

ABSTRACT
All viruses strategically alter the antiviral immune response to their benefit. The vaccinia virus (VACV) K1 protein has multiple immunomodulatory effects in tissue culture models of infection, including NF-κB antagonism. However, the effect of K1 during animal infection is poorly understood. We determined that a K1L-less vaccinia virus (vΔK1L) was less pathogenic than wild-type VACV in intranasal and intradermal models of infection. Decreased pathogenicity was correlated with diminished virus replication in intranasally infected mice. However, in intradermally inoculated ears, vΔK1L replicated to levels nearly identical to those of VACV, implying that the decreased immune response to vΔK1L infection, not virus replication, dictated lesion size. Several lines of evidence support this theory. First, vΔK1L induced slightly less edema than vK1L, as revealed by histopathology and noninvasive quantitative ultrasound technology (QUS). Second, infiltrating immune cell populations were decreased in vΔK1L-infected ears. Third, cytokine and chemokine gene expression was decreased in vΔK1L-infected ears. While these results identified the biological basis for smaller lesions, they remained puzzling; because K1 antagonizes NF-κB in vitro, antiviral gene expression was expected to be higher during vΔK1L infection. Despite these diminished innate immune responses, vΔK1L vaccination induced a protective VACV-specific CD8+ T cell response and protected against a lethal VACV challenge. Thus, vΔK1L is the first vaccinia virus construct reported that caused a muted innate immune gene expression profile and decreased immune cell infiltration in an intradermal model of infection yet still elicited protective immunity.
IMPORTANCE The vaccinia virus (VACV) K1 protein inhibits NF-κB activation among its other antagonistic functions. A virus lacking K1 (vΔK1L) was predicted to be less pathogenic because it would trigger a more robust antiviral immune response than VACV. Indeed, vΔK1L was less pathogenic in intradermally infected mouse ear pinnae. However, vΔK1L infection unexpectedly elicited dramatically reduced infiltration of innate immune cells into ears. This was likely due to decreased expression of cytokine and chemokine genes in vΔK1L-infected ears. As such, our finding contradicted observations from cell culture systems. Interestingly, vΔK1L conferred protective immunity against lethal VACV challenge. This suggests that the muted immune response triggered during vΔK1L infection remained sufficient to mount an effective protective response. Our results highlight the complexity and unpredictable nature of virus-host interactions, a relationship that must be understood to better comprehend virus pathogenesis or to manipulate viruses for use as vaccines.
KEYWORDS: K1L, poxvirus, vaccines, vaccinia virus, viral pathogenesis, virus-host interactions
INTRODUCTION
Vaccinia virus (VACV) is a large, double-stranded DNA virus that has approximately nearly 200 genes (1). VACV was used as the vaccine to eradicate smallpox. The continued study of VACV remains important because there are reports of VACV infections in animals and humans (2–4). Moreover, VACV derivatives are now being used as vectors for vaccines against other infectious diseases and as oncolytic virotherapy (5).
The vaccinia virus K1 protein is an intracellular 31-kDa protein encoded by the K1L gene. K1L was initially described as a host range factor because its expression is required for VACV replication in rabbit and hamster cell lines (6, 7). It is now known that K1 possesses other functions that antagonize the immune response. K1 is known to antagonize NF-κB (8, 9), PKR (10, 11), IRF1 (12), and type I interferon (IFN) (13) signal transduction pathways. There are several cellular and viral binding partners of K1, including the vaccinia virus C10 protein (14) and the cellular ACAP2 (15) and SAMD9 proteins (16). However, it is not clear how such interactions may aid in K1 immune evasion. K1 possesses 9 ankyrin repeat domains (ARDs), motifs that mediate protein-protein interactions (17). While poxviruses encode multiple ARD proteins (18), K1 lacks a C-terminal F box present in nearly all other poxvirus ARDs (18). This suggests that K1 may have physiological properties unique among poxviral ARDs.
Despite this vast knowledge of K1, only one publication has examined the role of K1 in pathogenesis (19). Liu et al. showed that deletion of the K1L gene attenuates VACV strain TianTan, as measured by weight loss and neurovirulence in intradermal and intracranial routes of infection (19). Unfortunately, the parental strain used by Liu et al. lacked additional genes involved in vaccinia virus immune evasion (A53R and C12L) or morphogenesis (A33L) (19), making it difficult to assess the specific contributions of K1 to VACV pathogenesis.
To fill this gap in knowledge, the contribution of the K1L gene to viral pathogenicity was examined here by deleting only the K1L gene (vΔK1L) from the VACV Western Reserve (WR) strain, the strain that is most often used for pathogenesis studies (20). We show here that a virus lacking the K1L gene was less pathogenic than the parental VACV strain WR (designated vK1L) using either the intranasal (i.n.) or intradermal (i.d.) ear pinna inoculation model of infection (21, 22). The i.d. model of infection was then further investigated. Interestingly, vΔK1L replicated to the same extent as vK1L, suggesting that pathogenicity was not due to diminished viral loads. Instead, the decreased pathogenicity of vΔK1L correlated with diminished leukocyte infiltration and muted immune gene expression response to infection in ears, as measured by flow cytometry and reverse transcription-quantitative PCR (RT-qPCR), respectively. These results suggested that the decreased pathogenicity of vΔK1L was due to a muted immune response, which in turn strengthens the concept that immunopathology plays a role in poxvirus-induced diseases. Despite this muted response by components of the innate immune response, vΔK1L infection still induced VACV-specific CD8+ T cells and protected against a lethal VACV challenge. Thus, vΔK1L had decreased pathogenicity that nevertheless stimulated protective immune responses. vΔK1L may be a valuable tool to further understand the relationship between viral pathogenesis, viral modulation of innate immunity, and stimulation of protective immune responses.
RESULTS
The K1 protein is not essential for replication in mouse cells in vitro.
The goal here was to identify the contribution of K1L to viral pathogenesis in a mouse model of infection. A vaccinia virus with the K1L gene deleted (vΔK1L) replicates to the same extent as wild-type vaccinia virus (vK1L) in several human and nonhuman primate cell lines (7, 23) but is compromised when replicating in a hamster and a rabbit cell line (e.g., BHK-21 and RK13) (23, 24). However, it is not known if K1L is a host range factor in mouse cells.
Since the goal was to determine the contribution of K1L to virus pathogenicity in a mouse model of infection, an important question to ask is if the K1L gene is required for virus replication in mouse cells. To answer this question, virus growth was characterized in mouse embryo fibroblasts (MEFs) (Fig. 1). MEFs were infected at a high multiplicity of infection (MOI) (10 PFU/cell) to examine virus replication in a one-step growth curve or at a low MOI (0.01 PFU/cell) to examine virus replication and spread in a multistep growth curve. Infectious viruses present in either cells or cell-free supernatants were quantified by using plaque assays (Fig. 1). In all cases, vΔK1L replicated to the same levels as vK1L using either one-step (Fig. 1A and B) or multistep (Fig. 1C and D) growth curve assays. A revertant virus in which the K1L gene was stably reinserted into vΔK1L (vK1Lrev) was also tested. As expected, the virus had growth properties similar to those of vK1L (Fig. 1). For all experiments, an additional virus lacking both the K1L and C7L genes (vΔK1LΔC7L) was examined as a control. vΔK1LΔC7L does not replicate in the murine NIH 3T3 cell line (12). In a similar manner, the same virus replicated to lower levels than viruses containing C7L in MEFs (Fig. 1). In summary, these data suggest that K1L is not a host range factor for murine cells.
FIG 1.

K1L is not essential for virus replication in vitro. MEFs were infected with vK1L, vΔK1L, vK1Lrev, or vΔK1LΔC7L at 0.01 or 10 PFU/cell in duplicate. After adsorption, cellular monolayers were washed thrice to remove unbound viruses. Cells (A and C) and cell-free supernatants (B and D) were collected separately at the indicated time points. Intracellular (A and C) or extracellular (B and D) virus titers were quantified by plaque assays using Vero cell monolayers. The data are expressed as the mean titer per sample ± standard error of the mean (SEM). The dashed lines represent limits of detection.
The K1L gene contributes to pathogenicity in an intranasal mouse model.
Liu et al. reported that the deletion of K1L from vaccinia virus strain TianTan (VTTΔK1L) decreased virus pathogenicity (19). However, it is difficult to appreciate the full impact of the K1L gene on pathogenicity using this virus background because VTTΔK1L has different properties than vΔK1L: VTTΔK1L replicates poorly in HeLa cells (19), while the WR-based vΔK1L does not (7, 23). This may be due to the fact that the parental virus and K1L-less construct used by Liu et al. lacked three additional genes involved in vaccinia virus immune evasion (A53R and C12L) and morphogenesis (A33L) (19), which may have unknown effects on virus pathogenicity. To overcome this problem, our approach was to use a WR strain of vaccinia virus in which only the K1L gene was absent (vΔK1L). This allowed examination of the pathogenic effect of the K1L gene without the confounding effects of the absence of other virus genes. WR was chosen as the vaccinia virus strain for study because it is most frequently used for pathogenesis studies in mice (20).
For wild-type vaccinia virus (vK1L), i.n. inoculation provides a system to evaluate viral pathogenesis in which weight loss and clinical signs of illness indicate the relative pathogenicity of a virus (25, 26). Typically, mice are inoculated with 104 PFU of wild-type VACV (vK1L) and examined daily. Under such conditions, vK1L caused weight loss and clinical signs of illness similar to the results reported by Alcami and Smith (26) (Fig. 2A). In contrast, vΔK1L-infected mice lost less weight (Fig. 2A). Statistically significant differences in weight between vK1L and vΔK1L were observed on days 5 to 7 postinfection (p.i.). Additionally, vΔK1L-infected mice, but not vK1L-infected mice, returned to their original weight by 14 days p.i., suggesting that mice fully recovered from vΔK1L infection. The reinsertion of K1L into vΔK1L (vK1Lrev) resulted in a virus with pathogenicity similar to that of vK1L, indicating that the K1L gene was responsible for this decreased pathogenicity (Fig. 2A). When data were charted as a Kaplan-Meir survival curve, all the mice infected with vK1L or vK1Lrev were euthanized due to weight loss by 8 days p.i. (Fig. 2B). In contrast, all vΔK1L-infected mice survived.
FIG 2.

Virus lacking K1L is less pathogenic when introduced by intranasal inoculation. Female BALB/c mice (n = 5 per group) were inoculated intranasally with 104 (A to C) or 105 (B to F) PFU of vK1L, vΔK1L, or vK1Lrev or PBS and monitored for 14 days. (A and D) Mice were weighed daily; the data are expressed as the percentage of weight loss from day 0. Mice were euthanized if more than 25% of their original body weight was lost. (B and E) A survival plot was generated based on data from panels A and D. (C and F) Clinical signs of illness were monitored daily for 14 days and scored. Statistical significance was determined by two-way ANOVA. The asterisks indicate the days on which the weight loss or signs of illness induced by vΔK1L were statistically significantly different from those induced by vK1L (P < 0.05).
Signs of illness were also examined daily and quantified based on a 5-point scoring system that is used to determine the extent of sickness caused by a poxvirus (27, 28). Notably, there was a 2-day delay before illness onset in mice infected with a virus devoid of K1L versus K1L-containing viruses. In addition, the signs of clinical illness were decreased in vΔK1L-infected mice compared to mice infected with K1L-containing viruses (Fig. 2C). As expected, mice inoculated with phosphate-buffered saline (PBS) showed no signs of weight loss or illness at any time point. Together, these data suggest that a virus devoid of the K1L gene is less pathogenic than its wild-type parental virus.
This experiment was repeated under more stringent conditions, namely, a 10-fold-higher dose (105 PFU) of virus (Fig. 2D to F). Again, vΔK1L was less pathogenic than K1L-containing viruses. Several differences were noticed when comparing the weights of vΔK1L-infected mice that received 105 versus 104 PFU (Fig. 2D). First, mice receiving 105 PFU vΔK1L lost more weight at earlier times p.i. than those inoculated with 104 PFU. Second, vΔK1L-infected mice did not return to their original weights by 14 days p.i. In addition, one vΔK1L-infected mouse was euthanized due to weight loss (Fig. 2D and E). These differences were likely due to the fact that an increased dose of virus increases the pathogenic effects of the virus (28). Indeed, when mice received the higher inoculum of 105 PFU, signs of illness occurred earlier in all the mice. However, signs of illness were significantly reduced in mice infected with vΔK1L versus K1L-containing viruses (Fig. 2F). Thus, the data in Fig. 2 show that K1L itself is a virulence factor in an i.n. model of infection. In summary, the data in Fig. 2 show that vΔK1L has decreased pathogenicity compared to wild-type vaccinia virus.
