

Abstract
Efavirenz is a widely prescribed medicine used to treat type 1 human immunodeficiency virus (HIV-1), the most prevalent pathogenic strain of the virus responsible for the acquired immune deficiency syndrome (AIDS) pandemic. Under prescribed dosing conditions, either alone or in combination therapy, efavirenz-induced CNS disturbances are frequently reported. Efavirenz was recently reported to interact in a similar concentration range with a number of receptors, transporters and ion channels including recombinant rat α1β2γ2 GABAA receptors whose actions were potentiated (Gatch et al., 2013; Dalwadi et al., 2016). Now we report on the molecular mechanism of efavirenz on GABAA receptors as a function of concentration and subunit composition via whole-cell recordings of GABA-activated currents from HEK293 cells expressing varying subunit configurations of GABAA receptors. Efavirenz elicited dual effects on the GABA response; it allosterically potentiated currents at low concentrations, whereas it inhibited currents at higher concentrations. The allosteric potentiating action on GABAA receptors was pronounced in the α1β2γ2, α2β2γ2 and α4β2γ2 configurations, greatly diminished in the α6β2γ2 configuration, and completely absent in the α3β2γ2 or α5β2γ2 configuration. In stark contrast, the inhibitory modulation of efavirenz at higher concentrations was evident in all subunit configurations examined. Moreover, efavirenz-induced modulatory effects were dependent on GABA concentration ([GABA]), with a pronounced impact on currents activated by low [GABA] but little effect at saturating [GABA]. Mutation of a highly-conserved threonine to phenylalanine in transmembrane domain 2 of the α1 subunit abolished the inhibitory effect of efavirenz in α1β2 receptors. Finally, mutations of any of the three conserved extracellular residues in α1/2/4 subunits to the conserved residues at the corresponding positions in α3/5 subunits (i.e., R84P, M89L or I120L) completely eliminated he potentiating effect of efavirenz in α1β2γ2 configuration. These findings demonstrate that efavirenz’s positive allosteric modulation of the GABAA receptor is mediated via a novel allosteric site associated with the extracellular domain of the receptor.
Keywords: recombinant GABAA receptors, adverse CNS side effects, picrotoxin, AIDS, antiretroviral
Graphical abstract
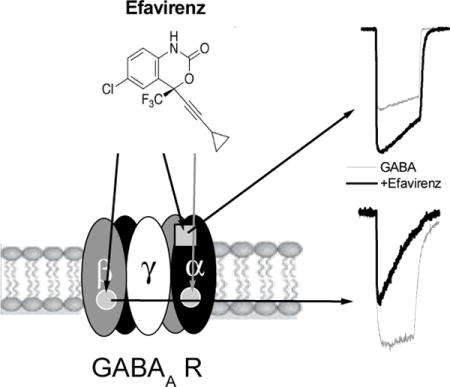
1. Introduction
The neurotransmitter γ-aminobutyric acid (GABA) inhibits neuronal activity in the mammalian CNS via activation of a chloride (Cl−) selective ligand-gated ion channel subtype called the GABAA receptor. The GABAA receptor is a known mediator of anxiety states, somnolence, and seizures/epilepsy. Diverse therapeutic and pharmacological agents, such as neurosteroids, carisoprodol, benzodiazepines, barbiturates, and anesthetics modulate GABAA receptor function (Huang et al., 2006; Gonzalez et al., 2009; Tan et al., 2011).
Efavirenz (Sustiva®, Stocrin®) is a non-nucleoside reverse transcriptase inhibitor (NNRTI) widely used in combination with other antiretrovirals for the treatment of HIV-1. However, efavirenz also known to exert neuropsychiatric adverse events in some patients (Barreiro et al., 2002; Treisman and Kaplin, 2002; Cespedes and Aberg, 2006; Sutterlin et al., 2006; Arendt et al., 2007; Treisman and Soudry, 2016). For instance, in a large clinical study (>1000 patients) CNS disturbances were reported in over half of all patients taking a standard dose of efavirenz including dizziness (28%), depression (19%), insomnia (16%), anxiety (9%), impaired concentration (8%), somnolence (7%), nervousness (7%), abnormal dreams (6%), and hallucinations (1.2%) (Sustiva package insert, 1998). Other reports indicated 40–70% of patients treated with efavirenz suffer from CNS symptoms (Blanch et al., 2001; Puzantian, 2002). The apparent psychoactivity of efavirenz may even be encouraging its diversion for recreational use (Gatch et al., 2013; Grelotti et al., 2014). In a previous report, GABAA receptors (α1β2γ2) were identified as one of several CNS targets of efavirenz (Gatch et al., 2013). In the present study we investigated the concentration and subunit-dependency of efavirenz on GABAA receptors in a molecular mechanistic context.
2. Materials and Methods
2.1. Expression of cloned receptors
Human embryonic kidney cell lines (HEK293) stably expressing recombinant human or rat α1β2γ2, α2β2γ2, α3β2γ2 or α6β2γ2 GABAA receptors were studied in the present investigation (short isoform of the γ2 subunit in all cases). Cells transiently expressing various wild type human or rat GABAA subunit configurations were also studied. A human HEK293 cell line was transiently transfected with recombinant receptor subunits using PolyJet™ DNA In Vitro transfection reagent (SignaGen Laboratories, Rockville, MD). Briefly, HEK293 cells were washed and placed in fresh Dulbecco’s modified eagle medium containing 10% FBS and antibiotics (penicillin 100 U/mL). For human or rat αx(x=1, 4, 5)β2γ2 GABAA receptors, a 1:1:3 ratio (total cDNA: 2.5 μg) of αx, β2 and γ2 subunits was added to cells growing exponentially on poly-L-lysine coated coverslips placed in a 35-mm culture dish. Transfected cells were used for electrophysiological analysis 24–48 h after the transfection.
2.2. Mutagenesis
Mutations of receptor cDNA were generated using a commercially available site-directed mutagenesis kit (QuickChange, Strategene, La Jolla, CA) and commercially produced mutagenic primers (MWG Biotech, NC). All mutants were verified by DNA sequencing (MWG Biotech, NC).
2.3. Electrophysiology
Whole-cell patch recordings were made at room temperature (22–25°C) at a holding potential of −60 mV. Patch pipettes of borosilicate glass (M1B150F, World Precision Instruments, Inc., Sarasota, FL) were pulled (Flaming/Brown, P-87/PC, Sutter Instrument Co., Novato, CA) to a tip resistance of 3–5 MΩ. The pipette solution contained (in mM): 140 CsCl, 10 EGTA, 10 HEPES, 4 Mg-ATP; pH 7.2. A coverslip containing cultured cells was placed in a small chamber (~1.5 mL) on the stage of an inverted light microscope (Olympus IMT-2) and superfused continuously (5–8 mL/min) with the following external solution containing (in mM): 125 NaCl, 5.5 KCl, 0.8 MgCl2, 3.0 CaCl2, 10 HEPES, 10 D-glucose, pH 7.3. GABA-evoked currents from the whole-cell configuration were obtained using a patch clamp amplifier (Axopatch 200A, Axon Instruments, Foster City, CA) equipped with a CV201A headstage. The currents were low-pass filtered at 5 kHz, monitored on an oscilloscope and a chart recorder (Gould TA240), and stored on a computer for subsequent analysis. To monitor the possibility that access resistance changed over time or during different experimental conditions, at the initiation of each recording we measured and stored the current response to a 5 mV voltage pulse on our digital oscilloscope. This stored trace was continually referenced throughout the recording. If a change in access resistance was observed throughout the recording period, the patch was aborted and the data were not included in the analysis. Current-voltage (I–V) relationship of GABA currents was assessed from a single cell using a ramp stimulus protocol. A transmembrane voltage ramped from −60 to +60 mV over 0.5 s time course was applied prior to (passive conductance phase) and during GABA application. The difference of these two current ramps (pA) represented the GABA-induced current, and was plotted as a function of applied holding potential (mV) to yield the I–V curve of GABA-induced currents.
2.4. Experimental protocol
GABA was prepared in extracellular solution and was applied to cells via gravity flow using a Y-shaped tube positioned near the target cell. With this system, the 10–90% rise time of the junction potential at the open tip is 60–120 ms (Huang and Dillon, 1999). Once a control response was determined, the effect of efavirenz was examined by co-applying it with a roughly EC30 concentration of GABA. Because recovery from the drug-induced effect was readily obtained upon wash out, the effects of multiple concentrations of efavirenz could generally be obtained from same cell. Efavirenz was tested at concentrations below 100 μM, because high concentrations resulted in a nonspecific cytotoxicity as indicated by loss of patch during drug application.
