria 2013. He is member of the Advisory Board of the Egyptian Society of Internal Medicine (ESIM) and its Journal and served as President of ESIM annual meeting, Cairo, 2014. He is the associate editor of the Journal of Advanced Research (JARE), the interdisciplinary publication of Cairo University, in charge of its medical branch. He won the prize of honour, Cairo University, for the great efforts in international publications, for the Year 2008. Member of the Egyptian National Committee for the management of Hepatitis C Virus (the extrahepatic manifestation) as well as the International Study Group of Extrahepatic Manifestations Related to Hepatitis C Virus infection (ISGEHCV). He is member of Geographical Variation in Rheumatoid Arthritis Group (GEO RA).

Dr. Mohsen Elshahaly is a Lecturer of Rheumatology, Physical medicine and Rehabilitation, Faculty of medicine-Suez Canal University since 2014 till now. He was born on the 3rd of March 1981 in Giza – Egypt. He graduated in 2004 from school of medicine – Suez Canal University. He got a master’s degree in Rheumatology, physical medicine and rehabilitation awarded by Suez Canal University. Also, he got a joint master’s degree in health professions eduction awarded by Maastricht University and Suez Canal University in 2011. He passed the specialty exam in rheumatology of the royal college of physicians and the British Society of Rheumatology in 2013. Dr. Elshahaly awarded M.D. degree in Rheumatology, Physical medicine and Rehabilitation from Suez Canal University and Newcastle University in 2014. He has also been awarded a musculoskeletal doctor of medicine degree by Newcastle University – UK in 2017 and passed the first two parts of the membership of royal college of physicians – UK. Dr. Elshahaly worked as a house officer in Suez Canal university hospitals from 1/3/2005 till 28/2/2006. He was assigned the job of demonstrator of Rheumatology, Physical medicine and rehabilitation, Suez Canal University from 23/6/2009 till 2/12/2009. He was promoted to assistant lecturer in 2/12/2009. He worked as a clinical research fellow in the musculoskeletal research group, Institute of cellular medicine, Newcastle University and The James cook university hospital, United Kingdom.

Thomas Bardin is a professor of Rheumatology at Paris Diderot University, a full time physician in the Department of Rheumatology at the Hôpital Lariboisière, Paris, France, and a member of the INSERM OSCAR unit. Dr. Bardin received his medical degree from the University Paris V in 1980, and he was accredited in rheumatology in 1981. He has been head of his department, president of the Société Française de Rhumatologie, chief editor of Revue du rhumatisme and Joint Bone Spine, associate editor of the Annals of the Rheumatic Diseases and a member of the editorial boards of Arthritis & Rheumatism, Clinical Experimental Rheumatology and Amyloid International. He is an international member of the American College of Rheumatology. Dr. Bardin has published over 290 articles in international peer-reviewed journals, and has been the editor or co-editor of more than 30 books in the field of rheumatology, including Therapeutics in Rheumatology and the rheumatology section of a French-language textbook on internal medicine. He has a strong interest in gout and was the convenor of the 2016 EULAR recommendations on the management of gout.
Footnotes
Peer review under responsibility of Cairo University.
References
- 1.Dalbeth N., Merriman T.R., Stamp L.K. Gout Lancet. 2016;388(10055):2039–2052. doi: 10.1016/S0140-6736(16)00346-9. [DOI] [PubMed] [Google Scholar]
- 2.Emmerson B.T. The management of gout. New Engl J Med. 1996;334(7):445–451. doi: 10.1056/NEJM199602153340707. [DOI] [PubMed] [Google Scholar]
- 3.Pascual E., Sivera F. Time required for disappearance of urate crystals from synovial fluid after successful hypouricaemic treatment relates to the duration of gout. Ann Rheum Dis. 2007;66(8):1056–1058. doi: 10.1136/ard.2006.060368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Singh J.A. Challenges faced by patients in gout treatment: a qualitative study. J Clin Rheumatol: Practical Rep Rheum Musculoskelet Dis. 2014;20(3):172–174. doi: 10.1097/RHU.0000000000000091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kuo C.F., Grainge M.J., Zhang W., Doherty M. Global epidemiology of gout: prevalence, incidence and risk factors. Nat Rev Rheumatol. 2015;11(11):649–662. doi: 10.1038/nrrheum.2015.91. [DOI] [PubMed] [Google Scholar]
- 6.McCarty D.J., Hollander J.L. Identification of urate crystals in gouty synovial fluid. Ann Intern Med. 1961;54:452–460. doi: 10.7326/0003-4819-54-3-452. [DOI] [PubMed] [Google Scholar]
- 7.Mandal A.K., Mount D.B. The molecular physiology of uric acid homeostasis. Annu Rev Physiol. 2015;77:323–345. doi: 10.1146/annurev-physiol-021113-170343. [DOI] [PubMed] [Google Scholar]
- 8.Kamei K., Konta T., Hirayama A., Suzuki K., Ichikawa K., Fujimoto S. A slight increase within the normal range of serum uric acid and the decline in renal function: associations in a community-based population. Nephrol, Dialysis, Transplant: official publication of the European Dialysis and Transplant Association – European Renal Association. 2014;29(12):2286–2292. doi: 10.1093/ndt/gfu256. [DOI] [PubMed] [Google Scholar]
- 9.Torres R.J., Puig J.G. Hypoxanthine-guanine phosophoribosyltransferase (HPRT) deficiency: Lesch-Nyhan syndrome. Orphanet J Rare Dis. 2007;2:48. doi: 10.1186/1750-1172-2-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reginato A.M., Olsen B.R. Genetics and experimental models of crystal-induced arthritis. Lessons learned from mice and men: is it crystal clear? Curr Opin Rheumatol. 2007;19(2):134–145. doi: 10.1097/BOR.0b013e328040c00b. [DOI] [PubMed] [Google Scholar]
