

ABSTRACT
The molecular constraints affecting Zika virus (ZIKV) evolution are not well understood. To investigate ZIKV genetic flexibility, we used transposon mutagenesis to add 15-nucleotide insertions throughout the ZIKV MR766 genome and subsequently deep sequenced the viable mutants. Few ZIKV insertion mutants replicated, which likely reflects a high degree of functional constraints on the genome. The NS1 gene exhibited distinct mutational tolerances at different stages of the screen. This result may define regions of the NS1 protein that are required for the different stages of the viral life cycle. The ZIKV structural genes showed the highest degree of insertional tolerance. Although the envelope (E) protein exhibited particular flexibility, the highly conserved envelope domain II (EDII) fusion loop of the E protein was intolerant of transposon insertions. The fusion loop is also a target of pan-flavivirus antibodies that are generated against other flaviviruses and neutralize a broad range of dengue virus and ZIKV isolates. The genetic restrictions identified within the epitopes in the EDII fusion loop likely explain the sequence and antigenic conservation of these regions in ZIKV and among multiple flaviviruses. Thus, our results provide insights into the genetic restrictions on ZIKV that may affect the evolution of this virus.
IMPORTANCE Zika virus recently emerged as a significant human pathogen. Determining the genetic constraints on Zika virus is important for understanding the factors affecting viral evolution. We used a genome-wide transposon mutagenesis screen to identify where mutations were tolerated in replicating viruses. We found that the genetic regions involved in RNA replication were mostly intolerant of mutations. The genes coding for structural proteins were more permissive to mutations. Despite the flexibility observed in these regions, we found that epitopes bound by broadly reactive antibodies were genetically constrained. This finding may explain the genetic conservation of these epitopes among flaviviruses.
KEYWORDS: Zika virus, genetic mapping, mutagenesis, transposons
INTRODUCTION
Despite progress in the development of effective vaccines (1, 2), Zika virus (ZIKV) remains a major public health problem due to the association of infections with serious birth defects and Guillain-Barré syndrome (3, 4). ZIKV belongs to the genus Flavivirus in the family Flaviviridae. These viruses have single-stranded positive-sense RNA genomes of about 11,000 nucleotides that are replicated by an error-prone RNA-dependent RNA polymerase (5). The high mutation rate of such enzymes allows RNA viruses to develop extraordinary sequence diversity and to rapidly adapt to selective pressures (5). However, the sequences of RNA virus genomes are optimized to encode the minimal set of functions required for the viral life cycle. The genetic compression of flaviviruses is highlighted by the fact that the mature proteins have multiple functions (6, 7). Therefore, despite the capacity to generate sequence diversity, genetic constraints incurred from genome optimization prevent regions of flaviviruses from tolerating mutations. Bioinformatic analysis of ZIKV strains demonstrates that certain regions of the genome are conserved, while other areas are highly variable (8). However, such historical sequence analysis may not reveal the full genetic potential of a virus.
In this study, transposon mutagenesis was combined with deep sequencing to assess the genetic constraints on the ZIKV genome. A 15-nucleotide transposon insertion was randomly integrated into the viral genome; this viral library was passaged in tissue culture and all tolerated insertion sites were identified. In previous studies, this methodology demonstrated that the genomic region encoding the major antigenic target (the hemagglutinin) of the antigenically diverse influenza A virus was tolerant of insertions (9). In contrast, such mutations were not well tolerated in the region encoding the conserved hemagglutinin of the serologically monotypic measles virus (10). Therefore, tolerance of an insertion in a ZIKV genomic region may be an indirect predictor of its genetic flexibility and adaptive potential.
The transposon mutagenesis screen revealed large differences in insertional tolerance between ZIKV regions. Most nonstructural (NS) genes involved in RNA replication were refractory to insertional mutagenesis. However, distinct patterns of insertional tolerance in the NS1 gene were observed at different stages of library selection. This result may reflect the multiple functions of NS1 at separate stages of the viral life cycle. The ZIKV structural proteins were more tolerant of insertions than the NS proteins, with particular mutational tolerance observed in the surface-exposed residues of the ZIKV envelope protein. Nevertheless, we did not detect mutants with insertions in regions of this protein that are bound by broadly neutralizing antibodies. This finding highlights the fact that cross-reactive epitopes are often under genetic constraints that make them highly conserved (9).
RESULTS
Screening of a ZIKV transposon library.
We recently reported the construction of a cDNA clone of the African lineage 1947 Uganda MR766 prototype ZIKV strain (11). This plasmid efficiently produces infectious viruses when transfected into permissive cells. MR766 cDNA clones were generated by randomly integrating a single 15-nucleotide insertion using a transposon-based method as previously described (9, 10) (Fig. 1A). This insertion did not carry stop codons in any open reading frame, thus resulting in a five-amino-acid insertion when integrated into a coding sequence. This library contained over 1 × 106 independent bacterial colonies, which is equivalent to ∼90-fold coverage of the viral genome (1 × 106 colonies/11,000 virus-derived nucleotides per infectious clone). The protocol utilized to generate this library was optimized to ensure that the majority of viral genomes carried only a single insertion (see Materials and Methods).
FIG 1.

ZIKV insertional mutant library screening protocol and deep sequencing results. (A) Representative images of individual mutants from the library of ZIKV cDNA clones (blue), each bearing a single 15-nucleotide insertion, represented by a colored line (not drawn to scale). (B) 293T cells were transfected with the mutant plasmid library to select for mutant genomes that could initiate RNA replication and virus release. (C) Forty-eight hours after transfection, the supernatant was collected and passaged on Vero cells to select mutants that retained the ability to spread. (D) At 48 h postinfection, the supernatant was passaged once more on Vero cells to select for the fittest mutants. At the time of supernatant collection, RNAs from all three of the cell populations were subjected to RT-PCR to amplify the viral genomes. The subsequent RT-PCR products were then deep sequenced. (E) Raw number of deep sequence reads with inserts at each location for the input plasmid library. The ZIKV genome schematic and bars in the graph show structural genes in red, nonstructural genes in blue, UTRs in black, and the intron used to stabilize the plasmid in bacteria in yellow. This intron was not present in the rescued viruses. Alternating gray and white shading within each graph differentiates the genomic regions. (F to H) Frequencies of insertions at each location following normalization to the representation in the input library after plasmid DNA transfection (F), infected cell passage 1 (G), and infected cell passage 2 (H), with a schematic of the genome shown below. Numbering along the x axis indicates the nucleotide position in the ZIKV genome, while the y axis gives the log raw number of reads (E) or the log fold change over input (F to H), with horizontal dotted lines representing the cutoff of the mutants enriched two standard deviations above the average. Values are averages for three independent screens (see Materials and Methods for details).
A pool of mutant viruses was rescued by transfecting 293T cells with the plasmid library (Fig. 1B). This and all subsequent selection steps were performed in triplicate to verify reproducibility. The titer of the rescued mutant library was 6.47 × 104 (± 2.72 × 104 [standard error of the mean]) 50% tissue culture infective doses (TCID50)/ml. This rescue titer is >1,000-fold lower than that typically recovered with the wild-type (WT) ZIKV plasmid and likely reflects the detrimental impact of the majority of transposon insertions (11). Supernatants collected 2 days after transfection were used to infect Vero cells to select for viral mutants based on their relative capacity to spread (Fig. 1C). To maintain library complexity, these cells were infected with 1.29 × 106 (± 5.43 × 105 [standard error of the mean]) TCID50 at a multiplicity of infection (MOI) of ∼0.05 infectious unit per cell. Two days following infection, supernatants were collected and passaged once more on Vero cells at an MOI of ∼0.5 (Fig. 1D). Because infected cells release progeny virus in less than 24 h, we estimate that each supernatant passage represented two or three rounds of the complete viral life cycle. This process allowed for progressive enrichment of the mutant genomes based on their relative fitness levels.
