

Abstract
Macrophages play important roles in HIV-1 pathogenesis as targets for viral replication and mediators of chronic inflammation. Similar to IFNγ-priming, HIV-1 primes macrophages, resulting in hyperresponsiveness to subsequent toll-like receptor (TLR) stimulation and increased inflammatory cytokine production. However, the specific molecular mechanism of HIV-1 priming and whether cells must be productively infected or if uninfected bystander cells also are primed by HIV-1 remains unclear. To explore these questions, human macrophages were primed by IFNγ or infected with HIV-1 before activation by TLR ligands. Transcriptome profiling by microarray revealed a gene expression profile for IFNγ-primed cells that was further modulated by the addition of lipopolysaccharide (LPS). HIV-1 infection elicited a gene expression profile that correlated strongly with the profile induced by IFNγ (r = .679, p = .003). Similar to IFNγ, HIV-1 enhanced TLR ligand-induced tumor necrosis factor (TNF) protein expression and release. Increased TNF production was limited to productively infected cells. Specific signal transducer and activator of transcription (STAT)1 and STAT3 inhibitors suppressed HIV-1-mediated enhancement of TLR-induced TNF expression as well as HIV-1 replication. These findings indicate that viral replication and inflammation are linked through a common IFNγ-like, STAT-dependent pathway and that HIV-1-induced STAT1 and STAT3 signaling are involved in both inflammation and HIV-1 replication. Systemic innate immune activation is a hallmark of active HIV-1 replication. Our study shows that inflammation may develop as a consequence of HIV-1 triggering STAT-IFN pathways to support viral replication.
Keywords: : HIV, macrophages, priming, transcriptome, STAT
Introduction
The underlying mechanisms of chronic monocyte/macrophage activation, a major factor contributing to the morbidity in individuals living with HIV-1 infection,1–5 remain unclear. For example, microbial translocation of gut microbial substances, such as lipopolysaccharide (LPS) into the blood, is associated with systemic innate immune activation,1,6–10 which in turn predicts non-AIDS mortality among HIV-infected individuals.11 Macrophages are partially activated by inflammatory substances, for example LPS through binding to pattern recognition receptor toll-like receptor 4 (TLR4). However, complete classical (M1) macrophage activation requires both priming by IFNγ and activation through proinflammatory receptors, such as TLRs, for expression of relatively high levels of inflammatory cytokines (e.g., TNF) compared with partial macrophage activation with TLR ligands alone.12 Similarly, HIV-1 infection leads to macrophage priming and subsequent enhancement of proinflammatory responses that mimic M1 polarization.13,14 Specific mechanisms involved in HIV-1-induced priming remain unknown, but may be related to HIV-1-induced activation of signal transducer and activator of transcription (STAT) signaling, a pathway also activated by IFNγ. We previously showed that HIV-1 infection of macrophages leads to the activation of STAT1 and STAT315 in a manner resembling IFNγ signaling primarily through STAT1 activation. HIV-1 may modulate components of IFN and/or cytokine signaling through STAT pathways to modify the cellular environment to favor viral replication and/or persistence.
HIV-1 priming of macrophages is dependent on the level of infection,14 but it is not clear if the priming effect is restricted to productively infected cells or if all cells in the culture become primed. HIV-1 infection of macrophages in vitro results in a heterogeneous mixture of productively infected, nonproductively infected, or uninfected bystander cell populations. Thus, phenotypic changes in macrophages may be due to the direct effects of HIV-1 in productively infected cells or the indirect effects on uninfected cells by host (IFNs or cytokines) or viral (free viruses, viral proteins, or viral nucleic acids) factors.16 Pathogen detection by innate immune cells requires extracellular, cytosolic or endosomal pattern recognition receptors, which for example bind viral nucleic acids and trigger downstream signaling pathways. For example, HIV-1 dsDNA resulting from reverse transcription is detected by the cytosolic protein IFI16 triggering expression of type I IFNs by CD4+ T cells.17 In contrast, HIV-1 infection of macrophages can directly induce the expression of interferon-stimulated genes (ISGs) without de novo type I IFN synthesis.18 Whether the IFN-like priming phenotype in macrophages is due to a direct effect of HIV-1-induced signal transduction or the secondary production of type I IFNs remains unclear.
In addition to integral roles in macrophage activation and induction of antiviral responses, signaling through STAT and interferon regulatory factor (IRF) pathways can directly modulate HIV-1 replication through transactivation of the interferon-stimulated regulatory element in the HIV-1 long terminal repeat (LTR) by IRF-1 and IRF-2.19–21 As IRF1 and IRF2 typically mediate activation of ISGs downstream of IFNγ-induced STAT1 signaling, the shared roles of IRF-mediated host inflammatory gene expression and activity of the HIV-1 LTR could explain why HIV-1 infection triggers systemic inflammation as a collateral consequence of the pathways required for full HIV-1 gene expression.
To identify the molecular mechanisms of HIV-1-induced IFNγ-like priming versus viral replication in the presence and absence of secondary stimuli, global gene expression responses by human macrophages to different types of activation, including priming with IFNγ, partial activation with LPS, or classical M1 activation with IFNγ priming plus LPS were assessed. The approach allowed the assembly of a reference data set of gene expression profiles for different activation states, which was then directly compared with the effect of HIV-1 infection in the presence or absence of LPS. Since productively infected and nonproductively infected macrophages could not be isolated without potentially modifying the transcriptome profile, the question regarding whether or not HIV-1 priming of macrophages is due to direct effects of HIV-1 in productively infected cells or also occurs in nonproductively infected or bystander cells was addressed separately by flow cytometry. Subsequently, the roles of STAT1 or STAT3 signaling in immune activation and virus replication were investigated in HIV-1-infected macrophages. The results provide evidence that HIV-1-induced macrophage priming is molecularly similar to IFNγ-induced priming and that proinflammatory priming and viral replication are linked. These findings support a model that systemic inflammation during chronic HIV-1 infection is a consequence of the molecular changes required for HIV-1 gene expression and replication.
Material and Methods
Differentiation of monocyte-derived macrophages
Peripheral blood monocytes were collected from healthy donors by countercurrent centrifugal elutriation at the laboratories of Dr. Howard Gendelman, University of Nebraska or Dr. Mark Wallet, University of Florida with approval for the use of human blood products granted by the institutional review boards at each institution. Monocytes were cultured for 7 days in DMEM containing 10% human type AB serum, l-glutamine, gentamicin, ciprofloxacin, and 1 ng/ml human macrophage colony-stimulating factor (PeproTech, Rocky Hill, NJ), as described.22
Microarray
Monocyte-derived macrophages (MDM) were cultured for 18 h in medium alone or with 10 ng/ml IFNγ (PeproTech). Some IFNγ-treated or untreated MDM cultures were subsequently activated for 30 min or 2 h with 10 ng/ml Escherichia coli LPS (strain O11:B4, Sigma-Aldrich, St. Louis, MO). Cells were washed with ice-cold phosphate-buffered saline (PBS) before preparing total RNA using the RNeasy Mini Kit (Qiagen, Valencia, CA). Affymetrix U133 Plus 2.0 gene arrays were used to assess human gene expression using the IVT Express Kit for labeling (Affymetrix, Santa Clara, CA). Chips were scanned using a GenePix 4400A microarray scanner. Intensity data were transformed to log base 2, normalized, and analyzed by a probe-by-probe comparison between each experimental and control probe group by a moderated t-test based on linear model for microarray data, which moderates standard errors across probes using a simple Bayesian model23 Gene expression fold changes were computed based on the normalized log-transformed signal intensity data.