- 11.Kanbara A., Seyama I. Effect of urine pH on uric acid excretion by manipulating food materials. Nucleosides, Nucleotides Nucleic Acids. 2011;30(12):1066–1071. doi: 10.1080/15257770.2011.596498. [DOI] [PubMed] [Google Scholar]
- 12.Towiwat P., Li Z.G. The association of vitamin C, alcohol, coffee, tea, milk and yogurt with uric acid and gout. Int J Rheum Dis. 2015;18(5):495–501. doi: 10.1111/1756-185X.12622. [DOI] [PubMed] [Google Scholar]
- 13.Mahmoud H.H., Leverger G., Patte C., Harvey E., Lascombes F. Advances in the management of malignancy-associated hyperuricaemia. Br J Cancer. 1998;77(Suppl 4):18–20. doi: 10.1038/bjc.1998.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Emmerson B. Hyperlipidaemia in hyperuricaemia and gout. Ann Rheum Dis. 1998;57(9):509–510. doi: 10.1136/ard.57.9.509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bedir A., Topbas M., Tanyeri F., Alvur M., Arik N. Leptin might be a regulator of serum uric acid concentrations in humans. Jpn Heart J. 2003;44(4):527–536. doi: 10.1536/jhj.44.527. [DOI] [PubMed] [Google Scholar]
- 16.Dessein P.H., Shipton E.A., Stanwix A.E., Joffe B.I., Ramokgadi J. Beneficial effects of weight loss associated with moderate calorie/carbohydrate restriction, and increased proportional intake of protein and unsaturated fat on serum urate and lipoprotein levels in gout: a pilot study. Ann Rheum Dis. 2000;59(7):539–543. doi: 10.1136/ard.59.7.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ichida K., Matsuo H., Takada T., Nakayama A., Murakami K., Shimizu T. Decreased extra-renal urate excretion is a common cause of hyperuricemia. Nature Commun. 2012;3:764. doi: 10.1038/ncomms1756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Enomoto A., Endou H. Roles of organic anion transporters (OATs) and a urate transporter (URAT1) in the pathophysiology of human disease. Clin Exp Nephrol. 2005;9(3):195–205. doi: 10.1007/s10157-005-0368-5. [DOI] [PubMed] [Google Scholar]
- 19.Bobulescu I.A., Moe O.W. Renal transport of uric acid: evolving concepts and uncertainties. Adv Chronic Kidney Dis. 2012;19(6):358–371. doi: 10.1053/j.ackd.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J., Liu Y., Rao F., Nievergelt C.M., O'Connor D.T., Wang X. Common genetic variants of the human uromodulin gene regulate transcription and predict plasma uric acid levels. Kidney Int. 2013;83(4):733–740. doi: 10.1038/ki.2012.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cho S.K., Kim S., Chung J.Y., Jee S.H. Discovery of URAT1 SNPs and association between serum uric acid levels and URAT1. BMJ Open. 2015;5(11):e009360. doi: 10.1136/bmjopen-2015-009360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tan P.K., Ostertag T.M., Miner J.N. Mechanism of high affinity inhibition of the human urate transporter URAT1. Sci Rep. 2016;6:34995. doi: 10.1038/srep34995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Phipps-Green A.J., Merriman M.E., Topless R., Altaf S., Montgomery G.W., Franklin C. Twenty-eight loci that influence serum urate levels: analysis of association with gout. Ann Rheum Dis. 2016;75(1):124–130. doi: 10.1136/annrheumdis-2014-205877. [DOI] [PubMed] [Google Scholar]
- 24.Kolz M., Johnson T., Sanna S., Teumer A., Vitart V., Perola M. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS Genet. 2009;5(6):e1000504. doi: 10.1371/journal.pgen.1000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu R., O'Connell M., Johnson K., Pritzker K., Mackman N., Terkeltaub R. Extracellular signal-regulated kinase 1/extracellular signal-regulated kinase 2 mitogen-activated protein kinase signaling and activation of activator protein 1 and nuclear factor kappaB transcription factors play central roles in interleukin-8 expression stimulated by monosodium urate monohydrate and calcium pyrophosphate crystals in monocytic cells. Arthritis Rheum. 2000;43(5):1145–1155. doi: 10.1002/1529-0131(200005)43:5<1145::AID-ANR25>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- 26.Cronstein B.N., Sunkureddi P. Mechanistic aspects of inflammation and clinical management of inflammation in acute gouty arthritis. J Clin Rheumatol: Practical Rep Rheumat Musculoskelet Dis. 2013;19(1):19–29. doi: 10.1097/RHU.0b013e31827d8790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Busso N., Ea H.K. The mechanisms of inflammation in gout and pseudogout (CPP-induced arthritis) Reumatismo. 2012;63(4):230–237. doi: 10.4081/reumatismo.2011.230. [DOI] [PubMed] [Google Scholar]
- 28.Ea H.K. Mechanisms of gout inflammation. Presse Med. 2011;40(9 Pt 1):836–843. doi: 10.1016/j.lpm.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 29.Dalbeth N., Lauterio T.J., Wolfe H.R. Mechanism of action of colchicine in the treatment of gout. Clin Ther. 2014;36(10):1465–1479. doi: 10.1016/j.clinthera.2014.07.017. [DOI] [PubMed] [Google Scholar]
- 30.Steiger S., Harper J.L. Mechanisms of spontaneous resolution of acute gouty inflammation. Curr Rheumatol Rep. 2014;16(1):392. doi: 10.1007/s11926-013-0392-5. [DOI] [PubMed] [Google Scholar]
- 31.Grassi W., De Angelis R. Clinical features of gout. Reumatismo. 2012;63(4):238–245. doi: 10.4081/reumatismo.2011.238. [DOI] [PubMed] [Google Scholar]
- 32.Gonzalez E.B. An update on the pathology and clinical management of gouty arthritis. Clin Rheumatol. 2012;31(1):13–21. doi: 10.1007/s10067-011-1877-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schlesinger N., Thiele R.G. The pathogenesis of bone erosions in gouty arthritis. Ann Rheum Dis. 2010;69(11):1907–1912. doi: 10.1136/ard.2010.128454. [DOI] [PubMed] [Google Scholar]