K1L contributes to virus replication in vivo.
Often, a decrease in virus pathogenicity correlates with decreased virus replication in mouse organs (29–34). The data in Fig. 3 show that this trend also occurred for vΔK1L in the i.n. infection route. For example, vΔK1L titers in lungs were approximately 10-fold lower than those of vK1L on day 2 p.i. and approximately 100-fold lower than those of vK1L on days 4 and 6 p.i. (Fig. 3A).
FIG 3.
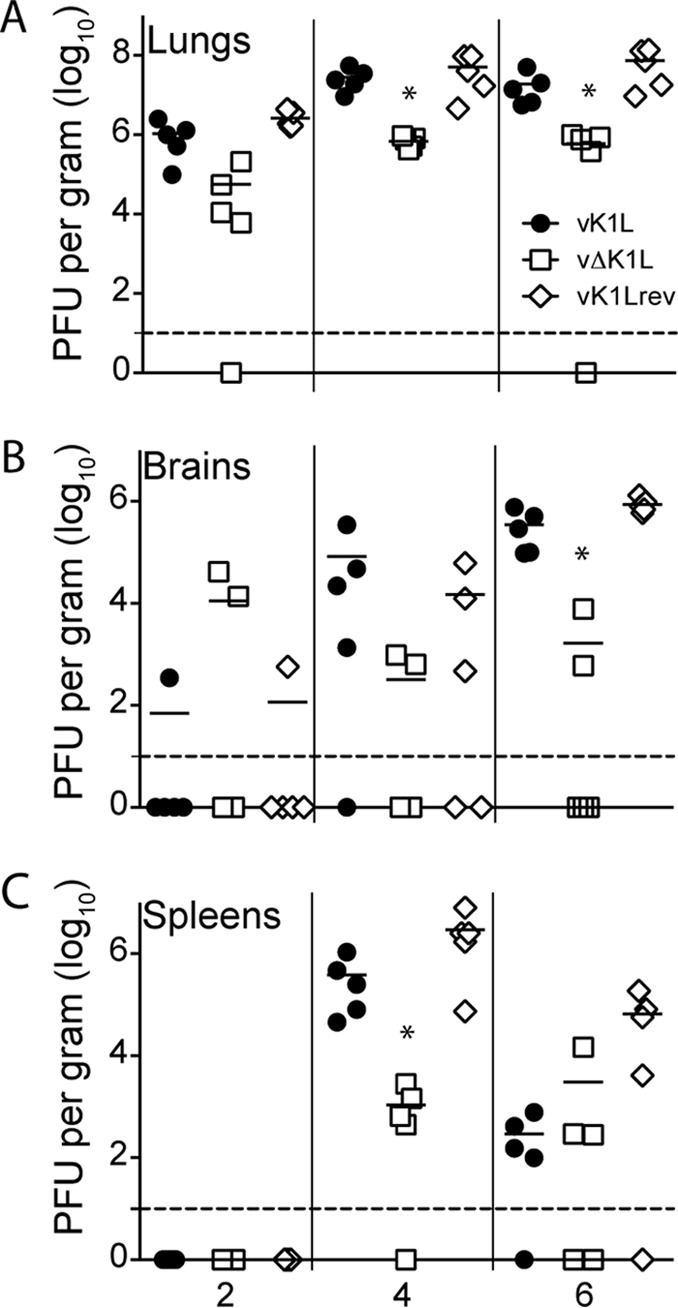
vΔK1L virus replication is lower than that of K1L-containing viruses in organs from intranasally infected mice. Female BALB/c mice (n = 5 per group) were infected intranasally with 104 PFU of vK1L, vΔK1L, or vK1Lrev. At the indicated times postinfection, lungs (A), brains (B), or spleens (C) were collected, and virus titers were determined by plaque assay on Vero cell monolayers. Each symbol represents the virus titer from an individual animal, and the mean titer is indicated by a line. Statistical significance was determined by Kruskal-Wallis test. The asterisks indicate data points at which titers from vΔK1L-infected mice were statistically significantly different from those from vK1L-infected mice (P < 0.05). The dashed lines represent the limits of detection.
During an i.n. infection, WR initially replicates in the lungs. Next, VACV disseminates to the brain and then spreads to distal organs, such as the spleen and ovaries (25, 28). Because vΔK1L titers were decreased in the lungs, it was not surprising that vΔK1L titers were also lower than those of vK1L in the brain (Fig. 3B). Note that only one mouse brain had detectable vK1L or vK1Lrev titers on day 2 p.i., while two mouse brains had vΔK1L viruses (Fig. 3B). This was expected, because VACV routinely is not detected in mouse brains until 3 days p.i. (25). By day 6 p.i., all mouse brains had detectable titers of vK1L or vK1Lrev. However, vΔK1L titers were 200-fold lower than those of vK1L. No viruses were detected in spleens at 2 days p.i. (Fig. 3C), reflective of the fact that wild-type VACV is detectable in spleens only as early as 3 days p.i. (25). Thus, it was not surprising that no viruses were detected in spleens at 2 days p.i. (Fig. 3C). While all virus titers increased rapidly by day 4 p.i. (Fig. 3C), vΔK1L titers were approximately 100-fold lower than vK1L titers. All virus titers were lower in spleens by day 6 p.i., which is similar to patterns observed by others (25, 26). Thus, in the i.n. model of infection, the reduced pathogenicity of vΔK1L correlated with a decrease in vΔK1L replication.
The K1 protein contributes to pathogenicity in an intradermal model of infection.
The i.d. inoculation of a mouse ear pinna provides a second model to evaluate vΔK1L pathogenicity (22, 35). The i.d. infection model has many strengths to study how VACV modulates the host immune response. It closely resembles the scarification route used during smallpox vaccinations and is most similar to the locale (skin) of infection for other poxviruses, including molluscum contagiosum virus and Orf (36, 37). Here, lesion formation and size are indicators of pathogenicity (38). Unlike the i.n. route of infection, i.d. inoculation is localized (22). Thus, there are no generalized clinical signs of illness like those observed during i.n. infections (e.g., weight loss or fur ruffling), and there is little to no virus detection in distal organs (22). Figure 4 shows the results using this model system. For all the viruses tested, gross lesions were observed starting on day 7 p.i. vK1L or vK1rev infection induced the largest lesions at days 12 and 8, respectively (Fig. 4). In contrast, lesions were smaller in vΔK1L-infected ears at all times examined. These differences were statistically significant on days 7 to 12 and 14 to 16 p.i. Thus, K1 also contributed to pathogenicity for the i.d. model of infection.
FIG 4.
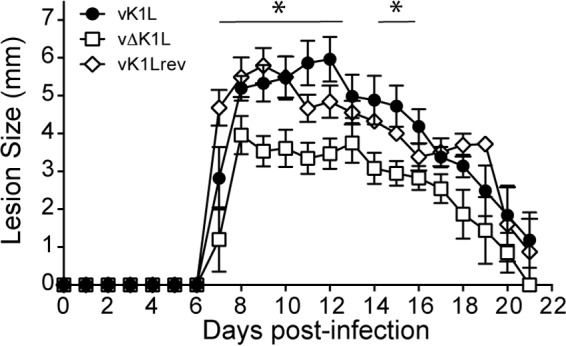
vΔK1L is less pathogenic during intradermal infection. C57BL/6 mice (n = 5 per group) were infected intradermally with 104 PFU of vK1L, vΔK1L, or vK1Lrev in the left ear pinna, and the sizes of the resulting lesions were measured daily for 21 days. The data are expressed as the means of lesion sizes ± SEM. Statistical significance was determined by two-way ANOVA. The asterisks indicate the days on which the lesion size caused by vΔK1L was statistically significantly different from that caused by vK1L (P < 0.05).
Decreased lesion size is not due to diminished virus replication in i.d. infection.
Data from the i.n. model of infection showed that decreased pathogenicity of vΔK1L was associated with its decreased replication in the lungs (Fig. 3). Thus, a logical assumption was that virus titers would be lower in vΔK1L-infected ears. Surprisingly, this premise did not hold true; Fig. 5 showed that there were no statistically significant differences in virus titers between viruses containing or lacking the K1L gene at any time point examined (Fig. 5). This included time points where there was no lesion formation (days 1 and 3 p.i.), just prior to visible lesion formation (day 5 p.i.), and times when lesions had formed and remained attached to ears (days 8 and 11 p.i.). The last two time points were chosen to minimize the possibility that, if there were a decrease in virus titers, it would not be due to a loss of virus encrusted in lesions. These data showed two important trends. First, lesion size did not correlate with virus replication, which was opposite to the results observed in the i.n. infection model. Second, since vΔK1L replicated to levels similar to those of vK1L in pinnae, K1L was not a host range gene in mouse ear pinnae.
FIG 5.

Virus replication in ear pinnae of intradermally inoculated mice. C57BL/6 mice (n = 5 per group) were infected with 104 PFU of vK1L, vΔK1L, or vK1Lrev as indicated. At the indicated days p.i., ears were collected, homogenized, and lysed, and viral titers of the lysates were determined by plaque assay. The data are expressed as the mean titer of virus (PFU) per gram of tissue.
Examination of edema during infection.
We continued studies with the i.d. infection system, but not with the i.n. infection system. This was because there are numerous examples of mutant VACVs that lack NF-κB antagonists where a decrease in viral pathogenesis correlates with a decrease in virus replication in the lungs and other organs (e.g., ΔA49, ΔC4, ΔE3, and ΔK7) (29, 31, 32, 39). In contrast, there is only one other published example of a mutant VACV that lacks the NF-κB inhibitor B7 (ΔB7R) where viral pathogenesis is not due to virus replication (40). Thus, the continued study of vΔK1L in ear pinnae would be more likely to uncover novel aspects of virus-host interactions.
The i.d. model of infection relies on the visual detection of lesion formation to assess virus pathogenicity (22). I.d. infection of ears causes inflammation (41–44). Thus, we were curious if edema, a classical sign of inflammation, was accelerated or delayed during vΔK1L infection. Commonly, histopathology is used to examine tissue damage; this method is semiquantitative (41–44). In contrast, ultrasound imaging successfully quantified inflammation for processes like fatty liver disease (45). A benefit of this technology is that it is noninvasive and allows scientists to quantitatively track progression of disease in the same mouse.
We chose to use ultrasound as a means of determining if early signs of inflammation (e.g., edema) were different in infected ears. Ultrasound was used to collect B-mode images for each ear (Fig. 6A, B-mode). The regions outlined in pink (fields of interest [FOI]) contain ear tissue, while the substrate used for ear placement is shown as a gray platform under the ear. The FOI was then used for quantitative ultrasound (QUS) backscatter coefficient (BSC) data assessment (Fig. 6A, BSC). As fluid concentration (edema) increases in an ear, the ultrasonic scattering magnitude decreases (46). This is quantified as a decrease in ultrasonic BSC, and the change can be quantified using QUS technology (46) and displayed in graphical form (Fig. 6B). The BSC color coding represents the quantitative magnitude of the gray-scale B-mode echo data. The color-coded scale on the right is in logarithmic units, in which red is related to the highest BSC value and the lowest fluid content, while dark blue is related to the lowest BSC value and the highest fluid content. Increase in fluid content for this study was edema, confirmed by histopathology. Edema is triggered by early inflammatory signaling. The BSC values from each mouse ear were determined, and the measurements from each ear were averaged (Fig. 6B) (46).
FIG 6.