HIV-1 infection
Stocks of the replication competent, macrophage-tropic, CCR5-using, molecular clones HIV-1AD824 or HIV-1NL4-3-BaL-HSA16 were prepared by transfection of 293T cells and titered on TZM-bl cells as described previously.25,26 MDM were infected with 100–5,000 tissue culture infectious dose 50% (TCID50) for 24 h. Cells were then washed and fresh medium added. HIV-1 Gag p24 in culture supernatants was measured by ELISA (Sino Biological, Daxing, China). In some experiments, MDM plated in UpCell culture dishes (Thermo Scientific Nunc, Rochester, NY) were infected by HIV-1NL4-3-BaL-HSA, which carries the gene for murine heat stable antigen (HSA), or CD24, expressed from an internal ribosome entry site.16 Following infection, MDM were removed from culture dishes, rested in cold PBS (4°C) for 20 min, and labeled with anti-murine CD24-biotin (eBiosciences, San Diego, CA) for enrichment using anti-biotin magnetic beads (StemCell Technologies, Vancouver, BC, CA). In addition, an HIV-1 luciferase-tagged reporter virus lacking Env (HIV-1 NL4-3.Luc.env-) was pseudotyped with the JR-FL envelop to generate virus particles as described previously.25 MDM infected with single-cycle, luciferase reporter HIV-1JRFL were lysed on post-infection day (PID) 4 and luciferase activity was measured according to the manufacturer's protocol using the luciferase assay system (Promega, Madison, WI) and the microplate luminometer Monolight 3096 (BD Biosciences, San Jose, CA).
STAT inhibitors
MDM were treated for 24 h with 100 nM STAT1 inhibitor (Fludarabine, Sigma Aldrich, St. Louis, MO) diluted in water, or 50 nM STAT3 inhibitor (LLL12, EMD Chemicals, Inc., Gibbstown, NJ) diluted in DMSO. On PID 1, cells were washed with PBS and fresh medium alone (control for STAT1 inhibitor), or medium with DMSO (control for STAT3 inhibitor), or STAT1 or STAT3 inhibitors were added. Additional treatments with the inhibitors or respective controls were conducted on PID 4 and 7, concomitant with a 50% medium change.
Quantification of viral RNA and DNA
MDM were cultured in medium alone or infected with 500 TCID50 HIV-1AD8. On PID 6, some samples were treated with 10 ng/ml IFNγ for 18 h. On PID 7, some samples were activated with 10 ng/ml LPS for 2 h. Total RNA was collected using an RNeasy Mini Kit (Qiagen, Germantown, MD) and cDNA was prepared using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems/Life Technologies, Grand Island, NY). Reverse transcriptase quantitative polymerase chain reaction (RT-qPCR) was performed using SYBR Select Master Mix (Applied Biosystems) on the StepOnePlus Real-Time PCR System (Applied Biosystems). Validated forward and reverse primers were purchased from Invitrogen (GBP1, GBP2, GBP3, GBP4, GBP5, GBP6, IL29, PTGS2, and TNF) or Qiagen (AKR1C3, DCLRE1B, HHEX, IL6, KITLG, MMP10, OLR1, P2RY14, and RGS3).
To measure intracellular HIV-1 DNA, MDM were pretreated with medium alone, DMSO, or respective STAT inhibitors for 24 h before infection with HIV-1AD8. Total DNA was extracted with the DNeasy Blood and Tissue Kit (Qiagen, Germantown, MD) and intracellular HIV-1 DNA was measured by quantifying gag and ApoB copies using a qPCR TaqMan assay as previously described.27
Integrated HIV-1 DNA was measured using a nested Alu-LTR qPCR assay previously published.28,29 All primers and probes were purchased from Integrated DNA Technology. Primers for the first-round PCR were: HIV LTR 5′-ATG CCA CGT AAG CGA AAC TCT GGG TCT CTC TDG TTA GAC-3′, Alu 1 5′–TCC CAG CTA CTG GGG AGG CTG AGG −3′, Alu 2 5′–GCC TCC CAA AGT GCT GGG ATT ACA G − 3′, ApoB Out Fwd 5′–AGG GAT CTG AAG GTG GAG GAC A-3′, and Apo Out Rev 5′–TGG CAG TGA TGG AAG CTG CGA −3′. The primers for the second-round qPCR were: Lambda 5′–ATG CCA CGT AAG CGA AAC T − 3′, UR2 5′-CTG AGG GAT CTC TAG TTA CC-3′, ApoB Inner Fwd 5′–TGA AGG TGG AGG ACA TTC CTC TA-3′, and ApoB Inner Rev 5′–CTG GAA TTG CGA TTT CTG GTA A −3′. Probes used in the second-round qPCR amplification were: HIV-1 5′–FAM/CAC TCA AGG CAA GCT TTA TTG AGG C/TAMRA-3 and ApoB 5′-Cy5/CGA GAA TCA CCC TGC CAG ACT TCC GT/RQS-3′.
Flow cytometry
MDM differentiated in UpCell culture dishes were treated with the medium alone, DMSO, STAT1, or STAT3 inhibitor, 24 h preinfection and on PID 1, 4, and 7. On PID 10, cells were treated with 3 μg/ml Brefeldin A (eBioscience, San Diego, CA) and activated with 1 μg/ml CL097 for 4–6 h. Intracellular levels of HIV-1 Gag p24 and TNF were measured using an LSRII flow cytometer (BD Biosciences, San Jose, CA) by permeabilizing the cells using the BD Cytofix/Cytoperm kit according to the manufacturer's instructions (BD Biosciences), staining with anti-p24 PE (Beckman Coulter, Indianapolis, IN) and anti-TNF APC (eBiosciences, San Diego, CA) antibodies. All data were analyzed using FlowJo (FlowJo, LLC, Ashland, OR).
MTS assay
Cell viability was evaluated using an MTS assay. MDM treated with medium, DMSO, STAT1, or STAT3 inhibitors 24 h pre- and postinfection and on PID 4 and 7 were incubated with CellTiter 96 AQueous One Solution (Promega) for 3 h on PID 10 before absorbance was measured using a universal microplate reader (BioTek Instrument, Inc., Winooski, VT). MDM treated with STAT1 or STAT3 inhibitor showed no significant difference in metabolic activity with an average activity of 93.5% and 103.5% relative to their specific control, respectively (data not shown).
Statistical analyses
Statistical analyses were performed using GraphPad Prism software (La Jolla. CA). Gene set comparisons between two groups were performed with Pearson correlation and simple linear regression. Comparisons between two groups were performed using unpaired T-test for normally distributed values and Mann–Whitney U-test for values that filed the Pearson omnibus normality test. Parametric or nonparametric two-way ANOVA was used based on the assessment of data distribution as well as repeated one-way ANOVA with Newman–Keuls correction to compare the effects of STAT inhibitors to respective control. Paired T-test was used where indicated. Differences were considered statistically significant when p < .05.
Results
Generation of macrophage activation transcriptome profiles
Total RNA was isolated from human MDM that were resting (medium only), partially activated (LPS only for 30 min or 2 h), primed with IFNγ (for 18 h) or classically (M1) activated (IFNγ for 18 h followed by LPS for either 30 min or 2 h) (Fig. 1A). Gene expression was assessed by Affymetrix gene arrays to generate transcriptome profiles. Genes that were significantly increased or decreased (≥2-fold increase or decrease with false discovery rate <.05) for any condition were used to generate a heat map organized by hierarchical clustering (Fig. 1B). Gene changes induced by 30 min LPS treatment alone or in combination with IFNγ revealed few qualitative differences from unstimulated or IFNγ only treated cells, respectively. In contrast, 2 h LPS treatment alone or in combination with IFNγ produced profiles that were markedly different from unstimulated or IFNγ only stimulated cells. Thus, only 2 h LPS treatment were considered for subsequent analyses. Overall, about 2,700 genes (1,446 upregulated and 1,263 downregulated) were altered by at least a twofold with treatment by IFNγ alone, 2 h LPS alone, or IFNγ+2 h LPS compared with untreated cells (Fig. 1C). Unique gene sets for LPS (187 upregulated and 71 downregulated), IFNγ (38 upregulated and 28 downregulated), and IFNγ+2 h LPS (363 upregulated and 729 downregulated) were used to assemble a panel of 17 signature genes, ranging from −4.1 to 8.6-fold change, which distinguished LPS activation from IFNγ priming or the classical M1 activation of MDM (Fig. 1D).