- 34.Kosmadakis G., Viskaduraki M., Michail S. The validity of fractional excretion of uric acid in the diagnosis of acute kidney injury due to decreased kidney perfusion. Am J Kidney Dis. 2009;54(6):1186–1187. doi: 10.1053/j.ajkd.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 35.Burns C.M., Wortmann R.L. Latest evidence on gout management: what the clinician needs to know. Therap Adv Chronic Dis. 2012;3(6):271–286. doi: 10.1177/2040622312462056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanbay M., Huddam B., Azak A., Solak Y., Kadioglu G.K., Kirbas I. A randomized study of allopurinol on endothelial function and estimated glomular filtration rate in asymptomatic hyperuricemic subjects with normal renal function. Clin J Am Soc Nephrol: CJASN. 2011;6(8):1887–1894. doi: 10.2215/CJN.11451210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luk A.J., Levin G.P., Moore E.E., Zhou X.H., Kestenbaum B.R., Choi H.K. Allopurinol and mortality in hyperuricaemic patients. Rheumatol (Oxford, England) 2009;48(7):804–806. doi: 10.1093/rheumatology/kep069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yokose C., Chen M., Berhanu A., Pillinger M.H., Krasnokutsky S. Gout and osteoarthritis: associations, pathophysiology, and therapeutic implications. Curr Rheumatol Rep. 2016;18(10):65. doi: 10.1007/s11926-016-0613-9. [DOI] [PubMed] [Google Scholar]
- 39.Mazzali M., Kanbay M., Segal M.S., Shafiu M., Jalal D., Feig D.I. Uric acid and hypertension: cause or effect? Curr Rheumatol Rep. 2010;12(2):108–117. doi: 10.1007/s11926-010-0094-1. [DOI] [PubMed] [Google Scholar]
- 40.Katsiki N., Papanas N., Fonseca V.A., Maltezos E., Mikhailidis D.P. Uric acid and diabetes: Is there a link? Curr Pharm Des. 2013;19(27):4930–4937. doi: 10.2174/1381612811319270016. [DOI] [PubMed] [Google Scholar]
- 41.Roddy E. Revisiting the pathogenesis of podagra: why does gout target the foot? J Foot Ankle Res. 2011;4(1):13. doi: 10.1186/1757-1146-4-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canoso J.J., Yood R.A. Acute gouty bursitis: report of 15 cases. Ann Rheum Dis. 1979;38(4):326–328. doi: 10.1136/ard.38.4.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Perez-Ruiz F., Castillo E., Chinchilla S.P., Herrero-Beites A.M. Clinical manifestations and diagnosis of gout. Rheum Dis Clin North Am. 2014;40(2):193–206. doi: 10.1016/j.rdc.2014.01.003. [DOI] [PubMed] [Google Scholar]
- 44.Pascual E., Batlle-Gualda E., Martinez A., Rosas J., Vela P. Synovial fluid analysis for diagnosis of intercritical gout. Ann Intern Med. 1999;131(10):756–759. doi: 10.7326/0003-4819-131-10-199911160-00007. [DOI] [PubMed] [Google Scholar]
- 45.Chhana A., Dalbeth N. The gouty tophus: a review. Curr Rheumatol Rep. 2015;17(3):19. doi: 10.1007/s11926-014-0492-x. [DOI] [PubMed] [Google Scholar]
- 46.Malik A., Schumacher H.R., Dinnella J.E., Clayburne G.M. Clinical diagnostic criteria for gout: comparison with the gold standard of synovial fluid crystal analysis. J Clin Rheumatol: Practical Rep Rheum Musculoskelet Dis. 2009;15(1):22–24. doi: 10.1097/RHU.0b013e3181945b79. [DOI] [PubMed] [Google Scholar]
- 47.Zhang W., Doherty M., Pascual E., Bardin T., Barskova V., Conaghan P. EULAR evidence based recommendations for gout. Part I: diagnosis. report of a task force of the standing committee for international clinical studies including therapeutics (ESCISIT) Ann Rheum Dis. 2006;65(10):1301–1311. doi: 10.1136/ard.2006.055251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Atdjian M., Fernandez-Madrid F. Coexistence of chronic tophaceous gout and rheumatoid arthritis. J Rheumatol. 1981;8(6):989–992. [PubMed] [Google Scholar]
- 49.Dincer H.E., Dincer A.P., Levinson D.J. Asymptomatic hyperuricemia: to treat or not to treat. Clevel Clin J Med. 2002;69(8):594. doi: 10.3949/ccjm.69.8.594. 7, 600-2 passim. [DOI] [PubMed] [Google Scholar]
- 50.Badulescu M., Macovei L., Rezus E. Acute gout attack with normal serum uric acid levels. Rev Med Chir Soc Med Nat Iasi. 2014;118(4):942–945. [PubMed] [Google Scholar]
- 51.Underwood M. Diagnosis and management of gout. BMJ (Clin Res Ed) 2006;332(7553):1315–1319. doi: 10.1136/bmj.332.7553.1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pascual E., Tovar J., Ruiz M.T. The ordinary light microscope: an appropriate tool for provisional detection and identification of crystals in synovial fluid. Ann Rheum Dis. 1989;48(12):983–985. doi: 10.1136/ard.48.12.983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phelps P., Steele A.D., McCarty D.J., Jr. Compensated polarized light microscopy. Identification of crystals in synovial fluids from gout and pseudogout. JAMA. 1968;203(7):508–512. doi: 10.1001/jama.203.7.508. [DOI] [PubMed] [Google Scholar]
- 54.Strasinger S.K., Di Lorenzo M.S. F. A. Davis Company; 2008. Urinalysis & body fluids. [Google Scholar]
- 55.Kramer H.M., Curhan G. The association between gout and nephrolithiasis: the National Health and Nutrition Examination Survey III, 1988–1994. Am J Kidney Dis. 2002;40(1):37–42. doi: 10.1053/ajkd.2002.33911. [DOI] [PubMed] [Google Scholar]
- 56.Fernandes EA, Bergamaschi SB, Rodrigues TC, Dias GC, Malmann R, Ramos GM, et al. Relevant aspects of imaging in the diagnosis and management of gout. Revista brasileira de reumatologia. 2016. [DOI] [PubMed]
- 57.Rettenbacher T., Ennemoser S., Weirich H., Ulmer H., Hartig F., Klotz W. Diagnostic imaging of gout: comparison of high-resolution US versus conventional X-ray. Eur Radiol. 2008;18(3):621–630. doi: 10.1007/s00330-007-0802-z. [DOI] [PubMed] [Google Scholar]
- 58.Namas R., Meysami A., Siegal D., Rubin B. Gout and ultrasound: the disease of kings and the queen of imaging. Gout Hyperuricaemia. 2014;1:94–100. [Google Scholar]
- 59.Bloch C., Hermann G., Yu T.F. A radiologic reevaluation of gout: a study of 2,000 patients. AJR Am J Roentgenol. 1980;134(4):781–787. doi: 10.2214/ajr.134.4.781. [DOI] [PubMed] [Google Scholar]
- 60.Malghem J, Vande Berg B, Lecouvet F. Goutte d’hier et d’aujourd’hui. Imagerie de l’appareil musculo-squlettique: Textes choisis. Montpellier (France): Sauramps Medical; 2011. p. 35–44.