Ultrasound imaging and quantitation of edema longitudinally in intradermally inoculated ears. C57BL/6 mice (day 3, n = 13 mice per experimental group; day 4, n = 10 mice per experimental group; day 5, n = 7 mice per experimental group) were either uninfected (UN) or inoculated i.d. with PBS or 104 PFU vK1L or vΔK1L in the left ear pinna. (A) Mice were anesthetized, and B-mode images (B-mode) were collected. The regions outlined in pink are the fields of interest, which contain ear tissue. The field of interest was used for quantitative ultrasonic data (BSC) assessment. The BSC color coding represents the quantitative magnitude of the gray-scale B-mode echo data. The color-coded scales on the right are in logarithmic units, with red representing the highest magnitude of the BSC and dark blue representing the lowest magnitude of the BSC. (B) BSC data quantitatively depicted as a function of time. The BSC values are expressed as the means ± SEM. Statistically significant differences between mice inoculated with PBS versus vK1L was determined by one-way ANOVA and is indicated by an asterisk (P < 0.05). (C) The thickness of the ear was selected from the spatially calibrated BSC image (Fig. 6A) for which the pixel dimensions were known (68 μm by 68 μm = 1 mm by 1 mm). The number of pixels was estimated in the region of the BSC image that depicted the thickest measure of the color-coded image. Statistically significant differences between inoculated mice were determined by one-way ANOVA and are indicated by asterisks (P < 0.05).
Uninoculated and PBS-inoculated ears are color coded in red, implying that there was little to no fluid infiltration (Fig. 6A), resulting in high BSC values (Fig. 6B). Infection with vK1L or vΔK1L showed an increase in edema longitudinally, with the most noticeable differences beginning on day 3 p.i. and continuing until day 5 p.i. (Fig. 6A), and BSC values decreased at these times (Fig. 6B). Overall, there was a statistically significant decrease in BSC between virus-infected ears and PBS-inoculated or uninoculated ears (Fig. 6B) at days 4 and 5 p.i. vΔK1L infection resulted in a slight decrease in BSC values compared to vK1L; however, it was not statistically significant (Fig. 6B). Thus, vΔK1L infection minimally affected edema.
The thickness of each ear was digitally measured as another means of assessing inflammation. The number of pixels was estimated in the region of the BSC image that depicted the thickest measurement of the color-coded image. The data are shown in Fig. 6C. Thicknesses of vΔK1L-infected ears were similar to those of vK1L-infected ears on days 1 to 3 p.i. The largest difference in ear thickness between vK1L and vΔK1L infections occurred on day 4 p.i., when there was an approximately 0.2-mm difference in thickness. However, this difference decreased by day 5 p.i., when vΔK1L-infected ears were nearly the same thickness as vK1L-infected ears. Thus, while vΔK1L-infected ears were less thick than vK1L-infected ears, these differences in ear thickness appear to be transient.
Some mouse ears used for ultrasound imaging were processed further for histological examination using hematoxylin and eosin (H&E) staining to evaluate tissue morphology (Fig. 7). Examination of the architecture of the ear tissue showed that, at day 3 p.i., both vΔK1L and vK1L infections resulted in mild edema. A single mouse of the vK1L infection group had a small focal region of more pronounced inflammation. By day 4 p.i., vΔK1L- and vK1L-infected ears exhibited similar pinna thickening, which was primarily due to moderate dermal edema. Ear thickening was also due to hyperplasia, with ballooning degeneration and pustules in the epidermis. By day 5 p.i., microscopic lesion formation was prominent in both vΔK1L- and vK1L-infected ears, and the lesions progressed in severity, with some ulceration and crusting. vΔK1L infection produced less extensive lesions in the pinna (Fig. 7). This correlates with the differences in lesion sizes shown in Fig. 6C. Overall, there was a moderate and transient decrease in edema in vΔK1L-infected ears versus vK1L-infected ears at all times examined. Thus, classical histopathology showed trends similar to those shown by quantitative ultrasound imaging, suggesting quantitative ultrasound technology is a new noninvasive technology to detect inflammation.
FIG 7.

Histological analysis of infected ear pinnae. C57BL/6 mice (n = 3 per group) were inoculated with PBS or 104 PFU vK1L or vΔK1L. At the indicated days postinfection, ears were collected, fixed in 10% neutral buffered formalin, processed, and stained with hematoxylin and eosin for examination by light microscopy (magnification, ×100). Representative images of the lesions observed in each group are shown. Bars, 500 μm.
vΔK1L infection results in decreased immune cell infiltration.
Figure 5 shows that, unlike the results of the i.n. infection model, the decreased lesion size observed in vΔK1L-infected ears was not due to decreased viral replication. Another mutant VACV that lacks the B14 NF-κB antagonist (vΔB14R) enhances the infiltration of immune cells compared to wild-type VACV during an i.d. infection (47). This suggested that an enhanced host inflammatory response may contribute to diminished lesion size.
To test if this also was the basis for reduced lesion sizes for vΔK1L-infected ears, we used flow cytometry to detect and quantify immune cell types that are known to infiltrate the ear upon VACV infection (44, 48). The presence of these innate immune cells was examined at day 5 postinfection. This time point was chosen because viral replication was robust, and it is a time where it is known that these types of innate immune cells would be detectable in infected ears (44). The results are shown in Fig. 8.
FIG 8.
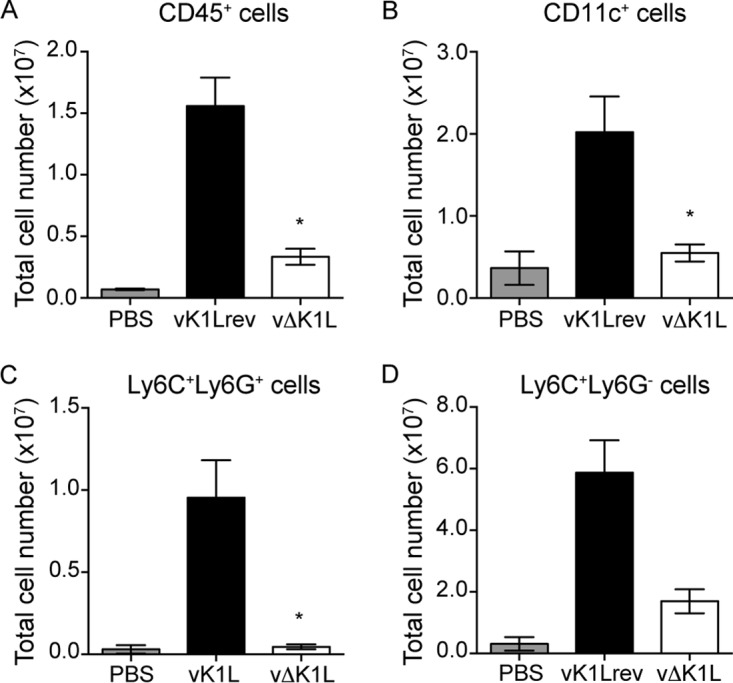
vΔK1L infection causes reduced infiltration of innate immune cells into ear pinnae. C57BL/6 mice were inoculated in the left ear with PBS (n = 10) or 104 PFU vK1Lrev (n = 7) or vΔK1L (n = 7). At 5 days p.i., ears were collected and processed. Single-cell suspensions were prepared from each ear. Flow cytometry analyses show the total numbers of CD45+ cells (leukocytes) (A), CD11c+ cells (B), Ly6C+ Ly6G+ cells (tissue-protective monocytes) (C), and Ly6C+ Ly6G− cells (inflammatory monocytes) (D) per ear. Statistical significance was determined by Kruskal-Wallis test (A to C) or one-way ANOVA (D). The asterisks indicate conditions where there was a statistically significant difference between vK1Lrev and vΔK1L (P < 0.05). The error bars indicate SEM.
The first striking result was that there were dramatically (4.3 times) fewer cellular infiltrates harvested from vΔK1L-infected versus vK1Lrev-infected ears (Fig. 8A). However, the number of infiltrates in vΔK1L-infected ears was 3 times higher than in PBS-inoculated ears. This implied that vΔK1L infection indeed drew some immune cells into the area of infection, but the number was diminished compared to vK1L-infected ears.
We were then interested in examining the numbers and types of infiltrating leukocytes in ears. Because the total numbers of cells varied between the groups of PBS-, vK1Lrev-, and vΔK1L-infected ears, we could not represent the data as a percentage of the total cell population. Instead, the data in Fig. 8A to D are presented as the total number of each cell population per ear to allow better comparisons. When examining CD45+ cell (leukocyte) infiltration, there were 4.7 times more CD45+ cells in vK1Lrev- than in vΔK1L-infected ears (Fig. 8A). The number of infiltrates in vΔK1L-infected ears was 3 times higher than in PBS-inoculated ears, indicating that vΔK1L infection was not inert.
We investigated the relative abundances of three subsets of CD45+ leukocytes that are reported to be detectable in infected ears 5 days p.i. (44, 48). The data are shown in Fig. 8B to D. They include tissue-protective monocytes (CD45+ CD11b+ CD11c− Ly6C+ Ly6G+) and inflammatory monocytes (CD45+ CD11b+ CD11c− Ly6C+ Ly6G−) (44, 48). CD45+ CD11c+ cells, which can include dendritic cells, NK cells, and activated T cells, were also investigated. There were dramatic decreases in all three cell types examined in vΔK1L- versus vK1Lrev-infected ears. Again, CD11c+, Ly6C+ Ly6G+, and Ly6C+ Ly6G− cell numbers were higher in vΔK1L- versus PBS-inoculated ears, suggesting that vΔK1L infection elicited moderate infiltration of leukocytes. These data showed a correlation between reduced lesion size and reduced immune cell infiltrates, implying that infiltrating immune cells may produce molecules that promote immunopathology.
vΔK1L infection results in a muted host immune gene expression profile.
In cell culture, vΔK1L infection resulted in increased transcription of an NF-κB-controlled gene (Tnf) compared to vK1L infection (9). Thus, one hypothesis was that the expression of immune genes under the control of NF-κB (e.g., cytokines and chemokines) would occur earlier and/or to a greater extent in vΔK1L- versus vK1L-infected ears. The results in Fig. 8 show that there was a decrease in infiltration of leukocytes during vΔK1L infection. Since leukocytes migrate to areas expressing chemokines and other cytokines, this raised an alternative hypothesis: that the expression of cytokines and chemokines would be decreased during vΔK1L infection. To test both hypotheses simultaneously, we evaluated the transcription profile of 24 host cell genes at days 1, 3, and 5 p.i. in ears inoculated i.d. with either PBS, vK1L, vΔK1L, or vK1Lrev using RT-qPCR. These time points were chosen for two reasons. First, it afforded the opportunity to examine if host immune genes were transcribed in vΔK1L-infected ears before vK1L-infected ears. Second, these time points were for ears harvested prior to lesion formation, thus minimizing potential RNA loss due to scab formation. Gene expression values from infected mice were normalized to those for PBS-inoculated mice euthanized on the same day. Statistical analyses were performed comparing PBS- to vK1L-infected or vK1L- to vΔK1L-infected ears.
The results from these studies are shown in Table 1. Several striking trends were observed. For example, there was no statistically significant difference in gene expression between vK1L- and vΔK1L-inoculated ears at 1 day p.i. These results contradicted the first hypothesis that, in the absence of an NF-κB antagonist, there would be an increase in the expression of NF-κB-controlled host genes during vΔK1L infection.
TABLE 1.