FIG. 1.

Gene array transcriptome analysis of macrophage activation states. (A) MDM were cultured for 7 days. On day 6, some samples were primed with 10 ng/mL IFNγ for 18 h. On day 7, MDM were left untreated or activated with 10 ng/mL LPS for 0.5 or 2 h. Total RNA was used to perform Affymetrix U133 Plus 2.0 gene arrays. (B) Normalized data were analyzed by hierarchical clustering to determine gene expression clusters. (C) The unique or shared expression of genes representing partial activation (2 h LPS), IFNγ priming, or classical M1 activation (IFNγ + 2 h LPS) that showed at least a twofold change in expression is represented by a Venn diagram. (D) Genes that uniquely identify different activation states were selected for further analyses. Experiment was conducted in three donors. MDM, monocyte-derived macrophages. Color images available online at www.liebertpub.com/aid
Differentiation of MDM activation profiles by the expression of signature genes
To compare transcriptome profiles of priming induced by IFNγ or by HIV-1, expression of the 17-gene panel was measured by RT-qPCR from MDM cultures infected by HIV-1AD8 or stimulated with IFNγ, alone or in combination with LPS activation. Hierarchical clustering of relative gene expression revealed that HIV-1 or IFNγ priming clustered together and was distinct from treatment conditions that included LPS (Fig. 2A). Although the magnitude of the effect by HIV-1 on the signature genes was reduced compared with IFNγ, a direct comparison between treatment and HIV-1 infection showed a significant correlation for the overall expression of the 17 genes (r = .679, p = .003) (Fig. 2B).
FIG. 2.

IFNγ- and HIV-1-induced macrophage-priming phenotype. (A) A panel of 17 macrophage activation genes was measured by RT-qPCR and fold change over unactivated MDM (medium only) was used to perform hierarchical clustering between the treatment groups. (B) Gene expression was compared between HIV-1-induced and IFNγ-induced MDM priming. The solid line represents the linear regression of all genes (r = .679, P = .003). Gene expression was compared between MDM (C) activated with LPS alone (2 h) and MDM primed with IFNγ and then activated with LPS (2 h) or (D) activated with LPS alone (2 h) and MDM infected with 500 TCID50 HIV-1AD8 and then activated with LPS (2 h). The red circles indicate genes modified in a similar direction (up or down) by both IFNγ and HIV-1 compared with LPS alone. The blue circles indicate genes enhanced by IFNγ, but suppressed or unaffected (ND, no difference) by HIV-1 infection compared with LPS alone. The fold change for each gene in comparison with LPS alone is indicated in parentheses. Experiment was conducted in three donors. RT-qPCR, reverse transcriptase quantitative polymerase chain reaction; TCID50, tissue culture infectious dose 50%. Color images available online at www.liebertpub.com/aid
To evaluate the effects of IFNγ or HIV-1 in combination with LPS, gene expression changes were first compared between classical (IFNγ/LPS) and LPS-activated macrophages (Fig. 2C). While six genes showed no change, classical activation enhanced mRNA levels of cytokines TNF, IL-6, and IL-29 as well as 5 GBPs and P2RY14, but suppressed levels of LPS-induced oxidized low-density lipoprotein receptor 1 (OLR1). Overall, HIV-1/LPS activation shared key characteristics with the IFNγ/LPS profile, including enhanced steady-state mRNA levels of TNF and the GBPs, and reduced levels of OLR1 expression (Fig. 2D). While the magnitude of a number of gene changes induced by HIV-1 was generally less than observed with IFNγ stimulation, IFNγ- or HIV-1-induced priming each enhanced TNF responses, which provided a basis to monitor the primed phenotype through released or cell-associated TNF protein.
HIV-1 infection directly primes macrophages for hyperactivation in response to extracellular inflammatory stimuli
MDM treated with IFNγ alone showed no change in TNF secretion relative to control (Fig. 3A). Treatment with LPS alone increased the levels of secreted TNF by approximately fivefold compared with control, whereas classic activation (IFNγ + LPS) significantly augmented TNF levels compared with LPS stimulation alone (p < .05) (Fig. 3A). Likewise, HIV-1 infection alone had no impact on TNF secretion, whereas the combination of virus and LPS increased TNF secretion (p < .05) (Fig. 3A).
FIG. 3.

HIV-1 infection directly primes MDM for hyperresponsiveness to TLR4 and TLR7/8-mediated activation. The effects of IFNγ stimulation versus HIV-1 infection on LPS- and CL097-induced TNF production were compared in MDM. (A) Untreated (medium alone), IFNγ-primed (10 ng/mL for 18 h), or HIV-1 infected MDM (50–5,000 TCID50 for 7 days) were left untreated or activated with 10 ng/mL LPS for 6 h. ELISA was used to measure supernatant TNF. Bars represent the mean ± SEM *p < .05 using two-way ANOVA with Bonferroni correction. (B) HIV-1-infected MDM were treated with medium alone or 10 ng/mL LPS for 4 h 7 days postinfection in the presence of 3 μg/mL Brefeldin A. Intracellular p24 and TNF were measured by flow cytometry. MFI of TNF in uninfected cells and HIV-1-infected MDM gated on either p24-negative or p24-positive populations in (B) to determine the effect of HIV-1 priming on activation by (C) LPS or (D) CL097. Bars in (C, D) represent the mean ± SEM MFI of TNF; *p < .05, ***p < .001 using two-way ANOVA with Bonferroni correction. MFI, mean fluorescence intensity; TLR, toll-like receptor; SEM, standard error of the mean.
To address whether HIV-1 priming requires direct infection of the MDM or if uninfected bystander cells are also primed by HIV-1, intracellular TNF was measured by flow cytometry in productively infected (p24-postive) or nonproductively infected (p24-negative) MDM in the same culture. While unstimulated MDM from HIV-1-uninfected cultures showed no TNF production, LPS stimulation increased intracellular TNF protein expression as expected (Fig. 3B, upper panels). In HIV-1-infected cultures, LPS stimulation increased TNF expression in both p24-negative and p24-positive MDM (Fig. 3B, lower panels). However, mean fluorescence intensity (MFI) for TNF expression was significantly higher in p24-positive compared with p24-negative cells and uninfected MDM (Fig. 3C). HIV-1 priming significantly enhanced LPS-induced intracellular TNF approximately 10-fold in p24-positive compared with p24-negative cells, even at LPS concentrations as low as 0.1 ng/ml (Supplementary Fig. S1; Supplementary Data are available online at www.liebertpub.com/aid).
In a previous study, we show increased proinflammatory cytokine production in HIV-1-infected cultures stimulated with several TLR ligands.13 Thus, to confirm that the phenotype observed in HIV-1 productively infected MDM stimulated with LPS are not restricted to LPS and TLR4 stimulation, MDM were infected or stimulated with IFNγ and then activated with the TLR7/8 agonist, CL097. CL097-induced TNF levels, measured by ELISA, were significantly enhanced (p < .05) in MDM cultures infected with HIV-1 compared with uninfected cultures stimulated with CL097 (data not shown). Flow cytometry analysis showed similar enhanced intracellular TNF expression in p24-positive cells upon challenge with CL097 as with LPS stimulation (data not shown) and the MFI of intracellular TNF was significantly greater (p < .001) in p24-positive MDM compared with p24-negative cells from the same culture or MDM from uninfected cultures treated with CL097 (Fig. 3D).