- 61.Omoumi P., Zufferey P., Malghem J., So A. Imaging in gout and other crystal-related arthropathies. Rheum Dis Clin North Am. 2016;42(4):621–644. doi: 10.1016/j.rdc.2016.07.005. [DOI] [PubMed] [Google Scholar]
- 62.Martel W. The overhanging margin of bone: a roentgenologic manifestation of gout. Radiology. 1968;91(4):755–756. doi: 10.1148/91.4.755. [DOI] [PubMed] [Google Scholar]
- 63.Gentili A. Advanced imaging of gout. Semin Musculoskelet Radiol. 2003;7(3):165–174. doi: 10.1055/s-2003-43227. [DOI] [PubMed] [Google Scholar]
- 64.Choi M.H., MacKenzie J.D., Dalinka M.K. Imaging features of crystal-induced arthropathy. Rheum Dis Clin North Am. 2006;32(2):427–446. doi: 10.1016/j.rdc.2006.04.001. viii. [DOI] [PubMed] [Google Scholar]
- 65.Gentili A. The advanced imaging of gouty tophi. Curr Rheumatol Rep. 2006;8(3):231–235. doi: 10.1007/s11926-996-0030-6. [DOI] [PubMed] [Google Scholar]
- 66.Durcan L., Grainger R., Keen H.I., Taylor W.J., Dalbeth N. Imaging as a potential outcome measure in gout studies: A systematic literature review. Semin Arthritis Rheum. 2016;45(5):570–579. doi: 10.1016/j.semarthrit.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 67.Dalbeth N., Clark B., McQueen F., Doyle A., Taylor W. Validation of a radiographic damage index in chronic gout. Arthritis Rheum. 2007;57(6):1067–1073. doi: 10.1002/art.22891. [DOI] [PubMed] [Google Scholar]
- 68.Nestorova R., Fodor D. Crystal-induced arthritis. In: El Miedany Y., editor. Musculoskeletal ultrasonography in rheumatic diseases. Springer International Publishing; Cham: 2015. pp. 137–167. [Google Scholar]
- 69.Grassi W., Gutierrez M., Filippucci E. Chapter 16 - crystal-associated synovitis A2 – Wakefield, Richard J. In: D'Agostino M.A., editor. Essential applications of musculoskeletal ultrasound in rheumatology. Content Repository Only!; Philadelphia: 2010. pp. 187–197. [Google Scholar]
- 70.Filippucci E., Di Geso L., Grassi W. Tips and tricks to recognize microcrystalline arthritis. Rheumatol (Oxford, England) 2012;51(Suppl 7):vii18–vii21. doi: 10.1093/rheumatology/kes332. [DOI] [PubMed] [Google Scholar]
- 71.Gutierrez M., Smith W., Thiele R., Keen H., Kaeley G., Naredo E. Defining elementary ultrasound lesions in gout. Preliminary results of Delphi consensus and web-exercise reliability. Ann Rheum Dis. 2014;73(Suppl 2):302. [Google Scholar]
- 72.Wright S.A., Filippucci E., McVeigh C., Grey A., McCarron M., Grassi W. High-resolution ultrasonography of the first metatarsal phalangeal joint in gout: a controlled study. Ann Rheum Dis. 2007;66(7):859–864. doi: 10.1136/ard.2006.062802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Filippucci E., Riveros M.G., Georgescu D., Salaffi F., Grassi W. Hyaline cartilage involvement in patients with gout and calcium pyrophosphate deposition disease. An ultrasound study. Osteoarthr Cartilage. 2009;17(2):178–181. doi: 10.1016/j.joca.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 74.Thiele R.G., Schlesinger N. Ultrasonography shows disappearance of monosodium urate crystal deposition on hyaline cartilage after sustained normouricemia is achieved. Rheumatol Int. 2010;30(4):495–503. doi: 10.1007/s00296-009-1002-8. [DOI] [PubMed] [Google Scholar]
- 75.Thiele R.G., Schlesinger N. Diagnosis of gout by ultrasound. Rheumatol (Oxford, England) 2007;46(7):1116–1121. doi: 10.1093/rheumatology/kem058. [DOI] [PubMed] [Google Scholar]
- 76.Naredo E., Uson J., Jimenez-Palop M., Martinez A., Vicente E., Brito E. Ultrasound-detected musculoskeletal urate crystal deposition: which joints and what findings should be assessed for diagnosing gout? Ann Rheum Dis. 2014;73(8):1522–1528. doi: 10.1136/annrheumdis-2013-203487. [DOI] [PubMed] [Google Scholar]
- 77.Abate M., Schiavone C., Salini V., Andia I. Occurrence of tendon pathologies in metabolic disorders. Rheumatol (Oxford, England) 2013;52(4):599–608. doi: 10.1093/rheumatology/kes395. [DOI] [PubMed] [Google Scholar]
- 78.Dalbeth N., Clark B., Gregory K., Gamble G., Sheehan T., Doyle A. Mechanisms of bone erosion in gout: a quantitative analysis using plain radiography and computed tomography. Ann Rheum Dis. 2009;68(8):1290–1295. doi: 10.1136/ard.2008.094201. [DOI] [PubMed] [Google Scholar]
- 79.McQueen F.M., Doyle A., Dalbeth N. Imaging in gout–what can we learn from MRI, CT, DECT and US? Arthritis Res Therapy. 2011;13(6):246. doi: 10.1186/ar3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gerster J.C., Landry M., Dufresne L., Meuwly J.Y. Imaging of tophaceous gout: computed tomography provides specific images compared with magnetic resonance imaging and ultrasonography. Ann Rheum Dis. 2002;61(1):52–54. doi: 10.1136/ard.61.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Dalbeth N., Clark B., Gregory K., Gamble G.D., Doyle A., McQueen F.M. Computed tomography measurement of tophus volume: Comparison with physical measurement. Arthritis Care Res. 2007;57(3):461–465. doi: 10.1002/art.22612. [DOI] [PubMed] [Google Scholar]
- 82.Perez-Ruiz F., Calabozo M., Pijoan J.I., Herrero-Beites A.M., Ruibal A. Effect of urate-lowering therapy on the velocity of size reduction of tophi in chronic gout. Arthritis Care Res. 2002;47(4):356–360. doi: 10.1002/art.10511. [DOI] [PubMed] [Google Scholar]
- 83.Gerster J.C., Landry M., Duvoisin B., Rappoport G. Computed tomography of the knee joint as an indicator of intraarticular tophi in gout. Arthritis Rheum. 1996;39(8):1406–1409. doi: 10.1002/art.1780390820. [DOI] [PubMed] [Google Scholar]
- 84.Omoumi P., Becce F., Ott J.G., Racine D., Verdun F.R. Optimization of radiation dose and image quality in musculoskeletal CT: emphasis on iterative reconstruction techniques (Part 1) Semin Musculoskelet Radiol. 2015;19(5):415–421. doi: 10.1055/s-0035-1569255. [DOI] [PubMed] [Google Scholar]
- 85.Omoumi P., Verdun F.R., Becce F. Optimization of radiation dose and image quality in musculoskeletal CT: emphasis on iterative reconstruction techniques (Part 2) Semin musculoskelet Radiol. 2015;19(5):422–430. doi: 10.1055/s-0035-1569254. [DOI] [PubMed] [Google Scholar]
- 86.Omoumi P., Becce F., Racine D., Ott J.G., Andreisek G., Verdun F.R. Dual-Energy CT: basic principles, technical approaches, and applications in musculoskeletal imaging (Part 1) Semin Musculoskelet Radiol. 2015;19(5):431–437. doi: 10.1055/s-0035-1569253. [DOI] [PubMed] [Google Scholar]
