

ABSTRACT
The herpes simplex virus (HSV) capsid is released into the cytoplasm after fusion of viral and host membranes, whereupon dynein-dependent trafficking along microtubules targets it to the nuclear envelope. Binding of the capsid to the nuclear pore complex (NPC) is mediated by the capsid protein pUL25 and the capsid-tethered tegument protein pUL36. Temperature-sensitive mutants in both pUL25 and pUL36 dock at the NPC but fail to release DNA. The uncoating reaction has been difficult to study due to the rapid release of the genome once the capsid interacts with the nuclear pore. In this study, we describe the isolation and characterization of a truncation mutant of pUL25. Live-cell imaging and immunofluorescence studies demonstrated that the mutant was not impaired in penetration of the host cell or in trafficking of the capsid to the nuclear membrane. However, expression of viral proteins was absent or significantly delayed in cells infected with the pUL25 mutant virus. Transmission electron microscopy revealed capsids accumulated at nuclear pores that retained the viral genome for at least 4 h postinfection. In addition, cryoelectron microscopy (cryo-EM) reconstructions of virion capsids did not detect any obvious differences in the location or structural organization for the pUL25 or pUL36 proteins on the pUL25 mutant capsids. Further, in contrast to wild-type virus, the antiviral response mediated by the viral DNA-sensing cyclic guanine adenine synthase (cGAS) was severely compromised for the pUL25 mutant. These results demonstrate that the pUL25 capsid protein has a critical role in releasing viral DNA from NPC-bound capsids.
IMPORTANCE Herpes simplex virus 1 (HSV-1) is the causative agent of several pathologies ranging in severity from the common cold sore to life-threatening encephalitic infection. Early steps in infection include release of the capsid into the cytoplasm, docking of the capsid at a nuclear pore, and release of the viral genome into the nucleus. A key knowledge gap is how the capsid engages the NPC and what triggers release of the viral genome into the nucleus. Here we show that the C-terminal region of the HSV-1 pUL25 protein is required for releasing the viral genome from capsids docked at nuclear pores. The significance of our research is in identifying pUL25 as a key viral factor for genome uncoating. pUL25 is found at each of the capsid vertices as part of the capsid vertex-specific component and implicates the importance of this complex for NPC binding and genome release.
KEYWORDS: HSV-1, cryo-electron microscopy, pUL25, CVSC, genome uncoating, nuclear pores
INTRODUCTION
Humans host eight herpesviruses that cause encephalitis, chicken pox, mononucleosis, congenital birth defects, shingles, Burkitt's lymphoma, Kaposi's sarcoma, and other diseases. Herpesviruses are ubiquitous in the population, with most people exposed to several of them during their lifetime. Few vaccines are available, and new avenues to effectively counteract herpesvirus infections are desperately needed, such as highly specific structural targets for small-molecule antivirals. Despite the very different pathologies produced by individual herpesviruses, viruses in this family have the same basic structure and recognizably similar patterns of replication (1).
The virion of all herpesviruses consists of an external membrane envelope, a proteinaceous layer called the tegument, and an icosahedral capsid containing the compressed double-stranded linear DNA genome. The outer envelope is arranged as a lipid bilayer anchoring multiple copies of approximately 11 viral glycoproteins that protrude externally and a small number of intrinsic membrane proteins (2). The envelope is obtained from the host cell and possesses lipid content similar to that found in the cellular cytoplasmic membrane (3, 4). The tegument layer occupies the space between the envelope and capsid, comprising approximately two-thirds of the virion volume and the majority of its mass. The tegument is a complex architecture that is composed of multiple copies of approximately 23 viral proteins but also contains cellular proteins as well as viral and cellular gene transcripts (5–7). Cryo-electron tomography revealed that the capsid is not centered within the herpes simplex virus 1 (HSV-1) virion but instead resides in close proximity to the envelope at one pole; at the opposite pole, the depth of tegument is approximately 35 nm between the envelope and the capsid (8).
During productive infection, over 80 proteins are expressed from the HSV-1 genome in a highly regulated manner. The capsid of HSV-1 is assembled using four of these proteins as building blocks: a major capsid protein, VP5, which forms the capsomeres (hexons and pentons), the triplex molecule found between the capsomeres, consisting of VP19C and VP23, and the scaffold protein (UL26.5 gene product). The tips of hexons bind the small VP26 protein that is dispensable for assembly. In addition, the dodecameric pUL6 portal complex occupies 1 of the 12 capsid vertices in place of a penton, and the capsid vertex-specific component (CVSC), a heterotrimer complex of pUL17, pUL25, and pUL36, binds specifically to the triplexes adjacent to each vertex (7, 9–11). The ∼200-MDa capsid assembles in the nucleus, whereupon the viral genome is packaged into the structure through the unique portal complex.
Nuclear pores represent gateways that incoming capsids of many viruses, including those belonging to the Herpesviridae, Hepadnaviridae, Adenoviridae, and Retroviridae families, must find and engage for successful infection (12). While little is known about the mechanisms involved in these early steps of infection, the incoming HSV capsid retains a subset of tegument proteins that are candidate effectors for genome delivery to nuclei (13–17). Upon docking at the NPC, capsids release their genomes into the nucleus, as evidenced by the predominance of empty capsids docked at NPCs (18). Genome release does not result from breakdown of the HSV capsid since the capsid remains intact on the cytoplasmic side of the nuclear pore complex (NPC) after the genome has been delivered into the nucleus (12). Electron microscopy (EM) reveals that capsids docked at NPCs are oriented with a capsid vertex facing into the pore channel (12). Whether a specific vertex is favored is not known, but it seems likely that aligning the portal vertex toward the NPC would favor efficient genome delivery into the nucleus. In addition, NPC binding requires importin beta and a functional RanGTP/GDP cycle (19). The packaged HSV DNA creates a pressure of tens of atmospheres within the capsid, and this pressure likely drives the initial translocation of the genome into the nucleus once release is triggered (20, 21). However, the trigger for release of the viral genome from the capsid is unknown. The HSV pUL36 and pUL25 proteins remain bound to the capsid after cell entry and are strong candidates as effectors of this process. First, they bind NPCs through their interaction with the NPC proteins Nup214 and Nup358 (22, 23). Second, HSV encoding temperature-sensitive mutations in pUL25 or pUL36 can dock at the NPC at nonpermissive temperatures but fail to release DNA (24–26). Finally, proteolytic cleavage of pUL36 is critical for DNA release from the NPC-bound capsid (27). Collectively, these findings indicate that the pUL25 and pUL36 proteins contribute to both capsid docking and DNA release at NPCs and that these roles can be separated (24–26). Unfortunately, the genes for these proteins cannot be deleted from HSV as they are essential for viral assembly, making analysis of their specific roles at the NPC challenging.
Studies of herpesvirus infection would benefit from the identification of mutants defective in genome release that are nonetheless amenable to biochemical and structural analysis. In the present study, we describe the isolation and analysis of an HSV pUL25 mutant that lacks the three C-terminal amino acids and can be propagated on UL25-complementing cells. When grown on noncomplementing cells, this mutant produces virions whose capsids enter cells and traffic to the nuclear membrane but fail to release their genomes efficiently. This mutant will be useful in biochemical and structural studies aimed at understanding the capsid-NPC interaction and what triggers release of the packaged genome after docking of the capsid at the NPC.
RESULTS
Isolation and characterization of HSV recombinant viruses.
O'Hara et al. (28) described a pUL25 HSV mutant lacking the C-terminal three amino acids that was unable to complement virus growth when provided in trans to a virus lacking pUL25. Nevertheless, the mutant produced enveloped virus particles that were readily detected in the cytoplasm and on the cell surface. The replication defect of this mutant was proposed to be at an early stage in infection, but detailed analysis was hampered by the limitations of the transient complementation assay that was used to characterize this mutant. We have isolated a virus harboring this pUL25 mutation (Fig. 1). The UL25 mutation was introduced by genetic manipulation of an HSV-1 (KOS) genome maintained in a bacterial artificial chromosome (BAC), as previously described (29). The deletion removed codons for the final three amino acids of the UL25 open reading frame and retained the stop codon. The BAC bFH518 was transfected into UL25-complementing 8-1 cells. The recovered mutant virus, vFH518, was plaque purified from 8-1 cells, and virus stocks were prepared on 8-1 cells. A fluorescent variant of the bFH518 BAC that encodes a pUL25/green fluorescent protein (GFP) fusion protein was also isolated by inserting the GFP gene in-frame between codons 50 and 51 of the UL25 open reading frame. The resulting BAC, bFH589, was transfected into 8-1 cells to isolate the fluorescent virus vFH589 (Fig. 1). This virus is similar to the previously described vFH422 virus, which expresses a full-length pUL25/GFP fusion protein and results in fluorescent capsids that allow imaging of individual particles in cells (29). Virus stocks were titrated on Vero and 8-1 cells to determine the effects of the UL25 mutation on viral replication (Fig. 1B). Compared to KOS and vFH422, which showed comparable growths on Vero and 8-1 cells, the vFH518 and vFH589 mutants did not produce plaques on Vero cells. When grown on noncomplementing Vero cells, the vFH518 mutant produced virions (described below) that did not produce plaques on Vero cells, although small plaques could be detected on 8-1 cells after several extra days of incubation (Fig. 1C). It should be noted that we have not detected wild-type revertants in any of the high-titer vFH518 stocks prepared on the pUL25-complementing 8-1 cells.