Gene expression during i.d. virus infection as detected by RT-qPCR
| Gene | Expressiona |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Day 1 p.i. |
Day 3 p.i. |
Day 5 p.i. |
|||||||
| vK1L | vΔK1L | vK1Lrev | vK1L | vΔK1L | vK1Lrev | vK1L | vΔK1L | vK1Lrev | |
| Chemokines | |||||||||
| Ccl4 | 0.47 | 0.33 | 0.54 | 1.90 | 0.62 | 1.98 | 55.97 | 4.51 | 49.82 |
| Cxcl2 | 0.48 | 0.30 | 0.53 | 1.44 | 0.60 | 1.15 | 10.41 | 1.99 | 8.83 |
| Cxcl5 | 0.17 | 0.14 | 0.18 | 2.71 | 0.44 | 8.92 | 29.76 | 5.62 | 39.96 |
| Ccl2 | 0.64 | 0.80 | 0.66 | 4.28 | 2.00 | 8.06 | 12.91 | 6.30 | 16.27 |
| Ccl5 | 0.73 | 0.69 | 0.75 | 0.54 | 0.49 | 0.89 | 7.41 | 1.84 | 6.84 |
| Cxcl9 | 1.94 | 0.87 | 1.07 | 3.41 | 0.95 | 8.22 | 307.96 | 87.92 | 360.41 |
| Cxcl10 | 0.91 | 0.52 | 0.64 | 9.03 | 1.02 | 22.95 | 302.86 | 193.49 | 419.48 |
| Cxcl1 | 2.68 | 1.52 | 2.16 | 18.23 | 10.46 | 26.78 | 13.95 | 16.10 | 20.90 |
| Cytokines | |||||||||
| Il1b | 0.35 | 0.24 | 0.45 | 1.57 | 0.45 | 1.09 | 12.47 | 2.67 | 11.38 |
| Il6 | 0.46 | 0.25 | 0.44 | 7.11 | 1.73 | 19.65 | 49.03 | 13.44 | 63.77 |
| Il10 | 0.54 | 0.42 | 0.59 | 2.28 | 0.94 | 4.12 | 9.76 | 2.93 | 12.76 |
| Il7 | 1.19 | 0.96 | 0.98 | 0.70 | 0.99 | 0.52 | 0.59 | 0.73 | 0.61 |
| Tnf | 1.04 | 0.89 | 0.91 | 1.16 | 0.94 | 0.97 | 0.86 | 0.97 | 0.78 |
| IFNs | |||||||||
| Ifng | 1.45 | 0.71 | 1.01 | 3.40 | 1.02 | 7.81 | 247.88 | 32.21 | 306.88 |
| Ifna4 | 1.41 | 1.40 | 1.29 | 0.60 | 0.85 | 0.85 | 0.19 | 0.51 | 0.22 |
| Ifnb | 1.67 | 1.41 | 1.42 | 1.23 | 1.02 | 2.24 | 0.91 | 1.20 | 1.17 |
| ISGsb | |||||||||
| Isg15 | 0.83 | 0.55 | 0.87 | 3.39 | 0.94 | 9.27 | 104.70 | 36.81 | 115.28 |
| Rnasel | 1.07 | 0.91 | 0.91 | 0.95 | 0.92 | 1.23 | 3.38 | 1.52 | 3.82 |
| Mx1 | 0.50 | 0.51 | 0.53 | 3.55 | 0.99 | 9.65 | 116.42 | 68.74 | 174.37 |
| Eif2ak2 | 1.12 | 0.95 | 1.10 | 1.29 | 1.07 | 1.56 | 2.64 | 1.91 | 3.02 |
| Keratinocyte stress response | |||||||||
| Krt6a | 0.40 | 0.34 | 0.45 | 1.61 | 0.54 | 2.12 | 32.94 | 13.01 | 37.82 |
| Krt16 | 0.52 | 0.47 | 0.52 | 2.21 | 0.87 | 2.84 | 15.39 | 6.54 | 18.60 |
| NF-κB pathway | |||||||||
| Nfkb2 | 1.17 | 1.04 | 1.02 | 1.08 | 1.12 | 0.93 | 0.52 | 0.93 | 0.57 |
| Nfkbia | 1.14 | 0.88 | 1.05 | 0.87 | 1.02 | 0.71 | 0.69 | 0.83 | 0.72 |
The data represent the average fold expression level of each gene (2−ΔΔCT) compared to that in PBS-inoculated ears that were harvested on the same day p.i. (n = 4 ears per group). The underlined values indicate a statistically significant difference compared to PBS-inoculated mice (P < 0.05). The boldface values indicate statistically significant differences between mice inoculated with vK1L and mice inoculated with vΔK1L (P < 0.05).
ISG, interferon-stimulated gene.
By day 3 p.i., there was a more robust host response to vK1L infection. Notably, there was a statistically significant induction of Cxcl1 and Il6 gene expression. Il10 mRNA levels were also statistically significantly increased, in agreement with data published by Cush et al. (49). While not statistically significant, there was a 9-fold increase in Cxcl10 gene expression in vK1L-infected ears, also in agreement with a previous publication (43). We observed that vK1Lrev-infected ears showed a slightly more robust response to virus infection in several instances for reasons that remain unclear. For example, Cxcl5, Isg15, Mx1, Eif2ak2 (PKR), and Krt6a mRNA levels were significantly higher in vK1Lrev-inoculated than in PBS-inoculated mice. Unexpectedly, vΔK1L-infected ears showed a different pattern of host gene expression. Nearly all the mRNAs examined were at lower levels in vΔK1L-infected ears than in vK1L-infected ears. Thus, in contrast to what was expected, removal of K1L led to a decrease in the expression of antiviral genes during infection.
vK1L or vK1Lrev infection continued to stimulate host gene transcription at day 5 p.i., and this increase was statistically significant compared to PBS-inoculated ears in most cases. As reported by others, we detected upregulation of Ccl4, Cxcl9, Ifng, Il1, Il6, and Mx1 in VACV-inoculated ears, suggesting these cytokines are important for detecting and resolving VACV infection (41, 43). The exceptions to this trend were for transcription of the Il7, Tnf, Ifna4, Ifnb, Nfkb2, and Nfkbia genes, which were either similar to or lower than values obtained for infections at day 1 p.i. Others have also reported the lack of tumor necrosis factor (TNF) and type I IFN gene expression during VACV infection (43). However, more recently, IFN gene expression has been detected by other researchers who have optimized RNA purification protocols (C. Norbury, personal communication).
When vΔK1L-infected ears were examined at day 5 p.i., there were several noticeable trends. Most of the host cell transcript levels were lower in vΔK1L- versus vK1L-infected ears. This result was surprising, because it was the opposite of our original hypothesis. The greatest reduction of expression was that of the Ccl4 gene, with a 12.4-fold reduction of mRNA for vΔK1L- versus vK1L-infected mice, followed by Ifng, with a 7.6-fold reduction. Other genes for which there was a statistically significant reduction of mRNA levels in vΔK1L- compared to vK1-infected mice were Cxcl2 and Cxcl5 (5.2-fold), Il1b (4.6-fold), Cxcl9 (3.5-fold), Il10 (3.3-fold), Rnasel (2.2-fold), and ccl2 (2.0-fold). There were several instances in which there were differences in gene expression, but these differences were not statistically significant due to variance among the mice within the groups. These genes included the chemokine Ccl5 and Cxcl10 genes, the Il6 gene, the interferon-stimulated Mx1 and Isg15 genes, and the keratinocyte stress response genes Krt6a and Krt16. As mentioned above, K1 antagonizes type I IFN (13) and NF-κB (8, 9). Interestingly, genes encoding a type I IFN (Ifnb) or NF-κB-controlled genes (Nfkbia and Tnf) (50–53) showed no K1L-dependent change in expression. However, other genes at least partially controlled by NF-κB (Ifna4, Il7, and Nfkb2) showed significant downregulation in the absence of K1L (54, 55). For Nfkb2, the mRNA level was slightly, but significantly, higher in vΔK1L- versus vK1L-infected mice. There was only one gene, Cxcl1, whose mRNA expression was slightly higher in vΔK1L- versus vK1L-infected mice; however, this difference was not statistically significant. Thus, there were two conclusions drawn from these data. First, the decreased expression of immune response genes correlated with a decrease in leukocyte infiltration and smaller lesion size. Second, vΔK1L counterintuitively dampened proinflammatory gene expression. Many of these proinflammatory molecules also induce immunopathology. Thus, vΔK1L-induced lesions may be smaller because of the diminished expression of these proinflammatory cytokines.
vΔK1L infection activates VACV-specific CD8+ T cells.
We observed that vΔK1L infection was less pathogenic. In addition, the infiltration of leukocytes and expression of immune response genes were lower in vΔK1L-infected than in vK1L-infected mice. We were curious if this decreased host response would also result in a diminished capacity to trigger a protective immune response. To answer this question, we examined vaccinia virus-specific activated CD8+ T cells from the spleens and lymph nodes of infected mice (Fig. 9A to D). In the spleens, virus infection significantly increased the number of activated (IFN-γ-expressing), vaccinia virus-specific CD8+ T cells compared to mice inoculated with PBS. Moreover, the magnitudes of the response were similar for vK1L and vΔK1L infections (Fig. 9A and B). Slightly different results were observed when examining lymph nodes of infected animals. While both viruses induced an increase in the percentages of VACV-specific CD8+ T cell populations, only vΔK1L-infected mice showed a statistically significant increase compared to PBS-inoculated mice. When the absolute numbers of VACV-specific CD8+ T cells in lymph nodes were evaluated (Fig. 9D), there was a greater number of CD8+ T cells in vΔK1L-infected versus vK1L-infected mice. However, both values were higher than those for PBS-inoculated mice. Thus, CD8+ T cell responses were similar despite the decrease in initial innate immune responses to vΔK1L infection.
FIG 9.

vΔK1L infection raises VACV-specific CD8+ T cell responses and protects against lethal VACV challenge. C57BL/6 mice (n = 5 mice per group) were inoculated i.d. with PBS or 104 PFU vK1L or vΔK1L. (A to D) At 10 days p.i., mice were euthanized, and spleens (A and B) or lymph nodes (C and D) were harvested. Single-cell suspensions were prepared from each sample and incubated with VACV B8R20–27 peptide and brefeldin A. The cells were stained for extracellular CD8 and intracellular IFN-γ, and activated VACV-specific CD8+ T cells were quantified by using flow cytometry. The data are expressed as means ± SEM. (A and C) Percentages of IFN-γ+ CD8+ T cells out of the total number of CD8+ T cells. (B and D) Total numbers of IFN-γ+ CD8+ T cells per organ. Statistical significance was determined by one-way ANOVA. The asterisks indicate conditions where there was a statistically significant increase between vK1L and vΔK1L (P < 0.05); ns, not statistically significant. (E) At 30 days p.i., mice were inoculated i.n. with 5 × 106 PFU vK1L. Individual animals were weighed daily for the next 7 days. Weight loss is expressed as the percentage ± SEM of the mean weight of the same group of animals on day 0. Mice were euthanized if more than 25% of their original body weight was lost.
vΔK1L protects against a lethal challenge by vaccinia virus.
Since vΔK1L infection raised VACV-specific CD8+ T cell responses equal to or greater than those with vK1L infection, we predicted that vΔK1L would be an effective vaccine against a lethal vaccinia virus challenge. To test this hypothesis, we used a previously described vaccination model in which mice were inoculated i.d. with PBS or with 104 PFU vK1L or vΔK1L (48). At day 30 p.i., mice were challenged i.n. with a lethal dose (5 × 106 PFU) of vK1L. We chose this dose because others have reported that doses of 105 or higher are sufficient to be lethal in the i.n. model of infection (48, 56). In addition, Jacobs et al. reported that i.d. vaccination protected mice when they were challenged with 5 × 106 PFU of wild-type vaccinia virus (48). Weight was recorded daily as a proxy for protection against the challenge (Fig. 9E). All PBS-vaccinated mice rapidly lost weight, and all the mice from this group were euthanized by day 6 p.i. vK1L-vaccinated mice were protected from the challenge. The mice did not lose as much weight as PBS-inoculated mice, and they nearly recovered their original weight by day 14 p.i. vΔK1L vaccination also protected mice against a lethal challenge. Interestingly, vΔK1L-vaccinated mice showed slightly less weight loss than vK1L-vaccinated mice, although these differences were not statistically significant. Thus, vΔK1L is a less pathogenic virus that still provides protection against a lethal virus challenge.
DISCUSSION
We report here that vΔK1L is less pathogenic in two models of infection. We went on to further examine the i.d. model of infection because results showed that the reduced pathogenicity was not due to a decrease in virus replication. Instead, reduced pathogenicity correlated with decreased innate immune cell infiltration and decreased inflammatory gene expression in ears. It is well known that the antiviral immune response can inadvertently damage the host to such a degree that it causes disease (57). Thus, we propose that there are two underlying mechanisms responsible for smaller vΔK1L-induced lesions. First, the diminished amount of chemokine expression in vΔK1L-infected ears, which is already observed at day 3 p.i., probably results in a decrease in infiltrating immune cells, which can express damaging cytokines. The second reason is that there are smaller amounts of proinflammatory cytokine gene expression at days 3 and 5 p.i. in ears. Since some of these cytokines are known to have immunopathogenic properties, this decreased expression in vΔK1L ears may result in smaller lesions. Despite these decreased innate immune responses, i.d. inoculation with vΔK1L still stimulates VACV-specific T cell responses and provides protection against a lethal VACV challenge equivalent to that provided by its parental virus. vΔK1L infection caused a reduction in immune cell infiltration and muted the expression of most host immune genes examined in the intradermal model of infection. Despite this decrease, vΔK1L still stimulated VACV-specific T cell responses and provided protection against a lethal VACV challenge equivalent to that provided by its parental virus. To our knowledge, this is the first vaccinia virus construct with this combination of characteristics.