2.5 Chemicals
GABA stock was made in doubly distilled H2O. Efavirenz, diazepam, picrotoxin and flumazenil were solubilized in dimethyl sulfoxide (DMSO) and added to cells at a final concentration of less than 0.05 % (v/v) DMSO. DMSO (0.05%) had no effect on GABA response. Efavirenz was purchased from Sequoia Research Products Limited (Pangbourne, UK) or provided by NIH via NIH AIDS Reagent Program (https://www.aidsreagent.org/). All other drugs were purchased from Sigma Aldrich or Tocris Biosciences (Minneapolis, MN).
2.6. Data analysis
All data were recorded on a chart recorder, and stored on a computer for subsequent off-line analysis (pClamp 6.0, Axon Instruments; and Origin 5.0, Microcal Software, Inc). GABA concentration–response profiles were fitted to the following equation: I/Imax =[GABA]n/(EC50n+[GABA]n), where I and Imax represent the normalized GABA-induced current at a given concentration and the maximal current induced by a saturating concentration of GABA, respectively, EC50 is 50% effective GABA concentration, and n is the Hill coefficient. Concentration-response profiles for the positive modulatory actions of efavirenz were generated using the equation I/Imax = [efavirenz]n/([efavirenz]n+EC50n), where I is the normalized current amplitude at a given concentration of efavirenz, Imax is the maximum GABA current induced by efavirenz, EC50 is the half-maximal effective concentration of efavirenz, and n is the Hill coefficient. The concentration-response relationship for the inhibitory action of efavirenz was fitted with the equation: I/I max = [efavirenz]n/([efavirenz]n + IC50n), where I is GABA current amplitude normalized to control, IC50 is the half-blocking concentration, and n is the Hill coefficient. Since efavirenz substantially enhanced current decay, during analysis of efavirenz-mediated inhibitory action, GABA current amplitudes with and without efavirenz were quantified at the end of the GABA application (end-application current).
All data are presented as mean ± SEM. The control current amplitude prior to efavirenz was designated 100% in all cases, and current amplitudes (initial or end-application current) were expressed as a percentage of that control. Student’s t-test (paired or unpaired) was used to determine statistical significance (p< 0.05).
3. Results
In our initial report (Gatch et al., 2013), we demonstrated that efavirenz potentiates recombinant rat GABAA α1β2γ2 receptor function. Here we provide evidence that efavirenz has dual modulatory effects on GABAA receptors that are concentration and subunit configuration dependent and further that these dual functions are mediated by efavirenz’s interaction with two distinct sites.
3.1. Efavirenz-mediated potentiation of GABA response
To ensure equipotent concentrations were used to elicit GABA responses, EC30 values from concentration-response data collected from various GABAA subunit configurations were calculated and used in the investigation of efavirenz modulatory effects (Table 1). Our results demonstrate that the α isoforms significantly impact the sensitivity of GABAA receptors to efavirenz. As shown in Figure 1, efavirenz potentiated the peak amplitude of GABA-gated currents in the αxβ2γ2 GABAA receptors (x=1, 2, 4) in a concentration-dependent and reversible manner when co-applied with EC30 GABA. Among three receptor subtypes, α1β2γ2 receptors were most sensitive to potentiation by efavirenz. The current amplitude was increased by 71% at 50 μM efavirenz and the average EC50 value was 13 ± 2.3 μM for efavirenz’s potentiation effect (n=5). In addition to enhancing peak current amplitude, efavirenz increased current decay at the higher concentrations (Fig. 1A). Consequently, 30 μM efavirenz reduced current amplitude at conclusion of GABA application to 46 ± 11% of control in α1β2γ2, 38 ± 14% in α2β2γ2 and 56 ±12% in α4β2γ2 (p<0.05, paired t-test), indicating possible co-existence of an inhibitory component mediated by efavirenz. We then examined the effect of pre-applied 30 μM efavirenz for 1 min on the subsequent response to efavirenz co-applied with 10 μM GABA in α1β2γ2 receptors. As shown in Fig. 2, compared to the response to co-application of efavirenz, pretreatment cells with efavirenz did not affect potentiation on initial GABA currents but considerably inhibited end-application currents (p<0.01, n=5, paired t-test). Pre-treatment with efavirenz reduced the relative end-application currents to 33 ± 7.9% of control compared to 132 ± 16% without pretreatment from the same cells (Fig. 2B).
Table 1.
GABA sensitivity of different GABAA receptor configurations
| Receptor configurations | EC50 (μM) | Hill coefficient | Sample size | EC30 (μM) |
|---|---|---|---|---|
| α1β2γ 2 | 34 ± 4.9 | 1.08 ± 0.15 | 8 | 10 |
| α2β2γ2 | 40 ± 2.3 | 1.48 ± 0.11 | 6 | 23 |
| α3β2γ21 | 33 ± 2.1 | 1.1 ± 0.03 | 4 | 20 |
| α4β2γ2 | 4.3 ± 0.2 | 1.19 ±0.05 | 5 | 2 |
| α5β2γ2 | 16 ± 1.0 | 1.27 ± 0.09 | 4 | 9 |
| α6β2γ21 | 1.6 ± 0.1 | 1.6 ± 0.11 | 4 | 1 |
| α1β22 | 1.6 ± 0.2 | 1.3 ± 0.10 | 4 | 0.5 |
| α1(T6′F)β22 | 1.7 ± 0.3 | 0.9 ± 0.10 | 4 | 0.5 |
| α1β2(T6′F)2 | 3.6 ± 0.5 | 1.1 ± 0.10 | 4 | 1.2 |
| α1(R84P)β2γ2 | 7.9 ± 1.2 | 1.54 ± 0.21 | 7 | 4 |
| α1(M89L)β2γ2 | 6.6 ± 0.5 | 1.50 ± 0.08 | 4 | 4 |
| α1(I120L)β2γ2 | 32 ± 1.0 | 1.17 ±0.04 | 5 | 10 |
| α1(R84P/M89L)β2γ2 | 4.1 ± 0.1 | 0.93 ± 0.03 | 5 | 1.7 |
| α1(R84P/M89L/I120L)β2γ2 | 5.3 ± 0.4 | 1.24 ± 0.09 | 4 | 2.5 |
1, 2. indicate the values reported in our previous publications:
Figure 1. Concentration-response relationship for efavirenz-mediated potentiation of α1β2γ2, α2β2γ2 and α4β2γ2 GABAA receptors.
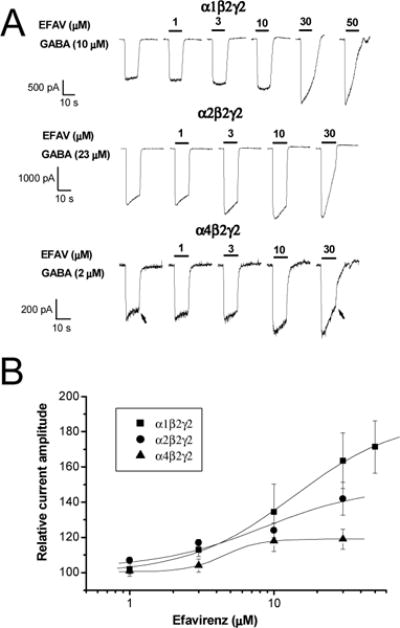
A, Representative traces showing whole-cell currents activated by EC30 GABA recorded from HEK293 cells expressing α1β2γ2 GABAA receptors. Efavirenz (1–50 μM) was co-applied with GABA for 10 sec. Arrows indicate the time at which end-application currents were measured. B, A plot of the efavirenz concentration-response relationship. All currents are normalized to the response to GABA without efavirenz. EC50 values for efavirenz response were 13 ± 2.3 μM, 8.4 ± 4.9 and 5.9 ± 1.3 μM for α1β2γ2, α2β2γ2 and α4β2γ2 GABAA receptors, respectively. Hill coefficients were 1.2 ± 0.2, 1.2 ± 0.0 and 1.2 ± 0.0 for α1β2γ2, α2β2γ2 and α4β2γ2 GABAA receptors, respectively. The curve shows the fit of the mean of at least 5 cells as described in the methods. EFAV=efavirenz.
Figure 2. Effect of pre-applied efavirenz on the response of α1β2γ2 receptors to efavirenz.