- 87.Nicolaou S. Invited commentary. Radio Graph. 2011;31(5):1376–1377. [Google Scholar]
- 88.Huppertz A., Hermann K.G., Diekhoff T., Wagner M., Hamm B., Schmidt W.A. Systemic staging for urate crystal deposits with dual-energy CT and ultrasound in patients with suspected gout. Rheumatol Int. 2014;34(6):763–771. doi: 10.1007/s00296-014-2979-1. [DOI] [PubMed] [Google Scholar]
- 89.Bongartz T., Glazebrook K.N., Kavros S.J., Murthy N.S., Merry S.P., Franz W.B., 3rd Dual-energy CT for the diagnosis of gout: an accuracy and diagnostic yield study. Ann Rheum Dis. 2015;74(6):1072–1077. doi: 10.1136/annrheumdis-2013-205095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Melzer R., Pauli C., Treumann T., Krauss B. Gout tophus detection-a comparison of dual-energy CT (DECT) and histology. Semin Arthritis Rheum. 2014;43(5):662–665. doi: 10.1016/j.semarthrit.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 91.McQueen F.M., Doyle A.J., Reeves Q., Gamble G.D., Dalbeth N. DECT urate deposits: now you see them, now you don't. Ann Rheum Dis. 2013;72(3):458–459. doi: 10.1136/annrheumdis-2012-202452. [DOI] [PubMed] [Google Scholar]
- 92.Glazebrook K.N., Guimaraes L.S., Murthy N.S., Black D.F., Bongartz T., Manek N.J. Identification of intraarticular and periarticular uric acid crystals with dual-energy CT: initial evaluation. Radiology. 2011;261(2):516–524. doi: 10.1148/radiol.11102485. [DOI] [PubMed] [Google Scholar]
- 93.Chowalloor P.V., Siew T.K., Keen H.I. Imaging in gout: A review of the recent developments. Therap Adv Musculoskelet Dis. 2014;6(4):131–143. doi: 10.1177/1759720X14542960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Toprover M., Krasnokutsky S., Pillinger M.H. Gout in the spine: imaging, diagnosis, and outcomes. Curr Rheumatol Rep. 2015;17(12):70. doi: 10.1007/s11926-015-0547-7. [DOI] [PubMed] [Google Scholar]
- 95.Zheng Z.F., Shi H.L., Xing Y., Li D., Jia J.Y., Lin S. Thoracic cord compression due to ligamentum flavum gouty tophus: a case report and literature review. Spinal Cord. 2015;53(12):881–886. doi: 10.1038/sc.2015.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nunes E.A., Rosseti A.G., Jr., Ribeiro D.S., Santiago M. Gout initially mimicking rheumatoid arthritis and later cervical spine involvement. Case Rep Rheumatol. 2014;2014:357826. doi: 10.1155/2014/357826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ahmad I., Tejada J.G. Spinal gout: a great mimicker. A case report and literature review. Neuroradiol J. 2012;25(5):621–625. doi: 10.1177/197140091202500518. [DOI] [PubMed] [Google Scholar]
- 98.Nygaard H.B., Shenoi S., Shukla S. Lower back pain caused by tophaceous gout of the spine. Neurology. 2009;73(5):404. doi: 10.1212/WNL.0b013e3181b04cb1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hsu C.Y., Shih T.T., Huang K.M., Chen P.Q., Sheu J.J., Li Y.W. Tophaceous gout of the spine: MR imaging features. Clin Radiol. 2002;57(10):919–925. doi: 10.1053/crad.2001.1001. [DOI] [PubMed] [Google Scholar]
- 100.Gongidi P., Gough-Fibkins S. Spondyloarthritis: a gouty display. J Radiol Case Rep. 2010;4(5):13–18. doi: 10.3941/jrcr.v4i5.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhao Z., Wang Y., Jin J., Deng X., Huang F. An analysis of abnormal magnetic resonance imaging of sacroiliac joints in patients misdiagnosed as spondyloarthritis. Zhonghua nei ke za zhi. 2014;53(9):724–729. [PubMed] [Google Scholar]
- 102.Kang H.J., Jung S.H., Yoon H.K., Hahn S.B., Kim S.J. Carpal tunnel syndrome caused by space occupying lesions. Yonsei Med J. 2009;50(2):257–261. doi: 10.3349/ymj.2009.50.2.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Chen C.K., Chung C.B., Yeh L., Pan H.B., Yang C.F., Lai P.H. Carpal tunnel syndrome caused by tophaceous gout: CT and MR imaging features in 20 patients. AJR Am J Roentgenol. 2000;175(3):655–659. doi: 10.2214/ajr.175.3.1750655. [DOI] [PubMed] [Google Scholar]
- 104.Godfrin-Valnet M., Godfrin G., Godard J., Prati C., Toussirot E., Michel F. Eighteen cases of crowned dens syndrome: Presentation and diagnosis. Neurochirurgie. 2013;59(3):115–120. doi: 10.1016/j.neuchi.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 105.Udayakumar D., Kteleh T., Alfata S., Bali T., Joseph A. Spinal gout mimicking paraspinal abscess: A case report. J Radiol Case Rep. 2010;4(6):15–20. doi: 10.3941/jrcr.v4i6.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chen C.H., Chen C.K., Yeh L.R., Pan H.B., Yang C.F. Intra-abdominal gout mimicking pelvic abscess. Skeletal Radiol. 2005;34(4):229–233. doi: 10.1007/s00256-004-0827-1. [DOI] [PubMed] [Google Scholar]
- 107.Grainger R., Dalbeth N., Keen H., Durcan L., Lawrence Edwards N., Perez-Ruiz F. Imaging as an outcome measure in gout studies: report from the OMERACT gout working group. J Rheumatol. 2015;42(12):2460–2464. doi: 10.3899/jrheum.141164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dalbeth N., Choi H.K., Terkeltaub R. Review: gout: a roadmap to approaches for improving global outcomes. Arthritis Rheumatol. 2017;69(1):22–34. doi: 10.1002/art.39799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Doherty M., Jansen T.L., Nuki G., Pascual E., Perez-Ruiz F., Punzi L. Gout: why is this curable disease so seldom cured? Ann Rheum Dis. 2012;71(11):1765–1770. doi: 10.1136/annrheumdis-2012-201687. [DOI] [PubMed] [Google Scholar]
- 110.Khanna D., Fitzgerald J.D., Khanna P.P., Bae S., Singh M.K., Neogi T. 2012 American College of Rheumatology guidelines for management of gout. Part 1: systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res (Hoboken) 2012;64(10):1431–1446. doi: 10.1002/acr.21772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Khanna D., Khanna P.P., Fitzgerald J.D., Singh M.K., Bae S., Neogi T. 2012 American College of Rheumatology guidelines for management of gout. Part 2: therapy and antiinflammatory prophylaxis of acute gouty arthritis. Arthritis Care Res (Hoboken) 2012;64(10):1447–1461. doi: 10.1002/acr.21773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Richette P., Doherty M., Pascual E., Barskova V., Becce F., Castaneda-Sanabria J. 2016 updated EULAR evidence-based recommendations for the management of gout. Ann Rheum Dis. 2017;76(1):29–42. doi: 10.1136/annrheumdis-2016-209707. [DOI] [PubMed] [Google Scholar]
