

SUMMARY
Poorly immunogenic tumor cells evade host immunity and grow even in the presence of an intact immune system, but the complex mechanisms regulating tumor immunogenicity have not been elucidated. Here, we discovered an unexpected role of the Hippo pathway in suppressing anti-tumor immunity. We demonstrate that, in three different murine syngeneic tumor models (B16, SCC7, and 4T1), loss of the Hippo pathway kinases LATS1/2 (large tumor suppressor 1 and 2) in tumor cells inhibits tumor growth. Tumor regression by LATS1/2 deletion requires adaptive immune responses, and LATS1/2 deficiency enhances tumor vaccine efficacy. Mechanistically, LATS1/2-null tumor cells secrete nucleic-acid-rich extracellular vesicles, which induce a type I interferon response via the Toll-like receptors-MYD88/TRIF pathway. LATS1/2 deletion in tumors thus improves tumor immunogenicity, leading to tumor destruction by enhancing anti-tumor immune responses. Our observations uncover a key role of the Hippo pathway in modulating tumor immunogenicity and demonstrate a proof of concept for targeting LATS1/2 in cancer immunotherapy.
Graphical abstract
INTRODUCTION
Cellular transformation, tumor growth, and metastasis constitute a multistep process that requires the continuous rewiring of signaling pathways and alterations of the reciprocal interaction between cancer cells and the tumor microenvironment, thereby allowing cells to acquire features to become fully neoplastic and eventually malignant (Hanahan and Weinberg, 2011). The Hippo pathway has gained great interest in recent years as being strongly involved in several of these key hallmarks of cancer progression (Harvey et al., 2013; Moroishi et al., 2015a) and, in general, serves important regulatory functions in organ development, regeneration, and stem cell biology (Johnson and Halder, 2014; Yu et al., 2015). The heart of the mammalian Hippo pathway is a kinase cascade involving mammalian STE20-like protein kinase 1 (MST1; also known as STK4) and MST2 (also known as STK3) (homologs of Drosophila Hippo), as well as two groups of MAP4Ks (mitogen-activated protein kinase kinase kinase kinases)—MAP4K1/2/3/5 (homologs of Drosophila Happyhour) and MAP4K4/6/7 (homologs of Drosophila Misshapen)—and the large tumor suppressor 1 (LATS1) and LATS2 (homologs of Drosophila Warts) (Meng et al., 2016). When the Hippo pathway is activated, MST1/2 or MAP4Ks phosphorylate and activate the LATS1/2 kinases, which, in turn, directly phosphorylate and inactivate Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ; also known as WWTR1), the two major downstream effectors that mediate transcriptional output of the Hippo pathway (Hansen et al., 2015). Activation of LATS1/2 kinases (and inactivation of YAP/TAZ) represents the major functional output of the Hippo pathway.
Previous studies have convincingly established the Hippo pathway as a suppressor signal for cellular transformation and tumorigenesis, though other studies revealed its oncogenic functions in certain contexts (Moroishi et al., 2015a; Wang et al., 2014). Deletion of MST1/2 in mouse liver results in tissue overgrowth and tumor development, demonstrating the tumor suppressor function of these kinases (Zhou et al., 2009). Complementarily, overexpression of YAP in mouse liver also promotes tissue overgrowth and tumorigenesis (Camargo et al., 2007; Dong et al., 2007). These studies have demonstrated an inhibitory role of the Hippo pathway in tumor initiation. However, effects of the Hippo pathway in tumor growth, especially in the context of reciprocal interactions between tumor cells and host anti-tumor immune responses, remain largely unknown. In the present study, we investigate the role of the LATS1/2 kinases in the growth of established tumors in the context of anti-tumor immunity. Surprisingly, inactivation of the “tumor suppressor” LATS1/2 in tumor cells strongly suppresses tumor growth in immune-competent, but not immune-compromised, mice due to the induction of host anti-tumor immune responses. Our data indicate a new paradigm for how tumor immunogenicity is regulated through the Hippo signaling pathway in tumor cells and also have implications for targeting LATS1/2 in cancer immunotherapy.