To confirm that HIV-1-induced priming was limited to productively infected cells, MDM were infected with HIV-1NL4-3-BaL-HSA and magnetically sorted based upon surface expression of murine CD24 before LPS stimulation. Independent of LPS stimulation, p24 was detected by western blot only in CD24-positive cells confirming infection (Fig. 4A). Enhanced supernatant levels of TNF were only seen in CD24-positive, LPS-treated MDM (Fig. 4B). Thus, HIV-1-induced priming was limited to productively infected cells, whereas uninfected cells in the same cultures were not primed.
FIG. 4.

Only productively infected (p24+) MDM are primed. (A) MDM were infected with 5000 TCID50 HIV-1NL4-3-BaL-HSA encoding the murine heat-stable antigen (HSA; CD24), which is expressed on the surface of productively infected cells. CD24+ and CD24− cells were sorted using magnetic beads and expression of HIV-1 gag p24 was measured by western blot to confirm efficient sorting of infected cells. (B) Uninfected and infected (CD24− and CD24+) MDM were replated and stimulated with 10 ng/mL LPS for 6 h. ELISA measured secreted TNF. The experiment was conducted in one donor. ND, not detectable.
HIV-1-induced priming is mediated through STAT1 and STAT3 and is required for HIV-1 replication in macrophages
The STAT signaling pathways are involved in a variety of immune and antiviral responses and different STAT pathways are induced by both type I and type II IFNs. While IFNγ signals primarily through STAT1, we previously showed that activation of both STAT1 and STAT3 occurs in response to HIV-1 infection of macrophages.15 To investigate the role of STAT1 and STAT3 in HIV-1 priming, MDM were treated with STAT1 or STAT3-specific inhibitors 24 h before infection with HIV-1AD8 and on PID 1, 4, and 7. Ten days postinfection, MDM were activated with CL097 and intracellular TNF and p24 levels were measured by flow cytometry. Although neither of the inhibitors had an effect on CL097-induced TNF in unprimed (p24-negative) cells, inhibitor treatment partially suppressed TNF expression in HIV-1-primed (p24-positive) cells (p < .01 and p < .05, respectively) (Fig. 5A, B), indicating a role for both STAT1 and STAT3 in HIV-1 priming of MDM.
FIG. 5.

STAT1 and STAT3 are involved in HIV-1-induced priming. MDM were treated with 100 nM Fludarabine (STAT1 inhibitor) or 50 nM LLL12 (STAT3 inhibitor) 24 h before infection and on PID 1, 4, and 7. Ten days postinfection, cells were activated with 1 μg/mL CL097 for 6 h. Intracellular p24 and TNF were measured by flow cytometry. Bars represent mean ± SEM MFI of TNF in uninfected cells and infected cells gated on either p24-negative or p24-positive populations; *p < .05, **p < .01 using repeated one-way ANOVA with Newman–Keuls correction. At least three donors were used in each experiment. STAT, signal transducer and activator of transcription.
Since a clear effect on HIV-1 priming was observed with STAT1 and STAT3 inhibitor treatment, we next looked at the effect by the same treatment on HIV-1 infection itself. Again, MDM were treated with STAT1 or STAT3 inhibitors 24 h before infection and on PIDs 1, 4, and 7. Treatment with either STAT1 or STAT3 inhibitors resulted in significantly reduced supernatant p24 levels over time (Fig. 6A, B) and significantly (p < .001 and p < .01, respectively) reduced the percentage of infected cells (Fig. 6C). With the frequency of p24-positive cells in control populations sat to 100%, the decrease in percentage of infected cells in the STAT1 and STAT3 inhibitor-treated populations was 86.7% and 69.6%, respectively.
FIG. 6.
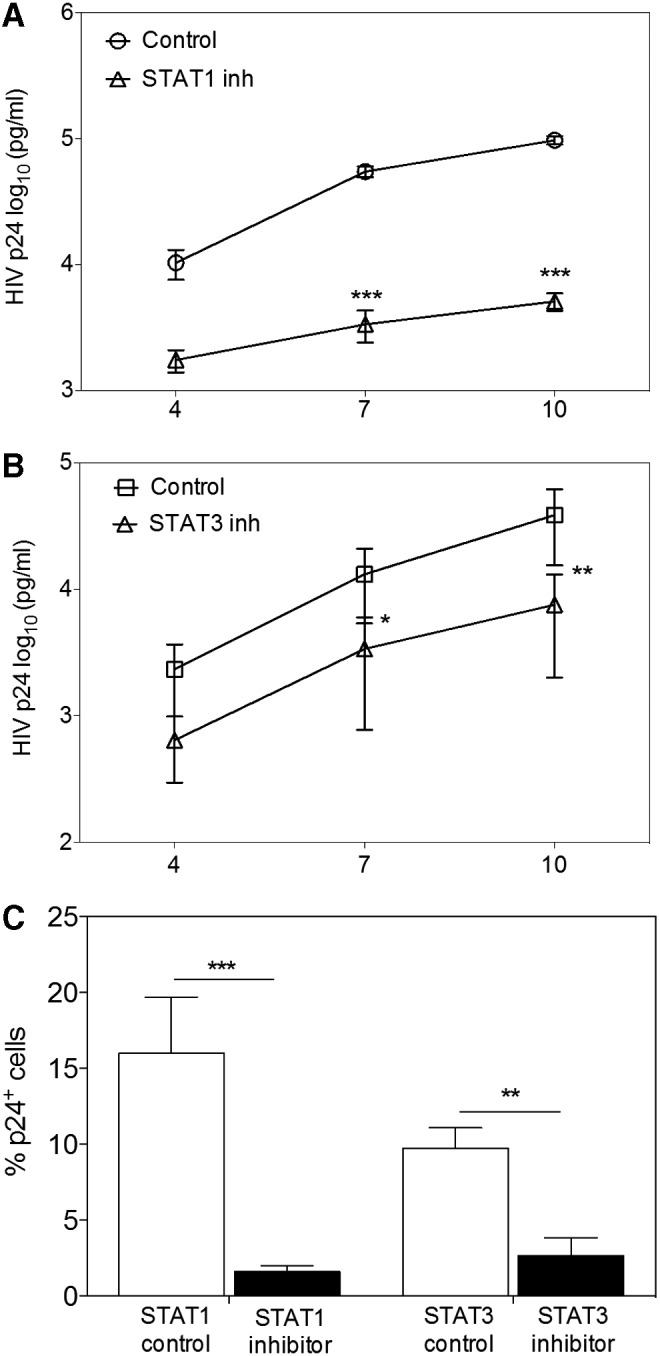
STAT1 or STAT3 inhibitors suppress HIV-1 infection of MDM. MDM were pretreated with 100 nM Fludarabine (STAT1 inhibitor) or 50 nM LLL12 (STAT3 inhibitor) 24 h before infection and on PID 1, 4, and 7. ELISA was used to measure supernatant p24 on PID 4, 7, and 10 in cultures treated with (A) Fludarabine and (B) LLL12. Bars represent mean ± SEM; *p < .05; **p < .01; ***p < .001 using two-way ANOVA with Bonferroni correction. (C) On PID 10, intracellular p24 was measured by flow cytometry and the percentage of productively infected MDM in Fludarabine- or LLL12-treated cultures was compared with medium alone (Fludarabine control) or DMSO (LLL12 control). Bars represent mean ± SEM MFI; **p < .01, ***p < .001 using paired T-test. At least three donors were used in the experiment.
