

Abstract
Proper signaling by G protein coupled receptors (GPCR) is dependent on the specific repertoire of transducing, enzymatic and regulatory kinases and phosphatases that shape its signaling output. Activation and signaling of the GPCR through its cognate G protein is impacted by G protein-coupled receptor kinase (GRK)-imprinted “barcodes” that recruit ß-arrestins to regulate subsequent desensitization, biased signaling and endocytosis of the GPCR. The outcome of agonist-internalized GPCR in endosomes is also regulated by sequence motifs or “barcodes” within the GPCR that mediate its recycling to the plasma membrane or retention and eventual degradation as well as its subsequent signaling in endosomes. Given the vast number of diverse sequences in GPCR, several trafficking mechanisms for endosomal GPCR have been described. The majority of recycling GPCR, are sorted out of endosomes in a “sequence-dependent pathway” anchored around a type-1 PDZ-binding module found in their C-tails. For a subset of these GPCR, a second “barcode” imprinted onto specific GPCR serine/threonine residues by compartmentalized kinase networks was required for their efficient recycling through the “sequence-dependent pathway”. Mutating the serine/threonine residues involved, produced dramatic effects on GPCR trafficking, indicating that they played a major role in setting the trafficking itinerary of these GPCR. While endosomal SNX27, retromer/WASH complexes and actin were required for efficient sorting and budding of all these GPCR, additional proteins were required for GPCR sorting via the second “barcode”. Here we will review recent developments in GPCR trafficking in general and the human ß1-adrenergic receptor in particular across the various trafficking roadmaps. In addition, we will discuss the role of GPCR trafficking in regulating endosomal GPCR signaling, which promote biochemical and physiological effects that are distinct from those generated by the GPCR signal transduction pathway in membranes.
Keywords: GPCR, signaling, trafficking, PKA, GRK, endosomes, SNX27
1. Introduction
The G protein-coupled receptors (GPCR) family constitutes the largest family of signaling molecules. This family recognizes several different varieties of chemical structures and transmit their stimuli through different signal transduction paradigms [1]. However, the number of second messengers that are generated by GPCR activation is rather limited (cyclic AMP/cyclic GMP, inositol triphosphate and Ca2+ etc.), yet these second messengers produce a plethora of diverse cellular responses. Compartmentalization of signaling is a well characterized paradigm for spatiotemporal regulation of signals generated by a given cellular GPCR. In its simplest form, compartmentalization is intended to enhance fidelity of signaling by limiting the diffusion of second messengers or by assembling higher order protein complexes, termed signalosomes [2]. These signalosomes bring together signaling networks, such as the GPCR-G protein and effector enzyme with regulatory proteins required for a specific cellular response [3].
Another mechanism for regulating the output of a given GPCR is through its desensitization and internalization to endosomes [4]. This mechanism was initially thought to be involved in regulating the density of the GPCR either by resensitization or by down-regulation [5, 6]. Lately, it has evolved as a major mechanism involved in a variety of physiological responses, such as MAP kinase activation and in sustained signaling by the internalized GPCR to generate unique physiological responses that are different from plasma membrane delimited signaling [7].
Desensitization of the GPCR is a cell-mediated response to enhanced stimulation of the GPCR by elevated levels of the agonist [8]. Sustained activation of the GPCR triggers its phosphorylation by a family of G protein-coupled receptor kinases (GRK)s [9]. GRK-mediated phosphorylation of the ß-AR, promotes the recruitment of ß-arrestins, which sterically hinder further ß-AR-G protein coupling and initiate a program of G protein-independent signaling [10, 11]. ß-arrestins are scaffolding proteins that play critical roles in: desensitizing the agonist-activated GPCR, linking the desensitized agonist-occupied GPCR to the endocytic machinery, sustaining the signaling output of endosomal GPCR, and in post-translational modifications (such as ubiquitination) of the GPCR [12]. In this review, we aim to show that compartmentalized signaling networks are also involved in regulating GPCR trafficking through complex cross-talk mechanisms centered around the primary GPCR trafficking barcodes. Where ever possible, we will illustrate these paradigms using the human ß1-AR as the model.
2. Determining the extent and mechanisms involved in GPCR sequestration
Nutrient membrane receptors, such as transferrin and LDL receptors are constitutively internalized via clathrin-coated vesicle (CCV)-mediated internalization [13]. GPCR differ from these receptors in that they undergo regulated endocytosis that is dependent on their activation by relatively high and sustained concentrations of their cognate agonists. Two major dynamin-dependent mechanisms were implicated in GPCR internalization from their membranous compartment to early endosomes [14, 15]. The first mechanism involved sequestration of the GPCR via clathrin-mediated endocytosis (CME); which is a complex process that uses arrestin bound to the agonist activated GPCR to tap into the endocytic machinery involved in this process [12]. CME involves the concentration of agonist-occupied transmembrane receptors into ‘coated pits’ that are formed on the plasma membrane by the assembly of clathrin with other cytosolic coat proteins [16]. Clathrin cage assembly “triskelion” requires assembly proteins such as AP2, which are involved in endocytic CCV formation. GPCR access to CCV is mediated primarily by the adaptor protein ß-arrestin that interacts with AP2 to facilitate CCV assembly and subsequent CME of the GPCR [17].
The other pathway involved caveolae that are flask-shaped cholesterol and sphingolipid-rich invaginations of the plasma membrane, in which many diverse signaling molecules are concentrated [18, 19]. The core components of caveolae are caveolin proteins, but their budding from the surface into the cytoplasm requires dynamin [20, 21]. Many reports have determined that, like CME, a majority of GPCR-bearing caveolae traffic to and fuse with early endosomal [EE] membranes [22, 23].
The extent and mechanism of GPCR internalization have been determined for many GPCR [24]. This review will focus primarily on ß1- and ß2-adrenergic receptors in order to illustrate the complexities involved in determining the extent and mechanism of GPCR internalization. The extent of GPCR translocation from the plasma membrane to the cytosol can be assessed by a variety of techniques, such as radioligand binding with hydrophilic ligands, confocal microscopy with fluorescently tagged GPCR, cell surface biotinylation and fluorescence cell sorting of GPCR [25-27]. Assessment of GPCR trafficking by confocal trafficking assays provides direct visual examination of this process, and if carried out properly, can provide a determination of the extent and rate of GPCR internalization. Using this technique along-side with radioligand binding assays, it was determined that ∼60% of ß2-AR were rapidly internalized in HEK-293 cells (EC50 ∼ 6min) by CME via a ß-arrestin-dependent mechanism [28-30]. Concerning the ß1-AR, its internalization from the cell membrane to endosomes and the extent of its sequestration varied from almost no internalization to significant amounts of internalization approaching those of the ß2-AR. Biotinylation of cell surface exposed proteins in cells followed by agonist treatment, consistently revealed significant >60% internalization of membranous ß1-AR in response to ß-agonists [27, 31]. Confocal microscopy measurements of ß1-AR internalization in response to agonists also revealed significant internalization of membranous ß1-AR approaching 50-70% [32]. Radioligand binding approaches have also detected significant ß1-AR translocation from the plasma membrane approaching 45% in HEK-293 cells [33]. Other studies have also demonstrated that ß-arrestin would translocate from cytosol to the membrane in response to activating the ß1-AR and that both CME and caveolae-based mechanisms were involved in this pathway [33, 34]. However, there are studies indicating that ß1-AR do not internalize in response to ß-agonists and do not recruit ß-arrestins from the cytosol to the membrane [35, 36]. The reasons for these discrepancies are unclear, but illustrate the necessary expertise needed to overcome inherent difficulties associated with determining and interpreting GPCR trafficking studies that are carried in different cells under a variety of trafficking conditions.
3. Role of agonist-promoted phosphorylation “barcoding” in regulating GPCR internalization or down-stream signaling
Continuous activation of GPCR by their cognate agonists promotes a sequence of events, termed “homologous desensitization” [8]. This process is initiated by phosphorylation of the agonist-activated GPCR by GRKs on unique serine and threonine residues in the intracellular loops and carboxy-tail of the GPCR [12]. These events initially uncouple the GPCR from the G protein and promote the binding of ß-arrestins [12, 30]. The GRK family is composed of 7 individual members and each of them generates a different phosphorylation repertoire onto the GPCR that is also influenced by the cell or organ under investigation [17]. Ubiquitously expressed GRK 2, 3, 5 and 6 are the prominent members of this class of kinases and each of them produces a distinct pattern of phosphorylation on a given GPCR in a given cell. These distinct patterns, establish “barcodes” that either recruit different ß-arrestins or impart distinct conformations to the recruited ß-arrestin. This hypothesis therefore has at least two connotations. The first, is that different GRKs phosphorylate distinct sets of S/T residues in a given GPCR to differentially regulate ß-arrestin binding patterns. The second is concerned with specific binding patterns or structural changes within the arrestin molecule that occur upon binding the phosphorylated GPCR, which will be addressed in section 13.
Nobles et al. [37], identified specific residues that are phosphorylated in the ß2-AR in response to agonist challenge by mass spectrometry and quantified the difference in the relative amounts of phosphorylation of each site with SILAC (stable isotope labeling with amino acids). These studies established that GRK2 and GRK6 differentially phosphorylated distinct sites in the ß2-AR. For example, depletion of GRK6 markedly reduced agonist-mediated phosphorylation of Ser261/262, while depletion of GRK2 increased their phosphorylation. Using phospho-specific antibodies, they determined that GRK2 and GRK6 targeted distinct sites in the carboxy-tail of the ß2-AR to establish unique signaling-specific phosphorylation barcodes. Similarly, Butcher et al. [38], noted that full and partial agonists to the M3-muscarinic receptor directed its phosphorylation at different sites, which might underlie a mechanism for biased signaling by different ligands. In a separate study, mapping the phosphorylation sites in the parathyroid hormone receptor 1 (PTH1R) indicated that two clusters of S/T residues were specifically responsible for PTH(1-34)-induced PTH1R phosphorylation. Mutational analysis within each cluster, indicated that Thr503 and Ser504 in the second cluster were primarily involved in recruiting arrestin3 [39]. Mutagenesis of the S/T residues involved in arrestin binding also prevented co-internalization of the PTH1R with arrestin upon agonist stimulation. Patterns of GPCR phosphorylation were also determined for a variety of GPCR such as gherlin receptor, chemokine receptor CXCR4 and M3 muscarinic receptor [37-42].
Based upon these findings, it would appear that the position of specific sites of GPCR phosphorylation or “barcode” is key for the interaction of the phosphorylated GPCR with arrestin. Depending on their position and abundance in the C-tail of a given GPCR, these sites would generate appropriate changes in ß-arrestins. (discussed later in section 13 of this review).
The effects of GRK-mediated phosphorylation on physiological outcomes of the muscarinic M3 acetyl choline receptor (M3-mACHR) were determined in transgenic M3-mACHR knock in mice. Mutagenesis of the phosphorylatable sites of the M3-mACHR, abolished arrestin-mediated internalization of the agonist-activated M3-mACHR, without interfering with downstream signaling by Gq [43]. These mutant mice manifested impairments in insulin secretion [43], hippocampal learning and memory [44] and bronchial contractions [45]. These block mutations provide a role for GRK-mediated phosphorylation in driving specific physiological responses, but mutant mice with deletions in specific phosphorylation sites will be required to dissect the role of specific kinases and their associated barcodes in regulating specific physiological outcomes.
4. Role of cis-acting sequences or “barcodes” in regulating the trafficking itinerary of the GPCR
A major advance in characterizing the outcome of the agonist-internalized GPCR was the discovery of the role of cis-acting sequences in imparting a recycling phenotype onto the agonist-internalized GPCR. GPCR containing a type-I PDZ binding motif (PBM) related to the sequence (X-S/T-X-Ø, where X is any amino acid, Ø is a hydrophobic amino acid while the amino acid at position -2 is either a serine or a threonine) in their carboxy-terminus recycled efficiently and were re-inserted into the cell membrane [46-48].
The sequences of recycling type-1 PBMs are fairly diverse and their effect on GPCR trafficking was reviewed by Romero et al. [49]. Mutational analysis using the human ß1-AR indicated that the identity of the “X” residues at positions -1 and -3 (X3-S/T-X1-ø) in this PBM, was not flexible because many substitutions at positions -1 and -3 inactivated the PDZ and prevented the recycling of the ß1-AR [48]. For example, substituting Lys (K) at position (P=) -1 or Glu (E) at P = -3 with Ala (A) and other small amino acids inhibited the recycling of the ß1-AR [49]. These studies also highlighted that a hydrophobic (V, I, L) amino acid at P = 0 was necessary and much preferred, but not essential for recycling of these GPCR mutants. For example, substituting the Val at P = 0 with Ser or other polar amino acids markedly reduced the recycling of these ß1-AR point mutants. Consequently, in this review we will refer to those PBM sequences that impart a recycling phenotype onto the ß1-AR as “recycling barcodes” to differentiate them from sequences related to the type-1 PBM that do not support the recycling of the ß1-AR.
Concerning the interchangeability of type-1 PBMs in regulating GPCR trafficking, it was determined that transplanting a PBM derived from the ß2-AR (DSLL) onto the C-tail on the non-recycling δ-opioid-R facilitated the recycling of the δ-opioid/ß2 chimeric receptor [50, 51]. These findings and many others compelled Gage et al [50] to propose that type-1 PBMs were sufficient to implant a recycling signal onto other recycling or non-recycling GPCR.
5. Role of post-translational modifications by compartmentalized signaling networks in regulating GPCR trafficking
i. Role of compartmentalized PKA in regulating the GPCR trafficking itinerary
An important distinction between the trafficking of the wild type ß1-AR and many other GPCR, is that the recycling of the agonist-activated ß1-AR was dependent on the activity of cyclic AMP-dependent protein kinase (PKA). Inhibition of PKA biochemically with H-89 or with the cell permeable myristoylated PKA inhibitory peptide (mPKI) or by global delocalization of PKA from its A-kinase anchoring protein (described later in section 6 of the review) inhibited the recycling of the human ß1-AR, but not that of the ß2-AR or other GPCR [27]. The preferred substrates for PKA are serine or threonine residues corresponding to the motifs RRX-S/T and/or RX-S/T. Two PKA-substrate serines were present in rodent ß1-AR, but a single substrate serine was located at position 312 (Ser312) in the third intracellular loop of the human ß1-AR. Inactivation of Ser312 in the human ß1-AR by mutagenesis to Ala ((S312A) ß1-AR), inhibited the recycling of the (S312A) ß1-AR mutant even though the PBM in its C-tail was still intact [27]. These findings suggested that an additional trafficking “barcode” unrelated to the type-1 PBM was apparently involved in recycling of the agonist-internalized ß1-AR.
Another instance for a role of PKA-mediated phosphorylation in regulating protein trafficking was its effect on aquaporin-2 (AQP2) water channels in renal collecting ducts [52]. In renal collecting duct cells, water channel forming AQP2 subunits were distributed in cytoplasmic punctate vesicles. These vesicles translocated to the apical membrane of renal collecting ducts in response to vasopressin-mediated activation of the Gs-coupled vasopressin receptor (V2R). Stimulation of V2R increased the production of cyclic AMP, which activated a “compartmentalized” PKA that phosphorylated AQP2 on Ser256 [53]. Phosphorylation of AQP2 on Ser256 promoted its translocation and insertion into the apical membrane to increase the permeability of collecting ducts to water. In addition, it appears that AQP2 contained a non-canonical type-1 PDZ, (ETKA) in its C-terminus, which if inactivated blocked the translocation of AQP2 to the apical membrane of renal collecting ducts in response to vasopressin [54].
ii. Role of ubiquitination in regulating the GPCR trafficking itinerary
Post translational modifications of the GPCR by membrane-associated signaling networks was also involved in regulating the down-stream trafficking itinerary of several GPCR. Many GPCR, contain ubiquitin target lysines in their 3rd intracellular loops or in their C-tails. For example, the 324SSLKILS330KGK333 motif in the C-tail of C-X-C chemokine receptor-4 (CXCR4) was critical for CXCL12-mediated internalization of CXCR4 and its lysosomal degradation [55]. Agonist-mediated activation of CXCR4 promoted the ubiquitination of CXCR4 on Lys327/331/333, which was a priori for its degradation in lysosomes [56]. Ubiquitination of the agonist-activated CXCR4 was dependent on Gi-mediated activation of PKCδ, which phosphorylated the CXCR4 in the membrane on Ser324/325 [57]. Mutagenesis or interference with either of these motifs altered the subsequent trafficking roadmap of CXCR4, suggesting that this receptor was pre-addressed to a specific endosomal roadmap prior to its internalization [58].