FIG 1.
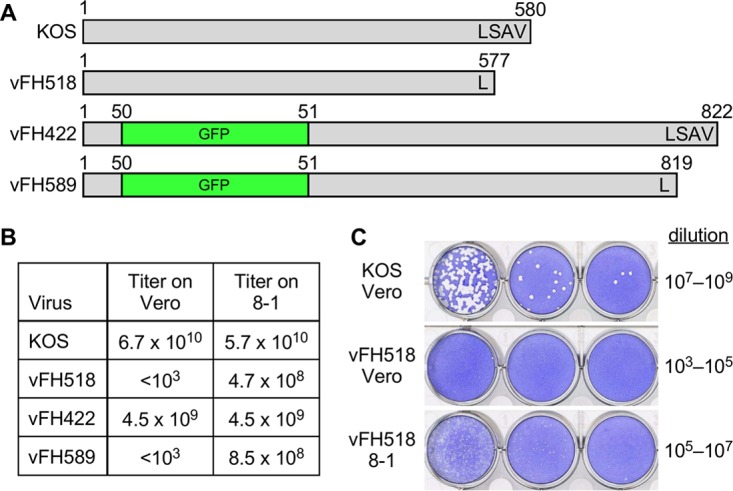
Recombinant viruses used in this study. (A) The pUL25 open reading frames (580 amino acids) from wild-type KOS and pUL25 recombinant viruses are shown, along with the location of the GFP insertion and the C-terminal three-amino-acid deletion. The virus names are indicated on the left of each construct. The amino acid numbers above each construct indicate protein lengths with the pUL25 C-terminal amino acids indicated. (B) The titers of viruses grown on Vero cells (KOS, vFH422) or UL25-complementing, 8-1 cells (vFH518, vFH589) were determined on the two cell lines. (C) KOS and vFH518 were grown on Vero cells, and their titers on Vero or 8-1 cells were determined. The plates were stained with crystal violet 3 days (KOS) or 5 days (vFH518) postplating.
Intracellular replication levels of parental KOS and the vFH518 mutant were compared in single-step growth curves for each virus in either Vero or 8-1 cells. Cells were infected with each virus at a multiplicity of infection (MOI) of 5 and were harvested at 0, 2, 4, 7, 10, 22, and 30 h postinfection (hpi). Cell-associated and cell-free viruses were assayed for infectious virus by plaque assay on 8-1 cells (Fig. 2). KOS produced similar amounts of virus on Vero and 8-1 cells, while vFH518 showed reduced virus propagation on both 8-1 and Vero cells, with titers down 1 to 2 log, respectively. There was a significant delay in the eclipse phase for the vFH518 virus grown on Vero cells, whereby the production of new virus was not detected until ∼10 hpi. This contrasts with KOS propagated on either cell line or vFH518 grown on 8-1 cells, where new virus was detected as early as 4 to 7 hpi.
FIG 2.
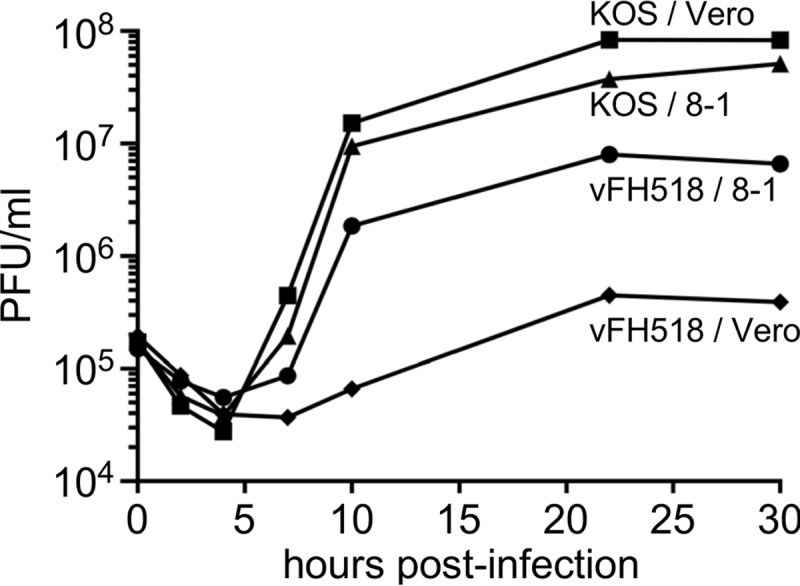
Single-step growth of KOS and UL25 mutants. Vero or 8-1 cells were infected with KOS or pUL25 mutant vHF518 at an MOI of 5 at 4°C for 1 h and incubated at 37°C to the indicated time points. Cells were lysed by multiple freeze-thaws, and viral progeny were quantified by plaque assay on 8-1 cells. The results shown are averages from duplicate experiments.
Isolation and analysis of pUL25 mutant extracellular viral particles.
To determine if the vFH518 and vFH589 mutants produce enveloped extracellular virus particles, Vero cells were infected with either KOS, vFH422, vFH518, or vFH589, and virions were isolated by sucrose gradient centrifugation from the medium of infected Vero cells (Fig. 3A). The virion band was collected with a syringe by side puncture of the gradient, and the virions were pelleted. The virions were resuspended in TNE buffer (10 mM Tris, 150 mM NaCl, and 1 mM EDTA [pH 7.5]), and their protein composition was determined on stained gels and by Western blotting. There was no discernible difference in the protein profiles for the KOS, vFH518, and vFH589 virions by SDS-PAGE analysis and Coomassie blue staining (Fig. 3B). Western blots of KOS, vFH422, vFH518, and vFH589 virions were probed with either pUL25 monoclonal antibody (MAb) 25E10 or a rabbit GFP polyclonal antibody (Fig. 3C). Wild-type pUL25 (65 kDa) was detected for KOS and vFH518, while the pUL25/GFP fusion, which is approximately 25 kDa larger than in the wild type, was detected in the virions for both vFH422 and vFH589. The UL25 products were not detected by Coomassie blue staining due to their low abundance in virions.
FIG 3.

Analysis of virions isolated from Vero cells infected with KOS or the UL25 mutant virus. (A) Vero cells infected with the indicated viruses at an MOI of 5 PFU per cell were harvested at 18 h postinfection, and virions were isolated from the medium on 20 to 60% sucrose gradients. (B) Equivalent amounts of virions were separated by SDS-PAGE, and the proteins were detected by Coomassie blue staining. The positions of the HSV virions proteins are indicated on the right. (C) Immunoblot analysis of virions with UL25 and GFP antibodies. The positions of the pUL25 and pUL25-GFP proteins are indicated on the right. Molecular mass standards are given in kilodaltons.
vFH518 and vFH589 produce defective virions on Vero cells.
The protein profiles of the virions isolated from Vero cells for both the vFH518 and vFH589 mutants demonstrated that they were nearly identical to that of wild-type virions. We measured the genome-to-PFU ratios for the two viruses to determine if the difference in viral titer for these mutants when grown on Vero cells or on complementing 8-1 cells was due to some of the virions lacking genomes. Virions were isolated from Vero cells separately infected with KOS, vFH422, vFH518, and vFH589 or from 8-1 cells infected with either vFH518 or vFH589. The genome copy number of the purified virions was determined by quantitative real-time PCR (qPCR), and the titer of each virion prep was determined by plaque assay on 8-1 cells (Table 1). For virion samples derived from Vero cells infected with vFH518 and vFH589, plaques were counted 5 to 6 days postplating compared to 3 days postplating for KOS and vFH422 in order for the plaques to be large enough to count. The total number of virion-associated genomes was similar for all four of the viruses isolated from Vero or 8-1 cells. The genome/PFU ratios for vFH518 and vFH589 virions isolated from Vero cells were approximately 90-fold and 75-fold higher, respectively, than for KOS isolated from Vero cells. vFH422 virions isolated from Vero cells had a genome/PFU ratio 4- to 5-fold higher than KOS. In contrast, the genome/PFU ratios for vFH518 and vFH589 virions isolated from 8-1 cells were both 5- to 10-fold higher than the KOS virions. The higher genome/PFU ratio with nearly equal numbers of total genomes for the vFH518 and vFH589 viruses compared to KOS demonstrate that the mutant virions isolated from Vero cells are intact and contain viral genomes but fail to efficiently initiate an infection.
