

Abstract
Genetic mosaic analyses represent an invaluable approach for the study of stem cell lineages in the Drosophila ovary. The generation of readily identifiable, homozygous mutant cells in the context of wild-type ovarian tissues within intact organisms allows the pinpointing of cellular requirements for gene function, which is particularly important for understanding the physiological control of stem cells and their progeny. Here, we provide a step-by-step guide to the generation and analysis of genetically mosaic ovaries using flippase (FLP)/FLP recognition target (FRT)-mediated recombination in adult Drosophila melanogaster, with a focus on processes of oogenesis that are controlled by diet-dependent factors.
Keywords: Oogenesis, clonal analysis, genetic mosaics, FLP/FRT, germline stem cell, follicle stem cell, follicle cell, Drosophila
1. Introduction
The ease of genetic mosaic generation in Drosophila melanogaster has allowed significant advances in understanding multiple aspects of stem cell biology and other processes during oogenesis [1,2]. Genetic mosaic analyses, which typically involve the generation of identifiable, genetically distinct clones of cells within the context of wild-type tissue, allow the tracing of cell lineages, determining exact cells in which gene function is required, and distinguishing between cell autonomous and non-cell autonomous roles for genes. Genetic mosaics afford the added advantage of circumventing the lethality of mutations in essential genes, thereby uncovering their roles in later developmental stages.
Methods for the generation of mosaic animals have evolved over the years from technically challenging experimental manipulations involving transplantation, to the use of sophisticated genetic tools that facilitate mitotic recombination. In the classic quail-chicken chimera example, cells transplanted from quail embryos were distinguished from those of the host chicken embryo by the dense regions of heterochromatin in their nuclei, permitting the mapping of their fate during development [3]. In Drosophila melanogaster, transplantation of pole cells allowed the removal of gene function exclusively from the germline [4], and transplantation of imaginal discs elucidated the tissue-autonomous and environmental factors influencing their developmental fate [5]. X-ray-induced mitotic recombination was useful in generating clones of mutant cells for the purpose of addressing cell autonomy of gene function [6]. With the advent of molecular tools for inducible, site-specific mitotic recombination taking advantage of the yeast-derived flippase (FLP)/FLP recognition target (FRT) system [7], the use of genetic mosaic analysis in Drosophila has become commonplace.
Genetic mosaic analyses are very versatile. Typically, genetic mosaics are generated in the context of heterozygous organisms that carry FRT sequences at the base of specific chromosomes arms. One chromosome arm carries a mutation of interest, while its homolog has a wild-type allele of the corresponding gene and a readily identifiable marker, such as a ubiquitously expressed transgene encoding green fluorescent protein (GFP) or β-galactosidase (β-gal). In addition, a transgene encoding FLP under the control of a heat-shock inducible or tissue-specific promoter is present in trans. Once FLP expression is induced – for example, by heat-shocking the organism at a specific point during development or adulthood – cells can undergo FLP-mediated mitotic recombination through homologous FRT sequences, potentially generating unequal sister chromatids (Fig. 1A). As sister chromatids segregate during mitosis, a homozygous mutant cell lacking the GFP (or β-gal) marker might be generated, forming a clone of GFP-negative mutant cells as it subsequently undergoes cell division rounds (Fig. 1B–G). It should be noted, however, that numerous variations of this technique have been developed, involving the generation of positively marked mutant clones, clones for overexpression of transgenes or RNA hairpins for RNA interference, or wild-type clones for lineage tracing analysis [8–10].
Fig. 1.

Confocal images of genetic mosaic ovarioles and germaria. (A) GFP- or β-gal-negative mutant cells can be generated as unequal sister chromatids, produced as a result of FLP/FRT-mediated mitotic recombination (grey dashed lines), segregate during mitosis. Mutant allele is indicated by pink box and asterisk. Marker (orange box) is a constitutively expressed transgene encoding GFP or β-gal. (B) Mosaic ovariole containing previtellogenic (asterisk) and vitellogenic (arrowhead) follicles with GFP-negative germline cysts. (C) In a mosaic germarium, a GFP-negative GSC (arrowhead) gives rise to GFP-negative progeny. (D) A GSC loss event. GFP-negative germline cysts are present, but the original GFP-negative GSC is absent. (E) The FSC is located immediately anterior to the 2a/2b border, and it is recognizable as the anterior-most cell (arrowhead) in a GFP-negative follicle cell clone. (In region 2a, individual 16-cell cysts do not fill entire diameter of germarium, whereas in region 2B, lens-shaped 16-cell cysts span the breadth of germarium.) (F) When the FSC is lost, GFP-negative follicle cells can be detected, but the most anterior follicle cells are far posterior to 2a/2b. (G) A transient clone (dashed line) in a follicle cell monolayer provides an indirect readout for follicle cell proliferation. Absence of GFP (green) indicates marker-negative cells; 1B1 (red) labels fusomes and follicle cell membranes; Lamin C (LamC, red) labels cap cell nuclear envelopes; DAPI (blue) labels nuclei. Scale bars represent 10 µM. Images in (C–F) are shown at the same magnification.