The trafficking of the internalized agonist-activated ß2-AR was also altered by its ubiquitination on specific lysine residues in its 3rd intracellular loop or in the C-tail [59]. The sequence of events was initiated by GRK-mediated phosphorylation of the agonist-occupied ß2-AR, followed by the binding of scaffold composed of ß-arrestin-2, which in turn recruited the ubiquitin ligase Nedd4-1 [60, 61]. Ubiquitination of the ß2-AR steered the receptor away from PBM-guided recycling towards the proteasomal degradative pathway even when its PBM was still intact [61]. In addition, ubiquitination was apparently involved in maintaining endosomal residence of the ubiquitinated ß2-AR and in prolonging its signaling in endosomes through additional protein-protein interactions that were mediated by the arrestin-ubiquitinated GPCR complex [62].
iii. Role of non-PDZ sequences in regulating GPCR trafficking
In addition to cis-acting barcodes, such as the PBM or p-Ser312, non-PDZ trafficking sequences were involved in trafficking of the agonist-internalized μ-opioid and dopamine-R1 receptors [63, 64]. Endosomal tubules containing GPCR with these motifs displayed faster budding kinetics than tubules that budded via the “sequence-dependent” pathway [63, 64]. Trafficking non-PDZ sequences transplanted onto either recycling or nonrecycling GPCR promoted their recycling. For example, replacement of the last 10 amino acids (including the PBM) in the ß1-AR with the core recycling sequences of the μ-opioid-R or domapine-R1 promoted the recycling of ß1-AR-[μ-opioid-R] and ß1-AR-dopamine-R1] chimeras, even when Ser312 was mutated to alanine [48]. Therefore, non-PDZ recycling “barcodes” represent a class of transplantable trafficking sequences that would enable the recycling of any GPCR. For example, there is a non-PDZ recycling “barcode” in the multiple cloning region of the mammalian expression vector pcDNA 3.0 [65], which would explain the trafficking of GFP fusions to the C-tail of the ß2-AR and other GPCR [66, 67].
IV. Role of constitutive activation of PKA-target serines on the GPCR trafficking roadmap
As outlined earlier, PKA-mediated phosphorylation of Ser312 in the 3rd intracellular loop of the ß1-AR was required for its recycling. But, what happens to the ß1-AR if this serine was mutated to its phospho-serine mimic aspartic acid (S312D) ß1-AR)? In this case, the (S312D) ß1-AR construct was expressed in the membrane, internalized normally in response to ß-agonists and recycled efficiently after the removal of the ß-agonist [27]. This construct however, recycled when PKA was inhibited and when the PBM was inactivated [68]. These finding indicate, that this point mutation altered the trafficking of the ß1-AR from a PBM/p-Ser312-dependent or “sequence-dependent” roadmap into a “sequence-independent” roadmap. Another example is that of AQP2 which is constitutively inserted into the apical membrane of collecting ducts upon mutating its PKA target Ser256 to aspartic acid [53]. Finally, mutating the two PKA-target serines in the C-tail of the ß2-AR to aspartic acid, altered its trafficking roadmap from that of the “sequence-dependent’ pathway into the “sequence-independent” pathway [69].
The findings described above indicate that the PBM and kinase compartmentalization played an essential role in regulating GPCR trafficking, but how was PKA compartmentalized and the role of the PBM in this required further elucidation.
6. Role of specific AKAPs in regulating the trafficking and signaling of the GPCR
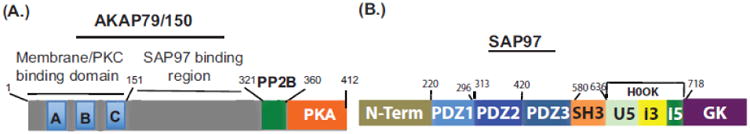
The PKA holoenzyme is targeted near potential substrates principally via RII subunit association with AKAPs [70]. Each AKAP contains a PKA binding site and a unique subcellular targeting domain that restricts its location within the cell either to unique cellular compartments or to specific substrates [70]. Interference with AKAP-PKA interactions inhibited the recycling of the agonist-internalized ß1-AR [27, 71]. A specific role for AKAP79/150 (AKAP5) in regulating the trafficking of the ß1-AR was elucidated by molecular and genetic approaches in AKAP5 null (AKAP5−/−) cells [Fig. 1A]. Depletion of AKAP5 by siRNAs or by genetic ablation of its gene had little effect on the membranous distribution of WT ß1-AR or their endocytosis in response to isoproterenol [72 and Fig. 1A]. Translocation of endosomal ß1-AR to the membrane was inhibited in AKAP5-depleted HEK-293 cells and in AKAP5−/− cardiac myocytes. Recycling of the ß1-AR was restored when full-length AKAP5 was introduced, indicating that this effect was specific to full-length [1-427]-AKAP5. Re-expression of AKAP5 lacking the membrane and PKC binding domains ([151-427]-AKAP5) or either the calcineurin- (AKAP5Δ321-360) or PKA-binding ([1-360]-AKAP5) domains, could not rescue the recycling of the ß1-AR recycling in AKAP5-null cells [71]. These results were expected because proper targeting of AKAK5 to the inner leaflet of the cell membrane and its interaction with PKA were essential for AKAP5 activity. What was unexpected however, was that expression of an AKAP5 construct with deleted calcineurin-binding motif (AKAP5Δ321-360) could not rescue the trafficking of the ß1-AR in AKAP5 null cells [72].
Fig. 1. A, Structure of A-kinase anchoring protein79/150 [AKAP5] and B, SAP97.
A role for PKA-AKAP interactions in AQP2 trafficking was discovered several years prior to the identification of their role in ß1-AR trafficking [73]. The identities of the specific AKAP that is involved in AQP2 trafficking is still under investigation, but there appears to be significant mechanistic similarities between its trafficking and that of the ß1-AR. These similarities include, translocation of these proteins in response to phosphorylation by PKA and the involvement of “an anchored AKAP” and perhaps a PBM in this phenomenon [54, 73]. In addition, calcineurin is involved because inhibition of calcineurin with cyclosprin A or FK-506 inhibited the recycling of endosomal ß1-AR and the translocation of AQP2 in response to vasopressin [73, 74].
These results would explain why the (AKAP5Δ321-360) construct, lacking the calcineurin-binding site could not rescue the trafficking of the ß1-AR, but what is the relationship between the AKAP5-PKA complex and the ß1-AR? This question was addressed by serial deletions within the ß1-AR, which indicated that AKAP5 was bound to the type-1 PBM of the ß1-AR. The identification of the PBM of the ß1-AR as a site for integration of AKAP5/PKA, expanded the role of these sequences from purely trafficking domains into signal compartmentalization domains [68].
7. Role of PDZ-containing proteins in regulating GPCR trafficking
The mechanism of PBM in regulating GPCR trafficking involves their binding to PDZ modules, which are modular 75-100 amino acid protein-interaction domains, named after the first proteins identified to contain this module [75]. PDZ containing proteins contain one or more PDZ module, which bind to select PBM sequences, and several other protein-protein interacting domains that would assemble multiprotein complexes [76]. In this review we will concentrate on PDZ proteins that have been shown to interact with class-1 PBMs in GPCR, even though class-II motifs corresponding to (X-Ø-X-Ø) were involved in recycling of the ionototropic GluR2 and 5-hydroxy tryptamine [5-HT] type4(e) receptor [77, 78].
The involvement of specific PDZ proteins in GPCR trafficking is well established for a small number of GPCR, most notably the ß-adrenergic receptor family. Affinity chromatography using a ß2-AR-derived carboxy-tail identified Na+/H+-exchange regulatory factors-1 and -2 (NHERF-1, -2) as binding partners for the ß2-AR PBM [79]. In addition, EBP50 (Ezrin binding phospho-protein of 50 kDa, which contains two PDZ domains was also identified by sequence analysis as a protein that binds to the PBM of the ß2-AR [46]. Two members of this PDZ family, namely NHERF-1 and NHERF-2 bind also to the cortical actin cytoskeleton through an Ezrin-radixin-moesin (ERM)-binding domain. Binding to cortical actin is a priori for ß2-AR recycling, suggesting that a NHERF-ERM-actin network was involved in recycling of the ß2-AR [46]. This hypothesis was substantiated by showing that connecting the δ-opioid-R (a non-recycling GPCR that does not contain a PBM) to the ERM-binding domain of NHERF-1 or directly to the actin-binding domain of Ezrin generated ß2-AR chimeras that recycled efficiently [80]. The dependence of ß2-AR PBM binding to NHERF1, 2 as a priori to recycling of the ß2-AR does not appear to be ironclad. Expression of siRNAs that down-regulated the protein levels of NHERF1 or NHERF2 or both did not inhibit either the internalization of the agonist-activated ß2-AR or its subsequent recycling from endosomes to the plasma membrane [81]. While these data might challenge the exclusivity of NHERF1,2 in regulating the trafficking of endosomal ß2-AR, they more likely suggest that there are other scenarios for connecting budding/or recycling ß2-AR bearing endosomes to the actin filament network.
Characterizing the repertoire of PDZ binding proteins that interacted with the ß1-AR BPM by yeast two-hybrid assays indicated that several proteins belonging to the MAGUK family, such as the post synaptic density protein PSD95, MAGI 1-3, GIPC (GAIP-interacting protein, carboxyl (C) terminus) and CAL (also known as PIST/GOPC) were binding partners to the ß1-AR PBM [82-86]. Some of these PDZ-binding proteins affected the sequestration or exerted negative effects on the trafficking of the ß1-AR. For example, overexpression of PSD-95, which binds avidly to the ß1-AR PBM, severely inhibited ß1-AR internalization in neonatal rodent cardiac myocytes by interfering with agonist-regulated concentration of the ß1-AR into coated-pits [68, 87]. Internalization of the ß1-AR in PSD-95 expressing cardiomyocytes was restored by reducing PSD-95-PBM interactions either by inactivation of the ß1-AR PBM or by overexpression of a peptide corresponding to the ß1-AR PBM [87]. Gardner et al [68], also determined that PSD-95 interfered with endocytosis and recycling of the wild type ß1-AR, suggesting that PSD-95 binding to the ß1-AR PBM retained the receptor on the cell surface and enhanced its signaling through the Gs-adenylyl cyclase axis to cyclic AMP [82]. Co-expression of PBM-binding proteins related to MAGI and GIPC with the ß1-AR profoundly impaired ß1-AR-mediated ERK1/2 activation without affecting its sequestration or its signaling to cyclic AMP [83-85]. Moreover, interaction of the ß1-AR PBM with the Golgi-associated protein CAL/GOPC/PIST increased the association of the ß1-AR with Golgi proteins, inhibited ß1-AR recycling and reduced its surface expression [86, 88].
These results indicate that the fate of the naive or the agonist-activated ß1-AR depends on the repertoire of PDZ-binding proteins expressed in a given cell. On the one hand, signaling by membranous ß1-AR was sustained in cells expressing PSD-95 because PSD-95 bound avidly to the ß1-AR and markedly reduced its internalization. On the other hand, signaling of the ß1-AR to MAPK and recycling of the agonist-internalized ß1-AR were markedly impaired in cells expressing PDZ-binding proteins related to MAGI/GIPC. Thus, the nature of the cell under study and its repertoire of PDZ-binding proteins acutely affected the signaling and trafficking outcomes of the ß1-AR and perhaps other GPCR.
However, as outlined earlier, a major difference in the trafficking of ß1-AR/AQP2 versus other GPCR was the dependence of ß1-AR/AQP2 trafficking on AKAP-PKA interactions. These findings suggested a role for SAP97, which unlike other MAGUK proteins is capable of complexing to an AKAP/PKA complex.
8. Role of compartmentalizing PDZ containing proteins, such as SAP97 in regulating signaling and trafficking of the GPCR
SAP97 is a MAGUK protein encoded by the DLG-1 gene [89, 90], and contains several protein-protein interacting domains (Fig. 1B). SAP97 is a modular protein composed of 3 PDZ-binding domains, which are preceded by an N-terminal L27 dimerization domain and followed by a src homology 3 (SH3) domain, then by an extensively spliced HOOK region (between (636-718) and finally with an inactive guanylate kinase like domain [91, and Fig. 1B]. SAP97 is expressed in post-synaptic densities of neurons and in peripheral tissues, such as the heart and kidney, where its function(s) are less defined [92]. The second PDZ of SAP97 was the major PDZ that bound to the ß1-AR PBM with an affinity EC50 of 0.5 ± 0.2 μM [93]. A unique feature of SAP97 is its HOOK domain, which contains an alternatively spliced highly basic i3 sequence (between amino acids 649-697) that binds to the AKAP5/PKA complex and to other proteins [90]. The involvement of SAP97 in ß1-AR trafficking was characterized by many methods. Depletion of SAP97 inhibited the recycling of ß1-AR and re-expression of siRNA- resistant SAP97 rescued the recycling of the ß1-AR [93]. However, re-expression of SAP97 lacking the i3-i5 domains in the HOOK region that were involved in binding SAP97 to AKAP5, could not rescue the recycling of the ß1-AR in SAP97-depleted cell. In addition, the effect of point mutations in the PBM of the ß1-AR on its recycling indicated that PBM mutations which abolished SAP97-PBM interactions also inhibited the recycling of these ß1-AR mutants; while PBM mutations that maintained SAP97 binding were associated with a recycling ß1-AR phenotype [48, 93]. These results indicate that SAP97 binding to the ß1-AR PBM was essential for its regulation of ß1-AR trafficking in mammalian cells. In addition, they show that bipartite binding of SAP97 simultaneously to the ß1-AR BPM via its PDZ2 and to AKAP5/PKA via its I3 domain regulated the trafficking of the ß1-AR [48, 93]. This complex is termed the ß1-AR receptosome [Fig. 2], and was shown to be involved in signaling [48, 72] and in trafficking of the ß1-AR [74].
Fig. 2. Organization of the ß1-adrenergic receptosome around the type-1 PBM in the carboxy-tail of the ß1-AR.