TABLE 1.
Genome-to-PFU ratios of virions isolated from Vero or 8-1 cellsa
| Virion | Virions from Vero cells |
Virions from 8-1 cells |
||
|---|---|---|---|---|
| No. of genomes/PFU | Total no. of genomes | No. of genomes/PFU | Total no. of genomes | |
| KOS | 41 | 4.0 × 1011 | NDb | ND |
| 27 | 1.7 × 1011 | |||
| vFH518 | 2,906 | 1.6 × 1011 | 151 | 1.1 × 1010 |
| 3,134 | 4.4 × 1010 | |||
| vFH589 | 2,313 | 2.5 × 1011 | 436 | 1.3 × 1011 |
| 2,710 | 1.2 × 1011 | |||
| vFH422 | 187 | 1.3 × 1011 | ND | ND |
Virions were isolated from Vero or 8-1 cells infected with the indicated virus. The KOS, vFH518, and vFH589 virions were isolated from two separate Vero cell infections.
ND, not done.
The structural organization at the vertices of the capsid is not modified by the 3-amino-acid truncation of pUL25.
In order to check for structural consequences of the pUL25 truncation, we collected 4,503 micrographs of vFH518 in the frozen hydrated state; from these micrographs, we selected 6,665 images of capsids inside intact virions, of which 4,864 were used in the final icosahedral reconstruction. Views of the vFH518 capsid structure, together with the previously reported HSV-1 wild-type capsids (9) at similar resolution and quality, are shown in Fig. 4. The density corresponding to pUL25 is colored in red, and the C-terminal atomic model (residues 133 to 580, PDB ID 2F5U) fits well into each map. The mutant capsid does not exhibit structural differences compared to the wild-type virion. The location of the pUL25 C terminus, as indicated on the HSV-1 wild-type view, is on a side of the subunit that is facing away from the adjacent vertex and does not appear to interface with any other capsid component. Interestingly, the 3 amino acids do not have corresponding density in the wild-type map, suggesting that in vivo the C terminus is indeed flexible, as predicted. However, the imposition of icosahedral symmetry masks the unique portal vertex structure, whereby the disposition of the CVSC subunits may differ from that around the penton vertices shown here.
FIG 4.
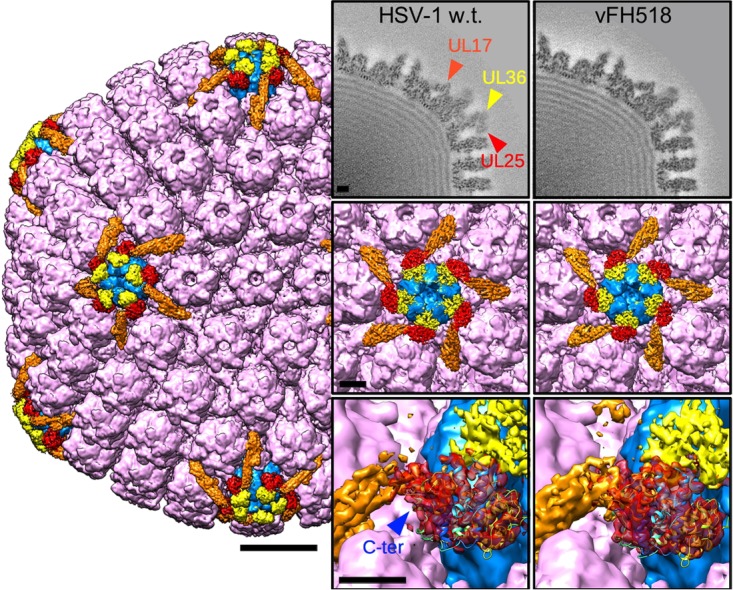
Cryo-EM density maps of capsids imaged inside virions. The HSV-1 wild-type (w.t.) capsid surface is viewed along the icosahedral 2-fold axis (EMD ID 6386 [9]). Hexons, triplexes, and the VP26 densities are all colored purple; pentons are blue; the CVSC molecule is orange in part and red for the density corresponding to the C-terminal atomic model of pUL25; PDB ID 2F5U (40); and in yellow is the putative pUL36 C-terminal domain. Bar, 200 Å. Panels on the right compare density maps from capsids of wild-type virus and vFH518 (EMD ID 8733): top row, central sections; center row, surface views of the penton vertex; bottom row, close-up views of the pUL25 density (red), including a fit of the atomic model with the C terminus (C-ter) marked. The original fit of this pUL25 fragment was made in the PRV map (9), and it appears to be identically located in the HSV-1 wild-type and vFH518 mutant capsids. Bars, 50 Å.
Viral protein expression is delayed during vFH518 and vFH589 infection.
To examine if the defect with the UL25 mutants was at an early or late step in infection, we measured the level of expression of representative viral proteins postinfection. Vero cells were infected (500 genomes/cell) with virions isolated from KOS-, vFH422-, vFH518-, and vFH589-infected Vero cells or with virions isolated from vFH518-infected 8-1 cells. Cell extracts were isolated at various times postinfection and then subjected to Western blot analysis (Fig. 5A). The blots were probed for the immediate early protein ICP4, the early protein ICP8, the late protein VP5, and the cell protein GAPDH (glyceraldehyde-3-phosphate dehydrogenase). ICP8, which is the only one of the three viral proteins that is not incorporated into the HSV virion, was detected at 4 and 7 hpi in vFH422- and KOS-infected cells, respectively (Fig. 5A). In contrast, expression of ICP8 was not detected in cells infected with virions isolated from Vero cells infected by vFH518 and vFH589, even as late as 12 hpi (Fig. 5A). ICP4 and VP5 were first detected at 2 and 4 hpi for KOS and vFH422, respectively, while the expression of both proteins was significantly delayed in Vero cells infected by vFH518 and vFH589. Both ICP4 and VP5 are components of virions, and the low levels of both proteins detected early in infection with the UL25 mutants most likely resulted from the input virions. When the time course was performed with vFH518 virions isolated from UL25-complementing 8-1 cells, the expressions of the three viral proteins showed kinetics similar to that seen in infection with KOS and vFH422. Infections done at a much higher genome multiplicity (1,000 KOS and 15,000 vFH518 genomes/cell) demonstrated that a small amount of ICP8 expression was detected at 16 h postinfection with the vFH518 mutant compared to KOS, where ICP8 was detected early in infection. The large amount of defective input vFH518 virions used in these infections was demonstrated by the constant amount of the two virion proteins, VP5 (major capsid protein) and ICP4, that were detected at all of the time points (Fig. 5B). This contrasts with wild-type KOS-infected cells, in which the input virus was low, as demonstrated by the lack of ICP4 and VP5 at early times postinfection.
FIG 5.

Comparison of protein expression from KOS or UL25 mutants. A time course assay of viral protein expression was carried out in Vero cells infected with KOS, vFH422, vFH518, and vFH589 virions isolated from Vero cells for all four viruses or with vFH518 virions isolated from 8-1 cells. Cells were incubated at 37°C, and at the indicated times postinfection the cell lysates were harvested. Western blot analysis of each time course lysate was carried out, and blots were probed with the indicated antibodies as described in Materials and Methods. Vero cells were infected with 500 genomes/cell for each virus (A) or with 1,000 (KOS) or 15,000 (vFH518) genomes/cell (B).
Virus entry into cells.
The failure of vFH518 and vFH589 viruses to synthesize viral proteins until late times postinfection could be due to a defect in (i) attachment/penetration of the host cell, (ii) postentry trafficking of capsids to the nucleus, or (iii) binding/release at the nuclear pore. pUL25 is required for microtubule-mediated transport of incoming capsids to the nuclear membrane through its interaction with viral tegument protein pUL36 (10). A detailed analysis of retrograde axonal trafficking was afforded by infecting primary sensory neurons with either vFH422 or vFH589. The dorsal root ganglion explant cultures generate axons, which radiate out from a central cell body mass; the narrow and elongated morphology of axons permits tracking of ingressing capsids. Incoming particles were imaged from 15 to 60 min postinfection, and velocities and run lengths were determined for >100 particles per virus (Fig. 6). The results demonstrate that vFH589 capsids undergo retrograde transport with wild-type kinetics. Therefore, the C-terminal three-amino-acid deletion does not interfere with the interaction of UL25 with pUL36, which is critical for retrograde capsid transport.