Our studies are the first to address the effects of K1L itself on viral pathogenesis. These data illustrate two concepts important to both the poxvirus and vaccinology fields. First, the antagonistic functions of K1 discovered during cell culture-based studies may not necessarily be replicated in vivo. For example, K1 is well known as an antagonist of NF-κB (8, 9). We reported that vΔK1L infection upregulated one NF-κB-controlled host gene (Tnf) in comparison to wild-type VACV in cell lines (9). Thus, our original expectation was that vΔK1L infection would increase NF-κB-controlled host gene transcripts more or earlier than vK1L-infected ear pinnae. However, we observed the opposite result: nearly all host gene transcripts were decreased in vΔK1L- compared to vK1L-infected ears at all times examined (Table 1). This implies that results obtained from cell culture models of infection may not mirror results obtained from in vivo models of infection. One other report supports this concept: van Buuren et al. reported that transcription of the NF-κB-controlled Tnf, Il1b, and Il6 genes was not altered in organs during intranasal infection with an ectromelia virus devoid of the EMV005 NF-κB antagonist (58). Data from this study and from that of van Buuren et al. demonstrate our lack of understanding of how these poxviral NF-κB inhibitors may truly impact viral pathogenesis.
The second important concept is that an inflammatory immune response triggered by wild-type VACV (vK1L) may be in excess of what is required to mount a protective immune response. Several recent publications suggest that limited inflammation is beneficial for CD8+ T cell activation, which requires signals from antigen, costimulation, and inflammatory cytokines (59). Others have suggested that the positive effect of inflammatory signals on T cell activation might compromise the quantity and quality of memory T cells (60). Recently, it was reported that sensing of a robust inflammatory response affects the transition of effector CD8+ T cells to memory T cells (61). In addition, chronic inflammation can disrupt epithelial integrity, which can affect T cell migration or access to pathogens (61). Thus, perhaps a VACV lacking K1L is one component of an improved vaccine vector.
One must keep in mind that K1 is a multifunctional protein, antagonizing not only NF-κB (8, 9), but also PKR (10, 11), IRF1 (12), and type I IFN (13) signal transduction pathways. Unfortunately, the K1 regions necessary for each function are not yet known. Until such mapping is performed, the specific K1 function that contributes to VACV pathogenesis remains to be determined. Thus, we cannot rule out the possibility that this decrease in inflammatory gene expression during vΔK1L infection may be due to other K1 functions.
VACV encodes at least 10 inhibitors of NF-κB, i.e., K1, A46, A49, A52, B14, C4, E3, K7, M2, and N1 (9, 29, 31, 62–67). Interestingly, the deletion of a single NF-κB antagonist results in an attenuated infection (29, 31, 32, 39, 68–70). It appears that K1L is unique among NF-κB antagonists studied to date in vivo. For example, viruses lacking B14R (ΔB14R) or N1L (ΔN1L) are each less pathogenic in the i.d. model of infection (47, 70). However, ΔB14R and ΔN1L each replicate to a lesser extent than wild-type VACV (47, 70), while vΔK1L replicates as well as VACV in the ear pinnae (Fig. 5). Interestingly, ΔB14R infection increases macrophage infiltrates in the ear, and this is suggested to be due to an increase in expression of chemokines and cytokines (47). However, vΔK1L infection is associated with a decrease in immune cell infiltration. These data highlight the fact that these poxviral NF-κB inhibitors are nonredundant in vivo and argue strongly in favor of a sophisticated multistep system used by VACV to master distinct cellular environments as it infects and is exposed to myriad organ-specific associated immune responses. Inflammatory gene expression profiles similar to the one performed here have identified host responses to infection with wild-type VACV (43, 71). To our knowledge, this is the first study to report a detailed analysis comparing the inflammatory gene responses to virus infection in wild-type VACV (vK1L) versus a VACV devoid of an immune evasion gene. We propose that the continued use of RT-qPCR or sequencing of the transcriptome to compare the immune gene expression profiles of ears when using mutant VACVs that lack different NF-κB antagonists is one approach that may identify how each VACV immune antagonist uniquely alters the host response to infection in vivo.
In agreement with our data, Liu et al. reported that deletion of the K1L gene attenuates VACV strain TianTan, and that T cell responses remain unaffected by removal of K1L. Liu et al. also observed that humoral responses were decreased when K1L was absent (19). Humoral responses for vΔK1L were not measured here. It is difficult to accurately predict if vΔK1L would give results similar to those of vTTΔK1L because the parental vector (vTT) lacks several genes other than K1L (19, 72). Nevertheless, we chose to examine only CD8+ T cell responses because others have reported that CD8+ T cells provide protection against VACV intranasal infection even in the absence of CD4+ T cells and B cells (71).
Investigation of VACV immune evasion molecules has identified important strategies viruses use to modulate the immune response. In turn, this information provides a greater understanding of how the host detects, responds to, and eliminates virus infection (20). VACV continues to be used as a vaccine vector and as an oncolytic therapy. Thus, information about virus-host interactions is also valuable for researchers who optimize current poxvirus-based vaccine vectors. A continuing challenge for researchers is to develop a VACV-based construct that is simultaneously efficacious and safe. VACV, while highly immunogenic, induces unacceptably high rates of morbidity and mortality. Thus, multiple research laboratories are now modifying VACV to develop vectors that retain immunogenicity and have improved safety profiles (73). One strategy used by researchers is to alter the expression of the immune evasion genes of a VACV-based vector (5) in an effort to retain VACV immunogenicity and improve safety. We show here that vΔK1L infection in mouse ear pinnae has decreased pathogenicity but no loss in its ability to activate an acquired immune response or protect against a lethal VACV challenge. Thus, vΔK1L may be a useful tool to further understand the relationship between viral modulation of innate immunity and development of immune memory, information that has practical applications for vaccine design and optimization.
MATERIALS AND METHODS
Viruses and cells.
Vaccinia virus strain Western Reserve (referred to as vK1L here) and vΔK1L were obtained from Bernard Moss (Laboratory of Viral Disease, National Institutes of Health). vΔK1L was described previously; it is a WR virus in which the K1L gene was replaced with the Escherichia coli gpt gene by homologous recombination (9). vK1Lrev is a recombinant virus in which the WR K1L gene was reinserted into vΔK1L at its natural locus. To create vK1Lrev, a DNA amplicon containing the entire K1L gene and flanking regions was PCR amplified from WR DNA using oligonucleotide primers 5′-GGGATATTAAGTGCATAGCC-3′ and 5′-CCGGAAGATCGCTATCG-3′. vK1Lrev was generated by homologous recombination, in which the K1L gene replaced the E. coli gpt gene. RK13 cells, which are nonpermissive for vΔK1L infection (23), were infected with vΔK1L and then transfected with the K1L-containing amplicon. Thus, only recombinant viruses stably expressing the K1L gene are expected to replicate in RK13 cells (74). Inoculated cellular monolayers were incubated for 48 h, and progeny viruses were isolated by plaque picking and subjected to four rounds of purification. The correct insertion of K1L was verified by PCR analysis of purified viral genomes from recombinant viruses, using two primer sets, 5′-CGGTTCCCATGATGAACGTAG-3′ and 5′-CCCATAGAACATCAGTCTCC-3′, and 5′-CATGCCGTCATGCTTAATG-3′ and 5′-TCACGCCATAACTACCACG-3′. In addition, verification of the presence of the K1L gene in vK1Lrev was done by PCR amplification using 5′-GGGATATTAAGTGCATAGCC-3′ and 5′-CCGGAAGATCGCTATCG-3′, and the resultant amplicon was sequence analyzed. vΔK1LΔC7L, which lacks both the K1L and C7L genes, was obtained from Yan Xiang (University of Texas Health Science Center) (75).
Vero, BSC-1, and RK13 cells and MEFs were cultured in Eagle's minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. Virus medium (MEM supplemented with 2.5% FBS) was used for virus adsorption phases and for virus infections. Viruses used for inoculation of mice were purified by sucrose density gradient centrifugation.
In vitro growth curves.
One-step and multistep growth curves were performed as follows. Confluent MEF monolayers were infected with vK1L, vΔK1L, vK1Lrev, or vΔK1LΔC7L at an MOI of 0.01 or 10 PFU/cell. After the initial adsorption phase, the cellular monolayers were washed three times to remove unattached viruses. At the indicated times postinfection, the cellular supernatants were removed to a new tube. The supernatants were subjected to centrifugation (800 × g for 10 min at 4°C), and the cell-free supernatants were transferred to new tubes. Cellular monolayers were collected by scraping into 1 ml virus medium. Samples were centrifuged at 800 × g for 10 min at 4°C. The cell pellets were resuspended in 1 ml of virus medium. For all samples, cells or supernatants were lysed by freeze-thawing three times. The samples were sonicated to release virions from cellular debris. Cellular lysates or supernatants were serially diluted, and 0.5 ml of each dilution was inoculated onto Vero cell monolayers in duplicate. Forty-eight hours later, plaques were visualized by staining cellular monolayers with a crystal violet dye-based solution. Data are expressed as the mean titer per sample ± SEM.
Murine intranasal and intradermal models of infection.
Female BALB/c and C57BL/6 mice (6 to 8 weeks old) were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed in AAALAC-approved animal biosafety level 2 (ABL-2) facilities at the University of Illinois at Urbana-Champaign (UIUC). All animal experiments and procedures were approved by the Institutional Animal Care and Use Committee at the UIUC.
For i.n. inoculations, BALB/c mice (n = 5 per group) were anesthetized by isoflurane (5% in O2) inhalation using a vaporizer unit (Vetland, Louisville, KY). The mice were inoculated with 104 or 105 PFU of vK1L, vΔK1L, or vK1Lrev in 20 μl (10 μl per nostril) or with PBS. The mice were weighed and monitored for signs of illness daily as a means to evaluate the severity of infection (76). Weight data were expressed as the percentage of the mean of each individual animal's weight loss from day 0. Mice that lost more than 25% of their original weight were euthanized. A clinical score for each mouse was assigned daily, based on a metric published by Berhanu et al. (27), to assess sickness. Briefly, the scoring was as follows: 0, normal; 1, slightly ruffled fur; 2, significantly ruffled fur; 3, score 2 combined with hunched posture and/or conjunctivitis; 4, score 3 combined with difficulty breathing, socializing, and/or moving; and 5, death. Clinical scores are expressed as the mean for each group.
For i.d. inoculations, anesthetized C57BL/6 mice (n = 5 per group) were inoculated i.d. in the left ear dorsal pinna with 104 PFU of vK1L, vΔK1L, or vK1Lrev in a 10-μl volume using a Hamilton (Reno, NV) syringe and a 27-gauge needle as described previously (22, 38). The infected ears were examined daily for the next 21 days for the presence of lesions. The lesion size was measured by using a 0.01-mm digital caliper (Neiko Tools, Homewood, IL). Lesion size is expressed as the mean for the group ± SEM.
In some cases, i.d.-inoculated mice were challenged with a lethal dose of vaccinia virus as a model for protective vaccination (48). Here, C57BL/6 mice (n = 5 per group) were inoculated i.d. as described above with PBS or 104 PFU of vK1L or vΔK1L. At 30 days postinfection, the mice were inoculated i.n. with vK1L (5 × 106 PFU) in a 20-μl volume and weighed daily for 7 days postinfection. Weight data were expressed as the percentage of the mean of each individual animal's weight loss from day 0. Mice that lost more than 25% of their original weight were euthanized.
Virus titers in tissues.