The focus of this chapter is how genetic mosaic analysis using adult-generated negatively-marked clones of cells in the germline or follicle cell lineage can be used to study a number of processes during Drosophila oogenesis that are known to be controlled by dietary conditions. Previous studies in our laboratory using this type of analysis have led to the identification of specific cells that require various nutrient-sensing or hormonal pathway components, allowing us to distinguish between direct versus indirect roles of systemic factors in controlling multiple distinct processes, including germline stem cell (GSC) and follicle stem cell (FSC) maintenance or proliferation, germline cyst growth and development, follicle cell proliferation, and vitellogenesis [11–17]. The described protocol represents a detailed guide to strain generation, FLP/FRT-mediated clonal induction, ovary dissection and immunostaining, and data analysis.
2. Materials
2.1 Drosophila strains and culture conditions
Suitable Drosophila strains (see Note 1), including mutant stock of interest, heat-shock inducible flippase (hs-Flp) (see Note 2), FRT insertion on appropriate chromosome arm (see Note 3), and a corresponding FRT insertion recombined to a ubiquitously expressed marker, such as ubi-GFP or arm-lacZ (for GFP or β-gal expression, respectively).
G418 (Sigma) diluted in water to appropriate concentration, according to specific FRT insertion (see Note 4).
Standard fly culture media in a plugged vial.
Dry active yeast, such as used in baking.
Wet yeast paste: ~20 g active dry yeast thoroughly mixed into ~35 mL of dH2O to the consistency of smooth peanut butter (see Note 5).
Water bath set at 37°C.
Vinyl-coated lead weight ring (or other weight of approximately 500 grams).
Kimwipes.
Plastic rack for fly vials.
Dissecting pin or thin spatula.
2.2 Dissection and immunostaining of ovaries
1.5 mL microfuge tubes (see Note 6).
Glass or plexiglass dissection dish.
Kimwipes.
Glass Pasteur pipette and bulb.
Phosphate buffered saline (PBS).
Grace’s Insect Medium (BioWhittaker).
3% Bovine serum albumin (BSA; Sigma) prepared in water.
Washing solution: 0.1% Triton X-100 in PBS (see Note 7).
Blocking solution: 5% BSA, 0.1% Triton X-100, 5% normal goat serum in PBS (see Note 8).
Fixation solution: 5.3% formaldehyde in Grace’s Insect Medium, prepared from 16% formaldehyde (Ted Pella) (see Note 9).
Primary antibodies: mouse anti-1B1 (Adducin-related protein; Developmental Studies Hybridoma Bank, DSHB), mouse anti-Lamin C (LC28.26; DSHB); chicken anti-GFP (Abcam) or chicken anti-β-gal (Abcam).
Secondary antibodies: anti-mouse Alexa Fluor 568 or 633 and anti-chicken Alexa Fluor 488 (Life Technologies).
Click-It Kit (Invitrogen), for EdU incorporation assay (see Note 10).
4',6-diamidino-2-phenylindole (DAPI), for staining DNA.
Microscope slides and coverslips.
Weights of approximately 120 grams, for flattening mounted samples.
Stereomicroscope.
2 pairs of sharpened forceps.
Tungsten needle and/or 27-gauge needle and syringe.
Nutator, for rotation of sample during fixation, washing, and immunostaining procedures.
2.3 Image acquisition and analysis
Confocal microscope, or equivalent microscopy set-up.
Image analysis software (such as ImageJ).
3. Methods
Overall, setting of the standard crosses to obtain control and experimental genotypes and performing the heat shock protocol described below take approximately two weeks if starting from expanded, healthy fly stocks. Following the final heat-shock, the timing of dissection for clonal analysis is a crucial variable for the appropriate interpretation of results, as discussed in section 3.3.
3.1 Drosophila strains and culture conditions
Generate a recombinant fly stock containing both the proximal FRT insertion and the mutant allele of interest on the same chromosome arm through standard crosses. FRT transgenes may carry different selection markers, but the majority include the neoR marker (see Notes 4 and 11). To select for flies carrying the neoR-containing FRT among progeny resulting from recombination cross (see Note 12), maintain the cross on food treated with G418 solution of appropriate concentration (see Note 13). Crosses should be transferred to fresh food every two days, such that the resulting progeny will be raised on G418 and thereby selected for the presence of the FRT insertion. Individual progeny should subsequently be screened for the presence of the mutant allele of interest for identification of flies carrying recombinant chromosome and balanced as a stock.