Deep sequencing revealed that insertional coverage throughout the ZIKV genome was high for the plasmid library (Fig. 1E). A total of 57% of the nucleotides and 86% of the codons were mutagenized in this ZIKV library. This is comparable to or higher than the mutational coverage used in previously published transposon libraries of other RNA viruses (9, 10, 12, 13). Thus, we believe that this level of mutational coverage is sufficient to survey the insertional tolerance of each genomic region of ZIKV. Comparison of the frequency of each insertional mutant in the plasmid library (Fig. 1E) and its frequencies within transfected (Fig. 1F) and serially infected (Fig. 1G and H) cells showed that regions of ZIKV displayed variable tolerance for insertions after selection. At 2 days posttransfection, over 54% of all viral mutants in the original plasmid library were no longer detected (Fig. 1F). The requirement to assemble infectious particles and infect new cells presented an even greater selection pressure, as over 94% of mutants did not survive passaging (Fig. 1G and H). The abundances and locations of insertions in the three independent replicate screens exhibited high degrees of reproducibility (Fig. 2). It is possible that mutant viruses acquired compensatory mutations required for the viability of a few insertion sites. However, spontaneous compensatory mutations are infrequent and therefore were unlikely to greatly influence the representation of these sites in the replicate screens.
FIG 2.

Demonstration of consistency between replicates of the screened library. Each graph shows a comparison of the frequencies of each mutant in the genome between replicates. Pairwise analysis of the representations of each insertion mutant between replicates of the screen for the transfection (A to C), passage 1 (D to F), and passage 2 (G to I) cell populations shows that there is a high and statistically significant degree of reproducibility. The x and y axes show the log fold enrichment of each insertion at each site normalized to the input library. The r and P values for Spearman's correlation are displayed.
The ZIKV genomic regions required for RNA replication are intolerant of transposon mutagenesis.
Figure 3A graphically depicts the progressive levels of selection observed during the transfection and passage stages of the screen. After supernatant passaging through Vero cells, insertions were more frequently enriched in the ZIKV structural proteins (Fig. 3A and B). Among the 105 mutations that were most highly represented (2 standard deviations above the average) in the passage 2 cell population (Fig. 1H, data above the horizontal dotted line), 58 of the sites were located in envelope (E), 14 in the membrane glycoprotein precursor (prM), 12 in the capsid (C), 10 in NS1, 7 in NS2A, 2 in NS3, 1 in NS5, and 1 in the 3′ untranslated region (UTR). No highly enriched insertions were detected in the 5′ UTR, NS2B, NS4A, the 2k peptide, or NS4B. None of the viable insertions were located in the sequences encoding the NS3 and NS5 enzyme active sites or the RNA binding domains. One permissive site was identified in the 3′ UTR, inside a region previously shown to be involved in flavivirus host adaptation and to be dispensable for replication (14). These results indicate that the nonstructural genes and the UTRs involved in RNA replication are particularly sensitive to transposon mutagenesis.
FIG 3.

Tolerance for insertions in the ZIKV genome following library transfection and passaging. (A) Viral mutant progression plots showing the relative abundances of individual insertion sites in cells following transfection and supernatant passage 2. Horizontal colored lines on the left side of the graph (genome portion) denote the location of each insertion in the ZIKV genome illustration, which is drawn to scale on the left. The thicknesses of the lines change in the center and the right of the graph to represent the proportions of each insertional mutant in the transfection and passage 2 cell populations, respectively. The schematic of the ZIKV genome to the right of the graph is distorted to show the relative abundance of insertions in each region after passaging. (B) To determine the insertional tolerance of each region of the ZIKV genome, the number of mutants selected at a level >2 standard deviations above average in passage 2 was divided by the total number of mutants in the genomic region in the input library. The values were then set relative to an arbitrary value of 1 for the region that best tolerated insertions in each replicate of the screen. The values displayed are the averages for the three replicate screens, while the error bars represent the standard deviations.
Viruses bearing individual insertion sites are recoverable and grow to high titers in multiple cell lines.
To validate the results of the above screen, 18 of the 105 highly enriched viruses from the second passage were individually reconstructed (Table 1). All of these viruses were recoverable, thus demonstrating that the screen specifically selected for viable mutants. Substantial insertional tolerance was observed in the structural proteins of ZIKV in this screen. For this reason, six mutant viruses bearing insertions in the structural proteins were selected for further study. First, the multicycle growth characteristics of these six mutants were assayed in Vero cells. These mutants grew to titers comparable to those of the WT virus (Fig. 4A). To test if the fitness of the mutants bearing insertions in structural proteins was mammalian cell line specific, the growth kinetics of the mutants were assayed on C6/36 and Aag2 mosquito cells (Fig. 4B and C). Again, all of the viruses displayed growth kinetics similar to those of the WT virus, indicating that the genetic flexibility observed in the structural proteins may not be specific to the mammalian cell lines utilized in this screen.
TABLE 1.
Rescued viruses bearing individual insertion sitesa
| Genomic region | Nucleotide preceding insertion | Codon preceding insertion | Translation of insert |
|---|---|---|---|
| C | 188 | 27 | VRPHL |
| 404 | 99 | VRPQK | |
| anC | 429 | 3 | TAAAD |
| 438 | 6 | GAAAI | |
| pr | 729 | 85 | GAAAK |
| 740 | 89 | LRPQR | |
| M | 777 | 8 | TAAAS |
| 789 | 12 | HAAAL | |
| E | 1445 | 156 | VRPHT |
| 1599 | 207 | NAAAN | |
| 1815 | 279 | AAAAG | |
| 1818 | 280 | NAAAA | |
| 2019 | 347 | DAAAV | |
| 2028 | 350 | TAAAQ | |
| NS1 | 3543 | 351 | AAAAT |
| NS2A | 3552 | 2 | TAAAS |
| NS3 | 4664 | 17 | MRPQE |
| NS5 | 8610 | 314 | HAAAT |
Individual insertional mutants were cloned, and viruses were recovered.
FIG 4.