To assess virus replication in organs after i.n. inoculations, organs (brains, lungs, and spleens) were harvested at 2, 4, and 6 days p.i. (n = 5 mice per experimental group). The organs were flash frozen in liquid nitrogen. Whole organs were thawed on ice, weighed, and then homogenized in 0.5 ml of virus medium using a 1-ml Dounce homogenizer (Wheaton, Millville, NJ). The homogenates were subjected to three freeze-thaw cycles and sonicated. The lysates were serially diluted, and 0.5 ml of a dilution was used to inoculate Vero cellular monolayers in duplicate. Forty-eight hours later, viral titers were detected by plaque assays. Virus titers are expressed as the amount of infectious virus per gram of tissue.
To determine virus replication during i.d. infections, ears were harvested at 1, 3, 5, 8, or 11 days postinfection (n = 5 mice per experimental group) and flash frozen in liquid nitrogen. The ears were thawed on ice, weighed, and placed in 2 ml RB sample tubes (Qiagen, Valencia, CA) with 0.5 ml of virus medium and a sterile 7-mm stainless steel bead (Qiagen, Valencia, CA). Samples were subjected to two homogenization rounds at 50 Hz for 3 min using a TissueLyser LT (Qiagen, Valencia, CA). The lysates were serially diluted, and 0.5 ml of a dilution was used to inoculate Vero cellular monolayers in duplicate. Forty-eight hours later, viral titers were detected by plaque assays. Virus titers were expressed as the amount of infectious virus per gram of tissue.
RT-qPCR analysis of host genes from infected ears.
Left ears were inoculated i.d. with vK1L, vΔK1L, vK1Lrev, or PBS (n = 4 mice per group) as described above. The ears were collected at 1, 3, or 5 days postinfection and flash frozen in liquid nitrogen. Each frozen ear was cut into two equal parts, and each piece was placed into a sterile tube that previously was incubated in a dry ice-ethanol bath for 15 min to minimize RNA degradation. Each tube also contained a sterile 7-mm stainless steel bead (Qiagen, Valencia, CA). Next, the tubes were placed in the TissueLyser LT (Qiagen, Valencia, CA) for 2 min at room temperature prior to adding 0.6 ml of RLT lysis buffer containing 1% 2-mercaptoethanol to each tube. Samples were homogenized using the TissueLyser LT at 50 Hz for 3 min at room temperature. The tubes were then mixed by inverting and subjected to another round of homogenization. The homogenates from each ear half were combined in one tube and centrifuged at 18,000 × g for 3 min. The supernatants were transferred to new tubes, and 1 volume of 70% ethanol was added. Total RNA was extracted from the samples using a Qiagen RNeasy kit (Qiagen, Valencia, CA). RNA was quantified, and the integrity of the RNA was assessed by using a 48 capillary fragment analyzer (Advanced Analytical Technologies, Ankeny, IA).
Two micrograms of RNA from each ear sample was reverse transcribed to cDNA as follows. Briefly, total RNA was incubated with 0.5 μg of oligo(dT) primer (IDT, Coralville, IA) and Moloney murine leukemia virus (M-MulV) reverse transcriptase (New England BioLabs, Ipswitch, MA) at 70°C for 5 min, followed by a 5-min incubation at 4°C, according to the manufacturer's instructions. cDNA was submitted to the Carver Biotechnology Center at the University of Illinois at Urbana-Champaign for further analyses. Briefly, all cDNA samples underwent standard target amplification, in which 1.25 μl of cDNA was incubated with 2.5 μl 2× TaqMan PreAmp master mix (Thermo Fisher) and 0.5 μl 0.2× TaqMan primer-probe mix (Thermo Fisher). The primer probes used were as follows, and the assay ID number for each primer set to amplify a specific gene is in parentheses: Ccl4 (Mm00443111_m1), Cxcl2 (Mm00436450_m1), Cxcl5 (Mm00436451_g1), Ccl2 (Mm00441242_m1), Ccl5 (Mm01302427_m1), Cxcl9 (Mm00434946_m1), Cxcl10 (Mm00445235_m1), Cxcl1 (Mm04207460_m1), Il1b (Mm00434228_m1), Il6 (Mm00446190_m1), Il10 (Mm01288386_m1), Il7 (Mm01295803_m1), Tnf (Mm00443258_m1), Ifng (Mm01168134_m1), Ifna4 (Mm00833969_s1), Ifnb (Mm00439552_s1), Isg15 (Mm01705338_s1), Rnase1 (Mm00712008_m1), Mx1 (Mm00487796_m1), Eif2ak2 (Mm01235643_m1), Krt6a (Mm00833464_g1), Krt16 (Mm01306670_g1), Nfkb2 (Mm00479807_m1), Nfkbia (Mm00477798_m1), and Gapdh (Mm99999915_g1). The reaction mixtures were incubated at 95°C for 10 min, followed by 14 cycles of 95°C for 15 s and 60°C for 4 min, using the MJ Research Tetrad thermal cycler (MJ Research, Waltham, MA). A portion of each reaction mixture was then incubated with TaqMan 6-carboxyfluorescein (FAM)-MGB probes specific for each mouse gene shown in Table 1 (Thermo Fisher Scientific). Aliquots (5 μl) of each sample were loaded onto a Fluidigm 48.48 dynamic array integrated fluidic circuit (IFC) chip (Fluidigm) according to the manufacturer's directions. qPCR was performed in a Biomark HD Real-Time PCR (Fluidigm) using the following thermal conditions: 70°C for 30 min, 25°C for 10 min, and 95°C for 1 min, followed by 35 cycles of 96°C for 5 s and 60°C for 20 s.
Analysis of data was performed using Fluidigm Real Time PCR Analysis version 4.1.3. The averaged mRNA expression (fold induction) per group was quantified by calculating the 2−ΔΔCT value for each ear and then averaging these values, with values from time-matched mice inoculated with PBS as the calibrator for each day and the GAPDH or HPRT1 gene (data not shown) as the reference gene. Values were compared between ears infected with vK1L versus vΔK1L, PBS versus vK1L, PBS versus vΔK1L, or PBS versus vK1Lrev.
Ultrasound analysis of i.d.-inoculated mouse ears.
C57BL/6 mice were inoculated i.d. with vK1L, vΔK1L, or PBS or not inoculated, as described above. All the mice were anesthetized using isoflurane, and ears were imaged with the VisualSonics Vevo2100 (VisualSonics, Toronto, CA) high-frequency ultrasonic imaging system, using the MS-550S linear array, in dorsal recumbency on the indicated days postinfection. The Vevo2100 acquired the raw radio frequency (RF) image data from which (off-line) B-mode images were processed from each ear, and from those images, the ear thicknesses were estimated. Also from each B-mode image, an FOI (outlined in pink in Fig. 6A) was manually segmented to denote the region where the BSC was estimated and represented as a color-coded BSC image. The BSC is a QUS measure of the B-mode echo strength. To generate the BSC image, the FOI was divided into 75%-overlapped sub-regions of interest (sub-ROIs) with dimensions of 193 μm by193 μm (equivalent to 4 by 4 wavelengths at 32 MHz). For each sub-ROI, a BSC value was estimated using the reference phantom technique (77, 78), yielding a BSC versus ultrasonic frequency curve. Each sub-ROI's BSC value was generated by averaging the BSC over the frequency range from 25 to 43 MHz, denoted BSC (25 to 43 MHz), with a color then assigned to each BSC (25 to 43 MHz) value. To represent each ear with a single BSC value, all the sub-ROI BSC image values within the FOI were averaged (averaged over space; spatial average); these single BSC values were averaged and then graphically represented. The thickness of the ear was selected from the spatially calibrated BSC image (Fig. 6A), for which the pixel dimensions were known (68 by 68 pixels = 1 mm by 1 mm). The number of pixels was estimated in the region of the BSC image that depicted the thickest measure of the color-coded image.
Histopathology.
C57BL/6 mice were inoculated i.d. with vK1L, vΔK1L, or PBS or not inoculated, as described above. Mice (n = 3 per group) were euthanized on day 3, 4, or 5 postinfection. Ears were collected and fixed in 10% neutral buffered formalin. Three sections of each ear were taken from the inoculation site and routinely processed, paraffin embedded, sectioned, deparaffinized, and stained with hematoxylin and eosin. Representative microscopy images are shown.
Analysis of immune cell infiltration in infected ears.
Female C57BL/6 mice were inoculated i.d. in the left ear with vK1Lrev (n = 7), vΔK1L (n = 7), or PBS (n = 10) as described above. The mice were euthanized on day 5 postinfection. Each inoculated ear was collected, and the dorsal and ventral sheets were separated with forceps and incubated with type I collagenase (3 mg/ml; Worthington Biochemicals, Lakewood, NJ) and DNase I (1.25 U/ml; Worthington Biochemicals, Lakewood, NJ) for 1 h at 37°C. Each sample was then passed through a 70-μm nylon cell strainer to produce single-cell suspensions. Cells from each ear were counted via trypan blue exclusion to determine the total number of cells per ear and to ensure cellular viability. Because so few immune cells were present from each PBS-inoculated ear, cells from PBS-inoculated ears were combined to have enough cells for sufficient analyses. For the rest of the ears, there were sufficient immune cells, and cells from each ear were kept separate. The pooled cells from PBS-inoculated ears or cells from each infected ear were incubated with Fc-block (Biolegend, San Diego, CA) for 15 min and then incubated in a solution of antibodies in the presence of Fc-block and 10% mouse serum (Jackson ImmunoResearch, West Grove, PA) for 1 h. The antibodies were CD45-eFluor450 (clone 30-F11; 1:400; eBioscience, Waltham, MA), CD11b-phycoerythrin (PE) (clone M1/70; 1:200; BioLegend, San Diego, CA), CD11c-Alexa 488 (clone N418; 1:100; BioLegend), Ly6C-Alexa 700 (clone AL-21; 1:200; BD Bioscience, San Jose, CA), and Ly6G-PECy7 (clone 1A8; 1:200; BD Bioscience). All sample acquisition and compensation were performed using a BD LSRII flow cytometer (BD Bioscience). Data were analyzed with FCS Express software version 5 (DeNovo Software, Glendale, CA). Events were gated on live cells using forward versus side scatter. Dead cells were excluded on the basis of atypical fluorescence. Fluorescence minus one (FMO) was used to aid in gating of appropriate cellular populations. Leukocytes were identified as CD45+. We defined tissue-protective monocytes as CD45+ CD11b+ CD11c− Ly6C+ Ly6G+. Inflammatory monocytes were defined as CD45+ CD11b+ CD11c− Ly6C+ Ly6G− cells. CD45+ CD11c+ cells were also investigated, including dendritic cells, NK cells, and activated T cells. To account for the fact that there were different total numbers of cellular infiltrates obtained from PBS- versus vK1Lrev- versus vΔK1L-infected ears, the percent populations of different immune cells were then multiplied by the total number of cells per ear to obtain the total number of each immune cell population examined per ear. Data were expressed as the mean number of cells ± SEM per ear.
Detection of VACV-specific, IFN-γ-expressing CD8+ T cells.
C57BL/6 mice were inoculated i.d. with vK1L, vΔK1L, or PBS (n = 5 mice per experimental group) as described above. At 10 days postinfection, the mice were euthanized, and spleens and livers were harvested. Single-cell suspensions of spleens and lymph nodes were prepared as follows. Spleens were forced through a 70-μm nylon mesh (Thermo Fisher Scientific, Waltham, MA), and then erythrocytes were removed by treating samples with ammonium-chloride-potassium (ACK) red cell lysis buffer (Sigma-Aldrich). Lymph nodes were homogenized by using a BioMasher II disposable microtube homogenizer (Research Products International, Mt. Prospect, IL).
Single-cell suspensions (106 cells) of spleens or lymph nodes from each animal were kept separate, and cells from each spleen or lymph node were either unstimulated or stimulated with VACV-specific peptide (TSYKFESV; B8R20–27 epitope) for 4 h in the presence of brefeldin A (5 μg/ml; Sigma, St. Louis, MO). Using this approach, only CD8+ T cells recognizing VACV peptides become activated. Then, the cells were stained with fluorescein isothiocyanate (FITC)-conjugated anti-CD8 antibody (clone 53-6.7; 1:100; BioLegend) in cell-staining buffer (BioLegend, San Diego, CA) for 30 min on ice to stimulate VACV-specific T cells. Next, samples were fixed for 20 min at room temperature and permeabilized on ice in fixation and intracellular staining permeabilization buffer (BioLegend, San Diego, CA), respectively. The cells were stained for intracellular IFN-γ as a marker of T cell activation by incubating fixed cells with Alexa 647-conjugated anti-IFN-γ (clone XMG1.2; 1:100; BioLegend, San Diego, CA) overnight at 4°C. Samples were washed in permeabilization buffer. Flow cytometry was performed with a BD Accuri C6 cytometer (BD Bioscience, San Jose, CA), and data were analyzed with FCS Express software version 5 (DeNovo Software, Glendale, CA). Events were gated for live lymphocytes on forward versus side scatter. Dead cells were excluded on the basis of atypical fluorescence. Data were expressed as the percentage of IFN-γ+ CD8+ T cells, cells that are considered VACV-specific and activated, out of all CD8+ T cells. In addition, data were expressed as the total number of IFN-γ+ CD8+ T cells per lymph node or spleen.