FIG 6.

The HSV pUL25 mutant undergoes retrograde transport with wild-type particle dynamics. Primary sensory neurons were infected with HSV UL25/GFP-expressing viruses vFH422 or vFH589, and pUL25/GFP capsids were imaged during ingress from 15 to 60 min postinfection. Viral particles moving in axons greater than 0.5 μm were tracked, and individual velocities (left) and run lengths (right) were measured (vFH422, n = 145; vFH589, n = 226). Gaussian curves (for velocities) or decaying exponential curves (for run lengths) were fit to histograms by nonlinear regression; for all experiments, curve-fitting produced R2 values of >0.95. Insets show average velocity (left) and run length (right); error bars indicate standard deviations.
Capsid binding at the nuclear membrane.
The fate of incoming vFH589 capsids was assessed via the capsid-bound pUL25/GFP tag. Vero cells were infected with vFH589 or vFH422, and cells were fixed and imaged at 4 and 12 h postinfection. The pUL25/GFP signal was detected in the cytoplasm and primarily as puncta lining the nuclear surface at 4 h postinfection for both the vFH589- and vFH422-infected cells (Fig. 7). By 12 h postinfection, the pattern of immunofluorescence was similar to that observed at 4 h postinfection for vFH589-infected cells, with most of the cells containing capsids concentrated near or over the nuclei, with the exception of one cell that contained a strong nuclear pUL25/GFP signal. In contrast, the pUL25/GFP signal in vFH422-infected cells at 12 h postinfection was found in the nucleus and abundantly throughout the cytoplasm, indicative of viral replication, de novo pUL25 expression, and capsid assembly and egress. Quantitative fluorescent imaging in live cells demonstrated that only a small fraction of the vFH589-infected cells showed intranuclear fluorescence (virus replication compartments) even at late times postinfection (Fig. 8A to C). In contrast, over 80% of the cells infected with vFH422 showed abundant pUL25/GFP fluorescence in the nucleus and diffuse signal in the cytoplasm at late times postinfection. When cells were scored for viral cytopathic effect, 100% of the wild-type (vFH422)-infected cells showed the typical morphology (cell rounding and/or spikelike cell extensions) of HSV-1-infected cells at a late time postinfection (Fig. 8D). In contrast, less than 30% of the cells infected with the pUL25 mutant (vFH589) appeared to be infected. Based on these findings, we conclude that vFH589 capsids enter cells and accumulate in the cytoplasm and at the nuclear membrane but fail to efficiently initiate infection as demonstrated by the absence of pUL25/GFP fluorescence in nuclei at late times postinfection.
FIG 7.

Localization of incoming capsids in cells. Vero cells were infected with vFH422 or vFH589 at 37°C, and cells were fixed at 4 and 12 h postinfection. Incoming capsids were detected by the capsid-bound pUL25/GFP fluorescent signal. Viral gene expression was assessed by diffuse pUL25/GFP signal (green) in the cytoplasm, and the nuclei were stained with DAPI (blue). The boxed areas in the upper panels are the regions enlarged in the corresponding lower panel. Bars, 20 μm (upper panels) and 10 μm (lower panels).
FIG 8.

The HSV pUL25 mutant exhibits delayed gene expression and cytopathology. Vero cells were infected with HSV UL25/GFP-expressing virus vFH422 or vFH589, and cells were imaged at 10.5 to 11.5 h postinfection or from 17 to 18 h postinfection. (A) Representative images of cells infected with the indicated viruses are shown by differential interference contrast (DIC) or pUL25/GFP fluorescence microscopy; bar, 30 μm. (B) The number of cells with intranuclear assemblies is reported as a percentage of the total number of cells. (C) Cells were scored for the presence of diffuse pUL25/GFP signal in the cytoplasm. The plot shows the percentage of total cells exhibiting diffuse cytoplasmic GFP signal. (D) Cells were scored on morphology (cell rounding and/or spikelike cell extensions). Shown is the percentage of cells exhibiting this cytopathic effect. For each time point in panels B to D, approximately 560 to 670 cells were counted per virus in total across two experiments. Error bars indicate standard deviations. Asterisks indicate statistically significant difference from vFH422 (**, P < 0.01; ***, P < 0.001).
DNA-containing capsids accumulate at nuclear pores of vFH518-infected Vero cells.
Transmission electron microscopy (TEM) of Vero cells infected with the vFH518 virions propagated on Vero cells revealed that the capsids docked at NPCs were primarily DNA filled (Fig. 9A, left panel). This is in marked contrast to Vero cells infected with vFH518 virions propagated on the pUL25-complementing cells, where the majority of the capsids engaged with NPCs were empty (Fig. 9A, right panel). Quantification of nucleus-associated capsids demonstrated that 92% contained the viral genome for vFH518 grown on Vero cells, while 93% of the capsids were empty when grown on the complementing cells (Fig. 9). Both the DNA-filled and empty capsids appear to be bound to the NPC as evidenced by the fact that they do not make direct contact with the nuclear membrane. Interestingly, we observed that capsids docked at the NPC were oriented with a vertex pointed toward the pore. The TEM data show that the pUL25 mutant capsid is compromised for genome release upon reaching the nucleus.
FIG 9.

Incoming HSV-1 capsids bind to NPCs. (A) Sections of Vero cells were infected with HSV-1 vFH518 virions isolated from Vero or 8-1 cells at an MOI of 3,000 genomes/cell. Cells were harvested at 4 h postinfection, fixed, and embedded in epon resin. Transmission electron microscopy of cells infected with the vFH518 virions propagated on Vero cells (top left panel) and Vero cells infected with vFH518 virions propagated on the pUL25-complementing 8-1 cells (top right panel). Bar, 100 nm. (B) Quantification of empty and DNA-containing NPC-bound capsids.
HSV-dependent activation of the cGAS DNA-sensing cascade is severely diminished in vFH518 and vFH589 infections.
During capsid delivery to the cytoplasm, transport to the nuclear pore, and uncoating, viral genomic DNA becomes exposed to cellular pattern recognition receptors (PRRs). For DNA viruses, activation of PRRs contributes to induction of a type I interferon (IFN) antiviral response. A prominent PRR involved in the HSV antiviral response is cyclic guanine adenine synthase (cGAS) (30). The binding of cytosolic cGAS to viral DNA stimulates production of cyclic adenine-guanine dinucleotides (cGAMP) that bind to the cytosolic adaptor protein STING. This cGAMP-activated STING establishes a platform for tank binding kinase 1 (TBK1) which undergoes autophosphorylation and mediates IFN regulatory factor 3 (IRF3) phosphorylation (30). The pUL25 mutants offered an opportunity to determine if cGAS activation was dependent upon genome injection into nuclei or alternatively could occur in the cytoplasm of infected cells, where incoming capsids accumulate. A viral dose-response infection (500, 1,500, and 4,500 genome equivalents/cell) was carried out in the highly responsive human macrophage-like THP1 cell line with KOS, vFH422, vFH518, and vFH589. Mock-treated cells and cells exposed to two serotypes of adenovirus (Ad5 and Ad35) provide negative and positive controls, respectively, for activation of the cGAS/STING antiviral cascade (31). At 4 h postinfection, cell lysates were harvested and characterized for activation of the cGAS/STING/TBK1/IRF3 DNA-sensing cascade by Western blotting (Fig. 10). Infection of THP1 cells with increasing genome equivalents of KOS or vFH422 generated a dose-dependent increase in pser172TBK1 and pser396IRF3. In comparison, cells infected with vFH518 and vFH589 were compromised in the induction of pser172TBK1 and pser396IRF3.
FIG 10.

Compromised antiviral recognition of pUL25 mutant HSV in differentiated THP1 cell lines. A dose-response antiviral activation assay with KOS, vFH422, vFH518, and vFH589 viral strains was carried out in human macrophage THP1 cells. Cells were incubated with the indicated genome equivalents (500, 1,500, or 4,500 genomes/cell) of each virus, and total cell lysates were harvested 4 h postinfection. Mock samples were treated with medium only as a negative control, and cells exposed to Ad5 and Ad35 viruses provide weak and strong positive controls for activation of the cGAS/STING antiviral cascade in THP1 cells (31). Western blot analysis of each lysate was carried out and probed with the indicated antibodies as described in Materials and Methods.
A consequence of activating the cGAS DNA-sensing cascade is induction of type I interferons. Through paracrine/autocrine signaling, interferon binding to type I IFN receptors activates the Jak-STAT pathway, resulting in phosphorylation of STAT1 and -2. Tyr701STAT1 phosphorylation provides a secondary response marker for autocrine/paracrine interferon signaling. Levels of pTyr701STAT1 in KOS- or vFH422-infected cells are consistent with a strong antiviral IFN response. However, levels of pTyr701STAT1 were greatly reduced in vFH518- and vFH589-infected cells. Overall, these data are consistent with the hypothesis that the pUL25 truncation mutant compromises capsid uncoating so that viral DNA remains sequestered from detection by the cGAS DNA-sensing PRR during the early stages of virus entry.