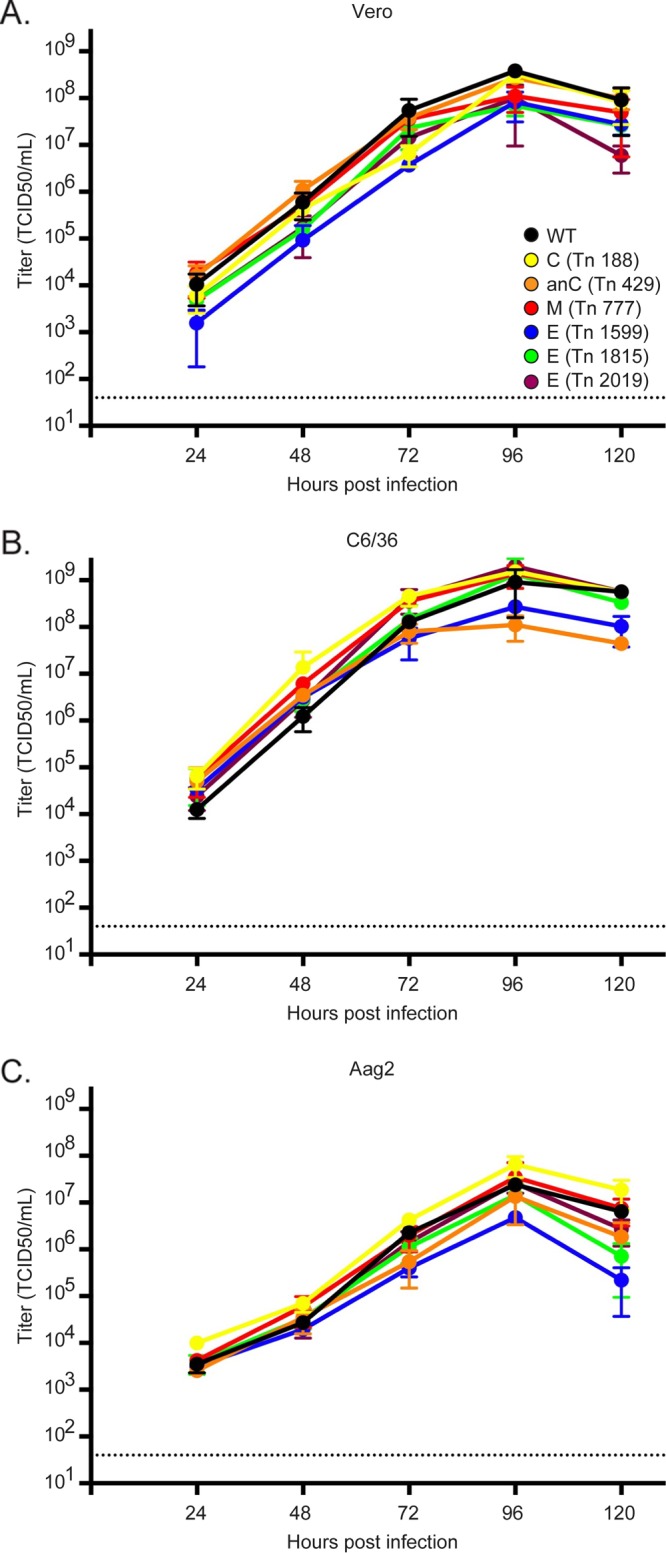
Viruses bearing insertions in the structural genes grow to high titers in mammalian and mosquito cells. Growth curves are shown for insertional mutants (colored lines) in comparison to the wild-type virus (black lines) in Vero (A), C6/36 (B), and Aag2 (C) cells. Cells were infected at an MOI of 0.01. Supernatants were collected at the indicated time points and titrated by TCID50 assays. Data points are the means for three replicates, while the error bars represent the standard deviations. The horizontal dashed line in each panel represents the limit of detection.
NS1 mutations are differentially selected in the transfection and passaging steps.
NS1 mutants exhibited a distinctive pattern of selection in the screen (Fig. 5). Clusters of mutations in both the wing domain and the β-ladder of NS1 were enriched in the transfected cell populations but were lost during passaging (Fig. 5A and B). Indeed, a mutant with an insertion after nucleotide 3011 of the genome (or after amino acid 174 of NS1) was enriched >650-fold in transfected cells. Four other sites in NS1 and seven sites in other regions (C, NS4B, and NS5) were enriched 10-fold in the transfected cell population (Fig. 1F). This selection pattern indicates that these insertions do not impair, and may even enhance, RNA replication but that they antagonize viral spread. Indeed, four of these sites in NS1 fall near dengue virus (DENV) single amino acid mutations known to disrupt interactions with C and NS4B (Fig. 5A, DENV motifs, and B, dimer structures) that are required for viral assembly (15, 16).
FIG 5.

NS1 insertional tolerance illustrates separate regions required for RNA replication and immune evasion. (A) Linear schematic of NS1 with the major structural domains labeled, with numbering corresponding to the NS1 amino acid number for each domain junction. Heat maps show the relative transposon insertion frequencies in cells following transfection and supernatant passage 2, and are colored from dark blue (0 reads with inserts) to yellow or red (2 standard deviations above average) to indicate the degree of selection at each insertion site. Tn 3011 indicates the location of the insertion mutant that was enriched 650-fold in transfected cells. Spheres below the heat maps indicate the positions of known DENV NS1 motifs, including NS4B-interacting residues (magenta; residues 10 and 11), C-interacting residues (green; residues 180 and 301), and the N-linked glycan (cyan; residue 207). Space-filling NS1 structures (Protein Data Bank [PDB] entry 5K6K) are color coded to indicate the frequencies of insertions in transfected (B) and passage 2 (C) cells (46). The heat map scale and colored spheres correspond to locations of known DENV motifs as shown in panel A. The antigenic domain of NS1 is also labeled in the dimer view in panel C. The 5K6K NS1 hexamer was aligned to the previously published ZIKV hexamer model (PDB entry 5IY3) (47).
Different sets of NS1 mutants in the β-ladder and wing domains became predominant following passaging (Fig. 5A and C, passage 2). These mutations occurred in two distinct clusters in NS1. One cluster of insertions, ZIKV NS1 amino acids 206 to 211, overlapped an N-linked glycosylation mark at amino acid position 207. A glycan at this position in DENV has been demonstrated to be required for secretion of the NS1 hexamer but is not essential for DENV RNA replication or growth in vitro (17–19). Secretion of the NS1 hexamer is thought to play a major role in DENV pathogenesis and immune evasion in vivo (20). The second enriched insertion cluster was located at the C terminus of NS1, on the surface of the tip of the β-ladder, a region implicated as a major antigenic domain of DENV NS1 (21). No mutations were selected in the β-roll of NS1, which is believed to be required for efficient RNA replication (Fig. 5A and C).
Insertions in the exposed surfaces of the envelope protein.
Viral glycoproteins are a major target of neutralizing antibodies and are often under strong selective pressures. However, the capacity to tolerate sequence diversity and to evade adaptive immune responses varies among RNA viruses. The screen revealed extensive genetic flexibility in the ZIKV E protein in the absence of these selective pressures. Over 55% of the most frequently selected mutants were located in the E protein. Furthermore, the ZIKV E gene was far more flexible than the antigenically monotypic measles virus hemagglutinin gene (Fig. 6A). All of the 58 selected E protein insertion sites were found in the ectodomain of this protein (Fig. 6B). Envelope domain I (EDI) contained 34 of the enriched sites. Only 4 insertions were found in envelope domain II (EDII), and none of these were near the highly conserved fusion loop in this region (Fig. 6B). The majority of these insertions were found near a glycan in the 150-loop of the E protein, which has been observed to be a highly variable domain in ZIKV as well as in flaviviruses in general (22, 23) (Fig. 7A and B). Envelope domain III (EDIII) contained 20 of these selected insertion sites. All of these recovered insertions occurred in solvent-exposed E residues in the ZIKV E crystal structure (Fig. 7B). Among these residues, 66% were located on the surface of the virus (Fig. 7C). The remaining 34% of enriched E protein mutants had insertions in the EDIII C-C′ loop (Fig. 7A and B, side view), which is not exposed on the surface of the mature virion but is a known binding site for ZIKV antibodies (24).
FIG 6.

The ZIKV E gene is tolerant of insertional mutagenesis. (A) Comparison of ZIKV E and measles virus hemagglutinin (MeV H) insertional tolerances. The graph shows the percentage of insertional mutants in each glycoprotein coding sequence in the input library that were enriched >2 standard deviations above average following passaging (see Materials and Methods for analysis details). (B) (Left) Crystal structure (PDB entry 5IRE) of the E ectodomain monomer (48). The protein domains EDI (orange), EDII (cyan), EDIII (green), and fusion loop (FL) (magenta) are indicated. (Right) Viral progression plots showing the selection of insertion sites in passage 2. The linear representation of the E protein to the left of the plot is drawn to scale, while the illustration to the right is distorted to match the relative insertion abundance in each domain at passage 2.
FIG 7.