RESULTS
LATS1/2 Deletion Enhances Anchorage-Independent Growth In Vitro
To elucidate the role of the Hippo pathway in anti-tumor immunity, we took advantage of murine syngeneic tumor models of three different cancer types in three different host genetic backgrounds; B16-OVA melanoma (B16F10 melanoma expressing ovalbumin [OVA]) in C57BL/6 mice, SCC7 head and neck squamous cell carcinoma in C3H/HeOu mice, and 4T1 breast cancer in BALB/c mice. These syngeneic allograft models have been well characterized and extensively used to study reciprocal interactions between tumor cells and host anti-tumor immune responses (Dranoff, 2011; Lei et al., 2016). We have recently shown that deletion of LATS1/2 almost completely abolished YAP/TAZ regulation by the Hippo pathway, while deletion of other components had only a partial or minor effect on YAP/TAZ activity (Meng et al., 2015). Therefore, we deleted LATS1/2 in B16-OVA melanoma cells using CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 genome-editing technology (Ran et al., 2013). We obtained multiple independent LATS1/2 double-knockout (dKO) clones verified by the lack of protein expression of both LATS1 and LATS2 (Figure 1A). Two different clones generated by two independent CRISPR guide sequences were used for this study. Because YAP is a direct substrate of LATS1/2, of which phosphorylation can be readily detected with a phospho-YAP antibody or by mobility shift on a phos-tag gel, we use YAP phosphorylation status as an indicator of LATS1/2 activity. We found that YAP phosphorylation levels were regulated in response to LATS1/2-activating signals in wild-type (WT) B16-OVA cells; however, loss of LATS1/2 abolished YAP phosphorylation (Figure 1A). Phosphorylation of YAP/TAZ by LATS1/2 is known to promote YAP/TAZ cytoplasmic localization and inactivation (Zhao et al., 2007). Indeed, YAP/TAZ localized in the cytoplasm in response to filamentous actin disruption (which activates LATS1/2) in WT B16-OVA cells, yet YAP/TAZ remains localized in the nucleus in LATS1/2 dKO cells under the same condition (Figure 1B). LATS1/2 inactivation or YAP/TAZ hyperactivation is known to promote cell growth (Zhao et al., 2008). Although LATS1/2 dKO B16-OVA cells showed identical growth on regular cell culture plates compared with WT cells (Figure S1A), LATS1/2 dKO B16-OVA cells showed a significant increase in anchorage-independent growth in comparison to WT cells, in terms of both colony number and colony size (Figure 1C). These observations indicate that the Hippo pathway is still operational in B16-OVA melanoma cells and, in addition, that LATS1/2 deficiency activates YAP/TAZ and can further potentiate anchorage-independent growth of B16-OVA cells in vitro.
Figure 1. LATS1/2 Deletion Enhances Anchorage-Independent Tumor Cell Growth In Vitro.