Generate flies of control and experimental genotypes (see Note 14) through standard crosses. At 0–2 days after eclosion (see Note 15), transfer females of appropriate genotypes along with sibling males to vials especially prepared for heat shock. These vials should include half of a folded Kimwipe directly covering the food surface to prevent the flies from sticking to it during heat shock.
Place flies in heat shock vials in a plastic rack, spreading vials out to allow easy water flow between them. Heat shock flies in the 37°C water bath, placing the weight on top of the vials to keep the rack underwater, and maintaining the appropriate water level to ensure that flies are confined to the submerged portion of the vial. Heat shock should be conducted for one hour at a time, twice daily (see Note 16), for three consecutive days (see Note 17).
Following the final heat shock, transfer flies to vials supplemented with wet yeast paste, adding new males to the vials if some have died during heat shock. Transfer flies to vials containing fresh wet yeast daily until dissection. When selecting time points for dissection, consider the perdurance of both the marker used (see Note 18) and the protein of interest. Dissection time points up to ten days after heat shock will include both transient and permanent clones [18] (see Note 19), which is an important consideration when interpreting the data. Multiple time points are typically included in the analyses.
3.2 Dissection and immunostaining of ovaries
Prepare Eppendorf tubes for dissected ovaries by filling them with 3% BSA solution (see Note 20).
Using a Pasteur pipette, transfer Grace’s Insect Medium to a dissection dish (see Note 21).
Anesthetize flies using CO2 and select females for dissection. Pick up females one at a time by gently pinching the thorax with sharp forceps.
Submerge each female in a dissection well filled with Grace’s medium under a stereomicroscope. While holding females by the thorax, use the second pair of forceps to carefully pinch and pull away the posterior of the abdomen (at approximately two segments from the end). Ovaries should come out easily; otherwise, they can be pushed out of the abdomen.
Tease apart the anterior halves of ovarioles using a sharp tungsten needle or a fine-gauge needle in a syringe (see Note 22). Immobilize ovaries by holding on to their posterior end using a pair of forceps and run the tungsten needle between ovarioles to tear the muscle sheath away from the anterior half.
Before transferring dissected ovaries to Eppendorf tubes, remove the BSA solution from Ependorf tubes using a Pasteur pipette, and discard the solution. This will also serve to coat the pipette with BSA and prevent ovaries from sticking to the glass. Use this coated pipette to transfer the dissected ovaries to the Eppendorf tube.
Repeat this process for all genotypes, minimizing the time between dissection and fixation. Ideally, the time between dissection and fixation should not exceed 30 minutes.
Fix ovaries in freshly prepared fixation solution for 13 minutes with rotation on a nutator at room temperature (see Note 23).
Rinse ovaries three times in washing solution by letting ovaries settle to bottom of the tube, then repeatedly changing the buffer. Wash four times for at least 15 minutes each on nutator at room temperature (see Note 24).
Block ovaries in blocking solution for at least three hours at room temperature or overnight at 4°C on nutator (see Note 25).
Stain ovaries with primary antibodies diluted in blocking solution: anti-1B1 (1:10), anti-Lamin C (1:100), and anti-GFP (1:2000). Primary antibody incubation times range from three hours at room temperature to overnight at 4°C on nutator.
Wash samples in washing solution four times for at least 15 minutes each on nutator (see Note 26).
Stain ovaries with secondary antibodies (anti-mouse Alexa 568 and anti-chicken Alexa 488) diluted 1:200 in blocking solution and protected from light with aluminum foil. Secondary antibody incubation times range from one hour to five hours at room temperature on nutator.
Stain sample with 0.5 µg/mL DAPI in washing solution for 10 minutes at room temperature, protected from light, on nutator.
Wash sample in washing solution four times for at least 15 minutes each at room temperature, protected from light, on nutator.
Remove washing solution and add a small volume of the mounting medium of choice. (We use either Vectashield or 90% glycerol containing 20 mg/ml n-propyl gallate). Gently and thoroughly mix ovarioles with their mounting medium using a Pasteur pipette. Samples will keep at 4°C in the dark in mounting medium for extended periods of time (see Note 27).
To mount samples, transfer samples mixed with mounting medium onto a glass slide under a stereomicroscope. Using a pair of tungsten needles, carefully separate large late stage egg chambers from ovarioles and remove them from the slide. (For details on the staging of ovarian follicles, see Ref. 19.) The presence of large egg chambers on the slide will prevent the germaria from being sufficiently flattened by the mounting process, making it difficult to image them. Using tungsten needles, gently distribute ovarioles away from each other prior to adding the coverslip.
Add glass coverslip, cover it with a Kimwipe, and apply gentle pressure to the sample using a weight. This will flatten the ovarioles to facilitate imaging (see Note 28). Seal the coverslip using nail polish. Sealed, mounted slides will keep for extended periods of time at 4°C in the dark.