Mapping of insertion sites and broadly protective epitopes on the ZIKV E protein. (A) Top (as if viewed from outside the virion) and side views of the ribbon structure of the ZIKV E dimer complex crystal structure (PDB entry 5IRE) (48). The different protein domains are labeled and highlighted (EDI, orange; EDII, cyan; EDIII, green; fusion loop, magenta; and transmembrane domain and cytoplasmic tail, gray). The E protein 150-loop, fusion loop, and C-C′ loops are indicated. (B) The same crystal structure views, but with the transposon insertion frequencies at passage 2 indicated in colors from dark blue (0 reads with inserts) to red (2 standard deviations above average) on the surface structure of the E dimer complex. (C) Insertion frequencies mapped on the surface view of the whole ZIKV virion (48). (D to F) E protein structures showing both the passage 2 insertion frequencies (using the same heat map as that in panel C) and the binding sites (yellow outlines) of the 4G2 (D), C8 (E), and C10 (F) E antibodies. Antibody binding sites, which were previously mapped on the DENV E protein, were inferred by comparisons of the ZIKV and DENV protein sequences (27, 29).
The patterns of E protein genetic flexibility may have important consequences for the ability of ZIKV to evade adaptive immune responses. Antibodies generated against other flaviviruses can bind and/or neutralize ZIKV (25). The conserved EDII fusion loop, which was intolerant of transposon inserts, has been reported to be the target of a pan-flavivirus antibody, 4G2 (Fig. 7D) (26, 27). DENV envelope dimer epitope 1 (EDE1) antibodies, such as the C8 or C10 antibody, potently neutralize a broad range of DENV and ZIKV isolates (28). The ZIKV E protein regions targeted by these EDE1 antibodies did not tolerate insertions (Fig. 7E and F) (29). The genetic constraints identified within these regions may contribute to the conservation of these epitopes in multiple flaviviruses. Thus, this ZIKV transposon screen likely identifies genetic constraints of the E protein that affect the breadth of antibody responses to these viruses.
DISCUSSION
The capacity for a virus to exhibit genetic diversity is determined by the balance between maintaining sequences required for replication and the need to adapt to selective pressures. Here we report a genome-wide transposon mutagenesis screen that revealed the genetic capacity of ZIKV to tolerate insertions. A transposon mutagenesis approach is one strategy to screen for genetic flexibility within a viral genome. An alternative approach is to create a library of mutant viruses carrying single amino acid mutations (30–35). However, not all amino acid substitutions will significantly impair protein function. In comparison, the five-amino-acid insertion incorporated by transposon mutagenesis is more likely to affect both protein interaction interfaces and secondary structure. Thus, tolerance of a transposon-mediated insertion is a more stringent indicator of genetic flexibility of a genomic or protein domain, although this mutation lacks the resolution provided by single amino acid mutations. The results from both types of screens can be overlapped to define both domains that tolerate insertions and single amino acid changes within those domains that affect protein function.
We found that insertions in a vast majority of sites in the ZIKV genome inhibited viral replication and spread. We believe that this reflects the constraints placed on viruses with small RNA genomes to package required functions into a minimal amount of genetic space. Indeed, similar transposon screens showed that a variety of genetically distant viruses exhibit comparable intolerances to insertional mutagenesis (9, 10, 12, 13, 36). Such screens have one underlying principle: they select for mutant viruses that have the ability to replicate and spread in vitro. Thus, mutants lost during screening are likely impaired for RNA replication, assembly, or infection. Furthermore, these screens are performed in the absence of adaptive immune pressures and with cell lines with impaired interferon response pathways. Under these conditions, the genetic flexibility of the regions involved in viral immune antagonism or pathogenesis can be measured. In line with these assumptions, we found that the nonstructural (NS) genes involved in the RNA replication complex and the majority of the untranslated regions of the ZIKV genome were mostly intolerant of transposon mutagenesis. However, the genomic regions involved in viral antagonism and immune recognition, such as those encoding the NS1 protein and the surfaces of the envelope glycoprotein, displayed various degrees of genetic flexibility.
Clusters of NS1 insertion mutants were greatly enriched in the transfected cell populations, suggesting that these insertions are neutral or may even enhance viral RNA replication (Fig. 1 and 3). However, these mutants were lost during passaging of the rescued viruses. While these insertions are in different domains of the NS1 structure, they do occur near mutations previously shown to enhance DENV RNA replication and to negatively affect virion assembly (15, 16). We hypothesize that the present transposon screen highlights a similar role for the ZIKV NS1 protein in the transition between viral RNA replication and virion assembly. Additionally, sites in the NS4B and NS5 proteins were positively selected >10-fold in the transfected cell populations but were lost during library passage. The insertions in these genes may provide a map for residues involved in controlling viral packaging, a step following RNA replication.
Distinct NS1 insertions were enriched after passaging. Viruses tolerated insertions in two clusters of the β-ladder of the NS1 protein important for flavivirus immune evasion and pathogenesis. The first cluster of insertions was located around the N-glycosylation site, at amino acid position 207 in the NS1 protein (Fig. 5). This motif is dispensable for DENV cell culture replication (17, 37) but is required for secretion of the NS1 hexamer (18). Secretion of the NS1 hexamer has been shown to be involved in host immune evasion and neurovirulence of DENV in mice (17, 20). The second cluster of NS1 insertions selected during passaging was located at the distal tip of the NS1 protein. This region contains several linear epitopes in NS1 of DENV that are targeted by the adaptive immune response (21). Although further characterization is required to precisely define the multiple functions of the ZIKV NS1 protein, our findings imply that regions involved in host immune evasion and pathogenesis are genetically flexible in the ZIKV genome.
Insertion sites in the structural proteins C, prM, and E were enriched after passaging of the mutant viruses. Specifically, the E protein, which sits on the virus surface, was highly tolerant of transposon mutagenesis (Fig. 3). This was unexpected, because ZIKV appears to be monotypic, with little antigenic variation since its discovery in 1947 (38). There are several explanations for these findings. First, the highly variable 150-loop of the E protein may be associated with poor immunogenicity and/or the antibodies targeting this area may not select for antigenic variants. Even though this domain would allow for genetic or antigenic variation, it does not result in serological variability. A second explanation for the insertional tolerance of the E protein is that genetic variability is needed by ZIKV because the virus requires a mosquito vector. Replication in the different tissues/organs of the mosquito may result in additional genetic bottlenecks (39, 40), and only viruses that have the genetic “leeway” to tolerate changes may effectively survive. We examined whether the insertion mutants showed differential growth characteristics in mosquito cell lines, but we were unable to identify differences in growth. All of the reconstructed insertion mutants that were enriched in the passage 2 population of our screen had growth kinetics comparable to that of the wild-type virus. Nevertheless, it cannot be excluded that the growth and spread of ZIKV in the tissues of two different hosts (mosquitoes and humans) require an extraordinary genetic adaptability. Therefore, we cannot rule out that screening the ZIKV library in insect cell lines may reveal different insertional tolerances in the genome.
Genetic restrictions and sites intolerant of transposon mutagenesis were also identified in the E protein. These sites tended to be epitopes targeted by broadly protective monoclonal antibodies generated against other flaviviruses (Fig. 7E and F). These conserved sites likely reflect functional restrictions in the E protein. Cross-reactive and weakly neutralizing antibodies can cause antibody-dependent enhancement (ADE) of flavivirus infection through IgG engagement of Fcγ receptors on host cells (6, 41). Convalescent-phase plasmas from both DENV- and West Nile virus-infected individuals have been demonstrated to enhance ZIKV infection (42). Conversely, antibodies generated to ZIKV can enhance DENV infection (43). Future studies are required to assay the specificity and effect of antibodies binding to the genetically constrained regions of ZIKV in order to reveal their roles in pathogenesis.
Transposon mutagenesis has previously been used to assay the relative capacities of RNA viruses to tolerate mutations in their antigenic targets. The fact that these screens were performed with different experimental parameters makes it difficult to precisely compare their results. However, each screen was conducted under highly permissive conditions in order to recover as many of the viable mutants as possible. Under these conditions, the monotypic measles virus does not tolerate transposon mutations in its surface glycoproteins (10). This may be due to functional restraints of these viral components at the amino acid sequence level. In contrast, the head domain of the influenza A virus hemagglutinin is remarkably tolerant of transposon mutagenesis, possibly reflecting the ability of the hemagglutinin of influenza viruses to undergo dramatic antigenic drift (9). However, the situation with ZIKV is more complex. Even though areas on the surface of the ZIKV E protein are tolerant of insertional mutagenesis, the virus does not appear to undergo antigenic drift. More work is needed to fully understand why ZIKV retains its antigenic anatomy.