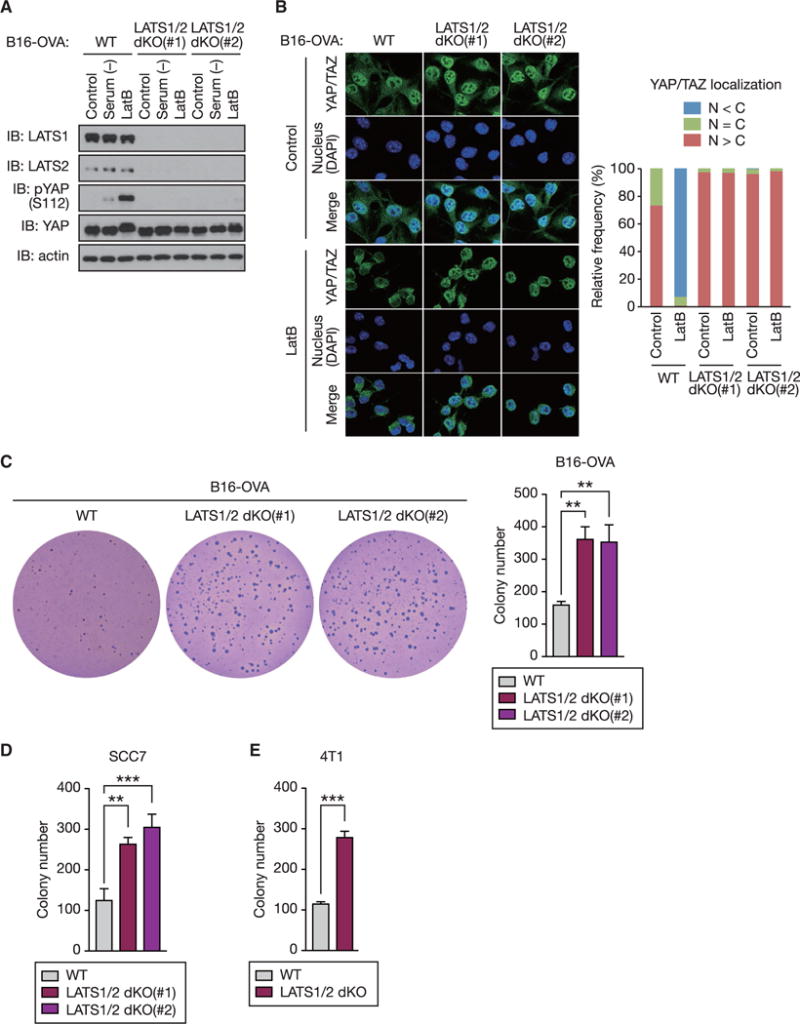
(A) Wild-type (WT) and two independent clones of LATS1/2 double knockout (dKO) B16-OVA melanoma cells were serum starved or treated with Latrunculin B (LatB) and then subjected to immunoblot (IB) analysis with antibodies to the indicated proteins.
(B) LatB-treated or non-treated (control) B16-OVA cells were subjected to immunostaining analysis. YAP/TAZ subcellular localization was determined by immunofluorescence staining for endogenous YAP/TAZ (green) along with DAPI for DNA (blue). Representative images are presented in the left panel. (right panel) Cells in five randomly selected views (~100 cells) were selected for the quantification of YAP/TAZ localization. N, nuclear; C, cytoplasmic.
(C) B16-OVA cells were subjected to soft-agar colony-formation assay, and the colonies were stained with crystal violet for quantification.
(D) Soft-agar colony-formation assay of SCC7 squamous cell carcinoma cells.
(E) Soft-agar colony-formation assay of 4T1 breast cancer cells.
Data are presented as means ± SD from three independent experiments (C–E). The p values were determined using a one-way ANOVA test followed by Tukey’s multiple comparison test (C and D) or an unpaired t test (E). **p < 0.01; ***p < 0.001. See also Figure S1.
We also deleted LATS1/2 in SCC7 squamous cell carcinoma cells and found that LATS1/2 deficiency almost completely blocked YAP phosphorylation (Figure S1B) and YAP/TAZ cytoplasmic localization (Figure S1C) in response to LATS1/2-activating signals. Notably, WT SCC7 cells showed high YAP phosphorylation and cytoplasmic localization of YAP/TAZ, even in the absence of LATS1/2-activating signals, suggesting high basal LATS1/2 activity in these cancer cells. Loss of LATS1/2 again increased anchorage-independent growth of SCC7 cells (Figures 1D and S1D). LATS1/2-dependent regulation of YAP phosphorylation (Figure S1E), YAP/TAZ subcellular localization (Figure S1F), and anchorage-independent cell growth (Figures 1E and S1G) were similarly observed in 4T1 breast cancer cells. Together, our data demonstrate that deletion of LATS1/2 in tumor cells promotes anchorage-independent tumor cell growth in vitro.