3.3 Image acquisition and analysis
Several general considerations in genetic mosaic analysis are crucial for accurate data interpretation. For example, perdurance of the protein of interest after removal of the cognate gene through mitotic recombination will depend on the stability of the protein and corresponding mRNA. Similarly, visualization of mutant cells will depend on the perdurance of GFP or β-gal markers. Finally, the marker expression level and the frequency of clone induction will vary depending on the specific marker and FRT insertions used for the experiments, respectively.
The types of images required vary depending on the type of analysis being conducted. We find it more efficient to acquire images for one type of analysis at a time rather than acquiring all types of images during the same microscopy sessions because the image acquisition mode may vary according to type of analysis. One should also be careful to avoid the analysis of damaged ovarioles (see Ref. 20) or those where immunostaining did not work well. The most common types of analyses performed in our lab are described below, starting with germline analyses involving ovarioles followed by those focused on the germarium, and ending with analyses of the follicle cell lineage.
Follicle growth and survival
The growth and survival of GFP- (or β-gal-) negative mutant germline cysts within developing follicles is assessed relative to flanking follicles containing GFP-positive cysts within the same ovariole (Fig. 1B, asterisk; Fig. 2A). In control mosaic ovarioles, GFP-negative follicles are larger than anterior and smaller than posterior flanking follicles. A deviation from this pattern in the experimental mosaics can reflect either a defect in cyst growth or premature death of the cyst. These two possibilities can be distinguished by co-staining ovaries with an apoptosis marker. Several dozen of ovarioles should be analyzed per genotype at 10 days after the last heat shock. (For more precise quantification of the extent of cyst growth delay or overgrowth, see Refs 16 and 17.)
Fig. 2.
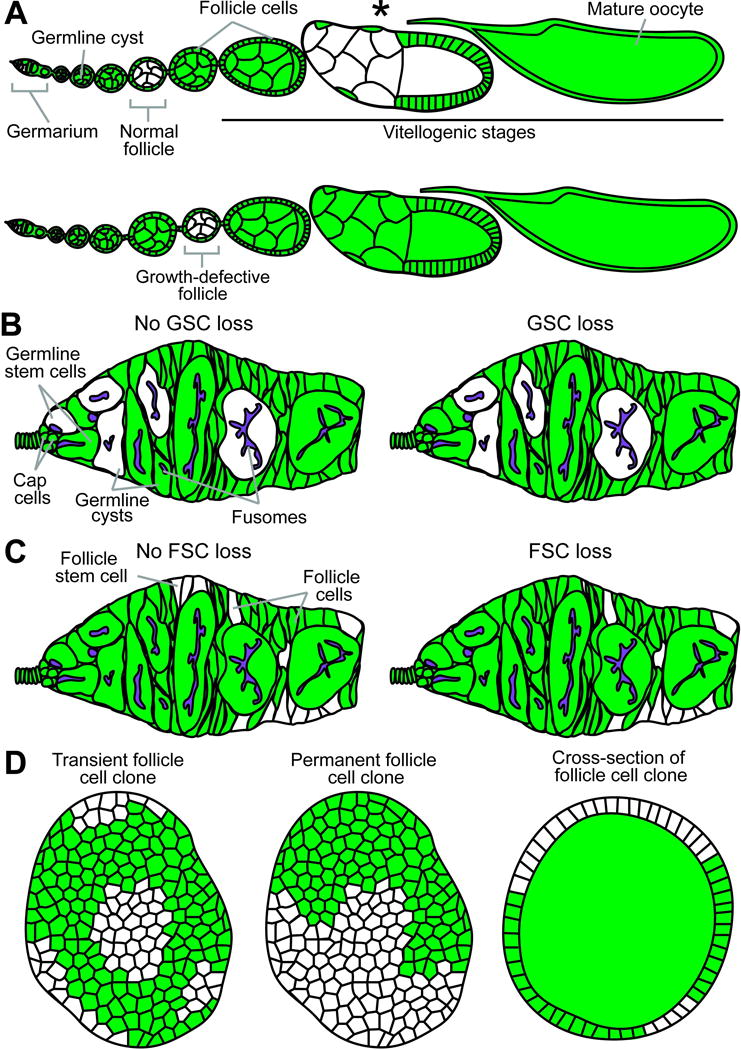
Diagrams of potential genetic mosaic analysis outcomes. (A) Top: Normal ovariole containing previtellogenic (“Normal follicle”) and vitellogenic (asterisk) follicles with GFP-negative germline cysts. Bottom: Follicle containing GFP-negative cyst shows a delay in growth, readily apparent in comparison to neighboring wild-type follicles. (B) A permanent clone derived from an identifiable GFP-negative GSC (left) populates the germarium (left). A recent GSC loss event is recognizable by the presence of GFP-negative germline cystoblasts/cysts within a mosaic germarium without the original GFP-negative mother GSC (right). (C) Permanent clones arising from an identifiable GFP-negative FSC in the germarium (left) or without a GFP-negative FSC (right), which indicates a loss event. (D) Transient (left) and permanent (middle) follicle cell clones are imaged in single planes for quantification of follicle cell proliferation by clone size or EdU incorporation frequency, respectively. Cross-sections of follicle cell clones (right) in the ovariole are often visible during germline cyst analyses.