DISCUSSION
Capsid-bound pUL25 has several functions: (i) stabilize capsids after DNA is packaged; (ii) trigger nuclear egress of the capsid once a stable DNA-containing capsid is formed; (iii) anchor pUL36, the major tegument protein (also called VP1/2), to capsids; and (iv) promote the delivery of genomes to nuclei upon entering cells (10, 26, 28, 32–35).
Together with pUL36 and pUL17, pUL25 forms a surface feature of the capsid called the capsid vertex-specific component (CVSC) (9, 35). Five copies of the CVSC bind to pairs of triplexes that surround each of the capsid pentons (36, 37). The location of the CVSC was initially observed in cryo-EM reconstructions of HSV nucleocapsids, and it was proposed to be a heterodimer of pUL17 and pUL25 (35). However, more-recent studies demonstrated that the pUL36 protein is required for the stable association of the CVSC with the capsid vertices. These studies demonstrated that pUL17 is present in the triplex-bridging part of the CVSC, with the major part of pUL25 occupying a position nearest the penton (Fig. 4) (9, 21, 38). Higher-resolution cryo-EM density maps of the CVSC revealed a group of helices covering the triplex bridge and extending toward the penton (9). Sections through this bundle indicated that it was composed of 4 or 5 helices that were predicted to originate from pUL17, pUL25, and pUL36. The essential roles in cleavage and stable packaging of the viral genome suggest that the pUL17 and pUL25 proteins also surround the unique portal vertex (7, 29, 32, 34, 39). The binding of pUL25 during assembly is copy controlled, with estimates of pUL25 copy number within the range of 60 to 75 copies, which is consistent with 5 copies of the CVSC around each of 12 vertices (7, 33).
In cells exposed to HSV-1, capsids deposited into the cytosol retain a subset of tegument proteins and are targeted to the nuclear pore, where the genome exits the capsid and is delivered into the nucleus. Capsid binding to the NPC occurs via a distinct orientation, with 1 of the 12 capsid vertexes facing the pore channel (12). It is not known if a specific vertex is favored, but it is likely that the portal vertex where DNA enters the capsid is also positioned for release of the viral genome. Studies of pUL25 and pUL36 demonstrated that both played critical roles in the binding of capsids to the nuclear pore (22, 23), while proteolytic cleavage of pUL36 was critical for DNA release from the NPC-bound capsid (27). Cells treated prior to HSV infection with the serine-cysteine protease inhibitor l-(tosylamido-2-phenyl) ethyl chloromethyl ketone (TPCK) prevented cleavage of pUL36 and release of DNA from capsids. The proteolytic cleavage of pUL36 was found to take place only after the capsid was attached to the nuclear pore, but the protease responsible for cleavage of pUL36 is not known, nor is it known how it specifically cleaves pUL36 on nuclear pore-bound capsids.
In this study, we present evidence that the C terminus of the pUL25 protein is required for triggering release of the viral genome from capsids docked at the nuclear pore. This region of pUL25 was initially identified in complementation assays using a plasmid expressing a C-terminally truncated pUL25 protein (28). A crystallographic structure of the HSV-1 pUL25 (C-terminal amino acids 134 to 580) reveals numerous flexible loops extending from a rigid core composed of several tightly packed alpha helices (40). Deletion of the three C-terminal amino acids removed one of the six unstructured loops (L6) (28), and we isolated this pUL25 mutant in the context of the virus. Our data show that the pUL25 C terminus is required for the efficient release of the HSV-1 genome into the nucleus from capsids, while it is fully dispensable for upstream events during initial infection. Despite the entry, intracellular transport, and cytoplasmic accumulation of capsids with the mutant viruses, activation of the cGAS DNA-sensing cascade did not occur. This finding indicates that the capsid effectively shelters the DNA genome from cellular pattern recognition receptors and furthermore has the interesting implication that during wild-type infection cGAS most likely detects herpesvirus DNA only after the capsid interacts with the NPC, and not before.
How might the pUL25 C-terminal deletion affect genome release from capsids docked at a nuclear pore? The defect does not appear to be due to improper assembly of virions, since similar amounts of pUL25 mutant virions containing viral genomes were recovered from Vero and pUL25-complementing cells (Table 1). In addition, the cryo-EM reconstructions of virion capsids did not detect any obvious differences in the location or structural composition of the CVSC subunits for the pUL25 mutant compared to wild-type HSV virion capsids (Fig. 4). The location of the pUL25 C terminus facing away from the vertex suggests that the truncation in vFH518 may not directly impede DNA release but might act indirectly. Since cleavage of pUL36 on nuclear pore-bound capsids is required for capsid uncoating, a plausible mechanism may involve the C terminus of pUL25 functioning to either activate or stimulate pUL36 cleavage. Indeed, we (9) and others (21) have noted that the pUL36 C-terminal region is likely an essential member of the CVSC that includes pUL17 and pUL25. Significantly, our model places the pUL25 C terminus in the proximity of the pUL36 C-terminal region (9), where it may contribute to the proteolysis of pUL36 that is necessary for DNA release. Visualizing the state of pUL36 on the pUL25 mutant capsids bound to nuclear pores may well shed light on the role of the pUL25 C terminus for pUL36 cleavage, and identifying the cysteine protease remains another important goal.
The genome release defect in the vFH518 mutant is the result of the deletion of the last 3 (serine, alanine, valine) amino acids of the 580-amino-acid pUL25 protein. The C terminus of pUL25 is proposed to form a small disordered loop region (28, 40). This region of pUL25 may engage viral or cell proteins to trigger uncoating once the capsid docks at a nuclear pore complex. HSV virions stripped of their envelope bind to nuclear pores in vitro, and this is inhibited to some degree by a nonspecific antibody to nucleoporins (NUPs) or by an antibody to importin β. Excess importin β also increases capsid binding to nuclei in vitro (19). The role of the NUPs and importin β can be examined by small interfering RNA (siRNA) knockdown of these proteins since NUP214 and NUP358 are required for NPC binding (22, 23).
In summary, we identified a small loop located at the C terminus of the HSV-1 pUL25 protein that is required for release of viral genomes from capsids upon reaching the nucleus following entry into cells. All herpesviruses encode a pUL25 analog, suggesting that understanding how pUL25 functions to trigger genome uncoating during HSV-1 infections will be relevant to the replication of other herpesviruses. We have previously shown that the HSV-1 pUL25 crystal structure can be fit into the pseudorabies virus (PRV) and Kaposi's sarcoma herpesviruses (KSHV) cryo-EM capsid density maps in the same peripentonal region of the CVSC molecule as HSV-1 (9, 38). However, in the case of KHSV, the orientation of the pUL25 analog is inverted relative to that in both HSV-1 and PRV (9), suggesting differences in mechanism despite a close similarity in structure. In addition, although the HSV-1 pUL25 protein can partially complement a PRV pUL25 null virus (41), there is a slight difference in the C-terminal amino acids of PRV and HSV-1 pUL25 (IPQFAAA and IPQYLSAV, respectively) suggesting that there may be a larger region of the pUL25 C terminus that is required for triggering uncoating. Sequence alignment of pUL25 homologs from several alphaherpesviruses located a stretch of highly conserved residues (bolded), DYDLLYFCLGFIPQYLSAV, located just prior to the C terminus of pUL25, that will be of interest to explore.
MATERIALS AND METHODS
Cells and viruses.
African green monkey kidney cells (Vero; CCL-81 from American Type Culture Collection, Rockville, MD) and UL25-transformed 8-1 cells were propagated as previously described (32). HSV wild-type KOS and the pUL25/GFP-expressing virus, vFH422, were previously described (29, 36). UL25 mutant viruses vFH518 and vFH589 were generated by recombination of a KOS (vFH518) or vFH422 (vFH589) genome contained in a bacterial artificial chromosome (BAC) as previously described (29, 42–44). Primers 5′-TACTTTTTATGTCTGGGGTTCATTCCACAGTACCTGTAGTGGGTGGTGGGCGAGTAGGGATAACAGGGTAATCGATTT-3′ and 5′-CCTAATGCCCCCTCCCCCCTCGCCCACCACCCACTACAGGTACTGTGGAATGAAGCCAGTGTTACAACCAATTAACC-3′ were used to amplify template sequences from plasmids p-EP-Kan-S that result in a deletion of the codons for the three C-terminal amino acids (SAV) of pUL25. To confirm mutations, viral DNA was amplified by PCR using primers that flanked the UL25 open reading frame, and the PCR product was sequenced.