Loss of LATS1/2 Inhibits Tumor Growth In Vivo
To investigate the role of the Hippo pathway in tumor growth in vivo, we subcutaneously transplanted equal numbers of WT or LATS1/2 dKO B16-OVA cells into the back flanks of C57BL/6 mice and monitored their growth. Unexpectedly, deletion of LATS1/2 in B16-OVA cells strongly inhibited tumor growth in vivo (Figures 2A and 2B). All mice died before day 22 in the WT B16-OVA-injected group, whereas injection with LATS1/2 dKO B16-OVA cells resulted in tumor-free survival in more than half of the mice (Figure 2C). We confirmed the growth-suppressive effect of LATS1/2 deletion with an independent clone of LATS1/2 dKO B16-OVA cells (Figure S2A). Next, we examined tumor growth of LATS1/2 dKO and WT SCC7 squamous cell carcinoma cells in syngeneic C3H/HeOu mice. All mice injected with the parental SCC7 cells showed aggressive tumor growth (Figures 2D and S2B), and 100% died before day 21 (Figure 2E). In contrast, none of the mice injected with LATS1/2 dKO SCC7 cells developed tumors, and all survived tumor free. In a 4T1 orthotopic allograft model, 4T1 breast cancer cells grow into solid tumors and can readily metastasize to the lung, liver, and brain when transplanted into the mammary fat pads of syngeneic BALB/c mice. Consistently, the parental 4T1 cells developed tumors and metastasized to the lung in BALB/c mice (Figures 2F, 2G, and S2C). On the other hand, LATS1/2 dKO 4T1 cells developed little tumors and had no metastasis when allografted in BALB/c mice. Thus, collectively, our observations indicate that loss of LATS1/2 in tumor cells dramatically inhibits tumor growth in vivo in multiple types of cancer in different host backgrounds. Based on the current dogma, these results are totally unexpected, as LATS1/2 kinases supposedly function as tumor suppressors.
Figure 2. Loss of LATS1/2 in Tumors Inhibits Tumor Growth In Vivo.
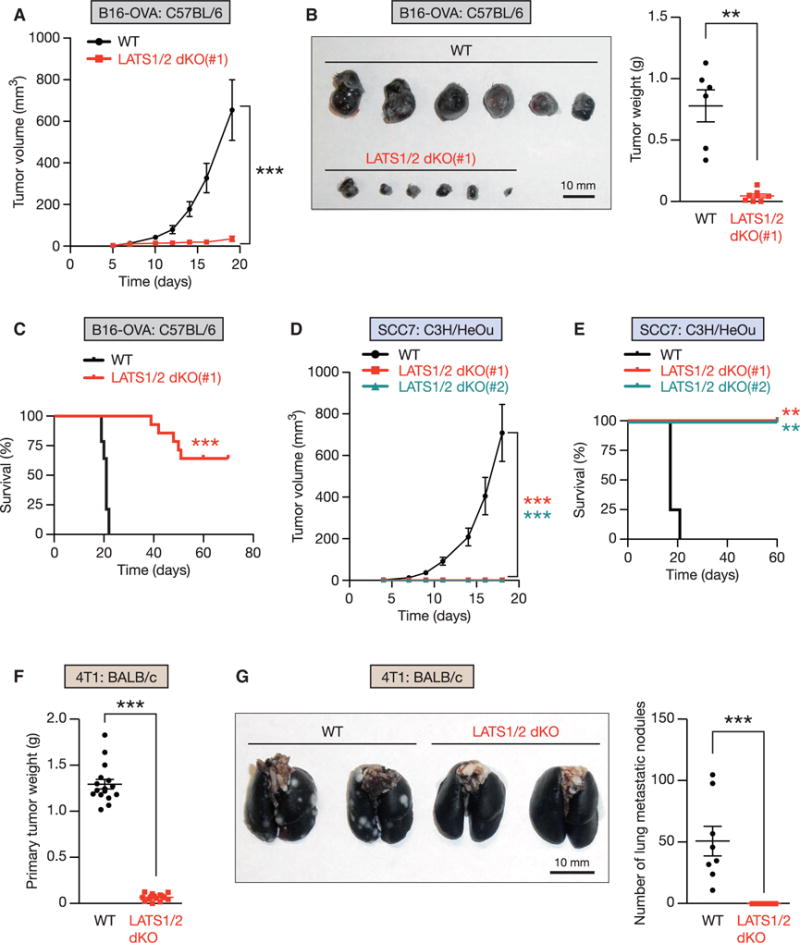
(A) Equal numbers of WT or LATS1/2 dKO B16-OVA cells were transplanted into C57BL/6 mice, and tumor growth was monitored after the indicated times. Data are presented as means ± SEM; n = 8 tumors for each group. ***p < 0.001, two-way ANOVA test.