Progression through vitellogenesis
Vitellogenesis begins at stage 8 of oogenesis [21]. To assess the progression of mutant cysts through vitellogenesis, we quantify the fraction of ovarioles that contain a GFP-negative vitellogenic cyst in control versus mutant mosaic ovarioles (Fig. 1B, arrowhead; Fig. 2A, asterisk). Do not include any “artificially truncated” ovarioles (i.e. in which vitellogenic cysts have been inadvertently removed from the ovariole during dissection or mounting) in the analysis. Although degenerating vitellogenic egg chambers with pyknotic nuclei may also be directly detected in mosaic ovarioles, it is not possible to reliably score such egg chambers as GFP-negative or –positive. The ideal number of ovarioles scored per genotype will depend on the penetrance of the phenotype, but, at a minimum, several dozen should be analyzed at 10 days after heat shock.
An alternative method for quantifying vitellogenesis block involves exclusively analyzing mosaic ovarioles in which the entire germline is homozygous mutant, and scoring what percentage of ovarioles have vitellogenic versus dying follicles, in relation to equivalent control mosaics. Samples sizes, however, will be inevitably small, given the rarity of mosaic ovarioles containing a fully mutant germline.
GSC maintenance
Method one: measuring the occurrence of directly observable GSC loss events. In germaria where all transient clones have exited the germaria [18], all GFP-negative cystoblasts and germline cysts will have arisen from a GFP-negative GSC (Fig. 1C, arrowhead; Fig. 2B). To quantify GSC loss, we count the number of germaria that contain GFP-negative GSCs along with their GFP-negative progeny (Fig. 1C and Fig. 2B, left), versus similar germaria in which the original GFP-negative GSCs have been lost (i.e. the presence of GFP-negative germline cysts/cystoblasts in the absence of a GFP-negative GSC indicates that the GSC was lost from the niche) (Fig. 1D and Fig. 2B, right) (see Note 29). The number of germaria showing a GSC loss event as a fraction of all germaria containing mosaic germline can be directly compared among different control and experimental mosaics. This approach provides a snapshot of GSC loss events, and a single time point (e.g. 7–10 days after the last heat-shock) can be informative when comparing control and mutant mosaic germaria. A subtle GSC loss phenotype may not become apparent unless many germline mosaic germaria germaria are analyzed, but approximately one hundred germaria per genotype represents a reasonable sample size.
Method two: calculating the fraction of ovarioles carrying GFP-negative GSCs over time. Quantify the number of germaria containing at least one GFP-negative GSC as a percentage of the total number of germaria in the sample (see Note 30). This proportion is sensitive to the recombination frequency of the FRT, so changes in the fraction of ovarioles containing GFP-negative GSCs should be tracked over time (e.g. 4–7 days, two weeks, three weeks, and four weeks after heat shock) in control and mutant mosaic germaria. Due to potential variability in the frequency of initial FLP/FRT-mediated recombination events, larger samples sizes (several hundred germaria per genotype per time point) allow more reliable measurements.
FSC maintenance
Currently, no reliable markers exist for FSCs, and they can only be unambiguously identified using a combination of criteria, including lineage tracing, morphology and position within germaria. Briefly, FSCs are the anterior-most somatic cells within follicle cell clones immediately anterior to the 2a/2b junction of the germarium (Fig. 1E, arrowhead; Fig. 2C, left). Follicle cells differ from more anteriorly located somatic cells, escort cells, by nuclear and cellular morphology [23]. The same general strategy described above to measure GSC loss can be used for FSCs (Fig. 1E,F, and Fig. 2C), with similar timing and sample size considerations.
Early cyst development
The number of early progeny of GFP-negative GSCs at different stages of development can be readily quantified in germaria containing at least one GFP-negative GSC. Germline cysts are staged by the morphology of their fusomes [24] (Fig. 1C; Fig. 2B). After counting the numbers of GFP-negative cystoblasts, and 2-, 4-, 8-, and 16-cell cysts present within each germarium, those numbers are normalized to the number of GFP-negative GSCs within that same germarium. By comparing the average number of different early GFP-negative GSC progeny present in control versus mutant mosaic germaria, it is possible to detect changes in the relative frequencies of various stages, which can be the result of stage-specific delay, arrest or death of germline cysts. Alternatively, the relative distribution of early germline stages can be compared between GFP-negative versus GFP-positive GSC progeny within the same population of germaria of a given genotype, which has the advantage of minimizing any potential influence of genotype background on the analyses. Analyzing several dozens of mosaic germaria per genotype at 7–10 days after heat shock should be sufficient to reveal differences in cyst distribution.