Adenoviruses Ad5 and Ad35 were obtained from ATCC. Viruses were purified by two rounds of CsCl banding and dialysis against 4% sucrose, 50 mM Tris (pH 8.0), and 2 mM MgCl2 and stored at −80°C. Viral particle numbers were quantified by spectrophotometric detection of intact virions at optical density at 260 nm (OD260) (1012 particles/OD260 unit).
HSV-1 virion isolation.
HSV-1 virions were isolated from the medium of infected Vero or 8-1 cells as previously described (9, 45). Briefly, 1.5 × 108 Vero or 8-1 cells were infected overnight (18 h at 37°C) at an MOI of 5 PFU per cell. Infected cells were scraped into the cell medium, and 5 M NaCl was added to a final concentration of 0.5 M NaCl. Cells were pelleted, and the medium was transferred to SW28 rotor tubes; virions were pelleted out of the medium by centrifugation at 20,000 rpm for 35 min. The resulting pellet was resuspended in 100 μl of phosphate-buffered saline (PBS) plus protease inhibitors. DNase I (Thermo-Fisher catalog number EN0521); the sample was incubated at room temperature for 30 min and then layered on top of a 20 to 50% sucrose gradient in TNE buffer (10 mM Tris, 150 mM NaCl, and 1 mM EDTA, pH 7.5; SW41 rotor at 24,000 rpm for 1 h). The virion band was collected with a syringe by side puncture of the gradient, transferred to an SW41 tube, and diluted 1:5 with TNE buffer, and the virions were pelleted. The virions were resuspended in TNE buffer, and the titer of each preparation was determined on Vero and 8-1 cells; the viral genome copy number was determined by qPCR.
Quantitation of viral genomes.
To measure the DNA quantity for the calculation of the genome copies per PFU, isolated (DNase-treated) HSV virions were diluted 1:100 in lysis buffer (10 mM Tris [pH 7.5], 10 mM KCl, 1 mM EDTA, 0.6% SDS) containing 1 mg/ml protease grade XIV (Sigma) and incubated overnight at 37°C. qPCR was carried out on the Applied Biosystems StepOnePlus real-time PCR system using Power SYBR green PCR master mix (Life Technologies catalog number 4367659) to determine the number of viral genomes using primers specific for the HSV-1 thymidine kinase (TK) gene. Standard curves were generated using purified KOS DNA. The protease-treated samples were tested over a range of 10-fold dilutions (10−4 to 10−8). Each dilution was analyzed in triplicate, and the number of copies of the HSV-1 TK gene in each well was determined based upon the KOS standard curve. The known sample volume and total DNA dilution for each well were used to calculate the total number of genomes produced. These values as well as the averages of all results for each virion preparation are reported in Table 1.
Immunofluorescence.
Vero cells were plated on 8-chamber slides (Nunc LabTek). The next day, the cells were infected with vFH422 or vFH589 at an MOI of 10. At 4 or 8 h postinfection, the cells were rinsed with PBS three times and fixed by submersion in 4% paraformaldehyde for 15 min. Cells were washed three times with PBS, and 0.1% Triton X-100 was added for 10 min to permeabilize the cells. After a final wash with PBS, Vectashield (Vector Laboratories) mounting medium with DAPI (4′,6-diamidino-2-phenylindole) was added to each of the chamber wells. A coverslip was added, and images were captured with an Olympus FluoView 1000 laser-scanning confocal microscope.
Western blotting.
Protein samples were separated on a 4 to 12% SDS-polyacrylamide gel, and proteins were transferred to nitrocellulose. The nitrocellulose was washed twice in Tris-buffered saline (TBS) and incubated overnight in Rockland Near Infra Red blocking buffer (catalog number MB-070-003; Rockland Immunochemicals, Inc., Gilbertsville, PA). UL25 mouse monoclonal antibody 25E10 (36), GFP rabbit polyclonal antibody (Invitrogen catalog number A-11122), VP5 rabbit polyclonal antibody NC1 (46), ICP8 mouse monoclonal antibody (Abcam catalog number ab20194), ICP4N rabbit polyclonal antibodies (Neal Deluca, University of Pittsburgh), and GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mouse monoclonal antibody (Ambion catalog number AM4300) were diluted 1:1,000, 1:5,000, 1:7,000, 1:1,000, 1:2,000, and 1:5,000, respectively. The diluted antibodies were reacted with the blocked nitrocellulose for 2 h at room temperature, washed five times in TBS with 0.5% Tween 20, and incubated with IRDye 800-conjugated secondary antibodies, goat anti-mouse (UL25, ICP8, GAPDH), or goat anti-rabbit (GFP, ICP4, VP5), from Rockland Immunochemicals diluted 1:15,000 in Rockland Near Infra Red blocking buffer with 0.1% Tween 20. The blots were washed and scanned using an Odyssey system (Li-Cor, Lincoln, NE).
Electron microscopy of thin sections.
Vero cells (2 × 106) were infected for 4 h at 37°C at an MOI of 3,000 genomes/cell. Cells were rinsed in PBS, fixed in 2.5% glutaraldehyde for 1 h at room temperature (RT), rinsed several times with PBS to remove the fixative, and harvested by scraping. The samples were stained (1% osmium tetroxide [OsO4]–1% potassium ferricyanide), sectioned, and mounted onto carbon grids for transmission electron microscopy by the Center of Biologic Imaging, University of Pittsburgh. Images were collected on an in-house FEI T12 electron microscope (FEI, Hillsboro, OR) equipped with a Gatan UltraScan 1000 charge-coupled-device (CCD) camera (Gatan, Pleasanton, CA).
Cryo-electron microscopy and image reconstruction of virion-embedded capsids.
Cryo-EM images of virions isolated from vFH518-infected Vero cells were collected as previously described for HSV-1 and pseudorabies virus (PRV) wild-type viruses (9). Briefly, 3.5 μl of virion sample was pipetted onto a freshly glow-discharged Quantifoil R2/1 EM grid (Quantifoil; Jena) and plunge-frozen into a liquefied mixture of ethane and propane (60:40 mix) using an FEI Vitrobot Mark III instrument (85% humidity; 4.5 to 8 s of blotting). Grids were imaged in an FEI Polara microscope (FEI, Hillsboro, Oregon) operating at 300 kV and controlled by the FEI “EPU” software package. Images were collected on an FEI Falcon 2 direct electron-detecting camera at a nominal magnification of ×59,000, with a postcolumn magnification factor of ×1.4, corresponding to 1.8 Å/pixel at the sample. Capsid images were picked manually using the ×3dpreprocess software (47), and defocus estimates were made with CTFFIND3 (48). The density map was determined with AUTO3DEM (49), and the resolution was estimated at 6.4 Å by the Fourier Shell Correlation (FSC) dropping to 0.3, comparable to the resolutions estimated for the HSV-1 and PRV wild-type capsid maps (9). Density maps were rendered as surfaces in UCSF Chimera (50).
Neuron culture, retrograde transport, and particle tracking.
Whole dorsal root ganglia (DRG) were isolated from embryonic chicks (embryonic day 8 [E8] to E10) as described previously (51, 52). Briefly, explants were cultured for 2 to 3 days on 22-mm2 no. 1.5 glass coverslips treated with poly-dl-ornithine and laminin and in Dulbecco's modified Eagle medium (DMEM)-F-12 (Invitrogen catalog number 11039-021) supplemented with a nutrient mix consisting of 0.08 g/ml bovine serum albumin fraction V powder (VWR), 0.4 mg/ml crystalline bovine pancreas insulin (Sigma-Aldrich), 0.4 μg/ml sodium selenite (VWR), 4 μg/ml transferrin (Intercell Technology), and 5 ng/ml nerve growth factor (NGF; Sigma-Aldrich) (53). Explants were infected with a final concentration of 1.2 × 109 particles/ml of either vFH422 or vFH589, and capsids were imaged during the first hour postinfection by time-lapse fluorescence microscopy with 100-ms streaming exposures on a Nikon Eclipse TE2000-U wide-field fluorescence microscope fitted with a 60× 1.49 numerical aperture (NA) objective and a Cascade II:512 camera. Individual velocities and run lengths for particles moving ≥0.5 μm were determined using the kymograph function of the MetaMorph software package (parameters: multiline tool, 20 pixel width, and average background subtraction) (54). The following number of particles were quantified: for vFH422, n = 145; for vFH589, n = 226. Gaussian (velocities) or decaying exponential (run lengths) curves were fit to histograms by nonlinear regression; for all experiments, curve fitting produced R2 values of >0.95.