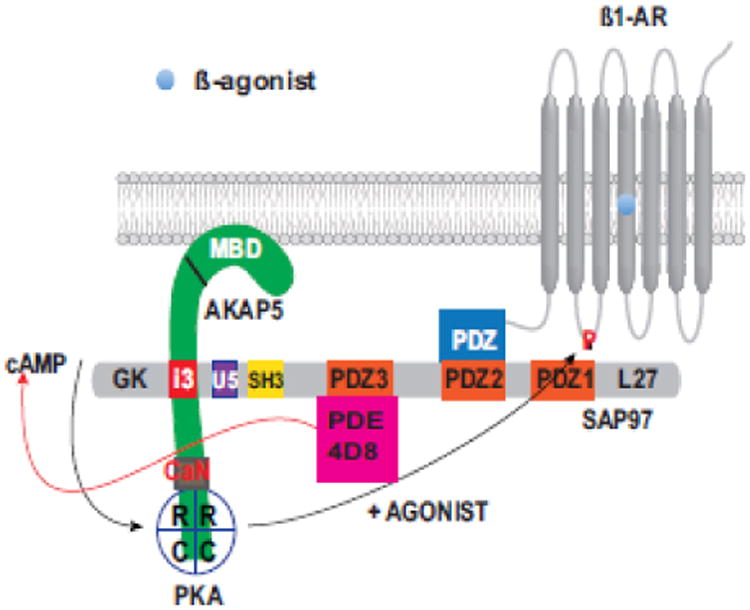
The figure illustrates bipartite binding of SAP97 simultaneously to the ß1-AR PBM (via its second PDZ) and to AKAP5 (via the I3 domain of SAP97). The figure indicates that PDE4D8 was bound to SAP97 as well. Activation of the ß1-AR signaling pathway in the membranes increases cyclic AMP, which activates PKA. Spatiotemporal activity of cyclic AMP is contained through its degradation by receptosomal PDE4D8. Activated PKA in turn, faithfully phosphorylates the ß1-AR on the serine at position 312 to imprint a recycling signal onto the ß1-AR prior to its sequestration.
The specific involvement of SAP97 in ß1-AR trafficking indicated that not all type-1 PBM sequences would be interchangeable in supporting GPCR trafficking [51]. This notion was substantiated by substitution studies in which the last 10 amino acids in the ß1-AR was substituted with other type-1 PBMs including the PBM of the ß2-AR, which does not bind SAP97 [49, 94]. These experiments determined that the PBM of the ß1-AR was exchangeable with PBMs that bound SAP97 such as GluR1 (ATGL) and aquaporin-2 (GTKA) PBMs, but not with the ß2-AR (DSLL) PBM [48]. Thus, even among the recycling barcodes there was a degree of specificity in supporting the trafficking of a given GPCR. However, irrespective of the GPCR trafficking mechanism, those GPCR that recycled were not degraded, while those that did not recycle were uniformly degraded [48]. Recycling receptors were re-inserted into the plasma membrane for another round of signaling as assessed by functional resensitization assays of recycled GPCR [93]. Therefore, recycling was critically involved in GPCR resensitization and in setting the overall GPCR signaling in mammalian cells [8, 95]. The latter effect will be discussed in more detail in section 14 of this review.
9. Role of cross-talk and feedback regulatory mechanisms in compartmentalized network-mediated regulation of GPCR trafficking
The findings described thus far indicate that a scaffold composed of SAP97-AKAP5/PKA that was tethered to the type-1 PBM of the ß1-AR was involved in regulating the trafficking of the ß1-AR [Fig. 2]. If so, then the pool of PKA anchored at the PBM would be selectively involved in regulating the trafficking of the ß1-AR through the phosphorylation of this GPCR on Ser312. Several complementary techniques were used to prove this notion. First, depletion of AKAP5 or SAP97 selectively inhibited the recycling of the agonist-internalized GPCR [93]. Second, cleavage of the 32P-phosphorylated ß1-AR with cyanogen bromide indicated the 3rd intracellular loop was selectively phosphorylated by the PBM-anchored pool of PKA [71]. Third, direct analysis of PKA-mediated phosphorylation of the ß1-AR on Ser312 by an α-PKA-site specific antibody, indicated that isoproterenol promoted the phosphorylation of Ser312 of the ß1-AR in cells bearing the ß1-AR, but not in cells bearing the ß1-ARΔPDZ mutant [96]. Therefore, cross talk between two separate domains, namely the PBM and Ser312 was involved in setting the trafficking itinerary of the ß1-AR. This hypothesis addressed many of the findings illustrated earlier, such as failure of the ß1-ARΔPDZ and (S312A) ß1-AR to recycle and inhibition of ß1-AR recycling by PKA inhibitors (H-89 and mPKI) and AKAP-PKA disruptors (st-Ht31) [96].
Another feature of the ß1-AR receptosome is that it addressed a problem encountered by all Gs-coupled GPCR, which is how to regulate the spatiotemporal effects of the diffusible mediator cyclic AMP. If Cyclic AMP was allowed to freely diffuse, it would indiscriminately activate soluble and compartmentalized pools of PKA. This problem is partly addressed by the ß1-AR through the recruitment of additional components to its receptosomal network. Sensitivity of the cell to cyclic AMP is regulated primarily by phosphodiesterases (PDE), which adjust the intensity and confine the three-dimensional geometry of the cyclic AMP signal to select subcellular compartments [97-100]. In the case of the ß1-AR, the concentration and diffusion of cyclic AMP generated by the ß1-AR signaling pathway was regulated at the level of the ß1-AR receptosome by phosphodiesterase 4D8 (PDE4D8), which binds to receptosomal SAP97 [101, 102]. The inclusion of PDE4D8 into the ß1-AR receptosome provides a powerful regulatory feedback enzyme that focuses the output of cyclic AMP to within the ß1-AR microenvironment [102, and Fig. 2]. This organization insures that low amounts of cyclic AMP are vectored towards the activation of those PKA pools within this boundary, such as the PBM-anchored pool of PKA. Therefore, faithful phosphorylation of the ß1-AR on Ser312 was mediated by a cross-talk mechanism between the PKA pool bound to the SAP97/AKAP5 complex tethered to the ß1-AR PBM and cyclic AMP generated by the ß1-AR signaling pathway, as illustrated in Fig 2.
10. New insights into intracellular trafficking mechanisms in early endosomes that are involved in targeted sorting of GPCRs to the recycling pathway
As discussed earlier, membranes containing the agonist internalized GPCR fuse with EE, which are a diverse set of endosomal vesicles involved in sorting out cargo based upon unique sequences within cargo proteins and on specific sets of sorting proteins in endosomes. The first set of sorting proteins to be characterized as regulators of GPCR trafficking was the endosomal rab GTPases family [103, 104]. This family consists of more than 60 proteins [104], which are expressed differentially in distinct types of endosomes to regulate vesicular sorting and trafficking [105, 106]. Early studies relied on co-localization fluorescence microscopy to determine the association of endosomal GPCR with the various Rab-GTPases [107]. Examining the association between the recycling wild type ß1-AR and the non-recycling (S312A) ß1-AR indicated that both constructs were initially internalized into the same subset of endosomes because they colocalized with Rab5a and EEA1 markers of EE [108]. In later time frames, the WT ß1-AR co-localized with the markers of sorting endosomes (Rab4a) and later with Rab11a in recycling endosomes; while the (S312A) ß1-AR co-localized instead with lysosomal markers (Rab9 and Lamp1), where it was eventually degraded [108]. These and many other studies indicated that recycling GPCR would bud out of EE into vesicles, which later matured into sorting and recycling endosomes by acquiring new sets of Rab GTPases [103]. These descriptive studies however could not elucidate the basic mechanisms for retention or sorting of GPCR cargo out of EE.
A major mechanism involved in endosomal sorting of cargo involves tabulation and budding of recycling cargo out of EE. Cargo destined for retention and eventual degradation bud instead into the lumen of these endosomes, which later mature to late endosomes/ lysosomes [13]. Using superb ultrastructural techniques, trafficking of GPCR cargoes out of EE was divided into “sequence-dependent” or PBM-mediated sorting versus “sequence-independent” sorting mechanisms [109]. GPCR that recycled in a PBM-dependent “i.e. sequence-dependent” manner colocalized with tubules that extended from the main body of EE and subsequently pinched off from EE, while GPCR that did not recycle were excluded from these tubules [109, 110]. An important distinction between these tubules and tubules involved in geometric/bulk or “sequence-independent recycling” of the transferrin receptor (Tfr) for example, was the involvement of the actin cytoskeleton [109, 110]. In this regard, budding tubules bearing TfR did not contain members of the actin cytoskeleton, while tubules bearing ß2-AR that trafficked in a PBM-dependent manner were enriched with actin and other cytoskeletal proteins. Actin-dependent concentration of receptors into budding endosomal tubules depended on the PBM sequence because ß2-AR with inactivated PBM did not enter the actin enriched compartment [110]. Therefore, specific set(s) of endosomal sorting proteins that interacted selectively with the PBM were involved in sorting the GPCR into this subset of tubules [80, 81].
11. Role of SNX27, retromer/WASH interactions in PBM-mediated or “sequence-dependent” GPCR trafficking
i. SNX27-mediated sorting of PBM-containing endosomal GPCR to the plasma membrane
Depletion of NHERF1/EBP50 and NHERF2 simultaneously using RNA interference produced small decreases in recycling of the ß2-AR that were not statistically significant [81]. These results challenged the hypothesis that NHERF1,2 represented critical PDZ recycling proteins and instituted a search for a distinct PDZ trafficking protein that associated with EE and possessed an affinity for type-1 PBMs. The search led to SNX27, which is an endosomal PDZ protein that was identified by affinity screen/mass spectrometry as a binding partner for the PBM of 5-HT type 4a receptor, Kir3.3 potassium channels and diacylglycerol Kinase ζ [78, 111-112]. SNX27 is an endosomal protein that contains a PDZ domain at its N-terminus, followed by a phosphoinositide-binding PX-domain and by a FERM-like domain (Fig. 3A). In addition to identifying a major role for SNX27 in cargo selection, these studies even suggested a role for SNX27 in endocytic recycling by showing its association with recycling Tfr in rab11 bearing endosomes [112]. A primary role for SNX27 in controlling the sorting of GPCR from EE to the plasma membrane was initially identified for the ß2-AR [113]. This process involved interactions between the ß2-AR PBM and the PDZ of SNX27, which subsequently controlled sorting of the GPCR from EE to the plasma membrane [81]. These findings were later substantiated for the parathyroid hormone receptor [114-115], ß1-AR [31, 96], AMPARs and NMDAR [116-118] and ion channels such as G protein-gated inwardly rectifying potassium channels [111, 119], among others. In these studies, selective depletion of SNX27 resulted in retention and subsequent degradation of GPCR that depended on the PBM for their recycling.
Fig. 3. Schematic illustrating the role of endosomal trafficking proteins in regulating the GPCR recycling by the one- or two barcode paradigms.
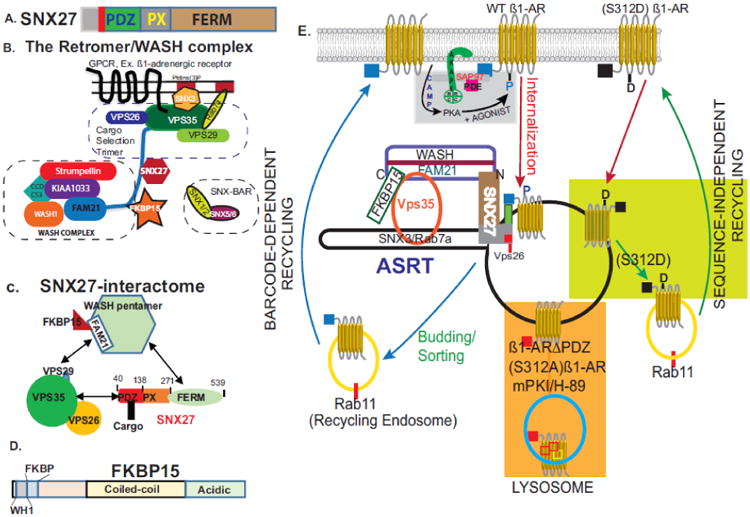
A, Organization of SNX27 into its three major protein binding domains. B. Structure of retromer and WASH complexes that are involved in endosomal trafficking. The retromer is composed of a cargo selection trimer (CST composed of Vps 35, 29 and 26) and BAR dimers. The retromer is recruited to endosomes via PtdIns(3)P-binders such as the GTPase Rab7a, and sorting nexin-3 (SNX-3). CST interacts with the WASH complex via the C-tail of FAM21 that also binds to SNX27 and FKBP15 to mediate endosome to plasma membrane trafficking. C, The SNX27 interactome. PBM-containing cargo binds to the PDZ of SNX27; while the PX and FERM domains of SNX27 are involved in recruiting SNX27 to endosomes and to the WASH complex respectively. In addition, a domain in SNX27 between 66-76 binds to Vps26 of the retromer. Adapted from [120]. D. Domain organization of FKBP15. E. Schematic of GPCR trafficking in endosomes. Activation of membranous ß1-AR promotes their internalization into early endosomes (red arrows). The type-1 PBM of the ß1-AR (blue) binds to the PDZ of SNX27 (green). SNX27 directs PBM containing cargoes into tubules that utilize actin/retromer/WASH complexes (termed ASRT) for budding out of early endosomes. This mode of trafficking is mediated by the PBM “barcode” and is sufficient to guide most GPCR out of early endosomes. However, tubulation and budding of WT ß1-AR out of endosomes required a second “barcode”, which was phospho-Ser312. This reversible “barcode” was imprinted by the ß1-adrenergic receptosome and is recognized by endosomal sorting proteins, such as FKBP15. Upon verifying its two barcodes, the ß1-AR is sorted from early endosomes into rab11 containing recycling endosomes and then re-inserted into the plasma membrane (blue arrows). Inhibition of PKA or inactivation of either barcode leads to retention of these cargoes within the body of early endomes (orange rectangle), which eventually mature into late endosomes/lysosomes. ß1-AR constructs with recycling nonPDZ sequences (black cube) or with a Ser312 to aspartic acid mutation are sorted out of early endosomes in a retromer/WASH independent pathway (light green rectangle).
ii. Characterization of the interactions between the GPCR PBM and the PDZ of SNX27
Measurement of the affinity of the interaction between a ß2-AR-derived tail sequence with an SNX27-derived PDZ domain by fluorescence polarization assays indicated that the affinity (Kd) of their binding was unexpectedly low at ∼ 17 μM [81]. Pull down assays between the C-tail of the ß1-AR and SNX27 indicated that the EC50 for their interaction was also low at ∼1.5 μM [96]. Detailed studies on the interaction between the PDZ of SNX27 with type-1 PBM derived for other GPCR or channels, indicated that the affinity of PBM binding to the SNX27 PDZ was apparently dependent on “an acidic clamp” imparted mainly by a pair of glutamic acids “E” in position -3 of the core PBM and at position -5 outside the core PBM [120]. For example, high affinity binding between PTHR PBM (E-5AE-3TVM0) or GIRK PBM (E-5SE-3SKV0) to the PDZ of SNX27 was attained when negatively charged amino acids (D or E) were present at positions -3 and -5 [119, 120]. The PBMs of ß1-AR (FA-5SE-3SKV0) is very similar to the PBM of GIRK, except for having a neutral Ala instead of a negatively charged glutamic acid at position -5. Whether this difference at position -5 is responsible for the relatively lower binding affinity of the ß1-AR PBM to the PDZ of SNX27 is not known.