GSC proliferation
To directly measure the frequency of GSCs in S phase, we quantify the total number of mutant, GFP-negative GSCs that have incorporated the thymidine analog EdU as a percentage of all GFP-negative GSCs observed (see Note 31). This number can be compared to either incorporation of EdU in neighboring, marker-positive GSCs, or in marker-negative GSCs in control mosaics. Although this is a labor-intensive process, we recommend scoring several hundreds of GFP-negative GSCs per genotype for reliable results, unless differences in proliferation rates are enormous and readily apparent.
An indirect (and less labor intensive) readout of GSC proliferation is the number of progeny per GSC present in each germarium. Comparing the number of germline cysts per GFP-negative versus GFP-positive GSCs is a relative measure of the number of GSC divisions in the recent past, as long as problems with cystoblast/cyst survival are ruled out (see “Early cyst development” heading above).
FSC proliferation
As for GSCs, FSC proliferation can be detected by EdU incorporation. In this case, lineage analysis is used to identify FSCs as described above, and the number of EdU-positive FSCs as a fraction of all GFP-negative FSCs is compared between mutant and control mosaic germaria. As for GSC proliferation analysis, samples sizes should be large.
Follicle cell proliferation
The proliferation of follicle cells can also be directly measured by quantifying the number of EdU-positive follicle cells as a fraction of all GFP-negative follicle cells analyzed during mitotic stages of follicle development (egg chamber stages 2–6; see Ref. 19). The percentage of EdU-positive follicle cells within the population of GFP-negative follicle cells can be compared to that of GFP-positive follicle cells within the same mutant mosaic ovarioles or to that of GFP-negative follicle cells in control mosaic ovarioles (Fig. 2D). Dozens of ovarioles should be scored at 10 days after heat shock.
Alternatively, transient follicle cell clone size (e.g. 3 days after heat shock) quantification may serve as a readout for follicle cell proliferation during mitotically dividing stages (egg chamber stages 2–6; [19]). GFP-negative clones should be compared in mutant and control mosaic ovarioles (Fig. 1G, dashed outline; Fig. 2D, left). One caveat of this approach, however, is that other factors (such as cell death or elimination) can also influence clone size. Dozens of clones should be analyzed per genotype.
Acknowledgments
We are grateful to members of the Drummond-Barbosa lab for critical comments during the preparation of this manuscript. This work was supported by National Institutes of Health (NIH) grant R01 GM069875 (D.D.B.). K.L. was supported by NIH training grant T32CA009110.
Footnotes
Many of the necessary strains can be obtained from the Bloomington Drosophila Stock Center at Indiana University (flystocks.bio.indiana.edu).
Rather than employing hs-Flp, one could drive a UAS-Flp transgene in a spatially restricted pattern using a Gal4 line with specific expression pattern [9], although this eliminates temporal control.
FRT insertion should map to same chromosome arm as the mutation of interest, which should be distal to the FRT.
The concentration of G418 is calculated based on the active concentration of the drug and the level of resistance conferred by expression of the neomycin resistance (neoR) transgene in different FRT insertion lines. G418 concentration should therefore be optimized for each specific FRT insertion, using appropriate positive and negative controls to ensure appropriate selection. For example, flies carrying one copy of the FRT82B insertion survive when raised on food treated with 30 mg/mL of active G418, while all control wild type flies die.
The consistency of yeast paste may change over time. We recommend storing prepared yeast paste at 4°C, covered with parafilm.
While 1.5 mL microtubes are usually used, smaller tubes may be used to conserve antibody, especially when ovary size is significantly reduced.
Immunostaining for the fusome marker 1B1 works best when Triton-X 100 is used, whereas for an alternate fusome marker, α-spectrin, we recommend Tween-20 instead.
The same detergent should be used in the washing and blocking solutions.
16% FA keeps for one week at 4°C after being opened, after which fixation quality deteriorates. Fixation conditions must be optimized for each antibody, but antibodies described in this protocol work reproducibly well under these fixation conditions.
If using EdU incorporation kit, the Alexa Fluor 633 secondary antibody should be used instead of Alexa Fluor 568, which has a similar emission spectrum to the Click-It conjugate. The manufacturer’s instructions should be used to visualize EdU.
Different FRT insertions vary in levels of neoR expression, which is controlled by a heat-shock inducible promoter. While the leakiness of the promoter is often sufficient for selection on G418-treated fly food at room temperature, it is sometimes necessary to periodically heat-shock flies at 37°C during the drug treatment for robust expression of neoR (e.g. FRT80B) and effective selection.