To determine the mechanism(s) involved in the suppression of HIV-1 infection in MDM by STAT1 or STAT3 inhibitor treatment, we investigated several steps in the viral life cycle. None of the inhibitors mediated a significant modulation of CD4 or CCR5 cell-surface expression (data not shown) or on the number of total or integrated proviral genomes detected 24 h postinfection (p = .4121 and p = .0859, respectively, Fig. 7A; and p = .5308 and p = .6633, respectively, Fig. 7B). To establish a potential effect of the inhibitors on viral gene expression, MDM were infected with single-cycle, luciferase reporter HIV-1JRFL. Four days postinfection, STAT1 or STAT3 inhibitor-treated cells showed significantly reduced luciferase activity compared with controls (p < .01) (Fig. 7C). Thus, when macrophages were treated with STAT1 or STAT3 inhibitor, HIV-1 replication appears blocked at the viral gene expression level. Together these data indicate a role for both STAT1 and STAT3 in HIV-1 priming and replication.
FIG. 7.
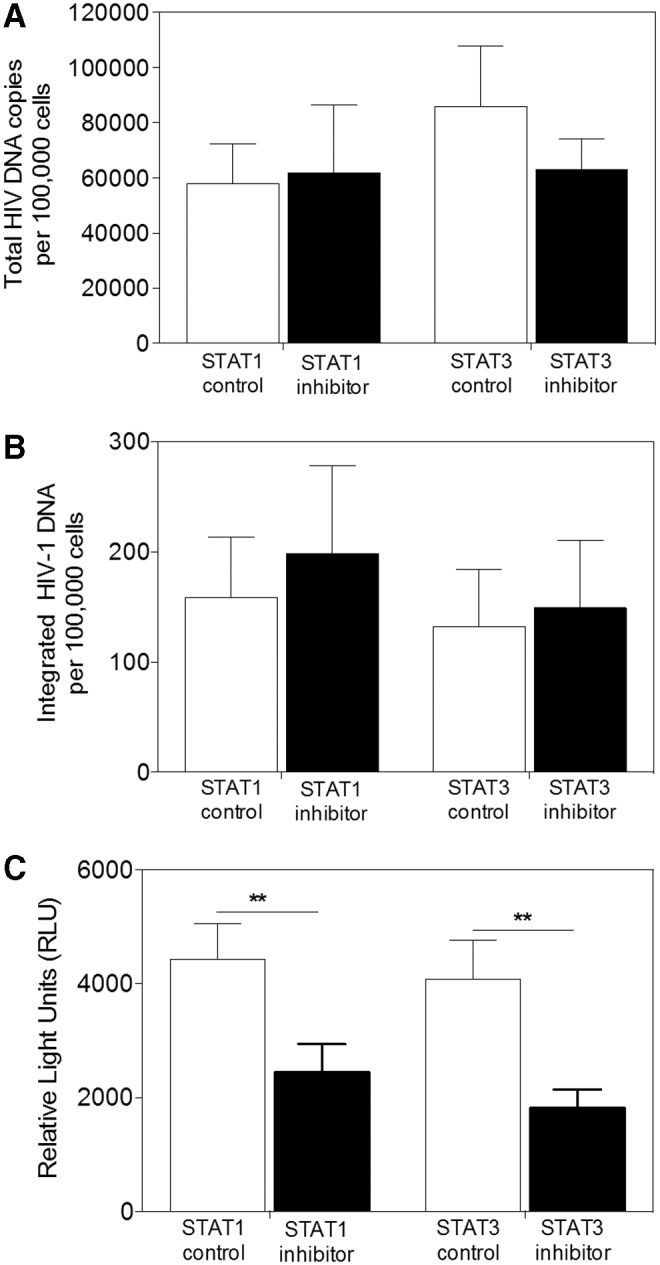
STAT1 and STAT3 are involved in HIV-1 replication. MDM were treated with 100 nM Fludarabine (STAT1 inhibitor) or 50 nM LLL12 (STAT3 inhibitor) 24 h before infection and total DNA was extracted on PID 1. Total (A) or integrated (B) HIV-1 proviral genomes were measured using qPCR, normalized to ApoB, and compared with medium alone (Fludarabine control) or DMSO (LLL12 control). Bars represent the mean ± SEM. (C) MDM treated with Fludarabine or LLL12, 24 h pre- and postinfection, with single-cycle luciferase reporter HIV-1JRFL were lysed on PID 4 and luciferase activity was measured. Bars represent mean ± SEM relative light units (RLU); **p < .01 using paired T-test. At least three donors were used in the experiment.
Discussion
In this study we investigate the molecular mechanisms causing the serious problem of persistent macrophage activation in HIV-1-infected individuals. Macrophages are primarily located in tissue making it difficult to conduct in vivo studies regarding HIV-1-induced macrophage activation. However, monocytes from peripheral blood can be stimulated to differentiate into macrophages and infected with HIV-1 ex vivo. Using this model, we demonstrate that productive HIV-1 infection of macrophages induces a hyperinflammatory state, where cells respond to inflammatory stimuli (LPS or CL097) with enhanced TNF expression, a phenotype that resembles classical macrophage activation (IFNγ and subsequent TLR stimulation). At the molecular level, HIV-1-induced priming resembles IFNγ-induced priming, raising the possibility that similar mechanisms are involved. However, we observed one major difference in the priming mechanism; whereas IFNγ-mediated signals are transduced primarily by STAT1, both STAT1 and STAT3 are required for HIV-1-induced priming. In addition, we show that STAT1 and STAT3 activation is involved in HIV-1 replication by regulating viral gene transcription. Thus, HIV-1-mediated STAT1 and STAT3 activation is important for HIV pathogenesis and plays a role in HIV-1-induced macrophage activation. This identifies both STAT1 and STAT3 as potential anti-inflammatory and antiviral targets in HIV-1 infection.
Infection by HIV-1 in vitro is heterogeneous and asynchronous, meaning that only some cells are infected and that these infected cells are at different stages of the viral life cycle due to the spreading nature of infection in culture. To avoid the pitfall of trying to identify HIV-1-induced gene changes in this complex population, we elected to first define the key macrophage gene expression profiles using soluble factors such as IFNγ and LPS, alone or in combination. This approach allowed us to identify the primary genes that distinguish various activation states (LPS alone, IFNγ alone, or IFNγ/LPS) upon which to focus our HIV-1 studies. Using this method, we were able to determine that HIV-1 infection induces a gene expression profile that shares hallmark characteristics with IFNγ-induced macrophage priming. When IFNγ-treated or HIV-1-infected macrophages were then treated with LPS, gene expression profiles emerged that were highly concordant, but not identical. Consequently, HIV-1 infection shares key characteristics with IFNγ, with differences in magnitude of gene expression and in the expression of some genes (e.g., P2RY14). Most likely, differences occur because HIV-1 alone fails to induce IFN expression (type I or type II) in macrophages,30–32 meaning that HIV-1 ISG expression is limited to genes that can be induced by alternate signaling pathways, such as the TBK1–IRF-3 or STAT1 and STAT3 pathways. In addition, the transcriptome profile generated was based on all MDM in an HIV-1-infected culture, which include both productively and unproductively infected cells. Based on the current study, only productively infected MDM are primed and it is possible that a higher magnitude of gene expression or a more distinct HIV-1 profile would be detected by enriching for p24-positive cells before measuring gene expression.