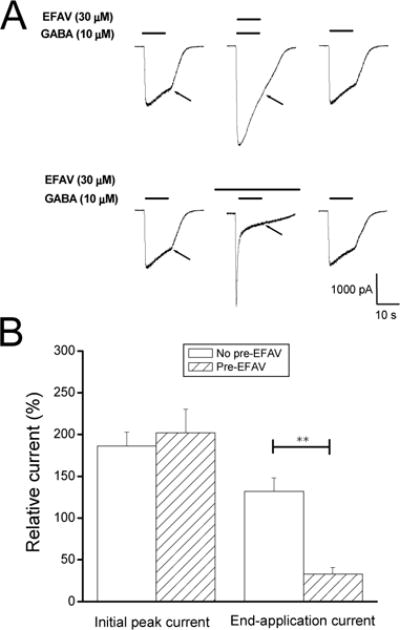
A, Representative traces showing whole-cell currents activated by 10μM GABA recorded from HEK293 cells expressing α1β2γ2 receptors. Efavirenz (30 μM) was co-applied with GABA for 10 sec to the cell without or with 1-min pre-treatment with efavirenz (30 μM). Note that end-application currents (indicated by arrows) were remarkably inhibited by pre-treatment with efavirenz compared to co-application only. B, Summary data on efavirenz-induced modulation of GABA response from the same cells without (No pre-EFAV) or with 1-min pretreated efavirenz (Pre-EFAV). **, p<0.01, unpaired t-test, n=5.
3.2. Efavirenz-mediated inhibition of GABA response
Further investigation of the subunit dependence of efavirenz modulatory effects uncovered an inhibitory action predominately present in α3β2γ2 and α5β2γ2 GABAA receptors. In contrast, to the prominent enhancement of GABA currents by efavirenz observed in α1/2/4β2γ2 configurations (Fig.1), efavirenz inhibited the peak current in a concentration-dependent manner (Fig. 3) with IC50 values of 14 ± 6.3 μM and 9.7 ±0.8 μM for α3β2γ2 and α5β2γ2, respectively (Fig. 3B). The end-application currents were almost completely inhibited by high concentrations of efavirenz (30 or 50 μM). In α6β2γ2, however, efavirenz elicited a biphasic effect: a slight potentiating effect on GABA currents (114 ± 5.1% of control, n=6, p<0.05) at 10 μM and a dominant inhibition at 30 μM (Fig. 3A&B). End-application amplitudes were reduced to 72 ± 7.2% of control and 46 ± 6.1% by 10 and 30 μM efavirenz, respectively (p<0.05, paired t-test, compared to control).
Figure 3. Efavirenz-mediated inhibitory response in α3β2γ2, α5β2γ2 and α6β2γ2 GABAA receptors.
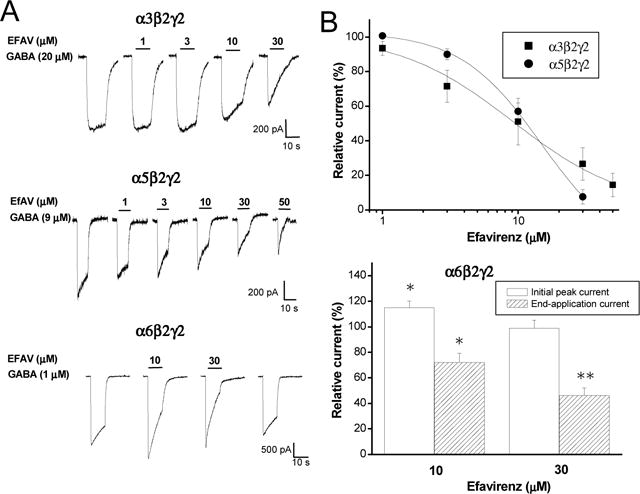
A, Representative traces showing whole-cell currents activated by EC30 GABA recorded from HEK293 cells expressing α3β2γ2, α5β2γ2 and α6β2γ2 GABAA receptors. Efavirenz (10–30 μM) concentration-dependently inhibited GABA currents without any potentiating effect in α3β2γ2 and α5β2γ2, and elicited slight enhancement on GABA initial peak currents but inhibited end-application currents in α6β2γ2. B, Summary data for effect of efavirenz on GABA response. IC50 values were 14 ± 6.3 (n=15) and 9.1 ± 7.7 μM (n=6) for α3β2γ2 and α5β2γ2, respectively. Hill coefficient were 1.4 ±0.5 and 1.0 ± 0.5 for α3β2γ2 and α5β2γ2, respectively. Biphasic effect of efavirenz on α6β2γ2. *, p<0.05; **, p<0.01, paired t-test. n=6.
3.3. Role of γ2 subunit and the benzodiazepine site
The γ2 subunit appears to be required for mature postsynaptic GABAA receptors and essential for GABAA receptor clustering during synaptogenesis (Essrich et al., 1998). From a pharmacological perspective, the γ2 subunit is also required for sensitivity to the benzodiazepine (BDZ) family of compounds (Sigel and Buhr, 1997). We thus examined the role of the γ2 subunit of the GABAA receptor on efavirenz-mediated modulation of GABAA currents. Experiments were performed on the receptors which lack γ2 subunits (i.e., α1β2) receptors. Currents activated using the GABA EC30 (0.5 μM) were recorded with co-application of varying concentrations of efavirenz. As shown in Figure 4, efavirenz was able to enhance the GABA response in the α1β2 configuration. The potentiation of initial peak current by 10 and 30 μM efavirenz was 41 ± 11% and 56 ± 14 % above the control, respectively in the α1β2 configuration, which was similar to the currents obtaining in the α1β2γ2 configuration (p>0.05, unpaired t-test). However, an inhibitory effect of efavirenz was more apparent in α1β2 compared to α1β2γ2 receptors. The end-application current was reduced to 56 ± 11% and 1.6 ± 1.7% of control by 10 and 30 μM efavirenz, respectively (n=8, p<0.05, unpaired t-test, compared to α1β2γ2 receptors).
Figure 4. Effect of efavirenz on α1β2 GABAA receptors.
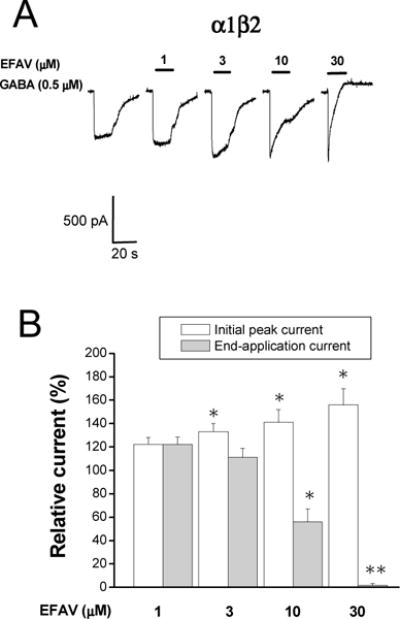
A, Representative traces showing whole-cell currents activated by EC30 GABA (0.5 μM) recorded from HEK293 cells transiently expressing α1β2 GABAA receptors. Efavirenz (1–30 μM) was co-applied with GABA for 10 sec. B, A Summary data showing the efavirenz concentration-response relationship. All currents are normalized to the response to 0.5 μM GABA. *, p<0.05; **, p<0.01, paired t-test, compared to the control, n=5–6.
As an additional assessment of potential involvement of the BDZ site in the actions of efavirenz, its effect was examined in the presence of a saturating concentration (3 μM) of flumazenil (FLU), a competitive antagonist of the BDZ site. As shown in Figure 5, 3 μM flumazenil was able to significantly block the potentiating effect of diazepam (1 μM) in α1β2γ2 receptors (n=9, p>0.05, paired t-test, compared between DZ and DZ+FLU); however, it failed to block the potentiating effect of efavirenz (n=13, p<0.05, paired t-test, compared between EFAV and EFAV+FLU). Flumazenil alone had a modest allosteric agonist actions at 3 μM (27 ± 6.3% increase in current amplitude compared to control, n=7, p<0.05, paired t-test), a phenomenon that has been reported previously (Weiss et al., 2002). This effect likely accounts for the additive potentiating effect when both efavirenz and flumazenil were co-applied with GABA (n=5, p<0.05, unpaired t test, Fig. 5).
Figure 5. Effect of flumazenil on efavirenz response in α1β2γ2 GABAA receptors.