In conclusion, the screen described here provides a framework to begin exploring the genetic restrictions that may affect the evolution of ZIKV. However, future studies are required to assess the full genetic potential of ZIKV during in vivo replication and transmission. Application of the transposon mutagenesis approach to other flaviviruses may be helpful in elucidating the evolutionary capabilities of these major pathogens.
MATERIALS AND METHODS
Cells.
African green monkey kidney cells (Vero; ATCC) and human embryonic kidney cells (293T) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco) containing 10% (vol/vol) fetal bovine serum (FBS) and penicillin-streptomycin (PS; Gibco) at 37°C with 5% CO2. The Aedes albopictus C6/36 cell line (a kind gift from Ana Fernandez-Sesma, Icahn School of Medicine at Mount Sinai) was grown in RPMI medium (Gibco) supplemented with 10% (vol/vol) FBS, sodium bicarbonate (Corning), 1 nM sodium pyruvate (Gibco), a nonessential amino acid solution (NEAA; Gibco), l-glutamine (Gibco), and PS at 33°C with 5% CO2. The Aedes aegypti Aag2 cell line (a kind gift from Ana Fernandez-Sesma) was maintained at 28°C without CO2 in Leibovitz's L-15 medium (Gibco) supplemented with 20% (vol/vol) FBS, 10% (vol/vol) tryptose phosphate broth (Gibco), NEAA, l-glutamine, and PS.
Generation of a ZIKV transposon library.
We previously reported the development of a ZIKV MR766 infectious clone termed pCDNA6.2 ATCCMR766 Intron3127 HDVr (11). From this plasmid, a polymerase II promoter and the hepatitis D virus ribozyme (HDVr) generated the authentic viral RNA 5′ and 3′ ends, respectively. The translation of a region in the MR766 genome that induces toxicity in bacteria was interrupted with an artificial intron. In mammalian cells, the viral RNA was spliced to recreate the authentic viral genome, which efficiently initiated viral RNA translation, replication, and infectious virus production.
The mutational strategy used in this paper was based on previously published protocols (9, 10). Transposon mutagenesis of a plasmid can generate insertions in the backbone of the plasmid, thereby resulting in a library containing a proportion of nonmutagenized WT viruses. To remove these WT genomes, the ZIKV clone was first mutagenized in a shuttle vector, and then only the mutant genomes were transferred into an expression vector. The following description outlines the cloning steps required to generate the shuttle and expression vectors utilized in the screen. The ZIKV cDNA was cloned into the shuttle vector by amplifying the genome from the initial infectious clone in two segments by use of the following primer sets: 5′-CCATGGCCGCGGGATGCTCTTCTCCGAGTTGTTGATCTGTGTGAGTC-3′ plus 5′-CTTGAGGGGTCTGTGAAGTG-3′ and 5′-CACAGACCCCTCAAGTATAGC-3′ plus 5′-GGCCGCACTAGTGATGCTCTTCTGCCAGAAACCATGGATTTCCCCAC-3′. The primers binding to the distal ends of the genome contained SapI sites directly flanking the viral UTRs. The viral genome contained three additional endogenous SapI sites that were silently mutated by amplifying the plasmid in three segments by use of the following primer pairs: 5′-GCGATCTAGAAGGGCCGTGACGCTCCCTTCTCAC-3′ plus 5′-GCTTTTCCCGGTCATCTTCTTAG-3′, 5′-ATGACCGGGAAAAGCATTCAACCGGAAAATCTGGAG-3′ plus 5′-GATTTCAAGTTCTTCACTGTGCCATGGCCCTTTC-3′, and 5′-GAAGAACTTGAAATCCGGTTTGAGG-3′ plus 5′-GCCCTTCTAGATCGCCGTG-3′. These three PCR products were cloned into the pGEM shuttle vector (Promega) by use of an In-Fusion HD kit (Clonetech), resulting in a pGEM clone of the entire MR766 cDNA flanked by, but not containing any, internal SapI sites. The pCDNA6.2 expression vector was modified to contain SapI sites directly after the cytomegalovirus (CMV) promoter and before the HDVr by amplifying the plasmid with primers 5′-AGATCTGGCTCTTCCGGCCGGCATGGTCCCAG-3′ and 5′-GGAAGAGCCAGATCTGGCTCTTCTCGGTTCACTAAACGAGCTCTG-3′. The subsequent PCR product was then recircularized with an In-Fusion HD kit. One additional SapI site in the pCDNA6.2 plasmid backbone was silently mutated by amplifying the plasmid backbone again with primers 5′-GCTGTTCCGCTTCCTCGC-3′ and 5′-AGGAAGCGGAACAGCGCC-3′, and then the plasmid was recircularized as described above. SapI subcloning of the modified MR766 cDNA from the pGEM vector into this pCDNA6.2 plasmid resulted in the removal of the SapI cloning sites and the recreation of the ZIKV junctions with the CMV promoter and HDVr found in the original infectious MR766 clone.
A total of 1.1 μg of the pGEM MR766 plasmid was subjected to in vitro mutagenesis in duplicate with a mutation generation system (Thermo Fisher Scientific) to randomly insert a kanamycin resistance-encoding transposon throughout the genome. The transposition reaction was performed under conditions that favor single insertions according to the manufacturer's protocol. The pool of mutagenized plasmids was then transformed into NEB Turbo competent Escherichia coli cells (New England BioLabs) and plated on 20 15-cm Luria broth (LB) agar plates containing both ampicillin and kanamycin to select for pGEM plasmids (ampicillin resistant) with transposon insertions (kanamycin resistant). These plates were incubated for 24 h at 30°C, and the resulting bacteria were then scraped and pooled for plasmid extraction with an Invitrogen HiPure maxiprep kit (Thermo Fisher Scientific). The mutagenized viral cDNA from this mixture was excised by SapI digestion, gel purified with a Qiaex II kit (Qiagen), and ligated into the SapI-digested modified rescue backbone with T4 ligase (New England BioLabs) for 16 h at 16°C. This plasmid pool was transformed into Turbo cells and grown on LB agar plates containing both ampicillin and kanamycin, and plasmid DNA was isolated as described above to create a library of pCDNA6.2 MR766 clones with transposons randomly integrated throughout the viral sequence. This plasmid pool was then digested with NotI to remove the majority of the insertion sequences from each transposon, including the kanamycin resistance genes. Plasmids containing a single insertion were linearized by this digestion. If multiple insertions occurred in the plasmid, this digestion would create deletions between the insertion sites and result in a truncated plasmid. Viral genomes containing single insertions were then isolated by agarose gel electrophoresis and purified with a Qiaex II kit (Qiagen). The resulting linearized plasmids were then self-ligated by use of T4 ligase for 16 h at 16°C. Finally, the ligated plasmids were transformed into NEB Turbo cells and propagated for 24 h at 30°C on 20 15-cm LB plates containing only ampicillin, and the final plasmid DNA library was isolated as described above. Although insertions in the NS1 intron were not removed from this library, the wild-type genome carried by these clones would be a minor proportion of the input library and therefore unlikely to greatly affect selection.
Rescue of the ZIKV transposon library.