The “recombination cross” is the cross between females carrying the FRT chromosome in trans to the mutation of interest and balancer males.
To prepare the fly food for G418 selection, etch a checkerboard pattern onto the surface of the pre-prepared food using a dissecting needle or thin spatula, then apply 200 µL of G418 solution. Dry food completely under a fume hood before transferring the crosses to the vials.
Experimental genotypes should carry the FRT insertion recombined to the mutant allele in trans to a corresponding wild-type FRT chromosome carrying a GFP or β-gal marker, in addition to the hs-Flp transgene on a separate chromosome. Control genotypes are virtually identical, with the exception that no mutant allele is present, such that marker-negative clones will be wild type.
To induce clones in the ovarian GSC niche, Drosophila should be heat shocked in the late larval and early pupal stages [14] rather than in adult stages.
Heat shocks should ideally be eight to twelve hours apart.
Between heat shocks, transfer flies to regular fly food supplemented with dry yeast.
For example, we find that perdurance of GFP makes the identification of negatively marked GSCs difficult until four days after the last heat shock.
Transient clones are derived from mitotic recombination occurring within individual dividing progeny of the stem cells (which further divide to form clones) and, as oogenesis progresses, they disappear. In contrast, permanent clones are derived from a stem cell, and tend therefore to be much longer lasting than transient clones.
Reagents and freshly dissected ovaries can be kept at room temperature or on ice, depending on which particular cellular proteins or structures will be visualized by immunostaining. For example, if EdU incorporation assay will be performed, all reagents and dissected ovaries should be kept at room temperature.
Placing a black background under the dissecting dish helps with visualization of the ovaries during dissection and mounting.
For assays conducted on unfixed tissue (e.g. EdU incorporation), or for the visualization of intact terminal filament structures, do not tease ovarioles apart at this stage. In these cases, ovarioles can be teased apart following fixation by returning them to the dissection plate with wash buffer.
Optimal fixation and staining conditions depend on the antibody being used and should be established prior to conducting this analysis. These conditions work well for the antibodies noted in this protocol, which are routinely used in our laboratory.
Once fixation solution has been thoroughly rinsed from the sample, washes are very flexible. Depending on the antigen being detected, the sample can remain in wash solution for up to 2 weeks at 4°C.
Samples can remain in blocking solution for extended periods of time at 4°C.
After samples have been stained with primary antibody, they can be stored in washing solution for extended periods of time at 4°C.
Labile epitopes and the Click-it reaction used to detect EdU incorporation are exceptions and should be imaged as soon as possible.
The extent to which ovarioles should be flattened varies depending on the type of analysis to be conducted. For example, to obtain good single-plane images of the follicle epithelium, additional weight (up to double) may be necessary. Conversely, samples lacking vitellogenic stages (such as those from flies on a poor diet) will be more easily flattened.
GSCs can be unambiguously identified by the presence of a stereotypically shaped, 1B1-positive fusome juxtaposed to the Lamin C-positive niche [22].
The percentage of ovarioles containing GFP-negative GSCs sometimes increase from early to later time points, possibly due to some GFP perdurance at early time points.
An increase in the percentage of EdU incorporation of GSCs could reflect either an increase in proliferation rates or a slower S phase. To distinguish between these possibilities, it is necessary to employ a secondary method of analysis (e.g., the use of a different cell cycle marker, such as the mitosis marker phosphorylated histone H3, or a direct comparison between the numbers of GSC progeny).