Previous studies of HIV-1 interactions with macrophages have conflicting results, with some reporting that HIV-1 promotes an IFN-like shift in the macrophage activation phenotype.13,14,33 For example, using a comparable model, Nottet et al. 1995 demonstrated that HIV-1 primes MDM in a similar way as IFNγ and that the priming is dependent on the level of HIV-1 infection in the MDM culture.14 In contrast, others describe an anti-inflammatory effect by HIV-1.34,35 One study even demonstrated that HIV-1 infection of MDM before type I IFN treatment results in the reduced expression of a subset of ISGs.36 Clearly, the relationship between IFNs and HIV-1 is context-dependent, with highly divergent outcomes when the IFN treatment of macrophages ex vivo occurs before or after HIV-1 infection.36 Parsing the in vivo effects of HIV-1 itself versus interferons is further complicated by the fact that IFNs produced by dendritic cells37 or NK cells38 during HIV-1 infection could prime macrophages for classical inflammatory activation. By using our ex vivo model, we determined that HIV-1-induced, IFN-like priming of macrophages is restricted to cells that are productively infected (p24+). This novel finding extends our knowledge regarding HIV-1 priming of MDM by showing that HIV-1 priming and subsequent response is intrinsic and can occur independent of secreted IFNs or other cytokines that modify LPS-induced TNF secretion by bystander cells through paracrine effects.
The result showing that HIV-1 priming only occurs in infected cells based on intracellular p24 expression, which can be measured as soon as 1 h postinfection,39 suggests that priming occurs early in the viral life cycle. Early induction of priming is also supported by our results showing involvement of STAT1 and STAT3 activation in HIV-1 priming of MDM. The first interaction between HIV-1 and MDM (binding of gp120 to CD4 receptor) activates the STAT signaling pathways.15 Why HIV-1 would trigger STAT1/3 activation, an outcome that might elicit antiviral immunity, likely reflects that both STAT1 and STAT3 activation are essential for HIV-1 replication. The HIV LTR contains an interferon-stimulated response element targeted by IRF-1 and IRF-2, downstream molecules of STAT activation.21,40 This supports the notion of STAT1 and STAT3 involvement in viral transcription. Additionally, STAT proteins associate with and enhance transcriptional activity of other cellular transcription factors important for HIV-1 replication, such as NF-κB and Sp1.41–44 Thus, blocking STAT1 or STAT3 interactions with, and activation of, other transcription factors might decrease and indirectly lead to suppressed HIV-1 replication.
STAT1 activation mediates a proinflammatory response, whereas STAT3 activation can induce a proinflammatory or an anti-inflammatory response depending on the cellular environment and activation signal.45,46 Gain-of-function mutations in STAT1 leads to increased sensitivity to cytokines, including IL-6, which normally activate STAT3.47 Similar, IFNγ priming of macrophages changes the way the cells respond to subsequent signaling events. For example, IL-10 stimulation, which normally activates STAT3 and anti-inflammatory responses, activates STAT1 in IFNγ-primed macrophages leading to enhanced proinflammatory response.12,48 Our data showing that STAT3 is involved in HIV-1 priming and augmentation of proinflammatory cytokine production, suggests that HIV-1 priming of macrophages induces STAT3 to act as a proinflammatory mediator. IFNγ-primed cells have increased STAT1/3 heterodimers, which correlate with decreased STAT3/3 homodimer activity.12 A similar shift in dimer formation may be responsible for the STAT3-dependent proinflammatory response in HIV-1-primed cells.
Dysfunctional STAT1 and STAT3 activation modifies the normal cytokine production and function. For example, enhanced STAT1 activation impairs the development of T helper 17 cells (Th17) important for maintaining the epithelial barrier in the gastrointestinal tract.47,49 Th17 cells are significantly decreased in HIV-1-infected individuals.49 Thus, HIV-1-induced STAT activation could contribute to the loss of Th17 cells, impairment of mucosal immunity, and subsequent microbial translocation leading to further activation of macrophages and HIV-1 pathogenesis.
From an antiviral perspective, HIV-1-mediated activation of STAT1 and STAT3 seems paradoxical, since most pathogens have developed various ways of modifying or inhibiting the STAT signaling pathways to evade antiviral cellular immunity. Several viruses, including human papilloma virus, ebola, and paramyxoviruses encode viral proteins, which in different ways prevent STAT1 activation or signaling leading to subsequent reduction in antiviral responses.50–54 In contrast, HIV-1 infection of macrophages directly activates STAT1 and STAT315 and downregulates STAT-dependent antiviral proteins, such as OAS1 and OAS3.36 This indicates that HIV-1 has developed mechanisms to evade STAT-induced antiviral cellular immunity, and instead uses the signaling pathway to support viral replication.55–57 Thus, HIV-1-associated inflammation and viral replication may be inextricably linked biological processes in macrophages.
In the current study, we used small inhibitory molecules to target STAT1 or STAT3. This method was chosen to mimic a pharmacological approach. With inhibitory molecules there is a risk for off-target effects. However, the STAT1 inhibitor, Fludarabine, interferes with STAT1 protein and mRNA expression, but does not affect activation of other STAT proteins (including STAT3), Jak1, Tyk2, or ERK1 at concentrations as high as 50 μM.58 Concentrations as high as 10 μM of the STAT3 inhibitor, LLL12, have no apparent adverse effects on STAT1 or ERK1/2 activity.59,60 Thus, at low concentrations of Fludarabine and LLL12 used in this study (100 nM and 50 nM, respectively), off-target effects are unlikely to contribute to the results.
Together our data support that HIV-1 infection of macrophages fundamentally alters the capacity of cells to respond to inflammatory stimuli. Increased sensitivity to TLR ligands could be a driving force behind chronic innate immune activation and pathogenic inflammation associated with morbidity and mortality. One intriguing possibility is that HIV-1-primed macrophages become hyperresponsive to endogenous TLR ligands as well as exogenous substances, such as bacterial LPS and CpG. For example, cerebrospinal fluid contains the endogenous TLR2/4 ligand hyaluronic acid (hyaluronan),61,62 and we have shown that IFNγ primes human macrophages to exert a proinflammatory response to hyaluronic acid through TLR4.63 HIV-1-induced priming of tissue macrophages, such as brain perivascular macrophages or microglia that are exposed to endogenous TLR-ligands, could contribute to HIV-1 neuropathogenesis. This hypothesis is supported by the study by Nottet et al. 1995 showing that HIV-1-primed MDM subsequently stimulated with LPS produces enhanced levels of TNFα, eicosanoids, and platelet-activating factor,14 molecules shown to contribute to neuronal death.64,65 Furthermore, Nottet et al. showed that interaction between astrocytes and HIV-1-infected MDM suppressed the production of proinflammatory molecules, indicating that astrocytes can regulate HIV-1 priming of brain MDM.14 Whether or not HIV-1 priming of MDM can be suppressed or regulated by other cell types in the body is currently unknown, although we showed previously that even though antiretroviral therapy diminishes T cell activation, MDM activation persist over basal levels as long as two years after initiation of antiretroviral treatment.7
Whether or not low-level viral replication or production persists in macrophages during prolonged antiretroviral therapy remains unknown, but episodic replication could provide a source for elevated levels of innate immune biomarkers that persist during suppressive therapy. The kinetics of the HIV-1-induced priming phenotype and whether or not priming persists when viral replication enters a quiescent or latent state in the macrophage remain to be elucidated. The current data show that macrophages must be productively infected (p24-positive) to be primed, but the possibility exists that the epigenetic reprogramming of HIV-1-infected macrophages results in a permanently hyperresponsive phenotype. The clinical relevance of understanding HIV-1-induced priming is based on the unmet need to identify cells and mechanisms responsible for HIV-1-associated chronic inflammation.
Authors' Contribution
K.S.A. and M.A.W. conceived, designed, and preformed experiments, and wrote the article. Due to equal contribution to the article, K.S.A. and M.A.W. are co-first authors. J.P.T. and M.N.C. executed experiments and contributed to finalizing the article. J.W.S. provided scientific input and edited the article. M.M.G. oversaw experimentation and article revisions.