Pools of mutant viruses were generated by transfection of 293T cells with the above-described mutant plasmid library in triplicate, utilizing modified methods as previously published (11). Four 6-well polylysine-coated plates of 80% confluent 293T cells in 1 ml fresh DMEM with 10% FBS (vol/vol) and PS were transfected with 5 μg DNA, 200 μl Opti-MEM (OM; Gibco), and 15 μl TransIT LT1 (Mirus Bio) per well. At 2 days posttransfection, the cells were harvested in 6 ml TRIzol (Thermo Fisher Scientific) for total RNA purification. The supernatants from these cultures were collected, centrifuged at 4,000 × g for 5 min to clarify the supernatant, and then used to infect 70% confluent Vero cells in 15-cm dishes for 1 h with frequent rocking. After the infection, the medium was replaced with 20 ml fresh DMEM with 10% FBS (vol/vol) and PS. After 48 h, the infected cells were collected in TRizol for RNA preparation as described above, and the cell supernatant was clarified and used to initiate a second round of Vero cell infections. After 48 h, these cells were lysed with TRIzol. At each step, 6 ml of TRIzol was used to extract RNA from the cells. Both the RNA and virus samples were frozen at −80°C. Infectious virus titers were determined for aliquots of the supernatant from each stage of passaging by 50% tissue culture infective dose (TCID50) assay as previously described (11). Briefly, the aliquots were serially diluted in 100 μl DMEM containing 2% FBS (vol/vol) and used to infect confluent Vero cells plated the day before in 96-well plates. Cytopathic effect (CPE) was scored at 6 days postinfection, and the Reed and Muench method was used to quantify the viral titer (44).
RT-PCR and deep sequencing.
Total RNA was extracted from the above-described TRIzol lysates by use of a TRIzol Plus RNA purification kit according to the manufacturer's protocol (Thermo Fisher Scientific). On-column PureLink DNase treatment was used to remove any potential plasmid contamination from the infected cells (Thermo Fisher Scientific). The viral RNA was then amplified in three segments by utilizing a SuperScript III RT-PCR Platinum Taq High Fidelity kit (Thermo Fisher Scientific). Primer sequences are available upon request. The reaction mixture with the infected cells was split in two: one half of the reaction mixture was run with the reverse transcription (RT) step according to the manufacturer's protocol, and the other half was run without the RT step to ensure that there was no DNA contamination. The input plasmid library was linearized by digestion with ScaI. The cDNAs and the restricted plasmid library were gel extracted with a Qiaex II kit. The cDNAs for the overlapping PCR products in the replicate passage were pooled in equal molecular ratios. Both the pooled passage cDNAs and the input library were sheared to 200-bp fragments by Covaris sonication and prepared for deep sequencing by use of an NEB Next Ultra II kit and multiplex oligonucleotides (New England BioLabs). Each passage replicate and the input library were given a unique index barcode, multiplexed, and sequenced on a MiSeq sequencer using a MiSeq v3 kit (Illumina).
Analysis of deep sequencing data.
Sequence reads containing the transposon sequence marker (TGCGGCCGCA; contained in all insertion sites) were filtered from the raw reads, trimmed, and mapped to the genome (GenBank accession number KX830960) by utilizing Bowtie2 as previously described (9, 10). The raw number of reads with inserts was counted at each genomic site (see Table S1 in the supplemental material). The total number of reads without inserts at each nucleotide position in the genome was also counted in order to determine the sequencing coverage (Table S1).
Although the sequencing coverage was high throughout the genome, the number of total reads at each position of the genome displayed variation due to bias incurred from RT-PCR and deep sequencing. Bias in total coverage depth can lead to certain sites in the genome having an artificially lower or higher frequency of detected reads with insertions. To correct for sequencing bias, the raw number of reads with insertions was divided by the total number of reads without insertions at that genomic position (Table S1). This ratio provides the relative detection of each insertion at every position in the genome normalized to sequencing coverage. A small proportion of sites were detected only in the passaged or infected cell population, not in the input library, at the sequencing depth obtained. Therefore, these sites must exist in the input library but were below the limit of detection. To correct for this technical issue, insertion sites not detected in the input library were given an arbitrary input value of 0.5 because they must have had an insertion frequency value between 0 (not present in the input library) and 1 (detected in the input library) prior to being divided by the total number of reads at that genomic position (Table S1).
The insertion ratio of each mutant in the screened populations was then divided by its insertion ratio in the input library (Table S1). This value provides the relative enrichment of each mutant from the input library. The data presented are averages for three replicate screens. The normalized sequence data for each replicate can be viewed in Table S1 in the supplemental material.
Although extensive efforts were made to remove WT genomes from the input library, analysis of the deep sequencing reads of this library suggested the presence of genomes without insertions. To determine the extent of this, the number of detected mutant genomes (88,769) was divided by the sum of all reads without insertions at each genomic position (1,629,044,287) to give the overall insertion ratio of 5.4 × 10−5 for all of the genomes in the input library. This ratio was multiplied by the length of the genome (10,807 nucleotides) to give an estimate that 59% of the genomes in the library contained insertions.
Viral proportion graphs were generated with the R ggplot2 package and are based on previously published viral progression plots (45). The PyMOL molecular graphics system, version 1.7.4.5, was utilized to analyze insertion sites in the crystal structure. The python scripts color_b.py and e2s (http://pldserver1.biochem.queensu.ca/~rlc/work/pymol/color_b.py and https://pymolwiki.org/index.php/Expand_To_Surface, respectively) were utilized to color the space-filling crystal structures based on transposon insertion frequency.
For the comparison of measles virus hemagglutinin to ZIKV E, the previously published measles virus data set was reanalyzed as outlined above for ZIKV (10). To determine the insertional tolerances of the ZIKV E and measles virus hemagglutinin glycoproteins, the number of glycoprotein insertional mutants selected at levels >2 standard deviations above average in passage 2 of each screen was divided by the total number of mutants in these coding sequences in each input library. A total of 58 insertional ZIKV E mutants were selected in the passage 2 population. This number was divided by 1,117, which was the total number of E protein mutants in the original library. This number was then multiplied by 100 to show that 5.19% of ZIKV E mutants were selected during passaging. Conversely, none of the 532 measles virus hemagglutinin mutants were selected during passaging, which equals a percentage of zero. This calculation provides the percentage of input sites selected after passaging and should compensate for differences in mutational coverage between the two screens.
Rescue and analysis of viruses bearing individual insertion sites.
Individual insertion sites were cloned by amplifying the viral cDNA clones by use of overlapping primer sets encoding the transposon sequence (primers available upon request). These PCR products were then cloned into the pCDNA6.2 plasmid by use of an In-Fusion HD kit. Wild-type and mutant viruses were individually recovered and titrated as described above. Growth curves were performed in triplicate by infecting 5 × 105 Vero cells or 1.5 × 106 C6/36 or Aag2 cells in 6-well plates at an MOI of 0.01 with either wild-type or mutant virus in 500 μl of infection medium (identical to each cell line's growth medium but containing only 2% [vol/vol] FBS). One hour after infection, the cells were washed with phosphate-buffered saline (PBS; Gibco), and 2 ml of fresh infection medium was added. Aliquots taken at the indicated time points were titrated by TCID50 assay as described above.
Accession number(s).
The original sequence files and the normalized dataset can be accessed with the NCBI Gene Expression Omnibus (GEO) series record GSE92546.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge Jennifer R. Hamilton and Mark J. Bailey for their help in setting up assays and Carina Storrs, Gayathri Vijayakumar, Jennifer R. Hamilton, and Mark J. Bailey for manuscript editing assistance. We thank Ana Fernandez-Sesma for the C6/36 and Aag2 cell lines used in this study. We also thank Maryline Panis and Benjamin R. tenOever for their help with deep sequencing the viral libraries.
This work was supported by the following grants: NIH grant R01DK095125 (M.J.E.), NIH grant 5U19AI109946-03 (P.P.), and CEIRS grant HHSN272201400008C (P.P.). M.J.E. holds an Investigators in Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.00698-17.