- 113.De Vera M.A., Marcotte G., Rai S., Galo J.S., Bhole V. Medication adherence in gout: a systematic review. Arthritis Care Res (Hoboken) 2014;66(10):1551–1559. doi: 10.1002/acr.22336. [DOI] [PubMed] [Google Scholar]
- 114.Terkeltaub R.A., Furst D.E., Bennett K., Kook K.A., Crockett R.S., Davis M.W. High versus low dosing of oral colchicine for early acute gout flare: Twenty-four-hour outcome of the first multicenter, randomized, double-blind, placebo-controlled, parallel-group, dose-comparison colchicine study. Arthritis Rheum. 2010;62(4):1060–1068. doi: 10.1002/art.27327. [DOI] [PubMed] [Google Scholar]
- 115.Terkeltaub R.A., Furst D.E., Digiacinto J.L., Kook K.A., Davis M.W. Novel evidence-based colchicine dose-reduction algorithm to predict and prevent colchicine toxicity in the presence of cytochrome P450 3A4/P-glycoprotein inhibitors. Arthritis Rheum. 2011;63(8):2226–2237. doi: 10.1002/art.30389. [DOI] [PubMed] [Google Scholar]
- 116.Boonmuang P., Nathisuwan S., Chaiyakunapruk N., Suwankesawong W., Pokhagul P., Teerawattanapong N. Characterization of statin-associated myopathy case reports in Thailand using the health product vigilance center database. Drug Saf. 2013;36(9):779–787. doi: 10.1007/s40264-013-0055-5. [DOI] [PubMed] [Google Scholar]
- 117.Kuncl R.W., Duncan G., Watson D., Alderson K., Rogawski M.A., Peper M. Colchicine myopathy and neuropathy. N Engl J Med. 1987;316(25):1562–1568. doi: 10.1056/NEJM198706183162502. [DOI] [PubMed] [Google Scholar]
- 118.Janssens H.J., Janssen M., van de Lisdonk E.H., van Riel P.L., van Weel C. Use of oral prednisolone or naproxen for the treatment of gout arthritis: a double-blind, randomised equivalence trial. Lancet. 2008;371(9627):1854–1860. doi: 10.1016/S0140-6736(08)60799-0. [DOI] [PubMed] [Google Scholar]
- 119.Man C.Y., Cheung I.T., Cameron P.A., Rainer T.H. Comparison of oral prednisolone/paracetamol and oral indomethacin/paracetamol combination therapy in the treatment of acute goutlike arthritis: a double-blind, randomized, controlled trial. Ann Emerg Med. 2007;49(5):670–677. doi: 10.1016/j.annemergmed.2006.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Rainer T.H., Cheng C.H., Janssens H.J., Man C.Y., Tam L.S., Choi Y.F. Oral prednisolone in the treatment of acute gout: a pragmatic, multicenter, double-blind, randomized trial. Ann Intern Med. 2016;164(7):464–471. doi: 10.7326/M14-2070. [DOI] [PubMed] [Google Scholar]
- 121.Richette P., Bardin T. Should prednisolone be first-line therapy for acute gout? Lancet. 2008;372(9646):1301. doi: 10.1016/S0140-6736(08)61548-2. author reply -2. [DOI] [PubMed] [Google Scholar]
- 122.Daoussis D., Antonopoulos I., Yiannopoulos G., Andonopoulos A.P. ACTH as first line treatment for acute gout in 181 hospitalized patients. Joint Bone Spine. 2013;80(3):291–294. doi: 10.1016/j.jbspin.2012.09.009. [DOI] [PubMed] [Google Scholar]
- 123.So A., De Smedt T., Revaz S., Tschopp J. A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther. 2007;9(2):R28. doi: 10.1186/ar2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ottaviani S., Molto A., Ea H.K., Neveu S., Gill G., Brunier L. Efficacy of anakinra in gouty arthritis: a retrospective study of 40 cases. Arthritis Res Ther. 2013;15(5):R123. doi: 10.1186/ar4303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schlesinger N., Alten R.E., Bardin T., Schumacher H.R., Bloch M., Gimona A. Canakinumab for acute gouty arthritis in patients with limited treatment options: results from two randomised, multicentre, active-controlled, double-blind trials and their initial extensions. Ann Rheum Dis. 2012;71(11):1839–1848. doi: 10.1136/annrheumdis-2011-200908. [DOI] [PubMed] [Google Scholar]
- 126.Qaseem A., Harris R.P., Forciea M.A. Management of acute and recurrent gout: a clinical practice guideline from the american college of physicians. Ann Intern Med. 2017;166(1):58–68. doi: 10.7326/M16-0570. [DOI] [PubMed] [Google Scholar]
- 127.Rees F., Jenkins W., Doherty M. Patients with gout adhere to curative treatment if informed appropriately: proof-of-concept observational study. Ann Rheum Dis. 2013;72(6):826–830. doi: 10.1136/annrheumdis-2012-201676. [DOI] [PubMed] [Google Scholar]
- 128.Dalbeth N., Wong S., Gamble G.D., Horne A., Mason B., Pool B. Acute effect of milk on serum urate concentrations: a randomised controlled crossover trial. Ann Rheum Dis. 2010;69(9):1677–1682. doi: 10.1136/ard.2009.124230. [DOI] [PubMed] [Google Scholar]
- 129.Zhu Y., Zhang Y., Choi H.K. The serum urate-lowering impact of weight loss among men with a high cardiovascular risk profile: the Multiple Risk Factor Intervention Trial. Rheumatology (Oxford) 2010;49(12):2391–2399. doi: 10.1093/rheumatology/keq256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Beyl R.N., Jr., Hughes L., Morgan S. Update on importance of diet in gout. Am J Med. 2016;129(11):1153–1158. doi: 10.1016/j.amjmed.2016.06.040. [DOI] [PubMed] [Google Scholar]
- 131.Azadbakht L., Mirmiran P., Esmaillzadeh A., Azizi T., Azizi F. Beneficial effects of a Dietary Approaches to Stop Hypertension eating plan on features of the metabolic syndrome. Diabetes Care. 2005;28(12):2823–2831. doi: 10.2337/diacare.28.12.2823. [DOI] [PubMed] [Google Scholar]
- 132.Kontogianni M.D., Chrysohoou C., Panagiotakos D.B., Tsetsekou E., Zeimbekis A., Pitsavos C. Adherence to the Mediterranean diet and serum uric acid: the ATTICA study. Scand J Rheumatol. 2012;41(6):442–449. doi: 10.3109/03009742.2012.679964. [DOI] [PubMed] [Google Scholar]
- 133.Kiltz U., Smolen J., Bardin T., Cohen Solal A., Dalbeth N., Doherty M. Treat-to-target (T2T) recommendations for gout. Ann Rheum Dis. 2016 doi: 10.1136/annrheumdis-2016-209467. [DOI] [PubMed] [Google Scholar]
- 134.Latourte A., Bardin T., Richette P. Prophylaxis for acute gout flares after initiation of urate-lowering therapy. Rheumatology (Oxford) 2014;53(11):1920–1926. doi: 10.1093/rheumatology/keu157. [DOI] [PubMed] [Google Scholar]
- 135.Nidorf S.M., Eikelboom J.W., Budgeon C.A., Thompson P.L. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. 2013;61(4):404–410. doi: 10.1016/j.jacc.2012.10.027. [DOI] [PubMed] [Google Scholar]