Another mechanism that would alter the affinity of the PBM in binding to SNX27 was phosphorylation of the GPCR. The affinity of the ß2-AR PBM ((407)S-6TND-3S(411)LL0) in binding to the PDZ of SNX27 increased significantly upon phosphorylation of Ser407 at position -6, suggesting that phosphorylation might assist in recycling of some proteins by SNX27-retromer pathways [120]. These data should be interpreted with caution because Ser407 and Ser411 are among several serines in the C-tail of the ß2-AR that are phosphorylated by GRK2/5 upon the activation of the ß2-AR [37, 121]. However, the outcomes of phosphorylating these serines on GPCR recycling are diametrically opposed, since phosphorylation of Ser411, at position -2 in the PBM would inactivate the PBM and inhibit GPCR recycling [47, 122].
iii. Role of other motifs in SNX27 in regulating its compartmentalization and its endosome to plasma membrane recycling
SNX27 associates with the actin polymerization machinery and with the retromer complex either directly or indirectly via the WASH multi-protein complex [123]. The retromer is composed of two major complexes, a stable cargo selection trimer (CST) consisting of VPS26-VPS29-VPS35 and SNX-BAR dimers that facilitate remodeling of endosomal membranes involved in transporting cargo from endosomes to the trans-Golgi network [124, 125, and Fig. 3B]. The complex of actin/sorting nexin/retromer tubule is termed “ASRT”, and is involved in “sequence-dependent” recycling of cargo via tubules that are biochemically distinct from the tubules involved in constitutive recycling of cargoes, such as Tfr and LDL receptors [13, 109]. In this regard, the versatile PDZ of SNX27 would be involved in sorting PBM-containing cargo into endosomes that mediate sequence-dependent sorting and budding of GPCR-containing membranes out of EE by the combined actions of retromer/WASH complexes [80, 113]. The Wiskott-Aldrich syndrome protein and SCAR homologue (WASH) complex is a large pentameric protein complex composed of strumpellin, SWIP, CCDC53, WASH1 and FAM21 [126, and Fig. 3B]. Two domains in SNX27 were independently involved in complexing SNX27 to the ASRT [Fig. 3C]. The first was a phosphoinositide binding (PX) domain in the middle region of SNX27, which promoted the association of SNX27 with the surface of endosomes. The second were multiple 4.1/ezrin/moesin/radixin (FERM)-like domains in the C-terminus of SNX27, which anchored SNX27 to the retromer and to endosomes [127-128, and Fig. 3C]. In addition to these well characterized protein binding domains, there was a stretch of 11-amino acids [67-77] upstream of the core PDZ pocket [87-135] of SNX27, which mediated the binding of SNX27 to VPS26 of the retromer [128]. This short motif apparently linked SNX27 to the retromer, while still permitted the PBM of cargoes destined for recycling to bind to the PDZ of SNX27. This short sequence might explain how SNX27 would function as a cargo selector for sorting PBM-containing proteins into retromer tubules of ASRT trafficking complexes [Fig. 3C].
iv. Role of FAM21 and its associated proteins in ASRT-mediated endosome to PM GPCR recycling
FAM21, a component of the WASH complex, has emerged as a major player in directing SNX27-retromer cargoes to the plasma membrane [129]. The C-tail of FAM21 mediates the association of the pentameric WASH complex with VPS35 of the CST [130] and Fig. 3C. This action is mediated in part, by the binding of the carboxy-tail of FAM21, which contains 21 (L-F-[D/E]3-10-L-F (LFa)) motifs to VPS35 of the retromer [130, 131]. In addition to VPS35, the C-tail of FAM21 binds to FERM domains in SNX27 and to FKBP15, a protein whose role in EE to plasma membrane trafficking is currently unknown [131, 132 and Fig. 3B and C].
The mechanism of FAM21 mediated effects on GPCR recycling is not known, but overexpression of the C-tail of FAM21 inhibited the recycling of GPCR that recycled by the ASRT pathway, such as ß1-AR and ß2-AR, without inhibiting the recycling of GPCR that trafficked by the “bulk” or constitutive pathway [96]. These results suggest that overexpression of the C-tail of FAM21 inhibited GPCR recycling by multiple mechanisms that include: competitive disruption of the association between the WASH complex and the retromer, interference with the binding SNX27/FKBP15 to the WASH complex, among other plausible culprits [Fig. 3B and C]. As described before, depletion of SNX27 inhibited the recycling of all those GPCR that recycled via their PBM through the ASRT pathway, but what about FKBP15? Depletion of endogenous FKBP15 had no effect on the internalization of ß1-AR or ß2-AR, but selectively inhibited the recycling of ß1-AR, without exerting an appreciable effect on the recycling kinetics of the ß2-AR [96]. An interpretation of these results would suggest that recycling of the ß2-AR depended on recruitment of SNX27 via the C-tail of FAM21 to the ASRT complex, while the recycling of endosomal ß1-AR depended on FAM21-mediated recruitment of SNX27 and FKBP15 to the endosomal ASRT [96].
12. Mechanism of Barcode-mediated sorting of the GPCR in early endosomes
i. One versus two barcodes for endosomal trafficking of GPCR through the ASRT pathway
As a way to explain these and other findings described earlier, a two-barcode hypothesis was proposed to describe the sequence of events for GPCR sorting/budding and subsequent recycling through the endosomal ASRT pathway [96]. This hypothesis stipulated that the sequence of events for sorting of ß1-AR and other GPCR in endosomes was initiated by recognition of the antecedent PBM barcode by the PDZ of SNX27 because inactivation of the PBM or depletion of SNX27 inhibited the recycling of all PBM-containing GPCR. Since inactivation of Ser312 inhibited the recycling of the PBM-containing ß1-AR, it was proposed that phospho-Ser312 would constitute a second barcode for sorting of endosomal ß1-AR via the ASRT pathway [96]. While the specific mechanism for recognition of phospho-Ser312 in endosomes is still obscure, a role for FKBP15 was proposed based upon its specific effect in trafficking of cargoes such as the ß1-AR and its presumed relationship to calcineurin or FKBP12 [96]. The significance of this latter relationship is unknown, but depletion of calcineurin or its inhibition with cyclosporin A or FK506 selectively inhibited the recycling of the ß1-AR without affecting the recycling of other PBM containing GPCR [74]. Similarly, inhibition of calcineurin interfered with the translocation of AQP2, which is mediated by the phosphorylation of Ser256 by an AKAP-anchored PKA [73].
ii. Identification of a role for FKBP15 in selective sorting of GPCR via their second barcode
FKBP15, also known as WAFL (WASP and FKBP-like protein), FKBP133/KIAA0674 [132, 133], encodes a 1216 amino acid putative trafficking protein that was up-regulated in inflamed mucosa of ulcerative colitis patients [134]. The FKBP15 protein consists of at least four domains, a N-terminal WASP homology 1 domain [69-171], FKBP-like [178-290], coiled coil [∼500-∼950] and C-terminal acidic end [∼985-1200] domains [132] and Fig. 3D. All bona-fide FKBPs possess peptidylprolyl isomerase (PPIase) activity that is inhibited by FK506 and other PPIase inhibitors, but FKBP15 failed to display PPIase activity or to bind to FK506 [133], thus its relationship to other FKBP proteins such as calcineurin requires further clarification.
Trafficking functions have been attributed to FKBP15 in endocytosis through its interaction with the AP2 complex that serves as a hub for CCV mediated endocytosis, and in endosomal trafficking through its association with strumpellin a prominent member of the WASH complex [134]. FKBP15 interacts with endogenous actin and with endosomes via its central coiled-coil domain and deletion of this region resulted in endosomal retention and transport of cargoes towards lysosomes [132]. Moreover, depletion of FKBP15 impaired the recycling of the agonist-internalized ß1-AR, suggesting that it may play a role in verifying the phospho-Ser312 barcode; which is a priori for budding of ß1-AR-containing vesicles out of EE [96]. Where does FKBP15 fit in the GPCR trafficking paradigms in unknown, but its selective involvement in ß1-AR trafficking through its second p-Ser312 barcode suggests a passive role for FKBP15 in trafficking of most GPCR, except for those that rely on a phospho-Ser “barcode” for budding out of EE. This subset of proteins is currently limited to the ß1-AR and perhaps AQP2. A search of the bank of human GPCRs that express a C-terminal type-1 PBM for PKA substrate serine or threonine motif (RRXS/T) in their 3rd IC loop indicated that the following GPCR might be influenced by the two-barcode hypothesis: calcitonin-R (NM_001164737 or AAA35640); serotonin 2B-receptor [5HT-2BR] (CAA85319) and the purinergic receptor [P2RY1] (AAN01278), in addition to many non-GPCR proteins.
13. Endocytic membrane trafficking and its role in modulating receptor signaling
i. Mechanism of ß-arrestin-mediated effects on GPCR localization and function
Endosomal signaling by the GPCR is dependent on the binding of the agonist-activated GPCR to ß-arrestin. As outlined earlier, GRK-mediated phosphorylation of the agonist-occupied GPCR leads to recruitment of cytosolic ß-arrestin to the membrane, where it binds to GRK-phosphorylated serine and threonine residues in the C-tail of a given GPCR. However, in the case of the human ß1-AR, it appears that activation of this receptor promotes the translocation of ß-arrestin to clathrin-coated particles and activation of MAPK without invoking a physical interaction between the ß1-AR and ß-arrestin [135].
ß-arrestin interaction with the phosphorylated GPCR leads to three separate events, namely: desensitization, internalization and endosomal signaling by the GPCR. The role of ß-arrestin in mediating the internalization of the GPCR was described in section 3. The specific mechanism involved in GPCR ß-arrestin interactions was initially elucidated by X-ray crystallography of a complex consisting of a phosphopeptide derived from the vasopressin type-2 receptor (V2R)-ß-arrestin1-Fab30 complex [136]. This study indicated that the phospho-peptide was inserted within a cleft in ß-arrestin that was created by the rotation of its amino and carboxy terminal domains relative to one another. This mode of interaction is referred to as the “hanging” model because it is mediated by the interaction of relatively few residues in arrestin with the phosphopeptide.
The structure of the activated GPCR with an arrestin was initially determined for the visual GPCR rhodopsin bound to visual arrestin [137]. In agreement with previous models [138], it revealed that the activation of rhodopsin was a two-step process. The first process involved the formation of “pre activated arrestin”, through the interaction of the phosphorylated C-tail of the GPCR with the N-terminus of visual arrestin. This interaction caused the displacement of the C-terminal tail and allowed a 20° rotation of the N- and C termini of visual arrestin. The second process was mediated by subsequent extensive interactions between arrestin and the transmembranal domains rhodopsin, which eventually led to “activated arrestin” [137].
Recruitment of ß-arrestin to the ß2-AR is particularly important because their interactions are involved in multiple events. The interaction between a modified ß2-AR chimeric construct with its C-terminus replaced by that of the V2R (ß2-V2R) with ß-arresrin was determined by a variety of ultrastructural methodologies [139]. Single particle EM indicated that the interaction of the ß2-V2R with ß-arrestin existed in two general confirmations, one involving the V2R phospho-peptide “hanging” or “tail” model”, which was closely equivalent to “pre-activated rhodopsin” mentioned earlier. This was followed by a set of extensive interactions between ß-arrestin and ß2-V2R helical interfaces that culminated in the insertion of the finger loop of ß-arrestin within the receptor core. This confirmation resulted in a horizontal arrangement of ß-arrestin with the receptor in the negatively-stained EM structures.
The significance of these distinct conformations of GPCR-ß-arrestin complexes on physiological structural interactions between the GPCR and ß-arrestin indicate that the “tail or hanging” confirmation mediated receptor internalization and promoted the creation of GPCR-ß-arrestin-c-SRC complexes that regulated multiple cellular functions downstream of the GPCR [140]. More extensive interactions between ß-arrestin and the transmembranal domains of the GPCR were apparently involved in ß-arrestin-mediated desensitization of the GPCR through displacement of the activated G protein from the GPCR by ß-arrestin or by attenuating the effect of the activated GPCR on the G protein.
ii. Visualization of interactions between different active GPCR conformation with ß-arrestin
Select members of the A and B families of Gs-coupled GPCR such as V2R [141], parathyroid hormone receptor (PTHR) [142], corticotropin releasing hormone receptor1 [143] and others [144], exhibited both membrane generated down-stream signaling and endosomal ligand-biased persistent signaling that was mediated by activated endosomal Gs and adenylate cyclase [24]. Direct evidence that an endosomal GPCR can activate G proteins to elicit a second wave of signaling was derived from distinct nanobody biosensors that could detect inactive vs. active states of ß2-AR or Gs [145]. Nanobody sensors for active ß2-AR or Gsα translocated from the cytoplasm to the membrane upon the activation of the ß2-AR by agonists, but did not associate with ß2-AR when they were clustered in clathrin-coated pits. These nanobodies re-associated with the active form of the ß2-AR in endosomal vesicles, indicating that GPCR-Gs interactions were reestablished in endosomes resulting in the generation of endosomal cyclic AMP [145]. The binding between ß-arrestin and class B GPCR, such as the V2R was not transient. Rather, ß-arrestin was tightly bound to the phosphorylated V2R in the membrane and was endocytosed with it into endosomes [146]. These results indicate that type A and type B GPCR differ in their propensity to form complexes between them and ß-arrestin. Class A GPCR, such as the ß1- and the ß2-AR, interacted transiently or very weakly with ß-arrestin, while class B GPCR, such as V2R and PTHR, interacted tightly with ß-arrestin [24]. Moreover, the specific phosphorylation barcodes of the GPCR that were imprinted by the different GRKs elicited distinct conformational changes in ß-arrestins. For example, activation of ß2-AR with the full agonist isoproterenol generated an increase in intramolecular interactions between the N- and C-termini of ß arrestin-2 [147, 37]. In GRK2-depleted cells, the effect of isoproterenol on intramolecular changes in ß-arrestin2 was significantly reduced, while depletion of GRK6 did not generate a conformational change in ß-arrestin-2 in response to isoproterenol [37]. Therefore, it would appear that modulation of the activation mechanism (i.e. rotation of the C- and N domains of arrestin) was related to differences in the selectivity of the arrestin to the phospho-barcode of the GPCR.
FRET and BRET biosensors that detected the activation of ß-arrestin by measuring intramolecular arrangement in (Fluorescin arsenical hairpin “FIAsH”) dual-fluorescently-labeled arrestin were developed [148, 149]. Activation of ß-arrestin by different GRK-phosphorylated GPCR indicate that distinct intramolecular signatures in ß-arrestin were generated by GPCR that form stable complexes with arrestin (such as the angiotensin1A-receptor and PTH1R) and that these signatures were different from those generated by GPCR that formed transient complexes, such as the ß2-AR and others [149]. Therefore, distinct GPCR phosphorylation barcodes may generate specific conformational signatures in arrestin that produce selective ß-arrestin-dependent signaling and trafficking outcomes [150].
iii. Role of ß-arrestin in Gs-mediated signaling of endosomal GPCR
Sustained endosomal signaling by the GPCR is emerging as a major signaling paradigm for those GPCR that bind tightly to ß-arrestin [144]. These activated GPCR internalize as a megacomplex bound to ß-arrestin and the G protein [151]. These megaplexes would more readily form at GPCR that interacted strongly with ß-arrestin bound to GRK-phosphorylated Ser/Thr clusters in their C-tail. While not all the serines (S) and threonines (T) in the C-tail of a given GPCR are phosphorylated by GRK [37], an accounting of the number of S/T residues in the C-tail of GPCR that generated robust second waves of signaling indicate that there was 4T and 13S in the C-tail of V2R, and 8T and 9S in the C-tail of PTHR. The C-tail of the V2R had a much greater propensity than the C-tail of the ß2-AR with 4T and 7S in eliciting prolonged Gs-mediated cyclic AMP signaling because exchanging the C-tail of the ß2-AR for that of the V2R, resulted in a ß2/V2R chimera that stably megaplexed ß-arrestin and Gs and generated far longer and more robust waves of endosomal cyclic AMP [151]. Species differences among a given GPCR should also be taken into account when determining endosomal signaling paradigms. For example, cells bearing mouse ß1-AR with 6T and 7S in its C-tail are expected to bind ß-arrestin, internalize and signal from endosomes to cyclic AMP at significantly higher levels than cells bearing the human ß1-AR with 1T and 6S in its C-tail.