REFERENCES
- 1.Pardi N, Hogan MJ, Pelc RS, Muramatsu H, Andersen H, DeMaso CR, Dowd KA, Sutherland LL, Scearce RM, Parks R, Wagner W, Granados A, Greenhouse J, Walker M, Willis E, Yu J-S, McGee CE, Sempowski GD, Mui BL, Tam YK, Huang Y-J, Vanlandingham D, Holmes VM, Balachandran H, Sahu S, Lifton M, Higgs S, Hensley SE, Madden TD, Hope MJ, Karikó K, Santra S, Graham BS, Lewis MG, Pierson TC, Haynes BF, Weissman D. 2017. Zika virus protection by a single low-dose nucleoside-modified mRNA vaccination. Nature 543:248–251. doi: 10.1038/nature21428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dowd KA, Ko S-Y, Morabito KM, Yang ES, Pelc RS, DeMaso CR, Castilho LR, Abbink P, Boyd M, Nityanandam R, Gordon DN, Gallagher JR, Chen X, Todd J-P, Tsybovsky Y, Harris A, Huang Y-JS, Higgs S, Vanlandingham DL, Andersen H, Lewis MG, De La Barrera R, Eckels KH, Jarman RG, Nason MC, Barouch DH, Roederer M, Kong W-P, Mascola JR, Pierson TC, Graham BS. 2016. Rapid development of a DNA vaccine for Zika virus. Science 354:237–240. doi: 10.1126/science.aai9137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mlakar J, Korva M, Tul N, Popović M, Poljšak-Prijatelj M, Mraz J, Kolenc M, Resman Rus K, Vesnaver Vipotnik T, Fabjan Vodušek V, Vizjak A, Pižem J, Petrovec M, Avšič Županc T. 2016. Zika virus associated with microcephaly. N Engl J Med 374:951–958. doi: 10.1056/NEJMoa1600651. [DOI] [PubMed] [Google Scholar]
- 4.Broutet N, Krauer F, Riesen M, Khalakdina A, Almiron M, Aldighieri S, Espinal M, Low N, Dye C. 2016. Zika virus as a cause of neurologic disorders. N Engl J Med 374:1506–1509. doi: 10.1056/NEJMp1602708. [DOI] [PubMed] [Google Scholar]
- 5.Lindenbach BD, Murray CJ, Thiel H-J, Rice CM. 2013. Flaviviridae, p 712–746. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 6.Pierson TC, Diamond MS. 2013. Flaviviruses, p 747–794. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE (ed), Fields virology, 6th ed Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 7.Belshaw R, Pybus OG, Rambaut A. 2007. The evolution of genome compression and genomic novelty in RNA viruses. Genome Res 17:1496–1504. doi: 10.1101/gr.6305707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu X, Vaughan K, Weiskopf D, Grifoni A, Diamond MS, Sette A, Peters B. 2016. Identifying candidate targets of immune responses in Zika virus based on homology to epitopes in other flavivirus species. PLoS Curr 8:ecurrents.outbreaks.9aa2e1fb61b0f632f58a098773008c4b. doi: 10.1371/currents.outbreaks.9aa2e1fb61b0f632f58a098773008c4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Heaton NS, Sachs D, Chen C-J, Hai R, Palese P. 2013. Genome-wide mutagenesis of influenza virus reveals unique plasticity of the hemagglutinin and NS1 proteins. Proc Natl Acad Sci U S A 110:20248–20253. doi: 10.1073/pnas.1320524110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fulton BO, Sachs D, Beaty SM, Won ST, Lee B, Palese P, Heaton NS. 2015. Mutational analysis of measles virus suggests constraints on antigenic variation of the glycoproteins. Cell Rep 11:1331–1338. doi: 10.1016/j.celrep.2015.04.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwarz MC, Sourisseau M, Espino MM, Gray ES, Chambers MT, Tortorella D, Evans MJ. 2016. Rescue of the 1947 Zika virus prototype strain with a cytomegalovirus promoter-driven cDNA clone. mSphere 1:e00246-16. doi: 10.1128/mSphere.00246-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arumugaswami V, Remenyi R, Kanagavel V, Sue EY, Ho TN, Liu C, Fontanes V, Dasgupta A, Sun R. 2008. High-resolution functional profiling of hepatitis C virus genome. PLoS Pathog 4:e1000182. doi: 10.1371/journal.ppat.1000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Beitzel BF, Bakken RR, Smith JM, Schmaljohn CS. 2010. High-resolution functional mapping of the Venezuelan equine encephalitis virus genome by insertional mutagenesis and massively parallel sequencing. PLoS Pathog 6:e1001146. doi: 10.1371/journal.ppat.1001146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Villordo SM, Carballeda JM, Filomatori CV, Gamarnik AV. 2016. RNA structure duplications and flavivirus host adaptation. Trends Microbiol 24:270–283. doi: 10.1016/j.tim.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Scaturro P, Cortese M, Chatel-Chaix L, Fischl W, Bartenschlager R. 2015. Dengue virus non-structural protein 1 modulates infectious particle production via interaction with the structural proteins. PLoS Pathog 11:e1005277. doi: 10.1371/journal.ppat.1005277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Youn S, Li T, McCune BT, Edeling MA, Fremont DH, Cristea IM, Diamond MS. 2012. Evidence for a genetic and physical interaction between nonstructural proteins NS1 and NS4B that modulates replication of West Nile virus. J Virol 86:7360–7371. doi: 10.1128/JVI.00157-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crabtree MB, Kinney RM, Miller BR. 2005. Deglycosylation of the NS1 protein of dengue 2 virus, strain 16681: construction and characterization of mutant viruses. Arch Virol 150:771–786. doi: 10.1007/s00705-004-0430-8. [DOI] [PubMed] [Google Scholar]
- 18.Somnuke P, Hauhart RE, Atkinson JP, Diamond MS, Avirutnan P. 2011. N-linked glycosylation of dengue virus NS1 protein modulates secretion, cell-surface expression, hexamer stability, and interactions with human complement. Virology 413:253–264. doi: 10.1016/j.virol.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Flamand M, Megret F, Mathieu M, Lepault J, Rey FA, Deubel V. 1999. Dengue virus type 1 nonstructural glycoprotein NS1 is secreted from mammalian cells as a soluble hexamer in a glycosylation-dependent fashion. J Virol 73:6104–6110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muller DA, Young PR. 2013. The flavivirus NS1 protein: molecular and structural biology, immunology, role in pathogenesis and application as a diagnostic biomarker. Antiviral Res 98:192–208. doi: 10.1016/j.antiviral.2013.03.008. [DOI] [PubMed] [Google Scholar]
- 21.Akey DL, Brown WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, DelProposto J, Ogata CM, Skiniotis G, Kuhn RJ, Smith JL. 2014. Flavivirus NS1 structures reveal surfaces for associations with membranes and the immune system. Science 343:881–885. doi: 10.1126/science.1247749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faye O, Freire CCM, Iamarino A, Faye O, de Oliveira JVC, Diallo M, Zanotto PMA, Sall AA. 2014. Molecular evolution of Zika virus during its emergence in the 20th century. PLoS Negl Trop Dis 8:e2636. doi: 10.1371/journal.pntd.0002636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heinz FX, Stiasny K. 2017. The antigenic structure of Zika virus and its relation to other flaviviruses: implications for infection and immunoprophylaxis. Microbiol Mol Biol Rev 81:e00055-16. doi: 10.1128/MMBR.00055-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao H, Fernandez E, Dowd KA, Speer SD, Platt DJ, Gorman MJ, Govero J, Nelson CA, Pierson TC, Diamond MS, Fremont DH. 2016. Structural basis of Zika virus-specific antibody protection. Cell 166:1016–1027. doi: 10.1016/j.cell.2016.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swanstrom JA, Plante JA, Plante KS, Young EF, McGowan E, Gallichotte EN, Widman DG, Heise MT, de Silva AM, Baric RS. 2016. Dengue virus envelope dimer epitope monoclonal antibodies isolated from dengue patients are protective against Zika virus. mBio 7:e01123-16. doi: 10.1128/mBio.01123-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Henchal EA, Gentry MK, McCown JM, Brandt WE. 1982. Dengue virus-specific and flavivirus group determinants identified with monoclonal antibodies by indirect immunofluorescence. Am J Trop Med Hyg 31:830–836. doi: 10.4269/ajtmh.1982.31.830. [DOI] [PubMed] [Google Scholar]