Live-cell imaging in Vero cells.
Vero cells were seeded onto flame-sterilized no. 1.5 glass coverslips and maintained in DMEM supplemented with 10% (vol/vol) bovine growth serum (BGS; HyClone). During infection, the BGS concentration was reduced to 2%. Cells were infected with ∼3 genomes/cell of either vFH422 or vFH589. At 10.5 to 11.5 h postinfection or from 17 to 18 h postinfection, infected cells were sealed into VALAB (a 1:1:1 mixture of Vaseline, beeswax, and lanolin) chambers containing cell culture medium. GFP emissions from pUL25/GFP were then imaged with 1,700-ms exposures on a Ti inverted microscope fitted with a 100× 1.45-NA objective (Nikon Instruments), a CSU-W1 confocal head (Yokogawa Electric Corporation), and a CascadeII:1024 EM-CCD camera (Photometrics), all housed in an environmental box at 37°C (InVivo Scientific). Illumination was provided by an Obis 488 laser (Coherent) and custom laser launch (Solamere Technology Group, Inc.). Approximately 600 cells were counted per virus across all experiments, and the following phenotypes were determined: morphology (cytopathic effect), diffuse pUL25/GFP signal in the cytoplasm, and intranuclear capsid clusters.
cGAS studies.
For experiments involving infection of THP1 cells, cells were plated at 7.5 × 105/well in 12-well plates and differentiated by treatment with 50 ng/ml phorbol 12-myristate 13-acetate (PMA) for 6 h. PMA-containing medium was removed, and cells were gently washed twice with PBS and allowed to recover for 48 h in fresh medium (1 ml RPMI, 10% fetal bovine serum [FBS], 1% penicillin-streptomycin) to reduce residual stress response from PMA treatment. For virus infections, virus stocks were diluted in Opti-MEM such that 50 μl of diluted virus introduced the appropriate number of genomes/cell/well. For mock samples, Opti-MEM alone was used. Following addition of virus dilutions, plates were gently rocked and incubated 4 h at 37°C in 5% CO2.
Whole-cell extracts were prepared by washing cells twice with ice-cold PBS and incubating them in lysis buffer (50 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1% NP-40) with addition of phosphatase inhibitor cocktails 1 and 2 (Sigma catalog numbers P2850 and P5726) and protease inhibitors (30 mM sodium fluoride, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, 10 μg/ml leupeptin, 1 μg/ml pepstatin, 1 mM benzamidine) for 30 min at 4°C on a rocking platform before scraping and transferring to tubes. The lysates were cleared by centrifugation at 13,000 × g for 20 min at 4°C, and protein quantification was performed with the DC protein assay kit (Bio-Rad Laboratories). For Western blot analysis, 20 μg total protein was separated using standard 10% SDS-polyacrylamide gels and transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon P; Millipore). All blots were blocked in 5% skim milk in TBS-Tween (0.1%) at room temperature for 1 h. Phospho-IRF3 (Ser396, catalog number 4947), phospho-STAT1 (58D6) (Tyr701, catalog number 9167), beta-actin (catalog number 4967), STING (catalog number 3337), pTBK1 (Ser172; catalog number 5483), and horseradish peroxidase (HRP)-linked anti-rabbit IgG (catalog number 7074) antibodies were from Cell Signaling. All primary antibodies were used at a dilution of 1:3,000 in 5% bovine serum albumin (BSA) in TBS. The HRP-linked secondary antibody was diluted 1:4,000 in 5% milk Tween-TBS. Detection was done with Luminata Crescendo Western HRP substrate (Millipore).
ACKNOWLEDGMENTS
We are grateful for assistance with cryo-electron microscopy from Matthijn Vos (FEI).
This work was supported by NIH grants R01 AI089803 to F.L.H. and J.F.C. and R01 AI056346 to G.A.S. G.R.D. was supported by the training program in the Cellular and Molecular Basis of Disease from the National Institutes of Health (T32 GM08061). Research reported in this publication was also supported by the Office of the Director, National Institutes of Health, under award number S10OD019995 (J.F.C.).
The content is solely our responsibility and does not necessarily represent the official views of the National Institutes of Health.
REFERENCES
- 1.Davison AJ, Eberle R, Ehlers B, Hayward GS, McGeoch DJ, Minson AC, Pellett PE, Roizman B, Studdert MJ, Thiry E. 2009. The order Herpesvirales. Arch Virol 154:171–177. doi: 10.1007/s00705-008-0278-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eisenberg RJ, Heldwein EE, Cohen GH, Krummenacher C. 2011. Recent progress in understanding herpes simplex virus entry: relationship of structure to function, p 131–152. In Weller SK. (ed), Alphaherpesviruses: molecular virology. Caister Academic Press, Norfolk, UK. [Google Scholar]
- 3.Spear PG, Roizman B. 1967. Buoyant density of herpes simplex virus in solutions of caesium chloride. Nature 214:713–714. doi: 10.1038/214713a0. [DOI] [PubMed] [Google Scholar]
- 4.van Genderen IL, Brandimarti R, Torrisi MR, Campadelli G, van Meer G. 1994. The phospholipid composition of extracellular herpes simplex virions differs from that of host cell nuclei. Virology 200:831–836. doi: 10.1006/viro.1994.1252. [DOI] [PubMed] [Google Scholar]
- 5.Loret S, Guay G, Lippe R. 2008. Comprehensive characterization of extracellular herpes simplex virus type 1 virions. J Virol 82:8605–8618. doi: 10.1128/JVI.00904-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sciortino MT, Suzuki M, Taddeo B, Roizman B. 2001. RNAs extracted from herpes simplex virus 1 virions: apparent selectivity of viral but not cellular RNAs packaged in virions. J Virol 75:8105–8116. doi: 10.1128/JVI.75.17.8105-8116.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bohannon KP, Jun Y, Gross SP, Smith GA. 2013. Differential protein partitioning within the herpesvirus tegument and envelope underlies a complex and variable virion architecture. Proc Natl Acad Sci U S A 110:E1613–E1620. doi: 10.1073/pnas.1221896110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grunewald K, Desai P, Winkler DC, Heymann JB, Belnap DM, Baumeister W, Steven AC. 2003. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 302:1396–1398. doi: 10.1126/science.1090284. [DOI] [PubMed] [Google Scholar]
- 9.Huet A, Makhov AM, Huffman JB, Vos M, Homa FL, Conway JF. 2016. Extensive subunit contacts underpin herpesvirus capsid stability and interior-to-exterior allostery. Nat Struct Mol Biol 23:531–539. doi: 10.1038/nsmb.3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Coller KE, Lee JI, Ueda A, Smith GA. 2007. The capsid and tegument of the alphaherpesviruses are linked by an interaction between the UL25 and VP1/2 proteins. J Virol 81:11790–11797. doi: 10.1128/JVI.01113-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leelawong M, Lee JI, Smith GA. 2012. Nuclear egress of pseudorabies virus capsids is enhanced by a subspecies of the large tegument protein that is lost upon cytoplasmic maturation. J Virol 86:6303–6314. doi: 10.1128/JVI.07051-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mettenleiter TC. 2016. Breaching the barrier—the nuclear envelope in virus infection. J Mol Biol 428:1949–1961. doi: 10.1016/j.jmb.2015.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Antinone SE, Zaichick SV, Smith GA. 2010. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J Virol 84:13019–13030. doi: 10.1128/JVI.01296-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Granzow H, Klupp BG, Mettenleiter TC. 2005. Entry of pseudorabies virus: an immunogold-labeling study. J Virol 79:3200–3205. doi: 10.1128/JVI.79.5.3200-3205.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Luxton GW, Haverlock S, Coller KE, Antinone SE, Pincetic A, Smith GA. 2005. Targeting of herpesvirus capsid transport in axons is coupled to association with specific sets of tegument proteins. Proc Natl Acad Sci U S A 102:5832–5837. doi: 10.1073/pnas.0500803102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abaitua F, Hollinshead M, Bolstad M, Crump CM, O'Hare P. 2012. A nuclear localization signal in herpesvirus protein VP1-2 is essential for infection via capsid routing to the nuclear pore. J Virol 86:8998–9014. doi: 10.1128/JVI.01209-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schipke J, Pohlmann A, Diestel R, Binz A, Rudolph K, Nagel CH, Bauerfeind R, Sodeik B. 2012. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J Virol 86:3682–3700. doi: 10.1128/JVI.06432-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rode K, Dohner K, Binz A, Glass M, Strive T, Bauerfeind R, Sodeik B. 2011. Uncoupling uncoating of herpes simplex virus genomes from their nuclear import and gene expression. J Virol 85:4271–4283. doi: 10.1128/JVI.02067-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ojala PM, Sodeik B, Ebersold MW, Kutay U, Helenius A. 2000. Herpes simplex virus type 1 entry into host cells: reconstitution of capsid binding and uncoating at the nuclear pore complex in vitro. Mol Cell Biol 20:4922–4931. doi: 10.1128/MCB.20.13.4922-4931.