(B) C57BL/6 mice were injected with WT or LATS1/2 dKO B16-OVA melanoma, and tumor weight was determined 20 days after transplantation. Data are presented as means ± SEM; n = 6 tumors for WT, and n = 8 tumors for LATS1/2 dKO (note that two of the tumors were completely rejected). **p < 0.01, Mann-Whitney test.
(C) Kaplan-Meier tumor-free survival curves for mice injected with WT or LATS1/2 dKO B16-OVA cells are shown (n = 14 mice for each group). ***p < 0.001, log-rank test.
(D) WT or two independent clones of LATS1/2 dKO SCC7 cells were transplanted into C3H/HeOu mice, and tumor growth was monitored after the indicated times. Data are presented as means ± SEM; n = 8 tumors for each group. The p values were determined using a two-way ANOVA test, comparing each group to the WT group. ***p < 0.001.
(E) Kaplan-Meier tumor-free survival curves for mice injected with WT or LATS1/2 dKO SCC7 cells are shown (n = 4 mice for each group). The p values were determined using a log-rank test, comparing each group to the WT group. **p < 0.01.
(F and G) In (F), BALB/c mice were injected with WT or LATS1/2 dKO 4T1 cells, and primary tumor weight was determined 28 days after transplantation. (G) WT or LATS1/2 dKO 4T1 cells were transplanted into the mammary fat pad of BALB/c mice, and lung metastasis of the primary tumor was determined 28 days after transplantation. Normal lung tissue was stained with black India ink, whereas tumor nodules remained white. The gross appearance of the lungs (left panel) and the tumor nodules on the lungs (right panel) were examined. Data are presented as means ± SEM; n = 16 tumors in (F), and n = 8 mice in (G) for each group. ***p < 0.001, Mann-Whitney test. See also Figure S2.
LATS1/2 Deletion Enhances Immunogenicity of Tumor Cells
Since LATS1/2 deletion exerts completely opposite effects on tumor cell growth in vitro and in vivo (Figures 1 and 2), we hypothesized that host factors may contribute to the apparent discrepancy between in vitro and in vivo phenotypes of LATS1/2 dKO tumor cells. Therefore, we examined the histopathology of tumors from allografted mice. We found massive infiltration of inflammatory cells in LATS1/2 dKO B16-OVA melanomas (Figure 3A), as well as in LATS1/2 dKO 4T1 breast cancers (Figure S3A), which were confirmed by staining with the pan-leukocyte marker CD45 (Figures 3B and S3B). These observations prompted us to hypothesize that immune cells infiltrate, and thereby eliminate, LATS1/2 dKO tumor cells. Both innate and adaptive immune responses work together to constitute host anti-tumor immunity, but the adaptive immune system plays a pivotal role in mediating robust and highly specific immune responses against tumors (Gajewski et al., 2013). Therefore, we examined host adaptive immune responses against tumor cells. We chose a B16-OVA melanoma model to explore this, because B16-OVA melanoma cells express a non-secreted form of chicken OVA as a surrogate tumor antigen that can be conveniently used to follow immune responses directed against the OVA antigen. In addition, B16-OVA has been extensively used to study cancer immunity, and many genetically altered syngeneic mouse C57BL/6 strains are available.
Figure 3. LATS1/2 Deficiency in Tumor Cells Induces Host Anti-tumor Immunity.