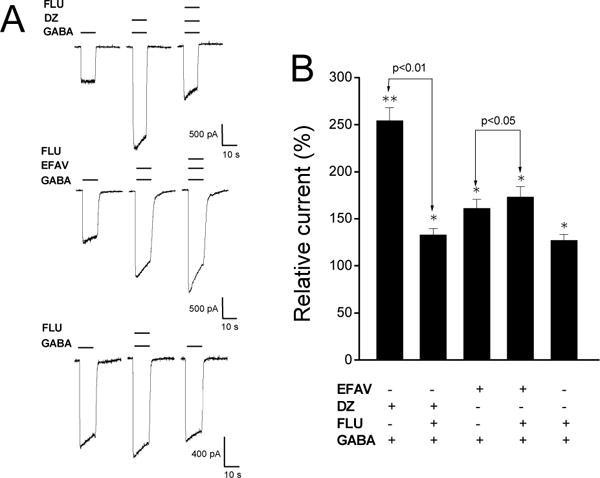
A, Representative traces showing whole-cell GABA-activated currents recorded from HEK293 cells expressing α1β2γ2 GABAA receptors. Flumazenil (3 μM) blocked potentiation caused by 1 μM diazepam but not by 10 μM efavirenz. Flumazenil (3 μM) alone potentiated the currents activated by 10 μM GABA. EFAV=efavirenz. DZ=diazepam. FLU=flumazenil. B, Summary data for effect of flumazenil on diazepam, efavirenz and GABA response. All currents were normalized to the control. *, p<0.05; **, p<0.01, paired t-test, compared to the control. n=7–13.
3.4. Effect of efavirenz on the GABA concentration-response relationship
Efavirenz may produce its modulatory action on GABA-activated currents by affecting the potency and/or the efficacy of GABA at the receptors. To distinguish between these possibilities, the effect of efavirenz on concentration-response curves was studied in α1β2γ2 receptors for its potentiating action and in α3β2γ2 for its inhibitory action, respectively. As shown in Fig. 6A, efavirenz’s enhancing effects appear to depend on [GABA]. Efavirenz (10 μM) had greater potentiating effect on the currents activated by low [GABA] but had little effect on the response to saturating [GABA] in α1β2γ2 receptors. As a result, EC50 for GABA values were significantly reduced by efavirenz (EC50: from 36 ± 6.0 to 24 ± 5.8 μM; Hill coefficient: from 1.5 ± 0.09 to 1.1 ± 0.09. n=10, p<0.01, paired t-test). In contrast, the maximal GABA current was not significantly changed by efavirenz (p>0.05, paired t-test, Fig. 6B). The inhibitory effect of efavirenz on α3β2γ2 receptors also displayed a dependence on [GABA] (Fig. 6C). Efavirenz significantly increased the EC50 for GABA from 34 ± 4.2 μM in the control to 89 ± 18 μM in the presence of 15 μM efavirenz (n=6, p<0.01, paired t-test) whereas the maximum current and Hill coefficient were not significantly altered (Fig. 6D). These data suggest that efavirenz blocks GABA currents in a competitive manner.
Figure 6. Effect of efavirenz on the GABA concentration-response curve recorded from α1β2γ2 (A&B) and α3β2γ2 (C&D) GABAA receptors stably expressed in HEK293 cells.
A&C, Representative traces showing whole-cell currents activated by different [GABA] in control and in the presence of efavirenz in α1β2γ2 (A) and α3β2γ2 (C). Note that Efavirenz modulated the response to a low [GABA] but did not affect the response to saturating [GABA] in both receptor configurations (A&C). B&D, Efavirenz significantly shifts the EC50 value for GABA without changing maximal currents. Currents are normalized to the maximal amplitude. EC50 values or Hill coefficients (in parentheses) were 36 ± 6.0 (1.5 ± 0.09) in control and 24 ± 5.8* μM (1.1 ± 0.09*) in presence of 10 μM efavirenz for α1β2γ2, and 34 ± 4.2 μM (0.96 ± 0.06) in control and in 89 ± 18* μM (0.84 ± 0.13) in the presence of 10 μM efavirenz for α3β2γ2. *, p<0.01, paired t-test, compared to the control, n=6–11.
3.5. Influence of the TM2 6′ amino acid on inhibitory effects of efavirenz
That efavirenz had inhibitory effects on all receptor subunit configurations examined suggested a conserved site for this action. A pore-lining threonine residue (T′6) within the second transmembrane domain (TM2), which is highly conserved among all GABAA subunits, confers sensitivity of GABAA receptors to picrotoxin (Gurley et al., 1995; Zhang et al., 1995), pentylenetetrazole (Dibas and Dillon, 2000), ethanol (Johnson et al., 2012) and 4′-ethynyl-4-n-propylbicycloorthobenzoate (EBOB) (Hisano et al., 2007). Thus, we examined whether this residue is also involved in efavirenz’s inhibitory effect. Since the inhibitory component is predominant in receptors lacking the γ2 subunit (Fig. 4), we assessed the efavirenz inhibitory modulation in α1β2 receptors. The effect of efavirenz on GABA currents activated by an EC30 concentration of GABA (Gonzales et al., 2008) was assessed in wild type α1β2, α1β2(T6′F) and α1(T6′F)β2 receptors. As shown in Figure 7, a point mutation of T6′F in either the α1 or β2 subunit eliminated sensitivity to picrotoxin, as reported previously (Sedelnikova et al., 2006). Similarly, the T6′F mutation in the β2 subunit significantly reduced the inhibitory effect of efavirenz, while the same mutation in the α1 subunit resulted in a complete abolishment of efavirenz-induced inhibition (Fig. 7). The average end-application current was 124 ± 10.6% and 95 ± 18% of control at 10 μM and 30 μM efavirenz, respectively (p>0.05, n=5–10, paired t-test, compared to control). In contrast, the T6′F mutation, when present in either the α1 or β2 subunit, did not influence the potentiating effect of efavirenz on peak current, suggesting that the threonine resides at the TM2 6′ position are not involved in allosteric potentiating effect of efavirenz. We were unable to test the efavirenz response in the α1(T6′F)β2(T6′F) configuration, because no GABA-activated current was detected, as reported previously (Gonzales et al., 2008). Our results indicate that the TM2 6′ position plays a key role in the inhibitory effect of efavirenz on GABAA receptors.
Figure 7. Effect of TM2 T6′F mutation on efavirenz sensitivity in α1β2 GABAA receptors.
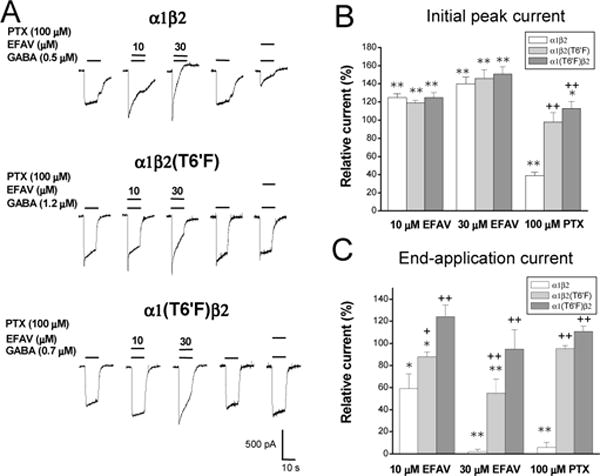
A, Representative traces showing EC30 GABA currents with or without co-application of efavirenz (EFAV, 10 or 30 μM) or picrotoxin (PTX, 100 μM) recorded from HEK293 transiently expressing α1β2, α1β2(T6′F) and α1(T6′F)β2 receptors. PTX-induced inhibition was abolished by the T6′F mutation in both α1β2(T6′F) or α1(T6′F)β2 receptors. Efavirenz-induced potentiation on initial current amplitude was not affected by TM2 T6′F mutations. In contrast, efavirenz-mediated inhibition on end-application current amplitude was greatly attenuated in α1β2(T6′F) and α1(T6′F)β2 receptors. B&C, Summary data for efavirenz and picrotoxin effect on initial (B) and end-application amplitude (C) activated by GABA in wild type α1β2, mutant α1β2(T6′F) and α1(T6′F)β2 receptors. Minimum of four cells were tested for efavirenz or PTX sensitivity. *, p<0.5; **, p<0.01, paired t-test, compared to control response without efavirenz or PTX. +, p<0.05; ++, p<0.01, unpaired t-test, compared to wild type.