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bauer DW, Huffman JB, Homa FL, Evilevitch A. 2013. Herpes virus genome, the pressure is on. J Am Chem Soc 135:11216–11221. doi: 10.1021/ja404008r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fan WH, Roberts AP, McElwee M, Bhella D, Rixon FJ, Lauder R. 2015. The large tegument protein pUL36 is essential for formation of the capsid vertex-specific component at the capsid-tegument interface of herpes simplex virus 1. J Virol 89:1502–1511. doi: 10.1128/JVI.02887-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Copeland AM, Newcomb WW, Brown JC. 2009. Herpes simplex virus replication: roles of viral proteins and nucleoporins in capsid-nucleus attachment. J Virol 83:1660–1668. doi: 10.1128/JVI.01139-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pasdeloup D, Blondel D, Isidro AL, Rixon FJ. 2009. Herpesvirus capsid association with the nuclear pore complex and viral DNA release involve the nucleoporin CAN/Nup214 and the capsid protein pUL25. J Virol 83:6610–6623. doi: 10.1128/JVI.02655-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abaitua F, Daikoku T, Crump CM, Bolstad M, O'Hare P. 2011. A single mutation responsible for temperature-sensitive entry and assembly defects in the VP1-2 protein of herpes simplex virus. J Virol 85:2024–2036. doi: 10.1128/JVI.01895-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Batterson W, Furlong D, Roizman B. 1983. Molecular genetics of herpes simplex virus. VIII. further characterization of a temperature-sensitive mutant defective in release of viral DNA and in other stages of the viral reproductive cycle. J Virol 45:397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Preston VG, Murray J, Preston CM, McDougall IM, Stow ND. 2008. The UL25 gene product of herpes simplex virus type 1 is involved in uncoating of the viral genome. J Virol 82:6654–6666. doi: 10.1128/JVI.00257-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jovasevic V, Liang L, Roizman B. 2008. Proteolytic cleavage of VP1-2 is required for release of herpes simplex virus 1 DNA into the nucleus. J Virol 82:3311–3319. doi: 10.1128/JVI.01919-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Hara M, Rixon FJ, Stow ND, Murray J, Murphy M, Preston VG. 2010. Mutational analysis of the herpes simplex virus type 1 UL25 DNA packaging protein reveals regions that are important after the viral DNA has been packaged. J Virol 84:4252–4263. doi: 10.1128/JVI.02442-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cockrell SK, Sanchez ME, Erazo A, Homa FL. 2009. Role of the UL25 protein in herpes simplex virus DNA encapsidation. J Virol 83:47–57. doi: 10.1128/JVI.01889-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li XD, Wu J, Gao D, Wang H, Sun L, Chen ZJ. 2013. Pivotal roles of cGAS-cGAMP signaling in antiviral defense and immune adjuvant effects. Science 341:1390–1394. doi: 10.1126/science.1244040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lam E, Stein S, Falck-Pedersen E. 2014. Adenovirus detection by the cGAS/STING/TBK1 DNA sensing cascade. J Virol 88:974–981. doi: 10.1128/JVI.02702-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McNab AR, Desai P, Person S, Roof LL, Thomsen DR, Newcomb WW, Brown JC, Homa FL. 1998. The product of the herpes simplex virus type 1 UL25 gene is required for encapsidation but not for cleavage of replicated viral DNA. J Virol 72:1060–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Newcomb WW, Homa FL, Brown JC. 2006. Herpes simplex virus capsid structure: DNA packaging protein UL25 is located on the external surface of the capsid near the vertices. J Virol 80:6286–6294. doi: 10.1128/JVI.02648-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stow ND. 2001. Packaging of genomic and amplicon DNA by the herpes simplex virus type 1 UL25-null mutant KUL25NS. J Virol 75:10755–10765. doi: 10.1128/JVI.75.22.10755-10765.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Trus BL, Newcomb WW, Cheng N, Cardone G, Marekov L, Homa FL, Brown JC, Steven AC. 2007. Allosteric signaling and a nuclear exit strategy: binding of UL25/UL17 heterodimers to DNA-Filled HSV-1 capsids. Mol Cell 26:479–489. doi: 10.1016/j.molcel.2007.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Conway JF, Cockrell SK, Copeland AM, Newcomb WW, Brown JC, Homa FL. 2010. Labeling and localization of the herpes simplex virus capsid protein UL25 and its interaction with the two triplexes closest to the penton. J Mol Biol 397:575–586. doi: 10.1016/j.jmb.2010.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toropova K, Huffman JB, Homa FL, Conway JF. 2011. The herpes simplex virus 1 UL17 protein is the second constituent of the capsid vertex-specific component required for DNA packaging and retention. J Virol 85:7513–7522. doi: 10.1128/JVI.00837-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dai X, Gong D, Wu TT, Sun R, Zhou ZH. 2014. Organization of capsid-associated tegument components in Kaposi's sarcoma-associated herpesvirus. J Virol 88:12694–12702. doi: 10.1128/JVI.01509-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Salmon B, Baines JD. 1998. Herpes simplex virus DNA cleavage and packaging: association of multiple forms of U(L)15-encoded proteins with B capsids requires at least the U(L)6, U(L)17, and U(L)28 genes. J Virol 72:3045–3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bowman BR, Welschhans RL, Jayaram H, Stow ND, Preston VG, Quiocho FA. 2006. Structural characterization of the UL25 DNA-packaging protein from herpes simplex virus type 1. J Virol 80:2309–2317. doi: 10.1128/JVI.80.5.2309-2317.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuhn J, Leege T, Klupp BG, Granzow H, Fuchs W, Mettenleiter TC. 2008. Partial functional complementation of a pseudorabies virus UL25 deletion mutant by herpes simplex virus type 1 pUL25 indicates overlapping functions of alphaherpesvirus pUL25 proteins. J Virol 82:5725–5734. doi: 10.1128/JVI.02441-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cockrell SK, Huffman JB, Toropova K, Conway JF, Homa FL. 2011. Residues of the UL25 protein of herpes simplex virus that are required for its stable interaction with capsids. J Virol 85:4875–4887. doi: 10.1128/JVI.00242-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tischer BK, Smith GA, Osterrieder N. 2010. En passant mutagenesis: a two step markerless red recombination system. Methods Mol Biol 634:421–430. doi: 10.1007/978-1-60761-652-8_30. [DOI] [PubMed] [Google Scholar]
- 44.Tischer BK, J von Einem Kaufer B, Osterrieder N. 2006. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- 45.Homa FL, Huffman JB, Toropova K, Lopez HR, Makhov AM, Conway JF. 2013. Structure of the pseudorabies virus capsid: comparison with herpes simplex virus type 1 and differential binding of essential minor proteins. J Mol Biol 425:3415–3428. doi: 10.1016/j.jmb.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cohen GH, Ponce de Leon M, Diggelmann H, Lawrence WC, Vernon SK, Eisenberg RJ. 1980. Structural analysis of the capsid polypeptides of herpes simplex virus types 1 and 2. J Virol 34:521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Conway JF, Steven AC. 1999. Methods for reconstructing density maps of “single” particles from cryoelectron micrographs to subnanometer resolution. J Struct Biol 128:106–118. doi: 10.1006/jsbi.1999.4168. [DOI] [PubMed] [Google Scholar]
- 48.Mindell JA, Grigorieff N. 2003. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol 142:334–347. doi: 10.1016/S1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 49.Yan X, Sinkovits RS, Baker TS. 2007. AUTO3DEM–an automated and high throughput program for image reconstruction of icosahedral particles. J Struct Biol 157:73–82. doi: 10.1016/j.jsb.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 51.Daniel GR, Sollars PJ, Pickard GE, Smith GA. 2015. Pseudorabies virus fast axonal transport occurs by a pUS9-independent mechanism. J Virol 89:8088–8091. doi: 10.1128/JVI.00771-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He Y, Baas PW. 2003. Growing and working with peripheral neurons. Methods Cell Biol 71:17–35. doi: 10.1016/S0091-679X(03)01002-1. [DOI] [PubMed] [Google Scholar]
- 53.Smith GA, Gross SP, Enquist LW. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc Natl Acad Sci U S A 98:3466–3470. doi: 10.1073/pnas.061029798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zaichick SV, Bohannon KP, Hughes A, Sollars PJ, Pickard GE, Smith GA. 2013. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 13:193–203. doi: 10.1016/j.chom.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]