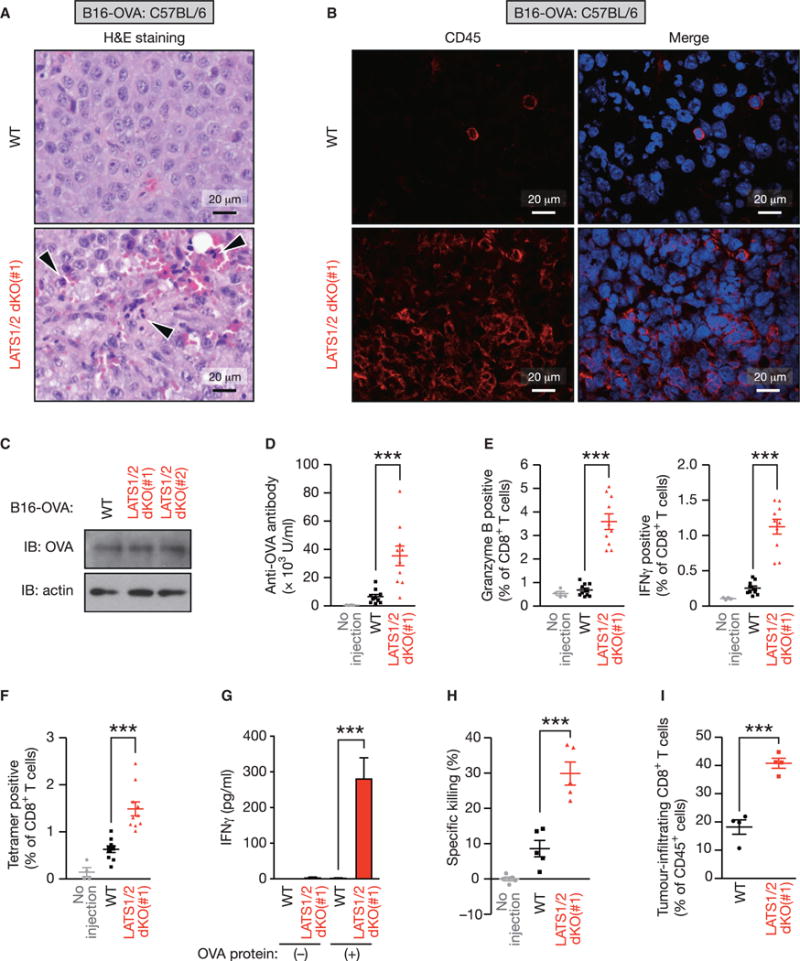
(A) WT or LATS1/2 dKO B16-OVA melanoma cells were injected into C57BL/6 mice. Tumors were paraffin embedded and stained with H&E 12 days after transplantation. Arrowheads indicate infiltration of inflammatory cells.
(B) Frozen sections from WT or LATS1/2 dKO B16-OVA melanomas were subjected to immunostaining analysis of CD45 (red) along with DAPI for DNA (blue).
(C) WT and two independent clones of LATS1/2 dKO B16-OVA melanoma cells were subjected to immunoblot (IB) analysis with antibodies to the indicated proteins.
(D–F) In (D), C57BL/6 mice were injected (or not injected) with WT or LATS1/2 dKO B16-OVA melanoma cells, and serum anti-OVA IgG concentrations were determined by ELISA 12 days after transplantation. (E) Splenocytes from C57BL/6 mice injected as in (D) were re-stimulated ex vivo with SIINFEKL peptide and then subjected to flow-cytometric analysis. SIINFEKL is an OVA-derived peptide being presented through the major histocompatibility complex class I (MHC class I) molecule H-2Kb. Frequency of CD8+ T cells expressing activation markers, Granzyme B or interferon γ (IFNγ), was determined. (F) Splenocytes from C57BL/6 mice injected as in (D) were subjected to flow-cytometric analysis. OVA-specific CD8+ T cells were quantified using Kb-SIINFEKL tetramers and plotted as a percentage of total CD8+ T cells. Data are presented as means ± SEM; n = 4 mice for the uninjected group, n = 10 mice for the WT-injected group, and n = 10 mice for the LATS1/2 dKO-injected group. ***p < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test.