3.6. Voltage-dependence of efavirenz modulation of GABAA receptors
Because a voltage-dependent effect of a drug may shed light on possible sites of action, we assessed the extent to which the effects of efavirenz on GABAA receptors may be influenced by membrane voltage. First, we tested efavirenz’s ability to modulate the current-voltage (I–V) relationship of GABA-induced currents in α1β2γ2 receptors. In the absence of efavirenz, the I–V relationship displayed outward rectification (Fig.8A1) consistent with our previous report (Huang et al., 2001). In the presence of 10 μM efavirenz, the reversal potential of GABA-activated current was not altered (−0.18 ± 1.83 mV at control, −0.24 ± 1.39 mV at efavirenz, n=5, p>0.05. 0.35 mV for calculated value with Nernst equation, Fig.8A1). However, the efavirenz-induced potentiation of the GABA response displayed an apparent voltage– dependence. Potentiation was more pronounced at negative membrane potentials, resulting in an I–V relationship that was close to linear in the presence of efavirenz (Fig.8A1). Moreover, the fraction of GABA-gated current potentiated by efavirenz was negatively correlated with membrane potential (R= −0.98, p<0.01, Fig.8A2). These observations indicate that efavirenz modulates GABAA receptors in a voltage-dependent manner. Considering the coexistence of potentiating and inhibitory actions of efavirenz on α1β2γ2 receptors, we conducted two additional experiments to characterize I–V relationships for efavirenz-induced inhibitory and potentiating components using α3β2γ2 receptors and α1(T6′F)β2 receptors, respectively. In α3β2γ2 receptors in which efavirenz has only an inhibitory effect, efavirenz did not alter the reversal potential for GABA-induced Cl− currents (1.48 ± 1.01 mV in the control, 2.81 ± 0.80 mV in the presence of efavirenz, n=9, p>0.05, Fig. 8B1). Moreover, inhibition of the GABA current by efavirenz was more pronounced at positive potentials and positively correlates with membrane potentials (R=0.96, p<0.01, Fig. 8B2). In α1(T6′F)β2 receptors, efavirenz potentiated GABA currents without altering reversal potential for Cl− ions (4.8 ± 1.3 mV in the control, 4.6 ± 1.4 mV in efavirenz, p>0.05, n=6. Fig.8C1). Figure 8C2 shows efavirenz-induced potentiation was dependent upon voltage in a manner similar to α1β2γ2 receptors (R=−0.79, p<0.01).
Figure 8. Current-voltage relationship for efavirenz effect on GABAA receptors.
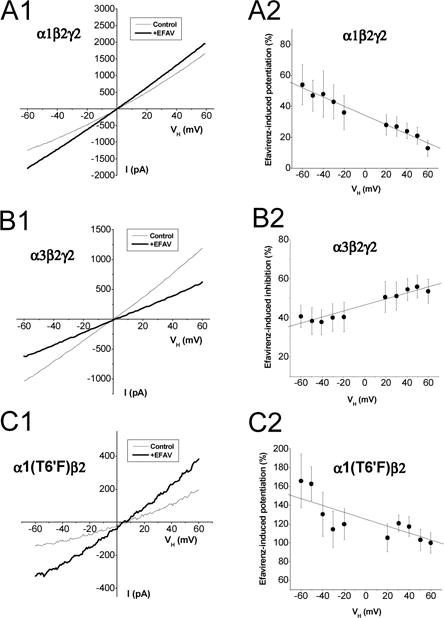
Whole-cell currents were recorded from single cells expressing α1β2γ2 (A), α3β2γ2 (B) or α1(T6′F)β2 receptors (C). Efavirenz (10 μM) was co-applied with EC30 GABA [10 μM for α1β2γ2, 20 μM for α3β2γ2 and 1 μM for α1(T6′F)β2]. A1&B1&C1, Average current response to a voltage ramp from −60 to +60 mV (0.24 mV/ms) at control and during efavirenz application. A2&B2&C2, Voltage-dependence of efavirenz effect on GABA currents. The currents are normalized to the control response at the same holding potential (VH). Solid line shows fit the mean of efavirenz-induced effect with a linear regression. α1β2γ2: n=5; α3β2γ2: n=9; α1(T6′F)β2: n=6.
3.7. Mapping the site for the potentiating effect of efavirenz
Efavirenz-mediated potentiation is present in αx(x=1,2,4)-containing configurations but absent in α3- or α5-containing receptors, indicating that the potentiating action is likely mediated via the residues that are conserved in α1/α2/α4 subunits but different from α3 or α5 subunits. Analysis on amino acid alignment of α subunit revealed three residues located in the position of 84, 89 and 120 of extracellular domain that meet such criteria (Table 2). Thus we assessed the role of these residues in efavirenz-mediated positive modulation in α1β2γ2 configuration using site-directed mutagenesis. Individual or combined mutation of the α1 subunit residues to the corresponding residues in the α3/5 isoforms, R84P, M89L or I120L, resulted in complete loss of potentiating effect of efavirenz on the currents activated by EC30 GABA (Table 1, Fig. 9). Compared to 76% potentiating effect induced by 30 μM efavirenz in wild type α1β2γ2, the average of relative GABA currents was 96 ± 2.2% of control in α1(R84P)β2γ2, 91 ± 4.9% in α1(M89L)β2γ2 and 108 ± 4.9% in α1(I120L)β2γ2 receptors (p>0.05, paired t-test, compared to the control. p<0.05, unpaired t-test, compared to WT). Furthermore, double (R84P/M89L) or triple (R84P/M89L/I120L) mutations did not yield additional change of efavirenz-induced modulation of GABA response compared to any single mutation (p>0.05, unpaired t-test. Fig. 9). Additionally, the receptors containing mutations of R84P and M89L in α1 subunit had a greatly reduced EC50 for GABA compared to wild type receptors (Table 1).
Table 2.
Amino acid sequence alignment of extracellular domain of GABAA receptor α(1–6) subunits.
| 83 | 84 | 85 | 86 | 87 | 88 | 89 | 90 | 91 | 92 | 118 | 119 | 120 | 121 | 122 | 123 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 α | L | R | L | N | N | L | M | A | S | K | L | R | I | T | E | D |
| 2 α | L | R | L | N | N | L | M | A | S | K | L | R | I | Q | D | D |
| 4 α | L | R | L | N | N | L | M | V | T | K | F | R | I | M | R | N |
| 3 α | L | P | L | N | N | L | L | A | S | K | L | R | L | V | D | N |
| 5 α | L | P | L | N | N | L | L | A | S | K | L | R | L | E | D | D |
| 6 α | L | S | L | N | N | L | M | V | S | K | F | R | I | M | Q | N |
The resides that are conserved in α1, α2 and α4 but not α3, α5 or α6 are marked in bold and underline
Figure 9. Effect of mutations of α1 R84P, M89L and I120L on efavirenz sensitivity in α1β2γ2 GABAA receptors.
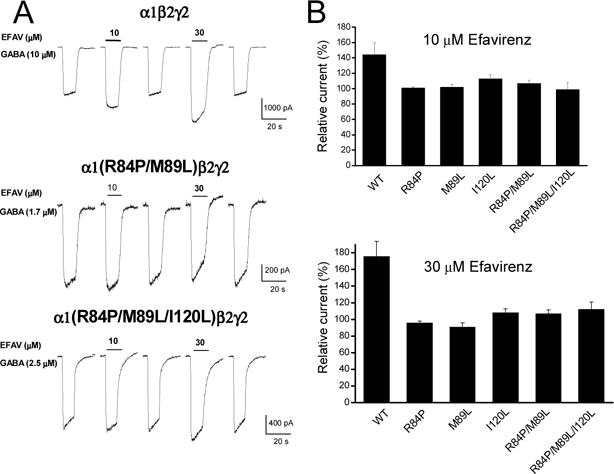
A, Representative traces showing EC30 GABA currents with or without co-application of efavirenz (10 or 30 μM) in wild type, α1(R84P/M89L)β2γ2 and α1(R84P/M89L/I120L)β2γ2 receptors. B, Summary data for efavirenz effect on wild type and mutant α1β2γ2 GABAA receptors. Minimum of five cells were tested for efavirenz. Note that efavirenz-mediated potentiation was completely lost in all mutant receptors. p>0.05, paired t-test, compared to control; p<0.05, unpaired t-test, compared to wild type.