Supplementary Material
Acknowledgment
The authors thank Drs. Keith W. Peden and Michel Tremblay for providing HIV-1AD8 and HIV-1NL4-3-BaL-HSA, respectively; Dr. Nicolas Chomont for sharing his protocol to measure integrated HIV-1 DNA; and the University of Florida Interdisciplinary Center for Biotechnology Research Cellomics Core Facility for providing flow cytometry. This study was supported in part by the National Institute on Drug Abuse [R01 DA031017 (MMG)] and the National Institute of Allergy and Infectious Diseases [K22 AI095015 and R56 AI108434 (MAW)]; by the Laura McClamma Endowment, the Stephany W. Holloway University Chair for HIV/AIDS Research, and the Center for Research in Pediatric Immune Deficiency.
Disclosure Statement
No competing financial interests exist.
References
- 1.Ancuta P, Kamat A, Kunstman KJ, et al. : Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS One 2008;3:e2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Armah KA, McGinnis K, Baker J, et al. : HIV status, burden of comorbid disease, and biomarkers of inflammation, altered coagulation, and monocyte activation. Clin Infect Dis 2012;55:126–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lien E, Aukrust P, Sundan A, Müller F, Frøland SS, Espevik T: Elevated levels of serum-soluble CD14 in human immunodeficiency virus type 1 (HIV-1) infection: Correlation to disease progression and clinical events. Blood 1998;92:2084–2092 [PubMed] [Google Scholar]
- 4.Nockher WA, Bergmann L, Scherberich JE: Increased soluble CD14 serum levels and altered CD14 expression of peripheral blood monocytes in HIV-infected patients. Clin Exp Immunol 1994;98:369–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ryan LA, Zheng J, Brester M, et al. : Plasma levels of soluble CD14 and tumor necrosis factor-alpha type II receptor correlate with cognitive dysfunction during human immunodeficiency virus type 1 infection. J Infect Dis 2001;184:699–706 [DOI] [PubMed] [Google Scholar]
- 6.Brenchley JM, Price DA, Schacker TW, et al. : Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 2006;12:1365–1371 [DOI] [PubMed] [Google Scholar]
- 7.Wallet MA, Rodriguez CA, Yin L, et al. : Microbial translocation induces persistent macrophage activation unrelated to HIV-1 levels or T-cell activation following therapy. AIDS 2010;24:1281–1290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang W, Lederman MM, Hunt P, et al. : Plasma levels of bacterial DNA correlate with immune activation and the magnitude of immune restoration in persons with antiretroviral-treated HIV infection. J Infect Dis 2009;199:1177–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klatt NR, Funderburg NT, Brenchley JM: Microbial translocation, immune activation, and HIV disease. Trends Microbiol 2013;21:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marchetti G, Tincati C, Silvestri G: Microbial translocation in the pathogenesis of HIV infection and AIDS. Clin Microbiol Rev 2013;26:2–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sandler NG, Wand H, Roque A, et al. : Plasma levels of soluble CD14 independently predict mortality in HIV infection. J Infect Dis 2011;203:780–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hu X, Ivashkiv LB: Cross-regulation of signaling pathways by interferon-gamma: Implications for immune responses and autoimmune diseases. Immunity 2009;31:539–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown J, Kohler J, Coberley C, Sleasman J, Goodenow M: HIV-1 activates macrophages independent of Toll-like receptors. PLoS One 2008;3:e3664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nottet HS, Jett M, Flanagan CR, et al. : A regulatory role for astrocytes in HIV-1 encephalitis. An overexpression of eicosanoids, platelet-activating factor, and tumor necrosis factor-alpha by activated HIV-1-infected monocytes is attenuated by primary human astrocytes. J Immunol 1995;154:3567–3581 [PubMed] [Google Scholar]
- 15.Kohler J, Tuttle D, Coberley C, Sleasman J, Goodenow M: Human immunodeficiency virus type 1 (HIV-1) induces activation of multiple STATs in CD4+ cells of lymphocyte or monocyte/macrophage lineages. J Leukoc Biol 2003;73:407–416 [DOI] [PubMed] [Google Scholar]
- 16.Imbeault M, Lodge R, Ouellet M, Tremblay MJ: Efficient magnetic bead-based separation of HIV-1-infected cells using an improved reporter virus system reveals that p53 up-regulation occurs exclusively in the virus-expressing cell population. Virology 2009;393:160–167 [DOI] [PubMed] [Google Scholar]
- 17.Thompson MR, Sharma S, Atianand M, et al. : Interferon γ-inducible protein (IFI) 16 transcriptionally regulates type i interferons and other interferon-stimulated genes and controls the interferon response to both DNA and RNA viruses. J Biol Chem 2014;289:23568–23581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nasr N, Maddocks S, Turville SG, et al. : HIV-1 infection of human macrophages directly induces viperin which inhibits viral production. Blood 2012;120:778–788 [DOI] [PubMed] [Google Scholar]
- 19.Crotti A, Lusic M, Lupo R, et al. : Naturally occurring C-terminally truncated STAT5 is a negative regulator of HIV-1 expression. Blood 2007;109:5380–5389 [DOI] [PubMed] [Google Scholar]
- 20.Selliah N, Zhang M, DeSimone D, et al. : The gammac-cytokine regulated transcription factor, STAT5, increases HIV-1 production in primary CD4 T cells. Virology 2006;344:283–291 [DOI] [PubMed] [Google Scholar]
- 21.Battistini A, Marsili G, Sgarbanti M, Ensoli B, Hiscott J: IRF regulation of HIV-1 long terminal repeat activity. J Interferon Cytokine Res 2002;22:27–37 [DOI] [PubMed] [Google Scholar]
- 22.Wallet MA, Reist CM, Williams JC, et al. : The HIV-1 protease inhibitor nelfinavir activates PP2 and inhibits MAPK signaling in macrophages: A pathway to reduce inflammation. J Leukoc Biol 2012;92:795–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smyth GK: Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol 2004;3:Article3 [DOI] [PubMed] [Google Scholar]
- 24.Theodore TS, Englund G, Buckler-White A, Buckler CE, Martin MA, Peden KW: Construction and characterization of a stable full-length macrophage-tropic HIV type 1 molecular clone that directs the production of high titers of progeny virions. AIDS Res Hum Retroviruses 1996;12:191–194 [DOI] [PubMed] [Google Scholar]
- 25.Tuttle D, Anders C, Aquino-De Jesus M, et al. : Increased replication of non-syncytium-inducing HIV type 1 isolates in monocyte-derived macrophages is linked to advanced disease in infected children. AIDS Res Hum Retroviruses 2002;18:353–362 [DOI] [PubMed] [Google Scholar]
- 26.Murthy KK, Cobb EK, el-Amad Z, et al. : Titration of a vaccine stock preparation of human immunodeficiency virus type 1SF2 in cultured lymphocytes and in chimpanzees. AIDS Res Hum Retroviruses 1996;12:1341–1348 [DOI] [PubMed] [Google Scholar]
- 27.Coberley CR, Kohler JJ, Brown JN, et al. : Impact on genetic networks in human macrophages by a CCR5 strain of human immunodeficiency virus type 1. J Virol 2004;78:11477–11486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chomont N, El-Far M, Ancuta P, et al. : HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 2009;15:893–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brussel A, Sonigo P: Analysis of early human immunodeficiency virus type 1 DNA synthesis by use of a new sensitive assay for quantifying integrated provirus. J Virol 2003;77:10119–10124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harman AN, Nasr N, Feetham A, et al. : HIV blocks interferon induction in human dendritic cells and macrophages by dysregulation of TBK1. J Virol 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gendelman HE, Friedman RM, Joe S, et al. : A selective defect of interferon alpha production in human immunodeficiency virus-infected monocytes. J Exp Med 1990;172:1433–1442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mosborg-Petersen P, Toth FD, Zachar V, et al. : Differential HIV replication and HIV-induced interferon production in mononuclear phagocytes: Relationship to cell maturation. Res Virol 1991;142:353–361 [DOI] [PubMed] [Google Scholar]