(G) C57BL/6 mice were injected as in (D), and the inguinal lymph nodes were cultured ex vivo with OVA protein. IFNγ levels in the culture supernatants were determined by ELISA. Data are presented as means ± SEM of triplicate cultures of pooled lymph node cells from 4 mice per group. ***p < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test.
(H) C57BL/6 mice were injected as in (D) and CD8+ T cells were isolated from splenocytes. T cell cytotoxicity assay was performed with CFSE (carboxyfluorescein succinimidyl ester)-labeled EL4 cells ex vivo and the percentage of specific killing was plotted. Data are presented as means ± SEM of five independent experiments with pooled CD8+ T cells from 3–4 mice per group. ***p < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test.
(I) WT or LATS1/2 dKO B16-OVA melanoma cells were injected into C57BL/6 mice, and tumors were subjected to flow-cytometric analysis 12 days after transplantation. Data are presented as means ± SEM of the percentage of CD8+ T cells infiltrating into tumors among total CD45+ cells; n = 4 tumors for each group. ***p < 0.001, unpaired t test. See also Figure S3.
Although WT and LATS1/2 dKO B16-OVA cells showed identical expression of OVA (Figure 3C), we detected significantly higher levels of serum anti-OVA antibody in mice injected with LATS1/2 dKO B16-OVA cells (Figure 3D), suggesting an enhanced tumor-specific humoral immune response in the LATS1/2 dKO B16-OVA-injected mice. Next, we examined cellular immune responses. CD8+ T cells isolated from the spleens of LATS1/2 dKO B16-OVA-injected mice produced multiple effector cytokines (Figures 3E and S3C), indicative of T cell activation. We observed significantly higher CD8+ T cell cross-priming when mice were injected with LATS1/2 dKO B16-OVA cells (Figures 3F and S3D), and in addition, lymph node cells isolated from draining lymph nodes of LATS1/2 dKO B16-OVA-injected mice showed a remarkably higher OVA-specific T cell response than did lymph node cells isolated from the parental B16-OVA-injected mice, as measured by interferon (IFN)γ production (Figure 3G). These observations suggest that tumor-specific cellular immune responses, particularly CD8+ T cell responses, are induced in mice injected with LATS1/2 dKO B16-OVA cells. Indeed, CD8+ T cells in LATS1/2 dKO B16-OVA-injected mice possessed OVA-specific cytotoxic activity ex vivo (Figures 3H and S3E) and infiltrated tumors in vivo (Figures 3I and S3F). Together, the aforementioned data demonstrate that LATS1/2 deletion in tumor cells stimulate tumor-specific humoral and cellular immune responses, leading to the establishment of robust anti-tumor immunity.
LATS1/2 Deficiency Enhances Tumor Vaccine Efficacy via Adaptive Immunity
Given that LATS1/2 deletion in tumor cells enhances host anti-tumor immune responses, we hypothesized that LATS1/2-null tumor cells, by stimulating anti-tumor immunity, may protect the host from challenge with the corresponding LATS1/2 WT tumor cells. To test this, we performed two sets of experiments: co-injection of LATS1/2 dKO and WT tumor cells into each side of the same mouse (Figure 4A) or immunization of mice with LATS1/2 dKO tumor cells prior to LATS1/2 WT tumor cell injection (Figures 4B and 4C). Strikingly, co-injection of LATS1/2 dKO B16-OVA cells significantly suppressed tumor growth of the co-injected WT B16-OVA cells (Figure 4A). Moreover, immunization of mice with irradiated LATS1/2 dKO B16-OVA cells, which were viable but unable to proliferate, strongly inhibited the corresponding LATS1/2 WT tumor growth, whereas immunization with irradiated parental B16-OVA cells produced a much weaker effect (Figure 4B). Notably, in our experimental setting, a single dose of tumor vaccination with irradiated WT B16-OVA cells was not sufficient to extend survival (Figure 4C). In contrast, a single dose of tumor vaccination with irradiated LATS1/2 dKO B16-OVA cells showed a significant delay in tumor growth and prolonged survival. Approximately 25% of the mice immunized with irradiated LATS1/2 dKO B16-OVA cells were tumor free when challenged with WT B16-OVA cells. The aforementioned observations suggest that LATS1/2 deficiency renders B16-OVA cells highly immunogenic and improves tumor vaccine efficacy. We further confirmed enhanced anti-tumor immunity by LATS1/2 deletion with a different syngeneic model. C3H/HeOu mice having rejected LATS1/2 dKO SCC7 cells were resistant to rechallenge with the parental SCC7 cells (Figure 4D), indicating that these animals have established immunological memory against the given tumor cells.