4. Discussion
Our original report was the first to demonstrate the ability of efavirenz to positively modulate α1β2γ2 GABAA receptors stably expressed in HEK293 cell lines (Gatch et al., 2013). Here we sought to gain a more molecular mechanistic understanding of its actions on GABAA receptors. First, the effects of efavirenz on GABA-currents were rapid (millisecond range) and reversible, suggesting that efavirenz acts directly on the GABAA receptors, likely at an extracellular site(s). Second, the present results demonstrate that efavirenz can exert both potentiating and inhibiting effects on the GABA response and the potentiating effect depends on the GABAA receptor subunit composition, being most prominent in α1β2γ2 configuration, greatly diminished in α1β2 and α6β2γ2 configurations, and absent in the α3β2γ2 and α5β2γ2 configuration. The inhibitory effect is readily recognized in the α3, α5 and α6-containing receptors in which both initial and end-application current were eventually inhibited by efavirenz. In α1, α2 and α4-containing receptors where the initial and end-application currents were oppositely affected by efavirenz, enhanced current decay (i.e., reduction of end-application) could reflect receptor desensitization or channel block caused by efavirenz. It is somehow difficult to completely dissect two actions that may occur simultaneously. However, our results especially from mutagenesis indicate that the efavirenz-induced current decay is uncoupled with its potentiating effect but associated with its inhibitory effect. First, the effect of efavirenz on current decay is more evident in α1β2 than α1β2γ2 while potentiation of initial current is comparable between these two receptors. At 30 μM efavirenz, the end-application currents were completely inhibited in α1β2 but were reduced to 46 ± 11% in α1β2γ2. In contrast, the potentiation elicited by 30 μM efavirenz was similar between the receptors (relative initial current: 156 ±14% in α1β2 and 164 ± 16% of control in α1β2γ2, p>0.05, unpaired t-test). Second, efavirenz-induced current decay is associated with its inhibitory action. The effect of efavirenz on current decay was diminished by a point mutation of T6′F in TM2 that conferred the sensitivity of pore blocker picrotoxin (PTX). Furthermore, as shown in Fig. 7C, the effect of the mutation on current decay was stoichiometrically similar between efavirenz and PTX. These data suggest that efavirenz shares similar action site and mechanism with PTX. More importantly, the ability of efavirenz to potentiate GABA response was not altered by the T6′F mutation (Fig. 7A). Our results suggest that efavirenz-induced current decay reflects its antagonistic action mediated by known PTX sites at TM2. Finally, pre-incubation of efavirenz for 1 min resulted in greater blockade on end-application currents but similar potentiation on initial peak current compared to coapplication. It appears that efavirenz is able to access and occupy its inhibitory site when the channels are closed, maximizing its inhibitory action without affecting potential action. Dual effects of efavirenz present in α1, α2 and α4-containing GABAA receptors are mediated by two distinct sites. In fact, similar dual modulation has also been demonstrated in other GABAA receptor modulators such as pentobarbital (Akk and Steinbach, 2000; Akk et al., 2004), lactones (Rho et al., 1997), carisoprodol (Kumar et al., 2015) and meprobamate (Kumar and Dillon, 2016). Dual modulatory effects of efavirenz on GABAA receptor is also dependent on [GABA]. Specifically, the potentiating effect is dependent on the concentration of GABA without apparently affecting maximum GABA currents in α1β2γ2 receptors. Efavirenz displayed only antagonistic modulation on α3β2γ2 receptors in a competitive manner.
While one might speculate that efavirenz acts at the BDZ site to exert its potentiating effect because efavirenz’s potentiating ability is less on BDZ-insensitive α6β2γ2 receptors, this is likely not the case as the BDZ site antagonist flumazenil failed to block efavirenz-induced potentiation in α1β2γ2 receptors. In addition, α3β2γ2 and α5β2γ2 receptors were completely insensitive to the potentiating effect of efavirenz, and the BDZ site is homologous to that in α1β2γ2 receptors (Buhr and Sigel, 1997; Hanson and Czajkowski, 2008; Hanson et al., 2008; Morlock and Czajkowski, 2011). For these reasons, the stimulatory effect of efavirenz does not appear to be due to it interacting at the BDZ site. Moreover, the lack of potentiation on α3β2γ2 and α5β2γ2 receptors also diminishes the likelihood that efavirenz interacts with the same sites as barbiturates (Thompson et al., 1996), neurosteroids (Lambert et al., 1990; Puia et al., 1993; Belelli et al., 2002), or carisoprodol (Kumar et al., 2015), as each of these ligands positively modulate these two receptors. The potentiating effect of efavirenz is highly dependent on α isoforms, being present in α1-, α2-, and α4-containing receptors but absent in α3- or α5-containing receptors. This unique subunit profile of positive modulation by efavirenz further led us to locate three homologous residues, R84, M89 and I120 on the extracellular domains in α1/2/4 subunits as the putative sites responsible for positive modulation of efavirenz. Mutating any one of the three residues was sufficient to disrupted the ability of efavirenz to potentiate α1β2γ2 receptors, suggesting that these residues likely mediate the potentiation effect of efavirenz in α1β2γ2 receptors. The presence of M89 and I120 in the α6 isoform, which retains a partial potential effect for efavirenz, seems consistent with their role in efavirenz’s action. However, how efavirenz interacts with GABAA receptors via these residues is not clear. While no crystal structure is available to disclose their three-dimensional location, we may gain some insights about the roles of these residues from analogies with 4 Å resolution of acetylcholine binding protein (AChBP) (Miyazawa et al., 2003; Unwin, 2005) and 3 Å resolution of GABAA receptor β3 crystal structures (Miller and Aricescu, 2014). The α1 R84 and M89 residues (equivalent to T82 and V87 in β3 subunit) are located between agonist binding loop D and A and I120 (equivalent to L118 in β3 subunit) at agonist binding loop E (Cromer et al., 2002). The interaction of R86 in the β3 subunit with its surrounding residues in the complementary subunit has been proposed to form the inter-subunit interface (Miller and Aricescu, 2014). Our data show that efavirenz increases GABA potency (i.e., reduces GABA EC50) without affecting efficacy. Consistent with this is the finding that the R84P and M89L also increases GABA potency. In addition, M is a polar and uncharged residue and contains sulfur, and usually participates in hydrogen bounds (Biswal et al., 2012). L has larger accessible surface than I as revealed by a study from a nonredundant bank of 587 3D structure proteins (Lins et al., 2003). We speculate that substitution of residue M or I with L may alter the secondary and tertiary structures governing the interaction of efavirenz with GABAA receptors, influencing GABA-binding and/or inter- subunit interface. However, whether efavirenz directly binds to these residues remains a topic of further investigation.
O’Toole and Jenkins (2012) reported that the outward rectification of GABAA receptors is inversely related to the degree of channel activation. As a result, positive modulators have less effect when Cl− ions are entering into the cell at positive membrane potentials (O’Toole and Jenkins, 2012). Consistent with their observation, the potentiating effect of efavirenz displays an apparent voltage-dependence with less potentiation at positive membrane potentials. The ability of efavirenz to inhibit all GABAA receptor compositions examined suggests a conserved site for its inhibitory action. The present results demonstrate that the inhibitory effects of efavirenz are influenced by membrane voltage, being greater at positive potentials. This type of voltage dependence is consistent with efavirenz binding within the electric field potential of the membrane, presumably within the ion channel pore. A similar pattern of voltage-dependence has been also reported for penicillin, an open channel blocker (Fujimoto et al., 1995). Penicillin interacts with the GABAA receptor channel at the same or overlapping sites for picrotoxin another channel pore blocker (Sugimoto et al., 2002; Bali and Akabas, 2007). Point mutation of a conserved threonine to phenylalanine at the pore lining 6′ position of the second transmembrane domain (TM2 T6′F) in either the α1 or β2 subunit weakened the antagonism by efavirenz, and implicating this residue in the inhibitory actions of efavirenz. In the case of picrotoxin, binding has been proposed to occur at the intracellular vestibule of the channel through a series of H-bonds formed with the 2′ position of the receptor (Zhorov and Bregestovski, 2000; Hibbs and Gouaux, 2011). While multiple sites of action for picrotoxin have been proposed (Buhr et al., 2001; Chen et al., 2006; Bali and Akabas, 2007), the replacement of threonine with the bulky benzyl side chain phenylalanine at the 6′ position sterically blocks picrotoxin access to its binding site from the extracellular side when the channel is in the open state (Gurley et al., 1995; Sedelnikova et al., 2006). This same mutation abolishes or significantly attenuates effects of several other ligands that block the pore of GABAA receptors, including pentylenetetrazole (Dibas and Dillon, 2000), ethanol (Johnson et al., 2012), and EBOB (Hisano et al., 2007) and carisoprodol (Kumar et al., 2011). Thus, additional studies will be needed to discern whether the TM2 T6′ threonine interacts directly with efavirenz, or is critical for receptor conformational changes elicited subsequent to efavirenz binding.