- 27.Crill WD, Chang G-JJ. 2004. Localization and characterization of flavivirus envelope glycoprotein cross-reactive epitopes. J Virol 78:13975–13986. doi: 10.1128/JVI.78.24.13975-13986.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dejnirattisai W, Wongwiwat W, Supasa S, Zhang X, Dai X, Rouvinsky A, Jumnainsong A, Edwards C, Quyen NT, Duangchinda T, Grimes JM, Tsai W, Lai C, Wang W, Malasit P, Farrar J, Simmons CP, Zhou ZH, Rey FA, Mongkolsapaya J, Screaton GR. 2015. A new class of highly potent, broadly neutralizing antibodies isolated from viremic patients infected with dengue virus. Nat Immunol 16:170–177. doi: 10.1038/ni.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rouvinski A, Guardado-Calvo P, Barba-Spaeth G, Duquerroy S, Vaney M-C, Kikuti CM, Navarro Sanchez ME, Dejnirattisai W, Wongwiwat W, Haouz A, Girard-Blanc C, Petres S, Shepard WE, Desprès P, Arenzana-Seisdedos F, Dussart P, Mongkolsapaya J, Screaton GR, Rey FA. 2015. Recognition determinants of broadly neutralizing human antibodies against dengue viruses. Nature 520:109–113. doi: 10.1038/nature14130. [DOI] [PubMed] [Google Scholar]
- 30.Thyagarajan B, Bloom JD. 2014. The inherent mutational tolerance and antigenic evolvability of influenza hemagglutinin. eLife 3:e03300. doi: 10.7554/eLife.03300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Doud MB, Bloom JD. 2016. Accurate measurement of the effects of all amino-acid mutations on influenza hemagglutinin. Viruses 8:155. doi: 10.3390/v8060155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haddox HK, Dingens AS, Bloom JD. 2016. Experimental estimation of the effects of all amino-acid mutations to HIV's envelope protein on viral replication in cell culture. PLoS Pathog 12:e1006114. doi: 10.1371/journal.ppat.1006114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ashenberg O, Padmakumar J, Doud MB, Bloom JD. 2017. Deep mutational scanning identifies sites in influenza nucleoprotein that affect viral inhibition by MxA. PLoS Pathog 13:e1006288. doi: 10.1371/journal.ppat.1006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu NC, Young AP, Al-Mawsawi LQ, Olson CA, Feng J, Qi H, Chen S-H, Lu I-H, Lin C-Y, Chin RG, Luan HH, Nguyen N, Nelson SF, Li X, Wu T-T, Sun R. 2014. High-throughput profiling of influenza A virus hemagglutinin gene at single-nucleotide resolution. Sci Rep 4:4942. doi: 10.1038/srep04942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fernandes JD, Faust TB, Strauli NB, Smith C, Crosby DC, Nakamura RL, Hernandez RD, Frankel AD. 2016. Functional segregation of overlapping genes in HIV. Cell 167:1762.e12–1773.e12. doi: 10.1016/j.cell.2016.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Remenyi R, Qi H, Su SY, Chen Z, Wu NC, Arumugaswami V, Truong S, Chu V, Stokelman T, Lo HH, Anders Olson C, Wu TT, Chen SH, Lin CY, Sun R. 2014. A comprehensive functional map of the hepatitis C virus genome provides a resource for probing viral proteins. mBio 5:e01469-14. doi: 10.1128/mBio.01469-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pryor MJ, Wright PJ. 1994. Glycosylation mutants of dengue virus NS1 protein. J Gen Virol 75:1183–1187. doi: 10.1099/0022-1317-75-5-1183. [DOI] [PubMed] [Google Scholar]
- 38.Dowd KA, DeMaso CR, Pelc RS, Speer SD, Smith ARY, Goo L, Platt DJ, Mascola JR, Graham BS, Mulligan MJ, Diamond MS, Ledgerwood JE, Pierson TC. 2016. Broadly neutralizing activity of Zika virus-immune sera identifies a single viral serotype. Cell Rep 16:1485–1491. doi: 10.1016/j.celrep.2016.07.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Forrester NL, Guerbois M, Seymour RL, Spratt H, Weaver SC. 2012. Vector-borne transmission imposes a severe bottleneck on an RNA virus population. PLoS Pathog 8:e1002897. doi: 10.1371/journal.ppat.1002897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lange A, Ferguson NM. 2009. Antigenic diversity, transmission mechanisms, and the evolution of pathogens. PLoS Comput Biol 5:e1000536. doi: 10.1371/journal.pcbi.1000536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang TT, Sewatanon J, Memoli MJ, Wrammert J, Bournazos S, Bhaumik SK, Pinsky BA, Chokephaibulkit K, Onlamoon N, Pattanapanyasat K, Taubenberger JK, Ahmed R, Ravetch JV. 2017. IgG antibodies to dengue enhanced for FcγRIIIA binding determine disease severity. Science 355:395–398. doi: 10.1126/science.aai8128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bardina SV, Bunduc P, Tripathi S, Duehr J, Frere JJ, Brown JA, Nachbagauer R, Foster GA, Krysztof D, Tortorella D, Stramer SL, García-Sastre A, Krammer F, Lim JK. 2017. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 356:175–180. doi: 10.1126/science.aal4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stettler K, Beltramello M, Espinosa DA, Graham V, Cassotta A, Bianchi S, Vanzetta F, Minola A, Jaconi S, Mele F, Foglierini M, Pedotti M, Simonelli L, Dowall S, Atkinson B, Percivalle E, Simmons CP, Varani L, Blum J, Baldanti F, Cameroni E, Hewson R, Harris E, Lanzavecchia A, Sallusto F, Corti D. 2016. Specificity, cross-reactivity, and function of antibodies elicited by Zika virus infection. Science 353:823–826. doi: 10.1126/science.aaf8505. [DOI] [PubMed] [Google Scholar]
- 44.Reed LJ, Muench H. 1938. A simple method of estimating fifty percent endpoints. Am J Hyg (Lond) 27:493–497. [Google Scholar]
- 45.Varble A, Albrecht RA, Backes S, Crumiller M, Bouvier NM, Sachs D, García-Sastre A, Tenoever BR. 2014. Influenza A virus transmission bottlenecks are defined by infection route and recipient host. Cell Host Microbe 16:691–700. doi: 10.1016/j.chom.2014.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brown WC, Akey DL, Konwerski JR, Tarrasch JT, Skiniotis G, Kuhn RJ, Smith JL. 2016. Extended surface for membrane association in Zika virus NS1 structure. Nat Struct Mol Biol 23:865–867. doi: 10.1038/nsmb.3268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Song H, Qi J, Haywood J, Shi Y, Gao GF. 2016. Zika virus NS1 structure reveals diversity of electrostatic surfaces among flaviviruses. Nat Struct Mol Biol 23:456–458. doi: 10.1038/nsmb.3213. [DOI] [PubMed] [Google Scholar]
- 48.Sirohi D, Chen Z, Sun L, Klose T, Pierson TC, Rossmann MG, Kuhn RJ. 2016. The 3.8 Å resolution cryo-EM structure of Zika virus. Science 352:467–470. doi: 10.1126/science.aaf5316. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.