14. Physiological significance of endosomal GPCR signaling and trafficking
Endosomal signaling by GPCR involves G-protein-dependent i.e. cyclic AMP-mediated signaling and G-protein-independent, such as ß-arrestin-mediated activation of MAPK kinase, which was observed in Gs-null cells [152]. Sustained endosomal signaling to cyclic AMP is involved in transcriptional activation of PTH and ß2-AR target genes, among others [153, 154]. Endosomal signaling of the ß2-AR to cyclic AMP markedly increased the transcription of cAMP response element binding protein (CREB) target genes, such as phosphoenol pyruvate carboxykinase, encoded by the PCK1 gene, while plasma-membrane localized cyclic AMP had no effect on this parameter [153]. Inhibition of PDE4, rendered plasma membrane localized cyclic AMP capable of triggering the activation of PCK1 gene transcription. Thus it appears that megaplexing the GPCR/Gs/ß-arrestin in endosomes away from the compartmentalized regulatory networks of the membrane might account for many of the physiological and pathological actions attributed to post-endocytic signaling by GPCR. These results might explain how the V2R would maintain the insertion of AQP2 water channels within the apical membrane of renal collecting ducts during dehydration [141, 151]. Activation of V2R, would result in rapid desensitization. But, their internalization as a stable V2R/Gs/ß-arrestin megaplex that signals persistently in endosomes, promoted sustained cyclic AMP generation [151]. Persistent cyclic AMP levels in the cytoplasm, activated pools of AKAP-anchored PKA that sustained the phosphorylation state of Ser256 in AQP-2 and retention of these water channels in the apical membrane of renal collecting ducts. In this system, ß-arrestin did not desensitize V2R-mediated activation of Gs, rather it increased the activation of Gs in plasma and in endosomal membranes [141, 155]. Termination of endosomal signaling by V2R and PTHR involved the acidification of EE vesicles to dissociate the ligand from the GPCR and to displace ß-arrestin from the megaplex [156]. These cargoes, then recruited the Vps subunits of the retromer to inhibit cyclic AMP signaling from V2R-bearing intracellular membranes and accelerate their degradation [156].
Another major role of endosomal trafficking involves GPCR resensitization, which is promoted through the re-insertion of the agonist-internalized and resensitized GPCR back into the plasma membrane [27].
15. A framework depicting the various GPCR trafficking roadmaps
The diverse trafficking paradigms related to the one- or two-barcode hypotheses were incorporated into a framework that illustrates their intracellular trafficking itineraries in EE and beyond. Firstly, both hypothesis stipulate that the PBM domain is the “antecedent“ barcode, which directs a given GPCR toward recycling via a “sequence-dependent” roadmap that utilizes the ASRT (actin/sorting nexin/retromer tubule) complex. Therefore, inactivation of these PBMs inhibits the trafficking of all GPCR that traffic via the ASRT complex. A major difference between these hypotheses concerns the function of their respective PBMs. The PBM of one-barcode trafficking GPCR is primarily a trafficking barcode that binds to PDZ-containing proteins, such as NHERF or SNX27, which link the GPCR to the actin cytoskeleton or to early endosomes, respectively. The PBM of two-barcode trafficking GPCR is involved in compartmentalized signaling as well as trafficking. For example, the PBM of the ß1-AR assembles a signaling complex composed of SAP97 and its binding partners AKAP5/PKA complexes, as well as PDE4D8 [72, 87, 93, 102 and Fig. 2]. These are represented within the gray rectangle in Fig. 3E. The function of this complex (receptosome) is to insure fidelity of signaling by PKA and to degrade cyclic AMP within the inner leaflet of the membrane [71, 87]. The receptosome imprints a recycling signal onto the ß1-AR by phosphorylating it on Ser312 in the 3rd intracellular loop by the PBM-anchored pool of PKA [71]. Therefore, type-1 PBMs that bind to SAP97 were effective in assembling this complex and in imparting a recycling phenotype onto the ß1-AR [68, 96].
As outlined in figure 3E, GPCR activation by their cognate agonists promotes their endocytosis (red arrows) into EE. EE are a diverse group of intracellular vesicles involved in sorting of diverse sets of soluble or membrane cargoes. Cargoes are sorted in EE by “ASRT” or by “bulk” sorting into tubules that bud to the outside of EE. Budding tubules form vesicles that proceed along a recycling roadmap to the trans-Golgi network, or to the plasma membrane. However, tubules in EE may bud into the lumen of EE (orange rectangle), in which they are retained and subsequently degraded as these endosomes mature into late endosomes/lysosomes [157]. Figure 3E indicates that the degradative pathway was trafficked by ß1-AR with an inactivated barcode (ß1-ARΔPDZ) or Ser312 ((S312A) ß1-AR), or when PKA was inhibited or SAP97 or AKAP5 were depleted [68, 71, 96].
Sorting of PBM containing cargo by the ASRT pathway was dependent on several factors. First, it was dependent on targeting the retromer and WASH complexes to the EE membrane [123, 158]. Targeting the retromer to EE membrane was essential for ASRT-guided recycling and was mediated by: (i) binding of rertomer components to phosphatidylinositol 3-phosphate (PtdIns(3)P)-binding proteins, such as Rab7a and SNX3, (ii) intermolecular and intramolecular associations between the retromer and WASH complexes [159]. Second, the primary node for these associations was provided by the C-tail of FAM21, which binds to Vps35 [130]. Third, SNX27 was intimately involved in associating the retromer and the WASH complex to EE and in sorting PBM-containing GPCR towards ASRT budding tubules [80, 113]. These associations were mediated by the region between 66-76, PX and FERM domains of SNX27 which bind to Vps26, PtdIns and to the WASH complex, respectively [129 and Fig. 3B]. The PDZ domain of SNX27 was essential for sorting GPCR with an active PBM toward ASRT budding tubules that promoted “sequence-dependent” recycling. Fourth, FKBP15, an EE associated membrane protein, was involved in sorting/budding of GPCR that depend on a “second barcode” into ASRT tubules [96]. Trafficking of vesicles down the ASRT pathway (blue arrows) required additional proteins such as Rab4 (sorting) and Rab11 with its associated MYO5 motors that travel along the actin cytoskeleton towards to plasma membrane [96, 108].
Within the light yellow rectangle in the right-hand side of Fig. 3E, we show the roadmap of GPCR that traffic in a “sequence-independent manner” (i.e. PBM- and ASRT-independent manner). These GPCR correspond to non-PDZ recycling sequences derived from the μ-opioid or dopamine1-R that are fused to the C-tail of the ß1-AR and include the (S312D) ß1-AR point mutant and the (Ser345/346 to Asp) ß2-AR double mutant [69, 160]. These GPCR or their modified point mutants or chimeras might traffic by geometric sorting via the “bulk” pathway involved in trafficking of Tfr and LDL receptors and others [69, 110], but that assumption still lacks experimental proof.
16. Physiological significance of the two-barcode hypothesis for GPCR trafficking
The two-barcode sorting hypothesis was forwarded to address peculiar trafficking paradigms of the ß1-AR and others such as AQP2. In both cases these GPCR contained an active PBM, but their trafficking also required concomitant phosphorylation of a critical serine residue by a pool of AKAP-anchored PKA. Confocal studies determined that intracellular endosomes bearing ß2-AR (which recycles solely by its PBM) were larger and did not significantly overlap with ß1-AR-bearing endosomes [36]. Moreover, these receptors trafficked to distinct populations of endosomes in cells depleted of the (PtdIns(3)P)-binding proteins Rab7a and SNX3 [96]. Where by, ß2-AR were retained in the perinuclear area, while ß1-AR were diffusely retained in the cytoplasm.
Another distinction between the two-barcode versus the single-barcode hypothesis is that the dual-barcode hypothesis incorporates alternative mechanism(s) to regulate the fate of the hyperstimulated GPCR. For example, the major regulatory mechanism in the single barcode hypothesis towards excessive activation of a given GPCR involves GRK-mediated phosphorylation of specific Ser/Thr residues in the C-tail of the GPCR. In the case of the single barcode trafficking ß2-AR, this would result in the phosphorylation of the ß2-AR on multiple Ser/Thr residues in its C-tail [37]. Phosphorylation of the ß2-AR on Ser/Thr at position -2 in the PBM, inactivates the PBM and directs the agonist-internalized ß2-AR towards the degradative pathway [122]. This is an important feed-back regulatory mechanism that was incorporated into the two-barcode GPCR trafficking hypothesis because it stipulated that the PBM provides the “antecedent” trafficking barcode. Phosphorylation of the ß2-AR on other Ser/Thr in the C-tail, enhances the binding of ß-arrestin, which promotes several functions, including: uncoupling and subsequent internalization of the ß2-AR, ubiquitination of the ß2-AR, and endosomal signaling, among other functions [37, 55-62, 141-145]. In addition, PKA-mediated phosphorylation of the ß2-AR switches its coupling from Gs to Gi [161]. These mechanisms provide the bulk of the response of the ß2-AR to high levels of catecholamines or adrenergic stress. Unlike, the ß2-AR, the ß1-AR does not switch appreciably from Gs to Gi signaling and is not ubiquitinated [162, 163], so it would require an alternative step(s) to regulate its expression and function in conditions associated with hyperadrenergic stress. The two-barcode hypothesis provides an alternative physiologically-relevant scenario to regulate the expression of this GPCR under these conditions. Specifically, increased hyperadrenergic stress increases cyclic AMP, which enhances the activity of “receptosomal” PKA that activates several feedback regulatory loops. On the one hand, phosphorylation of the SAP97-anchored pool of PDE4D8 by the AKAP-anchored PKA, increases its PDE activity in degrading cyclic AMP [100, 101]. On the other hand, phosphorylation of the ß1-AR on Ser312 by the AKAP5-anchored pool of PKA enhances its recycling and promotes the insertion of the “resensitized” ß1-AR into the cell membrane [71]. Under these conditions, p-Ser312 might serve as a signaling intensity sensor that would adjust the density of cellular levels of ß1-AR. The effect of this hypothesis is manifested in the observed effects of cardiotoxic insults on the densities of cardiac ß1-AR vs. their effects on cardiac ß2-AR [164]. In the unstimulated rodent heart, the density of the ß1-AR constituted ∼80% of the total ß-adrenergic receptor density [165, 166]. In chronic heart failure, ß-adrenoceptor number was reduced, presumably by selective down-regulation of cardiac ß1-AR by endogenous nor epinephrine that is elevated due to increased sympathetic activity [167-169]. Chronic infusions of ß-agonist, reduced the total density of cardiac ß-AR, but down-regulation of these receptors was confined to the ß1-AR subtype only [170]. Cardiotoxic and cardioprotective responses were mediated by ß1-AR and ß2-AR, respectively [171]. Therefore, selective up-regulation of signaling from cardiac ß2-AR might play a cardioprotective role in the pathogenesis of cardiomyopathy [172]; while selective down-regulation of ß1-AR might serve as an additional adaptation to protect the heart against cardiotoxicity from catecholamine overstimulation [169].
17. Concluding remarks
Post-translational modifications by kinases, such as GRK and PKA and by ubiquitination have emerged as important modifications in the regulation of GPCR trafficking and signaling in endosomes. These compartmentalized kinases are involved in promoting internalization, sustained endosomal signaling and subsequent recycling of endosomal GPCR. This review focused on the intricacies involved in insuring the fidelity of PKA-mediated GPCR phosphorylation by compartmentalizing PKA within the membrane and how this reversible modification might be involved in regulating the trafficking itinerary of the agonist-internalized GPCR. Future studies aimed at identifying the specific endosomal machinery involved in recognizing the PKA-imprinted signal and how single point mutations in these critical residues would alter the trafficking roadmap of the involved GPCR are key to our understanding of the mechanism of GPCR trafficking. Since GPCR trafficking and endosomal signaling are closely interlinked, it is likely that elucidating these mechanisms will lead to a greater understanding of the role that GPCR trafficking contributes to human health and disease.