5. Conclusion
The present results demonstrate that efavirenz has both stimulatory and inhibitory effects in GABAA receptors, and that the stimulatory effect is sensitive to subunit configuration. The positive allosteric modulatory effect of efavirenz that occurs at lower concentrations appears is sensitive to residues R84, M89 and I120 located on the extracellular domains of α1/2/4 subunits, which is a site distinct from the benzodiazepine, neurosteroid and barbiturate sites. The ability of efavirenz to inhibit GABAA receptors is sensitive to a channel-lining region of the second transmembrane domain of the α and/or β subunit(s), which is consistent with its channel blocking actions at higher concentrations. Brain accumulation of efavirenz in humans and rodents is expected to be in the tens of μM under normal dosing conditions (Dalwadi et al., 2016). Based on our present results, efavirenz may primarily act as an agonist or antagonist depending on efavirenz concentration and GABAA receptor subunit compositions expressed in the specific brain regions. Therefore, the CNS side effects of efavirenz related to its action on GABAA receptors could vary from depression (potentiation of the receptor function) to anxiety and nervousness (inhibition).
Highlights.
Efavirenz produces inhibitory and potentiating effects on recombinant GABAA receptors depending on concentration and/or receptor subunit configurations.
The two opposing modulatory effects of efavirenz are mediated via two distinctive sites on GABAA receptors.
Efavirenz’s allosteric potentiation of GABAA receptors via a new modulatory site at the extracellular domains.
Acknowledgments
We thank Ms. Cathy L Bell-Horner for preparing HEK293 cell lines and Dr. Eric Gonzales for performing T6′F mutagenesis and Mr. Rodney Ballard for conducting some preliminary studies.
Research reported in this publication was supported in part by the National Institutes of Health [Grant R01-MH063162 and R41-AG043243 (JAS); R01-DA022370 and U54GM1004912 (GHD)], ADDF Grant 20140803 (JAS), Intramural Grants [RI-6015 (JAS). RI-6146&RI-6031 (RQH)], and institutional funds [67673 (JAS)].
Abbreviations
- BDZ
benzodiazepine
- CNS
central nervous system
- DMSO
dimethyl sulfoxide
- DZ
diazepam
- EFAV
efavirenz
- FLU
flumazenil
- LGICs
ligand-gated ion channels
- HEK293
Human embryonic kidney cell line
- NNRTI
non-nucleoside reverse transcriptase inhibitor
- PTX
picrotoxin
- TM2
second transmembrane domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Chemical compounds: GABA (PubChem CID:119), Flumazenil (PubChem CID:3373), Efavirenz (PubChem CID: 64139), Diazepam (PubChem CID: 3013), Picrotoxin (PubChem CID: 518601)
References
- Akk G, Bracamontes J, Steinbach JH. Activation of GABA(A) receptors containing the alpha4 subunit by GABA and pentobarbital. The Journal of physiology. 2004;556:387–399. doi: 10.1113/jphysiol.2003.058230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akk G, Steinbach JH. Activation and block of recombinant GABA(A) receptors by pentobarbitone: a single-channel study. British journal of pharmacology. 2000;130:249–258. doi: 10.1038/sj.bjp.0703335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt G, de Nocker D, von Giesen HJ, Nolting T. Neuropsychiatric side effects of efavirenz therapy. Expert Opin Drug Saf. 2007;6:147–154. doi: 10.1517/14740338.6.2.147. [DOI] [PubMed] [Google Scholar]
- Bali M, Akabas MH. The location of a closed channel gate in the GABAA receptor channel. The Journal of general physiology. 2007;129:145–159. doi: 10.1085/jgp.200609639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreiro P, Garcia-Benayas T, Soriano V, Gallant J. Simplification of antiretroviral treatment–how to sustain success, reduce toxicity and ensure adherence avoiding PI use. AIDS Rev. 2002;4:233–241. [PubMed] [Google Scholar]
- Belelli D, Casula A, Ling A, Lambert JJ. The influence of subunit composition on the interaction of neurosteroids with GABA(A) receptors. Neuropharmacology. 2002;43:651–661. doi: 10.1016/s0028-3908(02)00172-7. [DOI] [PubMed] [Google Scholar]
- Bell-Horner CL, Dibas M, Huang RQ, Drewe JA, Dillon GH. Influence of subunit configuration on the interaction of picrotoxin-site ligands with recombinant GABA(A) receptors. Brain Res Mol Brain Res. 2000;76:47–55. doi: 10.1016/s0169-328x(99)00330-7. [DOI] [PubMed] [Google Scholar]
- Biswal HS, Gloaguen E, Loquais Y, Tardivel B, Mons M. Strength of NH…S Hydrogen Bonds in Methionine Residues Revealed by Gas-Phase IR/UV Spectroscopy. J Phys Chem Lett. 2012;3:755–759. doi: 10.1021/jz300207k. [DOI] [PubMed] [Google Scholar]
- Blanch J, Martinez E, Rousaud A, Blanco JL, Garcia-Viejo MA, Peri JM, Mallolas J, De Lazzari E, De Pablo J, Gatell JM. Preliminary data of a prospective study on neuropsychiatric side effects after initiation of efavirenz. Journal of acquired immune deficiency syndromes (1999) 2001;27:336–343. doi: 10.1097/00126334-200108010-00003. [DOI] [PubMed] [Google Scholar]
- Buhr A, Sigel E. A point mutation in the gamma2 subunit of gamma-aminobutyric acid type A receptors results in altered benzodiazepine binding site specificity. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:8824–8829. doi: 10.1073/pnas.94.16.8824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhr A, Wagner C, Fuchs K, Sieghart W, Sigel E. Two novel residues in M2 of the gamma-aminobutyric acid type A receptor affecting gating by GABA and picrotoxin affinity. The Journal of biological chemistry. 2001;276:7775–7781. doi: 10.1074/jbc.M008907200. [DOI] [PubMed] [Google Scholar]
- Cespedes MS, Aberg JA. Neuropsychiatric complications of antiretroviral therapy. Drug Saf. 2006;29:865–874. doi: 10.2165/00002018-200629100-00004. [DOI] [PubMed] [Google Scholar]
- Chen L, Durkin KA, Casida JE. Structural model for gamma-aminobutyric acid receptor noncompetitive antagonist binding: widely diverse structures fit the same site. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:5185–5190. doi: 10.1073/pnas.0600370103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromer BA, Morton CJ, Parker MW. Anxiety over GABA(A) receptor structure relieved by AChBP. Trends Biochem Sci. 2002;27:280–287. doi: 10.1016/s0968-0004(02)02092-3. [DOI] [PubMed] [Google Scholar]
- Dalwadi DA, Kim S, Amdani SM, Chen Z, Huang RQ, Schetz JA. Molecular mechanisms of serotonergic action of the HIV-1 antiretroviral efavirenz. Pharmacological research : the official journal of the Italian Pharmacological Society. 2016;110:10–24. doi: 10.1016/j.phrs.2016.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dibas MI, Dillon GH. The central nervous system convulsant pentylenetetrazole stimulates gamma-aminobutyric acid (GABA)-activated current in picrotoxin-resistant GABA(A) receptors in HEK293 cells. Neurosci Lett. 2000;285:193–196. doi: 10.1016/s0304-3940(00)01064-8. [DOI] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Luscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nature neuroscience. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Fujimoto M, Munakata M, Akaike N. Dual mechanisms of GABAA response inhibition by beta-lactam antibiotics in the pyramidal neurones of the rat cerebral cortex. British journal of pharmacology. 1995;116:3014–3020. doi: 10.1111/j.1476-5381.1995.tb15957.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatch MB, Kozlenkov A, Huang RQ, Yang W, Nguyen JD, Gonzalez-Maeso J, Rice KC, France CP, Dillon GH, Forster MJ, Schetz JA. The HIV antiretroviral drug efavirenz has LSD-like properties. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology. 2013;38:2373–2384. doi: 10.1038/npp.2013.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales EB, Bell-Horner CL, Dibas MI, Huang RQ, Dillon GH. Stoichiometric analysis of the TM2 6′ phenylalanine mutation on desensitization in alpha1beta2 and alpha1beta2gamma2 GABA A receptors. Neurosci Lett. 2008;431:184–189. doi: 10.1016/j.neulet.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez LA, Gatch MB, Taylor CM, Bell-Horner CL, Forster MJ, Dillon GH. Carisoprodol-mediated modulation of GABAA receptors: in vitro and in vivo studies. J Pharmacol Exp Ther. 2009;329:827–837. doi: 10.1124/jpet.109.151142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grelotti DJ, Closson EF, Smit JA, Mabude Z, Matthews LT, Safren SA, Bangsberg DR, Mimiaga MJ. Whoonga: potential recreational use of HIV antiretroviral medication in South Africa. AIDS Behav. 2014;18:511–518. doi: 10.1007/s10461-013-0575-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley D, Amin J, Ross PC, Weiss DS, White G. Point mutations in the M2 region of the alpha, beta, or gamma subunit of the GABAA channel that abolish block by picrotoxin. Receptors Channels. 1995;3:13–20. [PubMed] [Google Scholar]