- 136.Richette P., Perez-Ruiz F., Doherty M., Jansen T.L., Nuki G., Pascual E. Improving cardiovascular and renal outcomes in gout: what should we target? Nat Rev Rheumatol. 2014;10(11):654–661. doi: 10.1038/nrrheum.2014.124. [DOI] [PubMed] [Google Scholar]
- 137.Roberts C.J., Marshall A.J., Heaton S., Barritt D.W. Comparison of natriuretic, uricosuric, and antihypertensive properties of tienilic acid, bendrofluazide, and spironolactone. BMJ. 1979;1(6158):224–226. doi: 10.1136/bmj.1.6158.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Reach G. Treatment adherence in patients with gout. Joint Bone Spine. 2011;78(5):456–459. doi: 10.1016/j.jbspin.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 139.Greene M.L., Fujimoto W.Y., Seegmiller J.E. Urinary xanthine stones–a rare complications of allopurinol therapy. N Engl J Med. 1969;280(8):426–427. doi: 10.1056/NEJM196902202800806. [DOI] [PubMed] [Google Scholar]
- 140.Wright D.F., Duffull S.B., Merriman T.R., Dalbeth N., Barclay M.L., Stamp L.K. Predicting allopurinol response in patients with gout. Br J Clin Pharmacol. 2016;81(2):277–289. doi: 10.1111/bcp.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Roberts R.L., Wallace M.C., Phipps-Green A.J., Topless R., Drake J.M., Tan P. ABCG2 loss-of-function polymorphism predicts poor response to allopurinol in patients with gout. Pharmacogenomics J. 2016 doi: 10.1038/tpj.2015.101. [DOI] [PubMed] [Google Scholar]
- 142.Annemans L., Spaepen E., Gaskin M., Bonnemaire M., Malier V., Gilbert T. Gout in the UK and Germany: prevalence, comorbidities and management in general practice 2000–2005. Ann Rheum Dis. 2008;67(7):960–966. doi: 10.1136/ard.2007.076232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Delbarre F., Amor B., Auscher C., de Gery A. Treatment of gout with allopurinol. A study of 106 cases. Ann Rheum Dis. 1966;25(6 Suppl):627–633. doi: 10.1136/ard.25.suppl_6.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Becker M.A., Schumacher H.R., Jr, Wortmann R.L., MacDonald P.A., Eustace D., Palo W.A. Febuxostat compared with allopurinol in patients with hyperuricemia and gout. N Engl J Med. 2005;353(23):2450–2461. doi: 10.1056/NEJMoa050373. [DOI] [PubMed] [Google Scholar]
- 145.Schumacher H.R., Jr, Becker M.A., Wortmann R.L., Macdonald P.A., Hunt B., Streit J. Effects of febuxostat versus allopurinol and placebo in reducing serum urate in subjects with hyperuricemia and gout: a 28-week, phase III, randomized, double-blind, parallel-group trial. Arthritis Rheum. 2008;59(11):1540–1548. doi: 10.1002/art.24209. [DOI] [PubMed] [Google Scholar]
- 146.Becker M.A., Fitz-Patrick D., Choi H.K., Dalbeth N., Storgard C., Cravets M. An open-label, 6-month study of allopurinol safety in gout: The LASSO study. Semin Arthritis Rheum. 2015;45(2):174–183. doi: 10.1016/j.semarthrit.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 147.Reinders M.K., van Roon E.N., Jansen T.L., Delsing J., Griep E.N., Hoekstra M. Efficacy and tolerability of urate-lowering drugs in gout: a randomised controlled trial of benzbromarone versus probenecid after failure of allopurinol. Ann Rheum Dis. 2009;68(1):51–56. doi: 10.1136/ard.2007.083071. [DOI] [PubMed] [Google Scholar]
- 148.Tam S., Carroll W. Allopurinol hepatotoxicity. Am J Med. 1989;86(3):357–358. doi: 10.1016/0002-9343(89)90316-1. [DOI] [PubMed] [Google Scholar]
- 149.Greenberg M.S., Zambrano S.S. Aplastic agranulocytosis after allopurinol therapy. Arthritis Rheum. 1972;15(4):413–416. doi: 10.1002/art.1780150413. [DOI] [PubMed] [Google Scholar]
- 150.Gelbart D.R., Weinstein A.B., Fajardo L.F. Allopurinol-induced interstitial nephritis. Ann Intern Med. 1977;86(2):196–198. doi: 10.7326/0003-4819-86-2-196. [DOI] [PubMed] [Google Scholar]
- 151.Azulay J.P., Blin O., Valentin P., Abegg P., Pellissier J.F., Serratrice G. Regression of allopurinol-induced peripheral neuropathy after drug withdrawal. Eur Neurol. 1993;33(3):193–194. doi: 10.1159/000116934. [DOI] [PubMed] [Google Scholar]
- 152.Sidoroff A., Halevy S., Bavinck J.N., Vaillant L., Roujeau J.C. Acute generalized exanthematous pustulosis (AGEP)–a clinical reaction pattern. J Cutan Pathol. 2001;28(3):113–119. doi: 10.1034/j.1600-0560.2001.028003113.x. [DOI] [PubMed] [Google Scholar]
- 153.Bastuji-Garin S., Rzany B., Stern R.S., Shear N.H., Naldi L., Roujeau J.C. Clinical classification of cases of toxic epidermal necrolysis, Stevens-Johnson syndrome, and erythema multiforme. Arch Dermatol. 1993;129(1):92–96. [PubMed] [Google Scholar]
- 154.Kardaun S.H., Sidoroff A., Valeyrie-Allanore L., Halevy S., Davidovici B.B., Mockenhaupt M. Variability in the clinical pattern of cutaneous side-effects of drugs with systemic symptoms: does a DRESS syndrome really exist? Br J Dermatol. 2007;156(3):609–611. doi: 10.1111/j.1365-2133.2006.07704.x. [DOI] [PubMed] [Google Scholar]
- 155.Kim S.C., Newcomb C., Margolis D., Roy J., Hennessy S. Severe cutaneous reactions requiring hospitalization in allopurinol initiators: a population-based cohort study. Arthritis Care Res (Hoboken) 2013;65(4):578–584. doi: 10.1002/acr.21817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Yang C.Y., Chen C.H., Deng S.T., Huang C.S., Lin Y.J., Chen Y.J. Allopurinol use and risk of fatal hypersensitivity reactions: a nationwide population-based study in Taiwan. JAMA Intern Med. 2015;175(9):1550–1557. doi: 10.1001/jamainternmed.2015.3536. [DOI] [PubMed] [Google Scholar]
- 157.Hung S.I., Chung W.H., Liou L.B., Chu C.C., Lin M., Huang H.P. HLA-B∗5801 allele as a genetic marker for severe cutaneous adverse reactions caused by allopurinol. Proc Natl Acad Sci U S A. 2005;102(11):4134–4139. doi: 10.1073/pnas.0409500102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ng C.Y., Yeh Y.T., Wang C.W., Hung S.I., Yang C.H., Chang Y.C. Impact of the HLA-B(∗)58:01 allele and renal impairment on allopurinol-induced cutaneous adverse reactions. J Invest Dermatol. 2016;136(7):1373–1381. doi: 10.1016/j.jid.2016.02.808. [DOI] [PubMed] [Google Scholar]
- 159.Stamp L.K., Taylor W.J., Jones P.B., Dockerty J.L., Drake J., Frampton C. Starting dose is a risk factor for allopurinol hypersensitivity syndrome: a proposed safe starting dose of allopurinol. Arthritis Rheum. 2012;64(8):2529–2536. doi: 10.1002/art.34488. [DOI] [PubMed] [Google Scholar]