Statistical analysis.
To determine statistically significant differences between weight loss and lesion sizes during vK1L and vΔK1L infection, two-way analysis of variance (ANOVA), followed by Tukey's multiple-comparison test, was performed. For viral replication in the lungs, brain, and spleen, the nonparametric Kruskal-Wallis test, followed by Dunn's multiple-comparison test, was used to determine statistically significant differences between vK1L and vΔK1L. To determine statistically significant differences in immune cell infiltration between vK1Lrev and vΔK1L, one-way ANOVA or the Kruskal-Wallis test, followed by Tukey's or Dunn's multiple-comparison test, respectively, was performed. For the analyses of RT-qPCR assays, one-way ANOVA was used to determine if there were statistically significant differences between ears infected with vK1L versus vΔK1L, PBS versus vK1L, PBS versus vΔK1L, or PBS versus vK1Lrev. To determine if there were statistically significant differences in T cell responses between vK1L and vΔK1L, one-way ANOVA, followed by Tukey's multiple-comparison test, was performed. All analyses were executed using GraphPad (La Jolla, CA) Prism software.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (R37 EB002641). A.G.B.C. was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number T32GM070421 and the National Institute of Allergy and Infectious Diseases under awards AI117105, AI078876, and AI117105.
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
We acknowledge the technical assistance of Jamie R. Kelly. We thank Heather Hickman, David Tscharke, Christopher Norbury, Christopher Brooke, Brian Ferguson, Amy MacNeill, Geoffrey Smith, Barbara Pilas, and Angela Kouris for helpful discussions and advice.
REFERENCES
- 1.Moss B. 2013. Poxviridae, 6th ed, vol 2 Lippincott Williams & Wilkins, Hagerstown, MD. [Google Scholar]
- 2.Silva-Fernandes AT, Travassos CE, Ferreira JM, Abrahao JS, Rocha ES, Viana-Ferreira F, dos Santos JR, Bonjardim CA, Ferreira PC, Kroon EG. 2009. Natural human infections with vaccinia virus during bovine vaccinia outbreaks. J Clin Virol 44:308–313. doi: 10.1016/j.jcv.2009.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Kroon EG, Mota BE, Abrahao JS, da Fonseca FG, de Souza Trindade G. 2011. Zoonotic Brazilian vaccinia virus: from field to therapy. Antiviral Res 92:150–163. doi: 10.1016/j.antiviral.2011.08.018. [DOI] [PubMed] [Google Scholar]
- 4.Costa GB, Miranda JB, Almeida GG, Silva de Oliveira J, Pinheiro MS, Goncalves SA, Pimenta Dos Reis JK, Goncalves R, Ferreira PC, Bonjardim CA, Abrahao JS, Kroon EG, Trindade GS. 2017. Detection of vaccinia virus in urban domestic cats, Brazil. Emerg Infect Dis 23:360–362. doi: 10.3201/eid2302.161341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez-Sampedro L, Perdiguero B, Mejias-Perez E, Garcia-Arriaza J, Di Pilato M, Esteban M. 2015. The evolution of poxvirus vaccines. Viruses 7:1726–1803. doi: 10.3390/v7041726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drillien R, Koehren F, Kirn A. 1981. Host range deletion mutant of vaccinia virus defective in human cells. Virology 111:488–499. doi: 10.1016/0042-6822(81)90351-2. [DOI] [PubMed] [Google Scholar]
- 7.Perkus ME, Goebel SJ, Davis SW, Johnson GP, Limbach K, Norton EK, Paoletti E. 1990. Vaccinia virus host range genes. Virology 179:276–286. doi: 10.1016/0042-6822(90)90296-4. [DOI] [PubMed] [Google Scholar]
- 8.Bravo Cruz AG, Shisler JL. 2016. The vaccinia virus K1 ankyrin repeat protein inhibits NF-kB activation by preventing RelA acetylation. J Gen Virol 97:2691–2702. doi: 10.1099/jgv.0.000576. [DOI] [PubMed] [Google Scholar]
- 9.Shisler JL, Jin XL. 2004. The vaccinia virus K1L gene product inhibits host NF-kappaB activation by preventing IkappaBalpha degradation. J Virol 78:3553–3560. doi: 10.1128/JVI.78.7.3553-3560.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Willis KL, Patel S, Xiang Y, Shisler JL. 2009. The effect of the vaccinia K1 protein on the PKR-eIF2alpha pathway in RK13 and HeLa cells. Virology 394:73–81. doi: 10.1016/j.virol.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Willis KL, Langland JO, Shisler JL. 2011. Viral double-stranded RNAs from vaccinia virus early or intermediate gene transcripts possess PKR activating function, resulting in NF-kappaB activation, when the K1 protein is absent or mutated. J Biol Chem 286:7765–7778. doi: 10.1074/jbc.M110.194704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Meng X, Schoggins J, Rose L, Cao J, Ploss A, Rice CM, Xiang Y. 2012. C7L family of poxvirus host range genes inhibits antiviral activities induced by type I interferons and interferon regulatory factor 1. J Virol 86:4538–4547. doi: 10.1128/JVI.06140-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng XZ, Jiang CH, Arsenio J, Dick K, Cao JX, Xiang Y. 2009. Vaccinia virus K1L and C7L inhibit antiviral activities induced by type I interferons. J Virol 83:10627–10636. doi: 10.1128/JVI.01260-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McCraith S, Holtzman T, Moss B, Fields S. 2000. Genome-wide analysis of vaccinia virus protein-protein interactions. Proc Natl Acad Sci U S A 97:4879–4884. doi: 10.1073/pnas.080078197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bradley RR, Terajima M. 2005. Vaccinia virus K1L protein mediates host-range function in RK-13 cells via ankyrin repeat and may interact with a cellular GTPase-activating protein. Virus Res 114:104–112. doi: 10.1016/j.virusres.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 16.Sivan G, Ormanoglu P, Buehler EC, Martin SE, Moss B. 2015. Identification of restriction factors by human genome-wide RNA interference screening of viral host range mutants exemplified by discovery of SAMD9 and WDR6 as inhibitors of the vaccinia virus K1L-C7L-mutant. mBio 6:e01122. doi: 10.1128/mBio.01122-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Meng X, Xiang Y, Deng J. 2010. Structure function studies of vaccinia virus host range protein k1 reveal a novel functional surface for ankyrin repeat proteins. J Virol 84:3331–3338. doi: 10.1128/JVI.02332-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Herbert MH, Squire CJ, Mercer AA. 2015. Poxviral ankyrin proteins. Viruses 7:709–738. doi: 10.3390/v7020709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Z, Wang S, Zhang Q, Tian M, Hou J, Wang R, Liu C, Ji X, Liu Y, Shao Y. 2013. Deletion of C7L and K1L genes leads to significantly decreased virulence of recombinant vaccinia virus TianTan. PLoS One 8:e68115. doi: 10.1371/journal.pone.0068115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smith GL, Benfield CT, Maluquer de Motes C, Mazzon M, Ember SW, Ferguson BJ, Sumner RP. 2013. Vaccinia virus immune evasion: mechanisms, virulence and immunogenicity. J Gen Virol 94:2367–2392. doi: 10.1099/vir.0.055921-0. [DOI] [PubMed] [Google Scholar]
- 21.Williamson JD, Reith RW, Jeffrey LJ, Arrand JR, Mackett M. 1990. Biological characterization of recombinant vaccinia viruses in mice infected by the respiratory route. J Gen Virol 71:2761–2767. doi: 10.1099/0022-1317-71-11-2761. [DOI] [PubMed] [Google Scholar]
- 22.Tscharke DC, Smith GL. 1999. A model for vaccinia virus pathogenesis and immunity based on intradermal injection of mouse ear pinnae. J Gen Virol 80:2751–2755. doi: 10.1099/0022-1317-80-10-2751. [DOI] [PubMed] [Google Scholar]
- 23.Sutter G, Ramsey-Ewing A, Rosales R, Moss B. 1994. Stable expression of the vaccinia virus K1L gene in rabbit cells complements the host range defect of a vaccinia virus mutant. J Virol 68:4109–4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ramsey-Ewing AL, Moss B. 1996. Complementation of a vaccinia virus host-range K1L gene deletion by the nonhomologous CP77 gene. Virology 222:75–86. doi: 10.1006/viro.1996.0399. [DOI] [PubMed] [Google Scholar]
- 25.Reading PC, Smith GL. 2003. A kinetic analysis of immune mediators in the lungs of mice infected with vaccinia virus and comparison with intradermal infection. J Gen Virol 84:1973–1983. doi: 10.1099/vir.0.19285-0. [DOI] [PubMed] [Google Scholar]
- 26.Alcami A, Smith GL. 1996. A mechanism for the inhibition of fever by a virus. Proc Natl Acad Sci U S A 93:11029–11034. doi: 10.1073/pnas.93.20.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Berhanu A, King DS, Mosier S, Jordan R, Jones KF, Hruby DE, Grosenbach DW. 2009. ST-246 inhibits in vivo poxvirus dissemination, virus shedding, and systemic disease manifestation. Antimicrob Agents Chemother 53:4999–5009. doi: 10.1128/AAC.00678-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alcami A, Smith GL. 1992. A soluble receptor for interleukin-1-beta encoded by vaccinia virus—a novel mechanism of virus modulation of the host response to infection. Cell 71:153–167. doi: 10.1016/0092-8674(92)90274-G. [DOI] [PubMed] [Google Scholar]
- 29.Mansur DS, Maluquer de Motes C, Unterholzner L, Sumner RP, Ferguson BJ, Ren H, Strnadova P, Bowie AG, Smith GL. 2013. Poxvirus targeting of E3 ligase beta-TrCP by molecular mimicry: a mechanism to inhibit NF-kappaB activation and promote immune evasion and virulence. PLoS Pathog 9:e1003183. doi: 10.1371/journal.ppat.1003183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Colamonici OR, Domanski P, Sweitzer SM, Larner A, Buller RM. 1995. Vaccinia virus B18R gene encodes a type I interferon-binding protein that blocks interferon alpha transmembrane signaling. J Biol Chem 270:15974–15978. doi: 10.1074/jbc.270.27.15974. [DOI] [PubMed] [Google Scholar]
- 31.Ember SW, Ren H, Ferguson BJ, Smith GL. 2012. Vaccinia virus protein C4 inhibits NF-kappaB activation and promotes virus virulence. J Gen Virol 93:2098–2108. doi: 10.1099/vir.0.045070-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Benfield CT, Ren H, Lucas SJ, Bahsoun B, Smith GL. 2013. Vaccinia virus protein K7 is a virulence factor that alters the acute immune response to infection. J Gen Virol 94:1647–1657. doi: 10.1099/vir.0.052670-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferguson BJ, Benfield CT, Ren H, Lee VH, Frazer GL, Strnadova P, Sumner RP, Smith GL. 2013. Vaccinia virus protein N2 is a nuclear IRF3 inhibitor that promotes virulence. J Gen Virol 94:2070–2081. doi: 10.1099/vir.0.054114-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strnadova P, Ren H, Valentine R, Mazzon M, Sweeney TR, Brierley I, Smith GL. 2015. Inhibition of translation initiation by protein 169: a vaccinia virus strategy to suppress innate and adaptive immunity and alter virus virulence. PLoS Pathog 11:e1005151. doi: 10.1371/journal.ppat.1005151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin LC, Smith SA, Tscharke DC. 2012. An intradermal model for vaccinia virus pathogenesis in mice. Methods Mol Biol 890:147–159. doi: 10.1007/978-1-61779-876-4_9. [DOI] [PubMed] [Google Scholar]