- Hanson SM, Czajkowski C. Structural mechanisms underlying benzodiazepine modulation of the GABA(A) receptor. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2008;28:3490–3499. doi: 10.1523/JNEUROSCI.5727-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson SM, Morlock EV, Satyshur KA, Czajkowski C. Structural requirements for eszopiclone and zolpidem binding to the gamma-aminobutyric acid type-A (GABAA) receptor are different. Journal of medicinal chemistry. 2008;51:7243–7252. doi: 10.1021/jm800889m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E. Principles of activation and permeation in an anion-selective Cys-loop receptor. Nature. 2011;474:54–60. doi: 10.1038/nature10139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisano K, Ozoe F, Huang J, Kong X, Ozoe Y. The channel-lining 6′ amino acid in the second membrane-spanning region of ionotropic GABA receptors has more profound effects on 4′-ethynyl-4-n-propylbicycloorthobenzoate binding than the 2′ amino acid. Invertebrate neuroscience : IN. 2007;7:39–46. doi: 10.1007/s10158-006-0035-x. [DOI] [PubMed] [Google Scholar]
- Huang RQ, Bell-Horner CL, Dibas MI, Covey DF, Drewe JA, Dillon GH. Pentylenetetrazole-induced inhibition of recombinant gamma-aminobutyric acid type A (GABA(A)) receptors: mechanism and site of action. J Pharmacol Exp Ther. 2001;298:986–995. [PubMed] [Google Scholar]
- Huang RQ, Dillon GH. Effect of extracellular pH on GABA-activated current in rat recombinant receptors and thin hypothalamic slices. J Neurophysiol. 1999;82:1233–1243. doi: 10.1152/jn.1999.82.3.1233. [DOI] [PubMed] [Google Scholar]
- Huang RQ, Gonzales EB, Dillon GH. GABAA receptors" Structure, function and modulation. In: Arlas H, editor. Biolgical and Biophysical Aspects of Ligand-Gated Ion Channel Receptor Superfamilies. Research Signpost; Kerala: 2006. pp. 171–198. [Google Scholar]
- Johnson WD, 2nd, Howard RJ, Trudell JR, Harris RA. The TM2 6′ position of GABA(A) receptors mediates alcohol inhibition. The Journal of pharmacology and experimental therapeutics. 2012;340:445–456. doi: 10.1124/jpet.111.188037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Dillon GH. Assessment of direct gating and allosteric modulatory effects of meprobamate in recombinant GABA(A) receptors. European journal of pharmacology. 2016;775:149–158. doi: 10.1016/j.ejphar.2016.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Gonzalez LA, Dillon GH. Assessment of subunit-dependent direct gating and allosteric modulatory effects of carisoprodol at GABA(A) receptors. Neuropharmacology. 2015;97:414–425. doi: 10.1016/j.neuropharm.2015.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar M, Kumar M, Bell-Horner CL, Dillon G. Assessment of the molecular actions of carisoprodol on GABAA receptors. Soc Neurosci Abst. 2011;337 04/C27. [Google Scholar]
- Lambert JJ, Peters JA, Sturgess NC, Hales TG. Steroid modulation of the GABAA receptor complex: electrophysiological studies. Ciba Found Symp. 1990;153:56–71. doi: 10.1002/9780470513989.ch4. discussion 71–82. [DOI] [PubMed] [Google Scholar]
- Lins L, Thomas A, Brasseur R. Analysis of accessible surface of residues in proteins. Protein Sci. 2003;12:1406–1417. doi: 10.1110/ps.0304803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR. Crystal structure of a human GABAA receptor. Nature. 2014;512:270–275. doi: 10.1038/nature13293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- Morlock EV, Czajkowski C. Different residues in the GABAA receptor benzodiazepine binding pocket mediate benzodiazepine efficacy and binding. Molecular pharmacology. 2011;80:14–22. doi: 10.1124/mol.110.069542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Toole KK, Jenkins A. The apparent voltage dependence of GABAA receptor activation and modulation is inversely related to channel open probability. Molecular pharmacology. 2012;81:189–197. doi: 10.1124/mol.111.074476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puia G, Ducic I, Vicini S, Costa E. Does neurosteroid modulatory efficacy depend on GABAA receptor subunit composition? Receptors & channels. 1993;1:135–142. [PubMed] [Google Scholar]
- Puzantian T. Central nervous system adverse effects with efavirenz: case report and review. Pharmacotherapy. 2002;22:930–933. doi: 10.1592/phco.22.11.930.33624. [DOI] [PubMed] [Google Scholar]
- Rho JM, Donevan SD, Rogawski MA. Barbiturate-like actions of the propanediol dicarbamates felbamate and meprobamate. The Journal of pharmacology and experimental therapeutics. 1997;280:1383–1391. [PubMed] [Google Scholar]
- Sedelnikova A, Erkkila BE, Harris H, Zakharkin SO, Weiss DS. Stoichiometry of a pore mutation that abolishes picrotoxin-mediated antagonism of the GABAA receptor. The Journal of physiology. 2006;577:569–577. doi: 10.1113/jphysiol.2006.120287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigel E, Buhr A. The benzodiazepine binding site of GABAA receptors. Trends Pharmacol Sci. 1997;18:425–429. doi: 10.1016/s0165-6147(97)01118-8. [DOI] [PubMed] [Google Scholar]
- Sugimoto M, Fukami S, Kayakiri H, Yamazaki S, Matsuoka N, Uchida I, Mashimo T. The beta-lactam antibiotics, penicillin-G and cefoselis have different mechanisms and sites of action at GABA(A) receptors. British journal of pharmacology. 2002;135:427–432. doi: 10.1038/sj.bjp.0704496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutterlin S, Vogele C, Gauggel S. Neuropsychiatric complications of efavirenz therapy: suggestions for a new research paradigm. J Neuropsychiatry Clin Neurosci. 2006;22:361–369. doi: 10.1176/jnp.2010.22.4.361. [DOI] [PubMed] [Google Scholar]
- Tan KR, Rudolph U, Luscher C. Hooked on benzodiazepines: GABAA receptor subtypes and addiction. Trends in neurosciences. 2011;34:188–197. doi: 10.1016/j.tins.2011.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson SA, Whiting PJ, Wafford KA. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. British journal of pharmacology. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treisman GJ, Kaplin AI. Neurologic and psychiatric complications of antiretroviral agents. Aids. 2002;16:1201–1215. doi: 10.1097/00002030-200206140-00002. [DOI] [PubMed] [Google Scholar]
- Treisman GJ, Soudry O. Neuropsychiatric Effects of HIV Antiviral Medications. Drug Saf. 2016;39:945–957. doi: 10.1007/s40264-016-0440-y. [DOI] [PubMed] [Google Scholar]
- Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- Weiss M, Tikhonov D, Buldakova S. Effect of flumazenil on GABAA receptors in isolated rat hippocampal neurons. Neurochemical research. 2002;27:1605–1612. doi: 10.1023/a:1021674708556. [DOI] [PubMed] [Google Scholar]
- Zhang D, Pan ZH, Zhang X, Brideau AD, Lipton SA. Cloning of a gamma-aminobutyric acid type C receptor subunit in rat retina with a methionine residue critical for picrotoxinin channel block. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:11756–11760. doi: 10.1073/pnas.92.25.11756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhorov BS, Bregestovski PD. Chloride channels of glycine and GABA receptors with blockers: Monte Carlo minimization and structure-activity relationships. Biophys J. 2000;78:1786–1803. doi: 10.1016/S0006-3495(00)76729-4. [DOI] [PMC free article] [PubMed] [Google Scholar]