Acknowledgments
National Institutes of Health grant HL-085848 (SWB) supported this work.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Katritch V, Cherezov V, Stevens RC. Structure-function of the G protein-coupled receptor superfamily. Annu Rev Pharmacol Toxicol. 2013;53:531–556. doi: 10.1146/annurev-pharmtox-032112-135923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ellisdon AM, Halls ML. Compartmentalization of GPCR signalling controls unique cellular responses. Biochem Soc Trans. 2016;44:562–567. doi: 10.1042/BST20150236. [DOI] [PubMed] [Google Scholar]
- 3.Langeberg LK, Scott JD. Signalling scaffolds and local organization of cellular behaviour. Nat Rev Mol Cell Biol. 2015;16:232–244. doi: 10.1038/nrm3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hanyaloglu AC, von Zastrow M. Regulation of GPCRs by endocytic membrane trafficking and its potential implications. Annu Rev Pharmacol Toxicol. 2008;48:537–568. doi: 10.1146/annurev.pharmtox.48.113006.094830. [DOI] [PubMed] [Google Scholar]
- 5.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev. 2001;53:1–24. [PubMed] [Google Scholar]
- 6.Mohan ML, Vasudevan NT, Gupta MK, Martelli EE, Naga Prasad SV. G-protein coupled receptor resensitization-appreciating the balancing act of receptor function. Curr Mol Pharmacol. 2012;5:350–361. [PMC free article] [PubMed] [Google Scholar]
- 7.Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol. 2014;27:109–116. doi: 10.1016/j.ceb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefkowitz RJ, Pitcher J, Krueger K, Daaka Y. Mechanisms of β-adrenergic receptor desensitization and resensitization. Adv Pharmacol. 1998;42:416–420. doi: 10.1016/s1054-3589(08)60777-2. 1998. [DOI] [PubMed] [Google Scholar]
- 9.Claing A, Laporte SA, Caron MG, Lefkowitz RJ. Endocytosis of G protein-coupled receptors: roles of G protein-coupled receptor kinases and β-arrestin proteins. Prog Neurobiol. 2002;66:61–79. doi: 10.1016/s0301-0082(01)00023-5. [DOI] [PubMed] [Google Scholar]
- 10.Shenoy SK, Lefkowitz RJ. ß-arrestin-mediated receptor trafficking and signal transduction. Trends Pharmacol Sci. 2011;32:521–533. doi: 10.1016/j.tips.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kendall RT, Strungs EG, Rachidi SM, Lee MH, El-Shewy HM, Luttrell DK, Janech MG, Luttrell The ß-arrestin pathway-selective type 1A angiotensin receptor (AT1A) agonist [Sar1, Ile4, Ile8] angiotensin II regulates a robust G protein-independent signaling network. J Biol Chem. 2011;286:19880–19891. doi: 10.1074/jbc.M111.233080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferguson SSG, Downey WE, III, Calapierto AM, Barak LS, Menard L, Caron MG. Role of ß-arrestin in mediating agonist-promoted G-protein-coupled receptor internalization. Science. 1996;271:363–366. doi: 10.1126/science.271.5247.363. [DOI] [PubMed] [Google Scholar]
- 13.Maxfield FR, McGraw TE. Endocytic recycling. Nat Rev Mol Cell Biol. 2004;5:121–132. doi: 10.1038/nrm1315. [DOI] [PubMed] [Google Scholar]
- 14.Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol. 2005;6:112–126. doi: 10.1038/nrm1571. [DOI] [PubMed] [Google Scholar]
- 15.Gruenberg J. The endocytic pathway: a mosaic of domains. Nature Rev Mol Cell Biol. 2001;2:721–730. doi: 10.1038/35096054. [DOI] [PubMed] [Google Scholar]
- 16.Conner SD, Schmid SL. Regulated portals of entry into the cell. Nature. 2003;422:37–44. doi: 10.1038/nature01451. [DOI] [PubMed] [Google Scholar]
- 17.Miller WE, Lefkowitz RJ. Expanding roles for ß-arrestins as scaffolds and adapters in GPCR signaling and trafficking. Curr Opin Cell Biol. 2001;13:139–145. doi: 10.1016/s0955-0674(00)00190-3. [DOI] [PubMed] [Google Scholar]
- 18.Cheng ZJ, Singh RD, Marks DL, Pagano RE. Membrane microdomains, caveolae, and caveolar endocytosis of sphingolipids. Mol Membr Biol. 2006;23:101–110. doi: 10.1080/09687860500460041. [DOI] [PubMed] [Google Scholar]
- 19.Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. 2007;8:185–194. doi: 10.1038/nrm2122. [DOI] [PubMed] [Google Scholar]
- 20.Henley JR, Krueger EW, Oswald J, McNiven MA. Dynamin-mediated internalization of caveolae. J Cell Biol. 1998;141:85–99. doi: 10.1083/jcb.141.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oh P, McIntosh DP, Schnitzer JE. Dynamin at the neck of caveolae mediates their budding to form transport vesicles by GTP-driven fission from the plasma membrane of endothelium. J Cell Biol. 1998;141:101–114. doi: 10.1083/jcb.141.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pelkmans L, Burli T, Zerial M, Helenius A. Caveolin-stabilized membrane domains as multifunctional transport and sorting devices in endocytic membrane traffic. Cell. 2004;118:767–780. doi: 10.1016/j.cell.2004.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Pelkmans L, Zerial M. Kinase-regulated quantal assemblies and kiss-and-run recycling of caveolae. Nature. 2005;436:128–133. doi: 10.1038/nature03866. [DOI] [PubMed] [Google Scholar]
- 24.Pavlos NJ, Friedman PA. GPCR Signaling and Trafficking: The Long and Short of It. Trends Endocrinol Metab. 2016;S1043-2760(16):30146–1. doi: 10.1016/j.tem.2016.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hinkle PM, Puskas JA. Detection of G protein-coupled receptors by immunofluorescence microscopy. Methods Mol Biol. 2004;237:127–134. doi: 10.1385/1-59259-430-1:127. [DOI] [PubMed] [Google Scholar]
- 26.Marchese A. Monitoring chemokine receptor trafficking by confocal immunofluorescence microscopy. Methods Enzymol. 2016;570:281–292. doi: 10.1016/bs.mie.2015.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardner LA, Delos Santos NM, Matta SG, Whitt MA, Bahouth SW. Role of the cyclic AMP-dependent protein kinase in homologous resensitization of the ß1-adrenergic receptor. J Biol Chem. 2004;279:21135–21143. doi: 10.1074/jbc.M313652200. [DOI] [PubMed] [Google Scholar]
- 28.January B, Seibold A, Whaley B, Hipkin RW, Lin D, Schonbrunn A, Barber R, Clark RB. ß2-adrenergic receptor desensitization, internalization, and phosphorylation in response to full and partial agonists. J Biol Chem. 1997;272:23871–23879. doi: 10.1074/jbc.272.38.23871. [DOI] [PubMed] [Google Scholar]
- 29.Moore RH, Millman EE, Godines V, Hanania NA, Tran TM, Peng H, Dickey BF, Knoll BJ, Clark RB. Salmeterol stimulation dissociates ß2-adrenergic receptor phosphorylation and internalization. Am J Respir Cell Mol Biol. 2007;36:254–261. doi: 10.1165/rcmb.2006-0158OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodman OB, Jr, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. Beta-arrestin acts as a clathrin adaptor in endocytosis of the ß2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 31.Nakagawa T, Asahi M. β1-adrenergic receptor recycles via a membranous organelle, recycling endosome, by binding with sorting nexin27. J Memb Biol. 2013;246:571–579. doi: 10.1007/s00232-013-9571-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delos Santos NM, Gardner LA, White SW, Bahouth SW. Characterization of the residues in helix 8 of the human ß1-adrenergic receptor that are involved in coupling the receptor to G proteins. J Biol Chem. 2006;281:12896–12907. doi: 10.1074/jbc.M508500200. [DOI] [PubMed] [Google Scholar]
- 33.Rapacciuolo A, Suvarna S, Barki-Harrington L, Luttrell LM, Cong M, Lefkowitz RJ, Rockman HA. Protein kinase A and G protein-coupled receptor kinase phosphorylation mediates ß-1 adrenergic receptor endocytosis through different pathways. J Biol Chem. 2003;278:35403–35411. doi: 10.1074/jbc.M305675200. [DOI] [PubMed] [Google Scholar]
- 34.Hajjhussein H, Gardner LA, Fujii N, Anderson NM, Bahouth SW. The hydrophobic amino acid cluster at the cytoplasmic end of transmembrane helix III modulates the coupling of the ß1-adrenergic receptor to Gs. J Recept Signal Transduct Res. 2013;33:79–88. doi: 10.3109/10799893.2012.759590. [DOI] [PubMed] [Google Scholar]
- 35.Shiina T, Nagao T, Kurose H. Low affinity of ß1-adrenergic receptor for ß-arrestins explains the resistance to agonist-induced internalization. Life Sci. 2001;68:2251–2257. doi: 10.1016/s0024-3205(01)01013-x. 2001. [DOI] [PubMed] [Google Scholar]
- 36.Koliwer J, Park M, Bauch C, von Zastrow M, Kreienkamp HJ. The golgi-associated PDZ domain protein PIST/GOPC stabilizes the β1-adrenergic receptor in intracellular compartments after internalization. J Biol Chem. 2015;290:6120–6129. doi: 10.1074/jbc.M114.605725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nobles KN, Xiao K, Ahn S, Shukla AK, Lam CM, Rajagopal S, Strachan RT, Huang TY, Bressler EA, Hara MR, Shenoy SK, Gygi SP, Lefkowitz RJ. Distinct phosphorylation sites on the ß2-adrenergic receptor establish a barcode that encodes differential functions of β-arrestin. Sci Signal. 2011;4:ra51. doi: 10.1126/scisignal.2001707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Butcher AJ, Prihandoko R, Kong KC, McWilliams P, Edwards JM, Bottrill A, Mistry S, Tobin AB. Differential G-protein-coupled receptor phosphorylation provides evidence for a signaling bar code. J Biol Chem. 2011;286:11506–11518. doi: 10.1074/jbc.M110.154526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zindel D, Engel S, Bottrill AR, Pin JP, Prézeau L, Tobin AB, Bünemann M, Krasel C, Butcher AJ. Identification of key phosphorylation sites in PTH1R that determine arrestin3 binding and fine-tune receptor signaling. Biochem J. 2016;473:4173–4192. doi: 10.1042/BCJ20160740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butcher AJ, Hudson BD, Shimpukade B, Alvarez-Curto E, Prihandoko R, Ulven T, et al. Concomitant action of structural elements and receptor phosphorylation determines arrestin-3 interaction with the free fatty acid receptor FFA4. J Biol Chem. 2014;289:18451–18465. doi: 10.1074/jbc.M114.568816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem. 2010;285:7805–7817. doi: 10.1074/jbc.M109.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bouzo-Lorenzo M, Santo-Zas I, Lodeiro M, Nogueiras R, Casanueva FF, Castro M, et al. Distinct phosphorylation sites on the ghrelin receptor, GHSR1a, establish a code that determines the functions of β-arrestins. Sci Rep. 2016;6:22495. doi: 10.1038/srep22495. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kong KC, Butcher AJ, McWilliams P, Jones D, Wess J, Hamdan FF, Werry T, Rosethorne EM, Charlton SJ, Munson SE, Cragg AA, Smart AD, Tobin AB. M3-muscarinic receptor promotes insulin release via receptor phosphorylation/arrestin-dependent activation of protein kinase D1. Proc Natl Acad Sci U S A. 2010;107:21181–21186. doi: 10.1073/pnas.1011651107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Poulin B, Butcher A, Mc Williams P, Bourgognon JM, Pawlak R, Kong KC, Bottrill A, Mistry S, Wess J, Rosethorne EM, Charltonand SJ, Tobin AB. The M3-muscarinic receptor regulates learning and memory in a receptor phosphorylation/ arrestin-dependent manner. Proc Natl Acad Sci U S A. 2010;107:9440–9445. doi: 10.1073/pnas.0914801107. 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bradley SJ, Wiegman CH, Iglesias MM, Kong KC, Butcher AJ, Plouffe B, Goupil E, Bourgognon JM, Macedo-Hatch T, LeGouill C, Russell K, Laporte SA, Konig GM, Kostenis E, Bouvier M, Chung KF, Amrani Y, Tobin AB. Mapping physiological G protein-coupled receptor signaling pathways reveals a role for receptor phosphorylation in airway contraction. Proc Natl Acad Sci U S A. 2016;113:4524–4529. doi: 10.1073/pnas.1521706113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao TT, Deacon HW, Reczek D, Bretscher A, von Zastrow M. A kinase-regulated PDZ domain interaction controls endocytic sorting of the ß2-adrenergic receptor. Nature. 1999;401:286–290. doi: 10.1038/45816. 1999. [DOI] [PubMed] [Google Scholar]
- 47.Whistler JL, Enquist J, Marley A, Fong J, Gladher F, Tsuruda J, Murray SR, von Zastrow M. Modulation of postendocytic sorting of G protein-coupled receptors. Science. 2002;297:615–620. doi: 10.1126/science.1073308. [DOI] [PubMed] [Google Scholar]
- 48.Nooh MM, Chumpia MM, Hamilton TB, Bahouth SW. Sorting of ß1-adrenergic receptors is mediated by pathways that are either dependent or independent of Type-I PDZ, protein kinase A (PKA) and SAP97. J Biol Chem. 2014;289:2277–2294. doi: 10.1074/jbc.M113.513481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Romero G, von Zastrow M, Friedman PA. Role of PDZ proteins in regulating trafficking, signaling, and function of GPCRs: means, motif, and opportunity. Adv Pharmacol. 2011;62:279–314. doi: 10.1016/B978-0-12-385952-5.00003-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gage RM, Kim KA, Cao TT, von Zastrow M. A transplantable sorting signal that is sufficient to mediate rapid recycling of G protein-coupled receptors. J Biol Chem. 2001;276:44712–44720. doi: 10.1074/jbc.M107417200. [DOI] [PubMed] [Google Scholar]
- 51.Gage RM, Matveeva EA, Whiteheart SW, von Zastrow M. Type I PDZ ligands are sufficient to promote rapid recycling of G protein-coupled receptors independent of binding to NSF. J Biol Chem. 2005;280:2205–3313. doi: 10.1074/jbc.M406934200. [DOI] [PubMed] [Google Scholar]
- 52.Alpern RJ, Moe OW, Caplan M. Seldin and Giebisch's The Kidney. 5th. Elsevier Inc; NY, NY: 2013. Chapter 46: Polyuria and Diabetes Insipidus; pp. 1571–1600. [Google Scholar]
- 53.Fushimi K, Sasaki S, Marumo F. Phosphorylation of serine 256 is required for cAMP-dependent regulatory exocytosis of the aquaporin-2 water channel. J Biol Chem. 1997;272:14800–14804. doi: 10.1074/jbc.272.23.14800. 1997. [DOI] [PubMed] [Google Scholar]
- 54.Noda Y, Horikawa S, Furukawa T, Hirai K, Katayama Y, Asai T, Kuwahara M, Katagiri K, Kinashi T, Hattori M, Nimato N, Sasaki S. Aquaporin-2 trafficking is regulated by PDZ domain containing protein SPA-1. FEBS Lett. 2004;568:139–145. doi: 10.1016/j.febslet.2004.05.021. [DOI] [PubMed] [Google Scholar]
- 55.Orsini MJ, Parent JL, Mundell SJ, Marchese A, Benovic JL. Trafficking of the HIV coreceptor CXCR4. Role of arrestins and identification of residues in the c-terminal tail that mediate receptor internalization. J Biol Chem. 1999;274:31076–31086. doi: 10.1074/jbc.274.43.31076. [DOI] [PubMed] [Google Scholar]
- 56.Marchese A, Benovic JL. Agonist-promoted ubiquitination of the G protein-coupled receptor CXCR4 mediates lysosomal sorting. J Biol Chem. 2001;276:45509–45512. doi: 10.1074/jbc.C100527200. [DOI] [PubMed] [Google Scholar]
- 57.Busillo JM, Armando S, Sengupta R, Meucci O, Bouvier M, Benovic JL. Site-specific phosphorylation of CXCR4 is dynamically regulated by multiple kinases and results in differential modulation of CXCR4 signaling. J Biol Chem. 2010;285:7805–7817. doi: 10.1074/jbc.M109.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marchese A, Trejo JA. Ubiquitin-dependent regulation of G protein-coupled receptor trafficking and signaling. Cell Signal. 2013;25:707–716. doi: 10.1016/j.cellsig.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shenoy SK, McDonald PH, Kohout TA, Lefkowitz RJ. Regulation of receptor fate by ubiquitination of activated beta 2-adrenergic receptor and beta-arrestin. Science. 2001;294:1307–1313. doi: 10.1126/science.1063866. [DOI] [PubMed] [Google Scholar]