- 36.Randall CMH, Shisler J. 2013. Molluscum contagiosum virus: persistence pays off. Future Viro 8:561–573. doi: 10.2217/fvl.13.38. [DOI] [Google Scholar]
- 37.Spyrou V, Valiakos G. 2015. Orf virus infection in sheep or goats. Vet Microbiol 181:178–182. doi: 10.1016/j.vetmic.2015.08.010. [DOI] [PubMed] [Google Scholar]
- 38.Tscharke DC, Reading PC, Smith GL. 2002. Dermal infection with vaccinia virus reveals roles for virus proteins not seen using other inoculation routes. J Gen Virol 83:1977–1986. doi: 10.1099/0022-1317-83-8-1977. [DOI] [PubMed] [Google Scholar]
- 39.Brandt TA, Jacobs BL. 2001. Both carboxy- and amino-terminal domains of the vaccinia virus interferon resistance gene, E3L, are required for pathogenesis in a mouse model. J Virol 75:850–856. doi: 10.1128/JVI.75.2.850-856.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price N, Tscharke DC, Hollinshead M, Smith GL. 2000. Vaccinia virus gene B7R encodes an 18-kDa protein that is resident in the endoplasmic reticulum and affects virus virulence. Virology 267:65–79. doi: 10.1006/viro.1999.0116. [DOI] [PubMed] [Google Scholar]
- 41.Davies ML, Sei JJ, Siciliano NA, Xu RH, Roscoe F, Sigal LJ, Eisenlohr LC, Norbury CC. 2014. MyD88-dependent immunity to a natural model of vaccinia virus infection does not involve Toll-like receptor 2. J Virol 88:3557–3567. doi: 10.1128/JVI.02776-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hickman HD, Reynoso GV, Ngudiankama BF, Rubin EJ, Magadan JG, Cush SS, Gibbs J, Molon B, Bronte V, Bennink JR, Yewdell JW. 2013. Anatomically restricted synergistic antiviral activities of innate and adaptive immune cells in the skin. Cell Host Microbe 13:155–168. doi: 10.1016/j.chom.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hickman HD, Reynoso GV, Ngudiankama BF, Cush SS, Gibbs J, Bennink JR, Yewdell JW. 2015. CXCR3 chemokine receptor enables local CD8(+) T cell migration for the destruction of virus-infected cells. Immunity 42:524–537. doi: 10.1016/j.immuni.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fischer MA, Davies ML, Reider IE, Heipertz EL, Epler MR, Sei JJ, Ingersoll MA, Rooijen NV, Randolph GJ, Norbury CC. 2011. CD11b(+), Ly6G(+) cells produce type I interferon and exhibit tissue protective properties following peripheral virus infection. PLoS Pathog 7:e1002374. doi: 10.1371/journal.ppat.1002374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han AG, Erdman JW, Simpson DG, Andre MP, O'Brien WD. 2014. Early detection of fatty liver disease in mice via quantitative ultrasound. 2014 IEEE International Ultrasonics Symposium (IUS) doi: 10.1109/Ultsym.2014.0589:2363-2366._. [DOI] [Google Scholar]
- 46.Pohlhammer J, O'Brien WD Jr. 1981. Dependence of the ultrasonic scatter coefficient on collagen concentration in mammalian tissues. J Acoust Soc Am 69:283–285. doi: 10.1121/1.385349. [DOI] [PubMed] [Google Scholar]
- 47.Chen RA, Jacobs N, Smith GL. 2006. Vaccinia virus strain Western Reserve protein B14 is an intracellular virulence factor. J Gen Virol 87:1451–1458. doi: 10.1099/vir.0.81736-0. [DOI] [PubMed] [Google Scholar]
- 48.Jacobs N, Chen RA, Gubser C, Najarro P, Smith GL. 2006. Intradermal immune response after infection with vaccinia virus. J Gen Virol 87:1157–1161. doi: 10.1099/vir.0.81556-0. [DOI] [PubMed] [Google Scholar]
- 49.Cush SS, Reynoso GV, Kamenyeva O, Bennink JR, Yewdell JW, Hickman HD. 2016. Locally produced IL-10 limits cutaneous vaccinia virus spread. PLoS Pathog 12:e1005493. doi: 10.1371/journal.ppat.1005493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Haskill S, Beg AA, Tompkins SM, Morris JS, Yurochko AD, Sampson-Johannes A, Mondal K, Ralph P, Baldwin AS Jr. 1991. Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell 65:1281–1289. doi: 10.1016/0092-8674(91)90022-Q. [DOI] [PubMed] [Google Scholar]
- 51.Sun SC, Ganchi PA, Ballard DW, Greene WC. 1993. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science 259:1912–1915. doi: 10.1126/science.8096091. [DOI] [PubMed] [Google Scholar]
- 52.Shakhov AN, Kuprash DV, Azizov MM, Jongeneel CV, Nedospasov SA. 1990. Structural analysis of the rabbit TNF locus, containing the genes encoding TNF-beta (lymphotoxin) and TNF-alpha (tumor necrosis factor). Gene 95:215–221. doi: 10.1016/0378-1119(90)90364-W. [DOI] [PubMed] [Google Scholar]
- 53.Collart MA, Baeuerle P, Vassalli P. 1990. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol 10:1498–1506. doi: 10.1128/MCB.10.4.1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Libermann TA, Baltimore D. 1990. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol 10:2327–2334. doi: 10.1128/MCB.10.5.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lombardi L, Ciana P, Cappellini C, Trecca D, Guerrini L, Migliazza A, Maiolo AT, Neri A. 1995. Structural and functional characterization of the promoter regions of the NFKB2 gene. Nucleic Acids Res 23:2328–2336. doi: 10.1093/nar/23.12.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilcock D, Duncan SA, Traktman P, Zhang WH, Smith GL. 1999. The vaccinia virus A4OR gene product is a nonstructural, type II membrane glycoprotein that is expressed at the cell surface. J Gen Virol 80:2137–2148. doi: 10.1099/0022-1317-80-8-2137. [DOI] [PubMed] [Google Scholar]
- 57.Stanford MM, McFadden G, Karupiah G, Chaudhri G. 2007. Immunopathogenesis of poxvirus infections: forecasting the impending storm. Immunol Cell Biol 85:93–102. doi: 10.1038/sj.icb.7100033. [DOI] [PubMed] [Google Scholar]
- 58.van Buuren N, Burles K, Schriewer J, Mehta N, Parker S, Buller RM, Barry M. 2014. EVM005: an ectromelia-encoded protein with dual roles in NF-kappaB inhibition and virulence. PLoS Pathog 10:e1004326. doi: 10.1371/journal.ppat.1004326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Haring JS, Badovinac VP, Harty JT. 2006. Inflaming the CD8+ T cell response. Immunity 25:19–29. doi: 10.1016/j.immuni.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 60.Badovinac VP, Messingham KA, Jabbari A, Haring JS, Harty JT. 2005. Accelerated CD8+ T-cell memory and prime-boost response after dendritic-cell vaccination. Nat Med 11:748–756. doi: 10.1038/nm1257. [DOI] [PubMed] [Google Scholar]
- 61.Stelekati E, Shin H, Doering TA, Dolfi DV, Ziegler CG, Beiting DP, Dawson L, Liboon J, Wolski D, Ali MA, Katsikis PD, Shen H, Roos DS, Haining WN, Lauer GM, Wherry EJ. 2014. Bystander chronic infection negatively impacts development of CD8(+) T cell memory. Immunity 40:801–813. doi: 10.1016/j.immuni.2014.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bowie A, Kiss-Toth E, Symons JA, Smith GL, Dower SK, O'Neill LA. 2000. A46R and A52R from vaccinia virus are antagonists of host IL-1 and Toll-like receptor signaling. Proc Natl Acad Sci U S A 97:10162–10167. doi: 10.1073/pnas.160027697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen RA, Ryzhakov G, Cooray S, Randow F, Smith GL. 2008. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog 4:e22. doi: 10.1371/journal.ppat.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Myskiw C, Arsenio J, van Bruggen R, Deschambault Y, Cao J. 2009. Vaccinia virus E3 suppresses expression of diverse cytokines through inhibition of the PKR, NF-kappaB, and IRF3 pathways. J Virol 83:6757–6768. doi: 10.1128/JVI.02570-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schroder M, Baran M, Bowie AG. 2008. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J 27:2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gedey R, Jin XL, Hinthong O, Shisler JL. 2006. Poxviral regulation of the host NF-kappaB response: the vaccinia virus M2L protein inhibits induction of NF-kappaB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J Virol 80:8676–8685. doi: 10.1128/JVI.00935-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.DiPerna G, Stack J, Bowie AG, Boyd A, Kotwal G, Zhang Z, Arvikar S, Latz E, Fitzgerald KA, Marshall WL. 2004. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by Toll-like receptors. J Biol Chem 279:36570–36578. doi: 10.1074/jbc.M400567200. [DOI] [PubMed] [Google Scholar]
- 68.Stack J, Haga IR, Schroder M, Bartlett NW, Maloney G, Reading PC, Fitzgerald KA, Smith GL, Bowie AG. 2005. Vaccinia virus protein A46R targets multiple Toll-like-interleukin-1 receptor adaptors and contributes to virulence. J Exp Med 201:1007–1018. doi: 10.1084/jem.20041442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harte MT, Haga IR, Maloney G, Gray P, Reading PC, Bartlett NW, Smith GL, Bowie A, O'Neill LAJ. 2003. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Exp Med 197:343–351. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bartlett N, Symons JA, Tscharke DC, Smith GL. 2002. The vaccinia virus N1L protein is an intracellular homodimer that promotes virulence. J Gen Virol 83:1965–1976. doi: 10.1099/0022-1317-83-8-1965. [DOI] [PubMed] [Google Scholar]
- 71.Goulding J, Abboud G, Tahiliani V, Desai P, Hutchinson TE, Salek-Ardakani S. 2014. CD8 T cells use IFN-gamma to protect against the lethal effects of a respiratory poxvirus infection. J Immunol 192:5415–5425. doi: 10.4049/jimmunol.1400256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tartaglia J, Perkus ME, Taylor J, Norton EK, Audonnet JC, Cox WI, Davis SW, Vanderhoeven J, Meignier B, Riviere M, Languet B, Paoletti E. 1992. Nyvac—a highly attenuated strain of vaccinia virus. Virology 188:217–232. doi: 10.1016/0042-6822(92)90752-B. [DOI] [PubMed] [Google Scholar]
- 73.Moss B. 2013. Reflections on the early development of poxvirus vectors. Vaccine 31:4220–4222. doi: 10.1016/j.vaccine.2013.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Staib C, Drexler I, Ohlmann M, Wintersperger S, Erfle V, Sutter G. 2000. Transient host range selection for genetic engineering of modified vaccinia virus Ankara. Biotechniques 28:1137–1142, 1144–1136, 1148. [DOI] [PubMed] [Google Scholar]
- 75.Meng X, Xiang Y. 2006. Vaccinia virus K1L protein supports viral replication in human and rabbit cells through a cell-type-specific set of its ankyrin repeat residues that are distinct from its binding site for ACAP2. Virology 353:220–233. doi: 10.1016/j.virol.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 76.Symons JA, Alcami A, Smith GL. 1995. Vaccinia virus encodes a soluble type I interferon receptor of novel structure and broad species specificity. Cell 81:551–560. doi: 10.1016/0092-8674(95)90076-4. [DOI] [PubMed] [Google Scholar]
- 77.Lin SC, Heba E, Wolfson T, Ang B, Gamst A, Han A, Erdman JW Jr, O'Brien WD Jr, Andre MP, Sirlin CB, Loomba R. 2015. Noninvasive diagnosis of nonalcoholic fatty liver disease and quantification of liver fat using a new quantitative ultrasound technique. Clin Gastroenterol Hepatol 13:1337–1345 e6. doi: 10.1016/j.cgh.2014.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yao LX, Zagzebski JA, Madsen EL. 1990. Backscatter coefficient measurements using a reference phantom to extract depth-dependent instrumentation factors. Ultrasonic Imaging 12:58–70. [DOI] [PubMed] [Google Scholar]