References
- 1.Perrimon N. Creating mosaics in Drosophila. Int J Dev Biol. 1998;42:243–247. [PubMed] [Google Scholar]
- 2.Theodosiou NA, Xu T. Use of FLP/FRT system to study Drosophila development. Methods. 1998;14:355–365. doi: 10.1006/meth.1998.0591. [DOI] [PubMed] [Google Scholar]
- 3.Le Lievre CS, Le Douarin NM. Mesenchymal derivatives of the neural crest: analysis of chimaeric quail and chick embryos. J Embryol Exp Morphol. 1975;34:125–154. [PubMed] [Google Scholar]
- 4.Lehmann R, Nusslein-Volhard C. hunchback, a gene required for segmentation of an anterior and posterior region of the Drosophila embryo. Dev Biol. 1987;119:402–417. doi: 10.1016/0012-1606(87)90045-5. [DOI] [PubMed] [Google Scholar]
- 5.Hadorn E. Transdetermination in cells. Sci Am. 1968;219:110–114. doi: 10.1038/scientificamerican1168-110. passim. [DOI] [PubMed] [Google Scholar]
- 6.Simon MA, Bowtell DD, Dodson GS, Laverty TR, Rubin GM. Ras1 and a putative guanine nucleotide exchange factor perform crucial steps in signaling by the sevenless protein tyrosine kinase. Cell. 1991;67:701–716. doi: 10.1016/0092-8674(91)90065-7. [DOI] [PubMed] [Google Scholar]
- 7.Xu T, Rubin GM. Analysis of genetic mosaics in developing and adult Drosophila tissues. Development. 1993;117:1223–1237. doi: 10.1242/dev.117.4.1223. [DOI] [PubMed] [Google Scholar]
- 8.Lee T, Luo L. Mosaic analysis with a repressible cell marker (MARCM) for Drosophila neural development. Trends Neurosci. 2001;24:251–254. doi: 10.1016/s0166-2236(00)01791-4. [DOI] [PubMed] [Google Scholar]
- 9.Evans CJ, Olson JM, Ngo KT, Kim E, Lee NE, et al. G-TRACE: rapid Gal4-based cell lineage analysis in Drosophila. Nat Methods. 2009;6:603–605. doi: 10.1038/nmeth.1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Struhl G, Basler K. Organizing activity of wingless protein in Drosophila. Cell. 1993;72:527–540. doi: 10.1016/0092-8674(93)90072-x. [DOI] [PubMed] [Google Scholar]
- 11.Ables ET, Drummond-Barbosa D. The steroid hormone ecdysone functions with intrinsic chromatin remodeling factors to control female germline stem cells in Drosophila. Cell Stem Cell. 2010;7:581–592. doi: 10.1016/j.stem.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ables ET, Drummond-Barbosa D. Cyclin E controls Drosophila female germline stem cell maintenance independently of its role in proliferation by modulating responsiveness to niche signals. Development. 2013;140:530–540. doi: 10.1242/dev.088583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hsu HJ, Drummond-Barbosa D. Insulin levels control female germline stem cell maintenance via the niche in Drosophila. Proc Natl Acad Sci U S A. 2009;106:1117–1121. doi: 10.1073/pnas.0809144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsu HJ, Drummond-Barbosa D. Insulin signals control the competence of the Drosophila female germline stem cell niche to respond to Notch ligands. Dev Biol. 2011;350:290–300. doi: 10.1016/j.ydbio.2010.11.032. [DOI] [PubMed] [Google Scholar]
- 15.Hsu HJ, LaFever L, Drummond-Barbosa D. Diet controls normal and tumorous germline stem cells via insulin-dependent and -independent mechanisms in Drosophila. Dev Biol. 2008;313:700–712. doi: 10.1016/j.ydbio.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.LaFever L, Drummond-Barbosa D. Direct control of germline stem cell division and cyst growth by neural insulin in Drosophila. Science. 2005;309:1071–1073. doi: 10.1126/science.1111410. [DOI] [PubMed] [Google Scholar]
- 17.LaFever L, Feoktistov A, Hsu HJ, Drummond-Barbosa D. Specific roles of Target of rapamycin in the control of stem cells and their progeny in the Drosophila ovary. Development. 2010;137:2117–2126. doi: 10.1242/dev.050351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Margolis J, Spradling A. Identification and behavior of epithelial stem cells in the Drosophila ovary. Development. 1995;121:3797–3807. doi: 10.1242/dev.121.11.3797. [DOI] [PubMed] [Google Scholar]
- 19.Spradling AC. Developmental genetics of oogenesis. In: Bate M, editor. The Development of Drosophila melanogaster. Plainview, NY: Cold Spring Harbor Laboratory Press; 1993. [Google Scholar]
- 20.Haack T, Bergstralh DT, St Johnston D. Damage to the Drosophila follicle cell epithelium produces "false clones" with apparent polarity phenotypes. Biol Open. 2013;2:1313–1320. doi: 10.1242/bio.20134671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cummings MR, Brown NM, King RC. The cytology of the vitellogenic stages of oogenesis in Drosophila melanogaster. 3. Formation of the vitelline membrane. Z Zellforsch Mikrosk Anat. 1971;118:482–492. doi: 10.1007/BF00324615. [DOI] [PubMed] [Google Scholar]
- 22.Xie T, Spradling AC. A niche maintaining germ line stem cells in the Drosophila ovary. Science. 2000;290:328–330. doi: 10.1126/science.290.5490.328. [DOI] [PubMed] [Google Scholar]
- 23.Sahai-Hernandez P, Castanieto A, Nystul TG. Drosophila models of epithelial stem cells and their niches. Wiley Interdiscip Rev Dev Biol. 2012;1:447–457. doi: 10.1002/wdev.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Cuevas M, Spradling AC. Morphogenesis of the Drosophila fusome and its implications for oocyte specification. Development. 1998;125:2781–2789. doi: 10.1242/dev.125.15.2781. [DOI] [PubMed] [Google Scholar]