- 60.Han SO, Kommaddi RP, Shenoy SK. Distinct roles for ß-arrestin-2 and arrestin-domain-containing proteins in ß2-adrenergic receptor trafficking. EMBO Rep. 2013;14:164–171. doi: 10.1038/embor.2012.187. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shenoy SK, Xiao K, Venkataramanan V, Snyder PM, Freedman NJ, Weissman AM. Nedd4 mediates agonist-dependent ubiquitination, lysosomal targeting, and degradation of the beta2-adrenergic receptor. J Biol Chem. 2008;283:22166–22176. doi: 10.1074/jbc.M709668200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tian X, Irannejad R, Bowman SL, Du Y, Puthenveedu MA, von Zastrow M, Benovic JL. The α-Arrestin ARRDC3 Regulates the Endosomal Residence Time and Intracellular Signaling of the β2-Adrenergic Receptor. J Biol Chem. 2016;291:14510–14525. doi: 10.1074/jbc.M116.716589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanowitz M, von Zastrow M. A novel endocytic recycling signal that distinguishes the membrane trafficking of naturally occurring opioid receptors. J Biol Chem. 2003;278:45978–45986. doi: 10.1074/jbc.M304504200. [DOI] [PubMed] [Google Scholar]
- 64.Vargas GA, Von Zastrow M. Identification of a novel endocytic recycling signal in the D1 dopamine receptor. J Biol Chem. 2004;279:37461–37469. doi: 10.1074/jbc.M401034200. [DOI] [PubMed] [Google Scholar]
- 65.Nooh MM, Mancarella S, Bahouth SW. Identification of novel transplantable GPCR recycling motif for drug discovery. Biochem Pharmacol. 2016;120:22–32. doi: 10.1016/j.bcp.2016.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kallal L, Gagnon AW, Penn RB, Benovic JL. Visualization of agonist-inducedsequestration and down-regulation of a green fluorescent protein-tagged beta2-adrenergic receptor. J Biol Chem. 1998;273:322–328. doi: 10.1074/jbc.273.1.322. [DOI] [PubMed] [Google Scholar]
- 67.Kallal L, Benovic JL. Using green fluorescent proteins to study G-protein coupled receptor localization and trafficking. Trends Pharmacol Sci. 2000;21:175–180. doi: 10.1016/s0165-6147(00)01477-2. [DOI] [PubMed] [Google Scholar]
- 68.Gardner LA, Naren AP, Bahouth SW. Assembly of a SAP97-AKAP79-PKA scaffold at the type-1 PDZ motif of the human ß1-adrenergic receptor generates a receptosome involved in receptor recycling and networking. J Biol Chem. 2007;282:5085–5099. doi: 10.1074/jbc.M608871200. [DOI] [PubMed] [Google Scholar]
- 69.Bowman SL, Shiwarski DJ, Puthenveedu MA. Distinct G -protein coupled receptor recycling pathways allow spatial control of downstream G protein signaling. J Cell Biol. 2016;214:797–806. doi: 10.1083/jcb.201512068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD. Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science. 1996;271:1589–1592. doi: 10.1126/science.271.5255.1589. [DOI] [PubMed] [Google Scholar]
- 71.Gardner LA, Tavalin SA, Goehring A, Scott JD, Bahouth SW. AKAP79-mediated targeting of the cyclic AMP-dependent protein kinase to the ß1-adrenergic receptor promotes recycling and functional resensitization of the receptor. J Biol Chem. 2006;281:33537–33553. doi: 10.1074/jbc.M601809200. [DOI] [PubMed] [Google Scholar]
- 72.Li X, Nooh MM, Bahouth SW. Role of AKAP79/150 in ß1-adrenergic receptor trafficking and signaling in mammalian Cells. J Biol Chem. 2013;288:33797–33812. doi: 10.1074/jbc.M113.470559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jo I, Ward DT, Baum MA, Scott JD, Coghlan VM, Hammond TG, Harris HW. AQP2 is a substrate for endogenous PP2B activity within an inner medullary AKAP-signaling complex. Am J Physiol Renal Physiol. 2001;281:F958–965. doi: 10.1152/ajprenal.2001.281.5.F958. [DOI] [PubMed] [Google Scholar]
- 74.Li X, Matta SM, Sullivan RD, Bahouth SW. Carvedilol Reverses Cardiac Hypertrophy and Insufficiency in AKAP5 Knockout Mice by Normalizing the Activities of Calcineurin and Calmodulin Kinase-II. Cardiovascular Res. 2014;104:270–279. doi: 10.1093/cvr/cvu209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim E, Sheng M. PDZ domain proteins of synapses. Nat Rev Neurosci. 2004;5:771–781. doi: 10.1038/nrn1517. [DOI] [PubMed] [Google Scholar]
- 76.Zhu J, Shang Y, Zhang M. Mechanistic basis of MAGUK-organized complexes in synaptic development and signalling. Nat Rev Neurosci. 2016;17:209–223. doi: 10.1038/nrn.2016.18. [DOI] [PubMed] [Google Scholar]
- 77.Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- 78.Joubert L, Hanson B, Barthet G, Sebben M, Claeysen S, Hong W, Marin P, Dumuis A, Bockaert J. New sorting nexin (SNX27) and NHERF specifically interact with the 5-HT4a receptor splice variant: roles in receptor targeting. J Cell Sci. 2004;117:5367–5379. doi: 10.1242/jcs.01379. [DOI] [PubMed] [Google Scholar]
- 79.Hall RA, Premont RT, Chow CW, Blitzer JT, Pitcher JA, Claing A, Stoffel RH, Barak LS, Shenolikar S, Weinman EJ, Grinstein S, Lefkowitz RJ. The beta2-adrenergic receptor interacts with the Na+/H+-exchanger regulatory factor to control Na+/H+ exchange. Nature. 1998;392:626–630. doi: 10.1038/33458. [DOI] [PubMed] [Google Scholar]
- 80.Lauffer BE, Chen S, Melero C, Kortemme T, von Zastrow M, Vargas GA. Engineered protein connectivity to actin mimics PDZ-dependent recycling of G protein-coupled receptors but not its regulation by Hrs. J Biol Chem. 2009;284:2448–2458. doi: 10.1074/jbc.M806370200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Lauffer BE, Melero C, Temkin P, Lei C, Hong W, Kortemme T, von Zastrow M. SNX27 mediates PDZ-directed sorting from endosomes to the plasma membrane. J Cell Biol. 2010;190:565–574. doi: 10.1083/jcb.201004060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hu LA, Tang Y, Miller WE, Cong M, Lau AG, Lefkowitz RJ, Hall RA. ß1-adrenergic receptor association with PSD-95. Inhibition of receptor internalization and facilitation of ß1-adrenergic receptor interaction with N-methyl-D-aspartate receptors. J Biol Chem. 2000;275:38659–38666. doi: 10.1074/jbc.M005938200. [DOI] [PubMed] [Google Scholar]
- 83.Xu J, Paquet M, Lau AG, Wood JD, Ross CA, Hall RA. ß1-adrenergic receptor association with the synaptic scaffolding protein membrane-associated guanylate kinase inverted-2 (MAGI-2). Differential regulation of receptor internalization by MAGI-2 and PSD-95. J Biol Chem. 2001;276:41310–41317. doi: 10.1074/jbc.M107480200. [DOI] [PubMed] [Google Scholar]
- 84.Hu LA, Chen W, Martin NP, Whalen EJ, Premont RT, Lefkowitz RJ. GIPC interacts with the beta1-adrenergic receptor and regulates beta1-adrenergic receptor-mediated ERK activation. J Biol Chem. 2003;278:26295–26301. doi: 10.1074/jbc.M212352200. [DOI] [PubMed] [Google Scholar]
- 85.He J, Bellini M, Inuzuka H, Xu J, Xiong Y, Yang X, Castleberry AM, Hall RA. Proteomic analysis of beta1-adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem. 2006;281:2820–2827. doi: 10.1074/jbc.M509503200. [DOI] [PubMed] [Google Scholar]
- 86.He J, Bellini M, Xu J, Castleberry AM, Hall RA. Interaction with cystic fibrosis transmembrane conductance regulator-associated ligand (CAL) inhibits ß1-adrenergic receptor surface expression. J Biol Chem. 2004;279:50190–50196. doi: 10.1074/jbc.M404876200. [DOI] [PubMed] [Google Scholar]
- 87.Xiang Y, Devic E, Kobilka B. The PDZ binding motif of the ß1-adrenergic receptor modulates receptor trafficking and signaling in cardiac myocytes. J Biol Chem. 2002;277:33783–33790. doi: 10.1074/jbc.M204136200. [DOI] [PubMed] [Google Scholar]
- 88.Koliwer J, Park M, Bauch C, von Zastrow M, Kreienkamp HJ. The golgi-associated PDZ domain protein PIST/GOPC stabilizes the β1-adrenergic receptor in intracellular compartments after internalization. J Biol Chem. 2015;290:6120–6129. doi: 10.1074/jbc.M114.605725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wu H, Reuver SM, Kuhlendahl S, Chung WJ, Garner CC. Subcellular targeting and cytoskeletal attachment of SAP97 to the epithelial lateral membrane. J Cell Sci. 1998;111:2365–2376. doi: 10.1242/jcs.111.16.2365. [DOI] [PubMed] [Google Scholar]
- 90.Nikandrova YA, Jiao Y, Baucum AJ, Tavalin SJ, Colbran RJ. Ca2+/calmodulin-dependent protein kinase II binds to and phosphorylates a specific SAP97 splice variant to disrupt association with AKAP79/150 and modulate alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid-type glutamate receptor (AMPAR) activity. J Biol Chem. 2010;285:923–934. doi: 10.1074/jbc.M109.033985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fujita A, Kurachi Y. SAP family proteins. Biochem Biophys Res Commun. 2000;269:1–6. doi: 10.1006/bbrc.1999.1893. [DOI] [PubMed] [Google Scholar]
- 92.Muller BM, Kistner U, Veh RW, Cases-Langhoff C, Becker B, Gundelfinger ED, Garner CC. Molecular characterization and spatial distribution of SAP97, a novel presynaptic protein homologous to SAP90 and the Drosophila discs-large tumor suppressor protein. J Neurosci. 1995;15:2354–2366. doi: 10.1523/JNEUROSCI.15-03-02354.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nooh NM, Anjaparavanda PN, Xiang YK, Bahouth SW. SAP97 controls the trafficking and resensitization of the ß1-adrenergic receptor through its PDZ2 and i3 domains. pLos One. 2013;8:e63379. doi: 10.1371/journal.pone.0063379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Heydorn A, Søndergaard BP, Ersbøll B, Holst B, Nielsen FC, Haft CR, Whistler J, Schwartz TW. A library of 7TM receptor C-terminal tails. Interactions with the proposed postendocytic sorting proteins ERM-binding phosphoprotein 50 (EBP50), N-ethylmaleimidesensitive factor (NSF), sorting nexin 1 (SNX1), and G protein-coupled receptor-associated sorting protein (GASP) J Biol Chem. 2004;279:54291–54303. doi: 10.1074/jbc.M406169200. [DOI] [PubMed] [Google Scholar]
- 95.Sorkin A, von Zastrow M. Endocytosis and signalling: intertwining molecular networks. Nat Rev Mol Cell Biol. 2009;10:609–622. doi: 10.1038/nrm2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nooh MM, Bahouth SW. Two barcodes encoded by the type-1 PDZ and by phospho-Ser312 regulate retromer/WASH-mediated sorting of the ß1-adrenergic receptor from endosomes to the plasma membrane. Cell Signal. 2017;29:192–208. doi: 10.1016/j.cellsig.2016.10.014. [DOI] [PubMed] [Google Scholar]
- 97.Houslay MD. Underpinning compartmentalised cAMP signalling through targeted cAMP breakdown. Trends Biochem Sci. 2010;35:91–100. doi: 10.1016/j.tibs.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 98.Mc Cahill AC, Huston E, Li X, Houslay MD. PDE4 associates with different scaffolding proteins: modulating interactions as treatment for certain diseases. Handb Exp Pharmacol. 2008;186:125–166. doi: 10.1007/978-3-540-72843-6_6. 2008. [DOI] [PubMed] [Google Scholar]
- 99.Stangherlin A, Zaccolo M. Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2012;302:H379–390. doi: 10.1152/ajpheart.00766.2011. [DOI] [PubMed] [Google Scholar]
- 100.Conti M, Mika D, Richter W. Cyclic AMP compartments and signaling specificity: role of cyclic nucleotide phospho- diesterases. J Gen Physiol. 2014;143:29–38. doi: 10.1085/jgp.201311083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Richter W, Day P, Agrawal R, Bruss MD, Granier S, Wang YL, Rasmussen SG, Horner K, Wang P, Lei T, Patterson AJ, Kobilka B, Conti M. Signaling from ß1- and ß2-adrenergic receptors is defined by differential interactions with PDE4. EMBO J. 2008;27:384–393. doi: 10.1038/sj.emboj.7601968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fu Q, Kim S, Soto D, De Arcangelis V, Dipilato L, Liu S, Xu B, Zhang J, Shi Q, Xiang YK. A long-lasting ß1 adrenergic receptor stimulation of cAMP/PKA signals in cardiac myocytes. J Biol Chem. 2014;289:14771–14781. doi: 10.1074/jbc.M113.542589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zerial M, McBride H. Rab proteins as membrane organizers. Nat Rev Mol Cell Biol. 2001;2:107–117. doi: 10.1038/35052055. [DOI] [PubMed] [Google Scholar]
- 104.Pereira-Leal JB, Seabra MC. The mammalian Rab family of small GTPases: definition of family and subfamily sequence motifs suggests a mechanism for functional specificity in the Rab superfamily. J Mol Biol. 2002;301:1077–1087. doi: 10.1006/jmbi.2000.4010. [DOI] [PubMed] [Google Scholar]
- 105.Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nature Rev Mol Cell Biol. 2009;10:513–525. doi: 10.1038/nrm2728. [DOI] [PubMed] [Google Scholar]
- 106.Wandinger-Ness A, Zerial M. Rab proteins and the compartmentalization of the endosomal system. Cold Spring Harb Perspect Biol. 2014;6:a022616. doi: 10.1101/cshperspect.a022616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ehlers MD. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron. 2000;28:511–525. doi: 10.1016/s0896-6273(00)00129-x. [DOI] [PubMed] [Google Scholar]
- 108.Gardner LA, Hajjhussein H, Frederick-Dyer KC, Bahouth SW. Rab11a and its binding partners regulate the recycling of the ß1-adrenergic receptor. Cell Signal. 2011;23:46–57. doi: 10.1016/j.cellsig.2010.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Puthenveedu MA, Laufer B, Temkin P, Vistein R, Carlton P, Thorn K, Taunton J, Weiner OD, Parton RG, Von Zastrow M. Sequence-dependent sorting of recycling proteins by actin-stabilized endosomal microdomains. Cell. 2010;143:761–767. doi: 10.1016/j.cell.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Frederic JA, Bowersox S, Chen S, Beard G, Puthenveedu MA, Hanyaloglu AC. Spacially restricted G protein-coupled receptor activity via divergent endocytic compartments. J Biol Chem. 2013;289:3936–3977. doi: 10.1074/jbc.M113.526350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lunn ML, Nassirpour R, Arrabit C, Tan J, Mc Leod I, Arias CM, Sawchenko PE, Yates JR, 3rd, Slesinger PA. A unique sorting nexin regulates trafficking of potassium channels via a PDZ domain interaction. Nat Neurosci. 2007;10:1249–1259. doi: 10.1038/nn1953. [DOI] [PubMed] [Google Scholar]
- 112.Rincón E, Santos T, Avila-Flores A, Albar JP, Lalioti V, Lei C, Hong W, Mérida I. Proteomics identification of sorting nexin 27 as a diacylglycerol kinase zeta-associated protein: new diacylglycerol kinase roles in endocytic recycling. Mol Cell Proteomics. 2007;6:1073–1087. doi: 10.1074/mcp.M700047-MCP200. [DOI] [PubMed] [Google Scholar]