- 33.Chihara T, Hashimoto M, Osman A, et al. : HIV-1 proteins preferentially activate anti-inflammatory M2-type macrophages. J Immunol 2012;188:3620–3627 [DOI] [PubMed] [Google Scholar]
- 34.Noursadeghi M, Tsang J, Miller RF, et al. : Genome-wide innate immune responses in HIV-1-infected macrophages are preserved despite attenuation of the NF-kappa B activation pathway. J Immunol 2009;182:319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tsang J, Chain BM, Miller RF, et al. : HIV-1 infection of macrophages is dependent on evasion of innate immune cellular activation. AIDS 2009;23:2255–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wie SH, Du P, Luong TQ, et al. : HIV downregulates interferon-stimulated genes in primary macrophages. J Interferon Cytokine Res 2013;33:90–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marsili G, Remoli AL, Sgarbanti M, Perrotti E, Fragale A, Battistini A: HIV-1, interferon and the interferon regulatory factor system: An interplay between induction, antiviral responses and viral evasion. Cytokine Growth Factor Rev 2012;23:255–270 [DOI] [PubMed] [Google Scholar]
- 38.Vitale M, Caruso A, Licenziati S, et al. : Differential production of IFN-gamma, analyzed at the single-cell level, by specific subsets of human NK and T cells from healthy and HIV(+) subjects. Cytometry 2000;39:189–194 [DOI] [PubMed] [Google Scholar]
- 39.van ’t Wout AB, Lehrman GK, Mikheeva SA, et al. : Cellular gene expression upon human immunodeficiency virus type 1 infection of CD4(+)-T-cell lines. J Virol 2003;77:1392–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Van Lint C, Amella CA, Emiliani S, John M, Jie T, Verdin E: Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J Virol 1997;71:6113–6127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rohr O, Marban C, Aunis D, Schaeffer E: Regulation of HIV-1 gene transcription: From lymphocytes to microglial cells. J Leukoc Biol 2003;74:736–749 [DOI] [PubMed] [Google Scholar]
- 42.Shuai K: Modulation of STAT signaling by STAT-interacting proteins. Oncogene 2000;19:2638–2644 [DOI] [PubMed] [Google Scholar]
- 43.Yang XP, Irani K, Mattagajasingh S, et al. : Signal transducer and activator of transcription 3alpha and specificity protein 1 interact to upregulate intercellular adhesion molecule-1 in ischemic-reperfused myocardium and vascular endothelium. Arterioscler Thromb Vasc Biol 2005;25:1395–1400 [DOI] [PubMed] [Google Scholar]
- 44.Look DC, Pelletier MR, Tidwell RM, Roswit WT, Holtzman MJ: Stat1 depends on transcriptional synergy with Sp1. J Biol Chem 1995;270:30264–30267 [DOI] [PubMed] [Google Scholar]
- 45.Niemand C, Nimmesgern A, Haan S, et al. : Activation of STAT3 by IL-6 and IL-10 in primary human macrophages is differentially modulated by suppressor of cytokine signaling 3. J Immunol 2003;170:3263–3272 [DOI] [PubMed] [Google Scholar]
- 46.Hu X, Chakravarty SD, Ivashkiv LB: Regulation of interferon and Toll-like receptor signaling during macrophage activation by opposing feedforward and feedback inhibition mechanisms. Immunol Rev 2008;226:41–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu L, Okada S, Kong XF, et al. : Gain-of-function human STAT1 mutations impair IL-17 immunity and underlie chronic mucocutaneous candidiasis. J Exp Med 2011;208:1635–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrero C, Hu X, Li WP, et al. : Reprogramming of IL-10 activity and signaling by IFN-gamma. J Immunol 2003;171:5034–5041 [DOI] [PubMed] [Google Scholar]
- 49.Brenchley JM, Paiardini M, Knox KS, et al. : Differential Th17 CD4 T-cell depletion in pathogenic and nonpathogenic lentiviral infections. Blood 2008;112:2826–2835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hong S, Mehta KP, Laimins LA: Suppression of STAT-1 expression by human papillomaviruses is necessary for differentiation-dependent genome amplification and plasmid maintenance. J Virol 2011;85:9486–9494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reid SP, Valmas C, Martinez O, Sanchez FM, Basler CF: Ebola virus VP24 proteins inhibit the interaction of NPI-1 subfamily karyopherin alpha proteins with activated STAT1. J Virol 2007;81:13469–13477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kubota T, Yokosawa N, Yokota S, Fujii N: C terminal CYS-RICH region of mumps virus structural V protein correlates with block of interferon alpha and gamma signal transduction pathway through decrease of STAT 1-alpha. Biochem Biophys Res Commun 2001;283:255–259 [DOI] [PubMed] [Google Scholar]
- 53.Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM: STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol 2003;77:7635–7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez JJ, Parisien JP, Horvath CM: Nipah virus V protein evades alpha and gamma interferons by preventing STAT1 and STAT2 activation and nuclear accumulation. J Virol 2002;76:11476–11483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li W, Henderson LJ, Major EO, Al-Harthi L: IFN-gamma mediates enhancement of HIV replication in astrocytes by inducing an antagonist of the beta-catenin pathway (DKK1) in a STAT 3-dependent manner. J Immunol 2011;186:6771–6778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Magnani M, Balestra E, Fraternale A, et al. : Drug-loaded red blood cell-mediated clearance of HIV-1 macrophage reservoir by selective inhibition of STAT1 expression. J Leukoc Biol 2003;74:764–771 [DOI] [PubMed] [Google Scholar]
- 57.Magnani M, Paiardini M, Cervasi B, Serafini S, Fraternale A, Silvestri G: Fludarabine delivery by autologous red blood cells induces macrophage depletion in chronically SIV-infected Sooty Mangabeys. J Control Release 2006;116:e45–e47 [DOI] [PubMed] [Google Scholar]
- 58.Frank DA, Mahajan S, Ritz J: Fludarabine-induced immunosuppression is associated with inhibition of STAT1 signaling. Nat Med 1999;5:444–447 [DOI] [PubMed] [Google Scholar]
- 59.Lin L, Hutzen B, Li PK, et al. : A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia 2010;12:39–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ball S, Li C, Li PK, Lin J: The small molecule, LLL12, inhibits STAT3 phosphorylation and induces apoptosis in medulloblastoma and glioblastoma cells. PLoS One 2011;6:e18820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Laurent UB, Laurent TC, Hellsing LK, Persson L, Hartman M, Lilja K: Hyaluronan in human cerebrospinal fluid. Acta Neurol Scand 1996;94:194–206 [DOI] [PubMed] [Google Scholar]
- 62.Jiang D, Liang J, Noble PW: Hyaluronan as an immune regulator in human diseases. Physiol Rev 2011;91:221–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wallet M, Wallet S, Guiulfo G, Sleasman J, Goodenow M: IFNgamma primes macrophages for inflammatory activation by high molecular weight hyaluronan. Cell Immunol 2010;262:84–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gelbard HA, Nottet HS, Swindells S, et al. : Platelet-activating factor: A candidate human immunodeficiency virus type 1-induced neurotoxin. J Virol 1994;68:4628–4635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wahl LM, Corcoran ML, Pyle SW, Arthur LO, Harel-Bellan A, Farrar WL: Human immunodeficiency virus glycoprotein (gp120) induction of monocyte arachidonic acid metabolites and interleukin 1. Proc Natl Acad Sci U S A 1989;86:621–625 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.