- 160.Hande K.R., Noone R.M., Stone W.J. Severe allopurinol toxicity. Description and guidelines for prevention in patients with renal insufficiency. Am J Med. 1984;76(1):47–56. doi: 10.1016/0002-9343(84)90743-5. [DOI] [PubMed] [Google Scholar]
- 161.Chung W.H., Chang W.C., Stocker S.L., Juo C.G., Graham G.G., Lee M.H. Insights into the poor prognosis of allopurinol-induced severe cutaneous adverse reactions: the impact of renal insufficiency, high plasma levels of oxypurinol and granulysin. Ann Rheum Dis. 2015;74(12):2157–2164. doi: 10.1136/annrheumdis-2014-205577. [DOI] [PubMed] [Google Scholar]
- 162.Ko T.M., Tsai C.Y., Chen S.Y., Chen K.S., Yu K.H., Chu C.S. Use of HLA-B∗58:01 genotyping to prevent allopurinol induced severe cutaneous adverse reactions in Taiwan: national prospective cohort study. BMJ. 2015;351:h4848. doi: 10.1136/bmj.h4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Dalbeth N., Kumar S., Stamp L., Gow P. Dose adjustment of allopurinol according to creatinine clearance does not provide adequate control of hyperuricemia in patients with gout. J Rheumatol. 2006;33(8):1646–1650. [PubMed] [Google Scholar]
- 164.Stamp L.K., O'Donnell J.L., Zhang M., James J., Frampton C., Barclay M.L. Using allopurinol above the dose based on creatinine clearance is effective and safe in patients with chronic gout, including those with renal impairment. Arthritis Rheum. 2011;63(2):412–421. doi: 10.1002/art.30119. [DOI] [PubMed] [Google Scholar]
- 165.Juge P.A., Truchetet M.E., Pillebout E., Ottaviani S., Vigneau C., Loustau C. Efficacy and safety of febuxostat in 73 gouty patients with stage 4/5 chronic kidney disease: A retrospective study of 10 centers. Joint Bone Spine. 2016 doi: 10.1016/j.jbspin.2016.09.020. [DOI] [PubMed] [Google Scholar]
- 166.Chou H.Y., Chen C.B., Cheng C.Y., Chen Y.A., Ng C.Y., Kuo K.L. Febuxostat-associated drug reaction with eosinophilia and systemic symptoms (DRESS) J Clin Pharm Ther. 2015;40(6):689–692. doi: 10.1111/jcpt.12322. [DOI] [PubMed] [Google Scholar]
- 167.Bardin T., Chales G., Pascart T., Flipo R.M., Korng Ea H., Roujeau J.C. Risk of cutaneous adverse events with febuxostat treatment in patients with skin reaction to allopurinol. A retrospective, hospital-based study of 101 patients with consecutive allopurinol and febuxostat treatment. Joint Bone Spine. 2016;83(3):314–317. doi: 10.1016/j.jbspin.2015.07.011. [DOI] [PubMed] [Google Scholar]
- 168.MacDonald T.M., Ford I., Nuki G., Mackenzie I.S., De Caterina R., Findlay E. Protocol of the Febuxostat versus Allopurinol Streamlined Trial (FAST): a large prospective, randomised, open, blinded endpoint study comparing the cardiovascular safety of allopurinol and febuxostat in the management of symptomatic hyperuricaemia. BMJ Open. 2014;4(7):e005354. doi: 10.1136/bmjopen-2014-005354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Gutman A.B., Yu T.F. Benemid (p-di-n-propylsulfamyl)-benzoic acid) as uricosuric agent in chronic gouty arthritis. Trans Assoc Am Physicians. 1951;64:279–288. [PubMed] [Google Scholar]
- 170.Pui K., Gow P.J., Dalbeth N. Efficacy and tolerability of probenecid as urate-lowering therapy in gout; clinical experience in high-prevalence population. J Rheumatol. 2013;40(6):872–876. doi: 10.3899/jrheum.121301. [DOI] [PubMed] [Google Scholar]
- 171.Perez-Ruiz F., Alonso-Ruiz A., Calabozo M., Herrero-Beites A., Garcia-Erauskin G., Ruiz-Lucea E. Efficacy of allopurinol and benzbromarone for the control of hyperuricaemia. A pathogenic approach to the treatment of primary chronic gout. Ann Rheum Dis. 1998;57(9):545–549. doi: 10.1136/ard.57.9.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Perez-Ruiz F., Calabozo M., Fernandez-Lopez M.J., Herrero-Beites A., Ruiz-Lucea E., Garcia-Erauskin G. Treatment of chronic gout in patients with renal function impairment: an open, randomized, actively controlled study. J Clin Rheumatol. 1999;5(2):49–55. doi: 10.1097/00124743-199904000-00003. [DOI] [PubMed] [Google Scholar]
- 173.Bardin T., Keenan R.T., Khanna P.P., Kopicko J., Fung M., Bhakta N. Lesinurad in combination with allopurinol: a randomised, double-blind, placebo-controlled study in patients with gout with inadequate response to standard of care (the multinational CLEAR 2 study) Ann Rheum Dis. 2016 doi: 10.1136/annrheumdis-2016-209213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Saag K.G., Fitz-Patrick D., Kopicko J., Fung M., Bhakta N., Adler S. Lesinurad combined with allopurinol: a randomized, double-blind, placebo-controlled study in gout patients with an inadequate response to standard-of-care allopurinol (a US-based study) Arthritis Rheumatol. 2017;69(1):203–212. doi: 10.1002/art.39840. [DOI] [PubMed] [Google Scholar]
- 175.Takahashi S., Moriwaki Y., Yamamoto T., Tsutsumi Z., Ka T., Fukuchi M. Effects of combination treatment using anti-hyperuricaemic agents with fenofibrate and/or losartan on uric acid metabolism. Ann Rheum Dis. 2003;62(6):572–575. doi: 10.1136/ard.62.6.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Milionis H.J., Kakafika A.I., Tsouli S.G., Athyros V.G., Bairaktari E.T., Seferiadis K.I. Effects of statin treatment on uric acid homeostasis in patients with primary hyperlipidemia. Am Heart J. 2004;148(4):635–640. doi: 10.1016/j.ahj.2004.04.005. [DOI] [PubMed] [Google Scholar]
- 177.Richette P., Briere C., Hoenen-Clavert V., Loeuille D., Bardin T. Rasburicase for tophaceous gout not treatable with allopurinol: an exploratory study. J Rheumatol. 2007;34(10):2093–2098. [PubMed] [Google Scholar]
- 178.Sundy J.S., Baraf H.S., Yood R.A., Edwards N.L., Gutierrez-Urena S.R., Treadwell E.L. Efficacy and tolerability of pegloticase for the treatment of chronic gout in patients refractory to conventional treatment: two randomized controlled trials. JAMA. 2011;306(7):711–720. doi: 10.1001/jama.2011.1169. [DOI] [PubMed] [Google Scholar]
- 179.Vallon V., Thomson S.C. Targeting renal glucose reabsorption to treat hyperglycaemia: the pleiotropic effects of SGLT2 inhibition. Diabetologia. 2017;60(2):215–225. doi: 10.1007/s00125-016-4157-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Strauss M.B. Little Brown & Co.; USA: 1968. Familiar medical quotations. [PMC free article] [PubMed] [Google Scholar]