- 113.Temkin P, Lauffer B, Jager S, Cimermancic P, Krogan NJ, von Zastrow M. SNX27 mediates retromer tubule entry and endosome-to-plasma membrane trafficking of signaling receptors. Nature Cell Biol. 2010;13:715–721. doi: 10.1038/ncb2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chan AS, Clairfeuille T, Landao-Bassonga E, Kinna G, Ng PY, Loo LS, Cheng TS, Zheng M, Hong W, Teasdale RD, Collins BM, Pavlos NJ. Sorting nexin 27 couples PTHR trafficking to retromer for signal regulation in osteoblasts during bone growth. Mol Biol Cell. 2016;27:1367–1382. doi: 10.1091/mbc.E15-12-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Mc Garvey JC, Xiao K, Bowman SL, Mamonova T, Zhang Q, Bisello A, Sneddon WB, Ardura JA, Jean-Alphonse F, Vilardaga JP, Puthenveedu MA, Friedman PA. Actin-Sorting Nexin 27 (SNX27)-Retromer Complex Mediates Rapid Parathyroid Hormone Receptor Recycling. J Biol Chem. 2016;291:10986–11002. doi: 10.1074/jbc.M115.697045. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Hussain NK, Diering GH, Sole J, Anggono V, Huganir RL. Sorting Nexin 27 regulates basal and activity-dependent trafficking of AMPARs. Proc Natl Acad Sci USA. 2014;111:11840–11845. doi: 10.1073/pnas.1412415111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Loo LS, Tang N, Al-Haddawi M, Dawe GS, Hong W. A role for sorting nexin 27 in AMPA receptor trafficking. Nat Commun. 2014;5:3176. doi: 10.1038/ncomms4176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, Loo LS, Cai L, Thompson RC, Yang B, Chen Y, Johnson PF, Wu C, Bu G, Mobley WC, Zhang D, Gage FH, Ranscht B, Zhang YW, Lipton SA, Hong W, Xu H. Loss of sorting nexin-27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med. 2013;19:473–480. doi: 10.1038/nm.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Balana B, Maslennikov I, Kwiatkowski W, Stern KM, Bahima L, Choe S, Slesinger PA. Mechanism underlying selective regulation of G protein-gated inwardly rectifying potassium channels by the psychostimulant-sensitive sorting nexin 27. Proc Natl Acad Sci USA. 2100;108:5831–5836. doi: 10.1073/pnas.1018645108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Clairfeuille T, Mas C, Chan AS, Yang Z, Tello-Lafoz M, Chandra M, Widagdo J, Kerr MC, Paul B, Mérida I, Teasdale RD, Pavlos NJ, Anggono V, Collins BM. A molecular code for endosomal recycling of phosphorylated cargos by the SNX27-retromer complex. Nat Struct Mol Biol. 2016;23:921–932. doi: 10.1038/nsmb.3290. [DOI] [PubMed] [Google Scholar]
- 121.Fredericks ZL, Pitcher JA, Lefkowitz RJ. Identification of the G protein-coupled receptor kinase phosphorylation sites in the human ß2-adrenergic receptor. J Biol Chem. 1996;271:13796–13803. doi: 10.1074/jbc.271.23.13796. [DOI] [PubMed] [Google Scholar]
- 122.Hu LA, Chen W, Premont RT, Cong M, Lefkowitz RJ. G protein-coupled receptor kinase 5 regulates ß1-adrenergic receptor association with PSD-95. J Biol Chem. 2002;277:1607–1613. doi: 10.1074/jbc.M107297200. [DOI] [PubMed] [Google Scholar]
- 123.Harbour ME, Breusegem SY, Antrobus R, Freeman C, Reid E, Seaman MN. The cargo-selective retromer complex is a recruiting hub for protein complexes that regulate endosomal tubule dynamics. J Cell Sci. 2010;123:3703–3717. doi: 10.1242/jcs.071472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Carlton J, Bujny M, Peter BJ, Oorschot VM, Rutherford A, Mellor H, Klumperman J, McMahon HT, Cullen PJ. Sorting nexin-1 mediates tubular endosome-to-TGN transport through coincidence sensing of high- curvature membranes and 3-phosphoinositides. Curr Biol. 2004;14:1791–1800. doi: 10.1016/j.cub.2004.09.077. [DOI] [PubMed] [Google Scholar]
- 125.Seaman MNJ. Cargo-selective endosomal sorting for retrieval to the Golgi requires retromer. J Cell Biol. 2004;165:111–122. doi: 10.1083/jcb.200312034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Derivery E, Sousa C, Gautier JJ, Lombard B, Loew D, Gautreau A. The Arp2/3 activator WASH controls the fission of endosomes through a large multi protein complex. Dev Cell. 2009;17:712–723. doi: 10.1016/j.devcel.2009.09.010. [DOI] [PubMed] [Google Scholar]
- 127.Mc Gough JJ, Steinberg F, Gallon M, Yatsu A, Ohbayashi N, Heesom KJ, Fukuda M, Cullen PJ. Identification of molecular heterogeneity in SNX27-retromer-mediated endosome-to-plasma-membrane recycling. J Cell Sci. 2014;127:4940–4953. doi: 10.1242/jcs.156299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Steinberg F, Gallon FM, Winfield M, Thomas EC, Bell AJ, Heesom KJ, Tavare JM, Cullen PJ. A global analysis of SNX27- retromer assembly and cargo specificity reveals a function in glucose and metal ion transport. Nat Cell Biol. 2013;15:461–471. doi: 10.1038/ncb2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lee S, Chang J, Blackstone C. FAM21 directs SNX27-retromer cargoes to the plasma membrane by preventing transport to the Golgi apparatus. Nat Commun. 2016;7:10939. doi: 10.1038/ncomms10939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Harbour ME, Breusegem SY, Seaman NJ. Recruitment of the endosomal WASH complex is mediated by the extended “tail” of FAM21 binding to the retromer protein Vps35. Biochem J. 2012;442:209–220. doi: 10.1042/BJ20111761. [DOI] [PubMed] [Google Scholar]
- 131.Jia D, Gomez TS, Billadeau DD, Rosen MK. Multiple repeat elements within FAM21 tail link the WASH actin regulatory complex to the retromer. Mol Biol Cell. 2012;23:2352–2361. doi: 10.1091/mbc.E11-12-1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Viklund IM, Aspenström P, Meas-Yedid V, Zhang B, Kopec J, Agren D, Schneider G, D'Amato M, Olivo-Marin JC, Sansonetti P, et al. WAFL, a new protein involved in regulation of early endocytic transport at the intersection of actin and microtubule dynamics. Exp Cell Res. 2009;315:1040–1052. doi: 10.1016/j.yexcr.2008.12.004. [DOI] [PubMed] [Google Scholar]
- 133.Nakajima O, Nakamura F, Yamashita N, Tomita Y, Suto F, Okada T, Iwamatsu A, Kondo E, Fujisawa H, Takei K, Goshima Y. FKBP133: a novel mouse FK506-binding protein homolog alters growth cone morphology. Biochem Biophys Res Commun. 2006;346:140–149. doi: 10.1016/j.bbrc.2006.05.113. [DOI] [PubMed] [Google Scholar]
- 134.Pan YF, Viklund IM, Tsai HH, Pettersson S, Maruyama IN. The ulcerative colitis marker protein WAFL interacts with accessory proteins in endocytosis. Int J Biol Sci. 6:163–171. doi: 10.7150/ijbs.6.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Eichel K, Jullie D, von Zastrow M. β-Arrestin drives MAP kinase signalling from clathrin-coated structures after GPCR dissociation. Nat Cell Biol. 2016;18:303–310. doi: 10.1038/ncb3307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Shukla AK, Manglik A, Kruse AC, et al. Structure of active ß-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497:137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kang Y, Zhou XE, Gao X, et al. Crystal structure of Rhodopsin bound to arrestin by femtosecond X-ray laser. Nature. 2015;523:561–567. doi: 10.1038/nature14656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gurevich VV, Gurevich EV. The structural basis of arrestin-mediated regulation of G-protein-coupled receptors. Pharmacol Ther. 2006;110:465–502. doi: 10.1016/j.pharmthera.2005.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shukla AK, Westfield GH, Xiao K, et al. Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature. 2014;512:218–222. doi: 10.1038/nature13430. 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cahill TJ, 3rd, Thomsen AR, Tarrasch TJ, et al. Distinct conformations of GPCR-β-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc Natl Acad Sci U S A. 2017;114:2562–2567. doi: 10.1073/pnas.1701529114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Feinstein TN, Yui N, Webber MJ, Wehbi VL, Stevenson HP, King JD, Jr, Hallows KR, Brown D, Bouley R, Vilardaga JP. Noncanonical control of vasopressin receptor type2 signaling by retromer and arrestin. J Biol Chem. 2013;288:27849–27860. doi: 10.1074/jbc.M112.445098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ferrandon S, Feinstein TN, Castro M, Wang B, Bouley R, Potts JT, Gardella TJ, Vilardaga JP. Sustained cyclic AMP production by parathyroid hormone receptor endocytosis. Nat Chem Biol. 2009;5:734–742. doi: 10.1038/nchembio.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Inda C, Dos Santos Claro PA, Bonfiglio JJ, Senin SA, Maccarrone G, Turck CW, Silberstein S. Different cAMP sources are critically involved in G protein coupled receptor CRHR1 signaling. J Cell Biol. 2016;214:181–195. doi: 10.1083/jcb.201512075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Vilardage JP, Jean-Alphonse FG. Endosomal generation of cAMP in GPCR signaling. Nat Chem Biol. 2014;10:700–706. doi: 10.1038/nchembio.1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Irannejad R, Tomshine JC, Tomshine JR, Chevalier M, Mahoney JP, Steyaert J, Rasmussen SG, Sunahara RK, El-Samad H, Huang B, von Zastrow M. Conformational biosensors reveal GPCR signalling from endosomes. Nature. 2013;495:534–538. doi: 10.1038/nature12000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Oakley RH, Laporte SA, Holt JA, Barak LS, Caron MG. Association of β-arrestin with G protein-coupled receptors during clathrin-mediated endocytosis dictates the profile of receptor resensitization. J Biol Chem. 1999;274:32248–32257. doi: 10.1074/jbc.274.45.32248. [DOI] [PubMed] [Google Scholar]
- 147.Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in ß-arrestin report biased agonism at seven-transmembrane receptors. Proc Natl Acad Sci USA. 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Nuber S, Zabel U, Lorenz K, Nuber A, Milligan G, Tobin AB, Lohse MJ, Hoffmann C. β-Arrestin biosensors reveal a rapid, receptor-dependent activation/deactivation cycle. Nature. 2016;531:661–664. doi: 10.1038/nature17198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lee MH, Appleton KM, Strungs EG, Kwon JY, Morinelli TA, Peterson YK, Laporte SA, Luttrell LM. The conformational signature of β-arrestin2 predicts its trafficking and signalling functions. Nature. 2016;531:665–668. doi: 10.1038/nature17154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Yang F, Yu X, Liu C, Qu CX, Gong Z, Liu HD, Li FH, Wang HM, He DF, Yi F, Song C, Tian CL, Xiao KH, Wang JY, Sung JP. Selective mechanisms of arrestin conformations and functions revealed by unnatural amino acid incorporation and (19)F-NMR. Nat Commun. 2015;6:8202. doi: 10.1038/ncomms9202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Thomsen AR, Plouffe B, Cahill TJ, 3rd, Shukla AK, Tarrasch JT, Dosey AM, Kahsai AW, Strachan RT, Pani B, Mahoney JP, Huang L, Breton B, Heydenreich FM, Sunahara RK, Skiniotis G, Bouvier M, Lefkowitz RJ. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell. 2016;166:907–919. doi: 10.1016/j.cell.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Shenoy SK, Drake MT, Nelson CD, Houtz DA, Xiao K, Madabushi S, Reiter E, Premont RT, Lichtarge O, Lefkowitz RJ. ß-arrestin-dependent, G protein-independent ERK1/2 activation by the ß2-adrenergic receptor. J Biol Chem. 2006;281:1261–1273. doi: 10.1074/jbc.M506576200. [DOI] [PubMed] [Google Scholar]
- 153.Tsvetanova NG, von Zastrow M. Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat Chem Biol. 2014;10:1061–1066. doi: 10.1038/nchembio.1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chan AS, Clairfeuille T, Landao-Bassonga E, Kinna G, Ng PY, Loo LS, Cheng TS, Zheng M, Hong W, Teasdale RD, Collins BM, Pavlos NJ. Sorting nexin 27 couples PTHR trafficking to retromer for signal regulation in osteoblasts during bone growth. Mol Biol Cell. 2016;27:1367–1382. doi: 10.1091/mbc.E15-12-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wehbi VL, Stevenson HP, Feinstein TN, Calero G, Romero G, Vilardaga JP. Noncanonical GPCR signaling arising from a PTH receptor-arrestin-Gβγ complex. Proc Natl Acad Sci U S A. 2013;110:1530–1535. doi: 10.1073/pnas.1205756110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Feinstein TN, Wehbi VL, Ardura JA, Wheeler DS, Ferrandon S, Gardella TJ, Vilardaga JP. Retromer terminates the generation of cAMP by internalized PTH receptors. Nat Chem Biol. 2011;7:278–284. doi: 10.1038/nchembio.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Huotari J, Helenius A. Endosome maturation. EMBO J. 2011;30:3481–3500. doi: 10.1038/emboj.2011.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Seaman MN. The retromer complex-endosomal protein recycling and beyond. J Cell Sci. 2012;125:4693–4702. doi: 10.1242/jcs.103440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hartenik M, Port F, Lorenowicz MJ, McGough IJ, Silhankova M, Betist MC, van Weering JR, van Heesbeen RG, Middlekoop TC, Basler K, Cullen PJ, Korswagen HC. A SNX3 dependent retromer pathway mediates retrograde transport of the Wnt sorting receptor Wntless and is required for Wnt secretion. Nat Cell Biol. 2011;13:914–923. doi: 10.1038/ncb2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Vistein R, Puthenveedu MA. Reprogramming of G protein-coupled receptor recycling recycling and signaling by a kinase switch. Proc Natl Acad Sci U S A. 2013;110:15289–15294. doi: 10.1073/pnas.1306340110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Daaka Y, Luttrell LM, Lefkowitz RJ. Switching of the coupling of the ß2-adrenergic receptor to different G proteins by PKA. Nature. 1997;390:88–91. doi: 10.1038/36362. [DOI] [PubMed] [Google Scholar]
- 162.Martin NP, Whalen EJ, Zamah MA, Pierce KL, Lefkowitz RJ. PKA-mediated phosphorylation of the beta1-adrenergic receptor promotes Gs/Gi switching. Cell Signal. 2004;16:1397–1403. doi: 10.1016/j.cellsig.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 163.Liang W, Fishman PH. Resistance of the human beta1-adrenergic receptor to agonist-induced ubiquitination: a mechanism for impaired receptor degradation. J Biol Chem. 2004;279:46882–46889. doi: 10.1074/jbc.M406501200. [DOI] [PubMed] [Google Scholar]
- 164.Anthonio RL, Brodde OE, van Veldhuisen DJ, Scholtens E, Crijns HJ, van Gilst WH. ß-adrenoceptor density in chronic infarcted myocardium: a subtype specific decrease of ß1-adrenoceptor density. Int J Cardiol. 2000;72:137–141. doi: 10.1016/s0167-5273(99)00181-3. [DOI] [PubMed] [Google Scholar]
- 165.Molinoff PB. Alpha- and beta-adrenergic receptor subtypes properties, distribution and regulation. Drugs. 1984;28:1–15. doi: 10.2165/00003495-198400282-00002. [DOI] [PubMed] [Google Scholar]
- 166.Molenaar P, Summers RJ. Characterization of beta-1 and beta-2 adrenoceptors in guinea pig atrium: functional and receptor binding studies. J Pharmacol Exp Ther. 1987;241:1041–1047. [PubMed] [Google Scholar]
- 167.Bristow MR, Ginsburg R, Minobe W, Cubicciotti RS, Sageman WS, Lurie K, Billingham ME, Harrison DC, Stinson EB. Decreased catecholamine sensitivity and beta-adrenergic-receptor density in failing human hearts. N Engl J Med. 1982;307:205–211. doi: 10.1056/NEJM198207223070401. [DOI] [PubMed] [Google Scholar]
- 168.Bristow MR, Minobe WA, Raynolds MV, Port JD, Rasmussen R, Ray PE, Feldman AM. Reduced ß1-receptor messenger RNA abundance in the failing human heart. J Clin Invest. 1993;92:2737–2745. doi: 10.1172/JCI116891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Brodde OE. Beta-adrenoceptors in cardiac disease. Pharmacol Ther. 1993;60:405–430. doi: 10.1016/0163-7258(93)90030-h. [DOI] [PubMed] [Google Scholar]
- 170.Bristow MR, Hershberger RE, Port JD, Minobe W, Rasmussen R. Beta1- and beta2-adrenergic receptor-mediated adenylate cyclase stimulation in nonfailing and failing human ventricular myocardium. Mol Pharmacol ( 1989:295–303. [PubMed] [Google Scholar]
- 171.Fajardo G, Zhao M, Powers J, Bernstein D. Differential cardiotoxic/cardioprotective effects of ß-adrenergic receptor subtypes in myocytes and fibroblasts in doxorubicin cardiomyopathy. J Mol Cell Cardiol. 2006;40:375–383. doi: 10.1016/j.yjmcc.2005.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Bernstein D, Fajardo G, Zhao M, Urashima T, Powers J, Berry G, Kobilka BK. Differential cardioprotective/cardiotoxic effects mediated by beta-adrenergic receptor subtypes. Am J Physiol Heart Circ Physiol. 2005;289:H2441–H2449. doi: 10.1152/ajpheart.00005.2005. [DOI] [PubMed] [Google Scholar]