Figure 4. LATS1/2 Deletion in Tumors Stimulates Host Adaptive Immunity and Enhances Tumor Vaccine Efficacy.
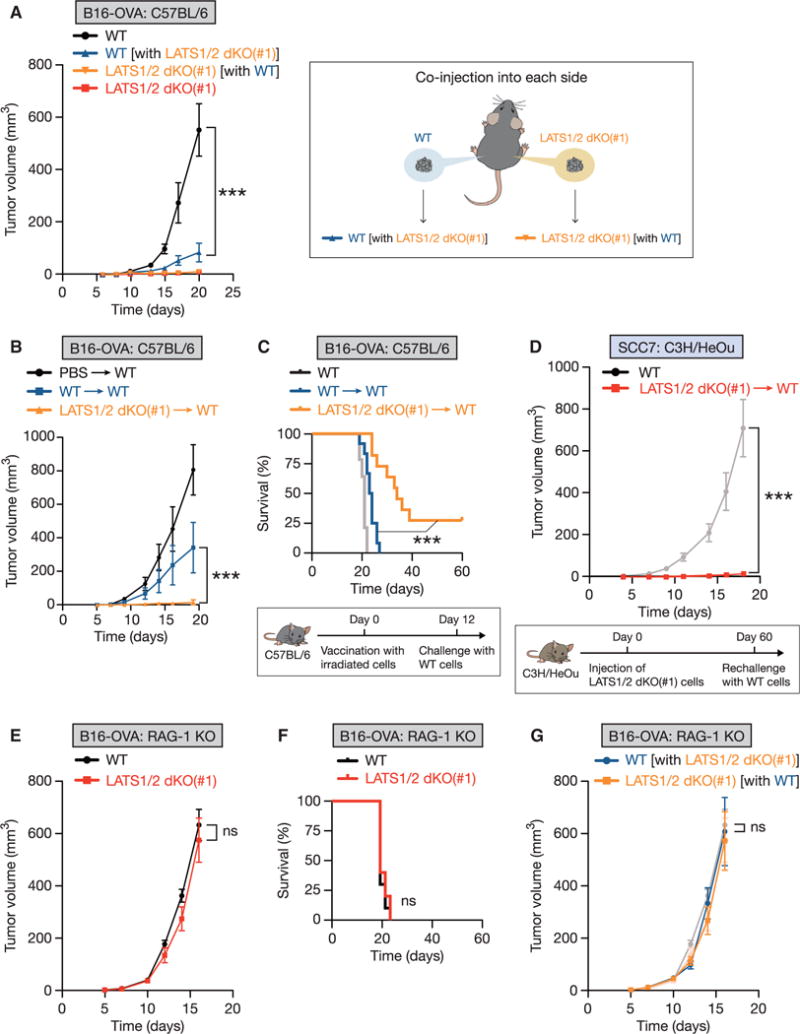
(A) WT or LATS1/2 dKO B16-OVA melanoma cells were injected into C57BL/6 mice, and tumor growth was monitored after the indicated times (left panel). For co-injection experiments, WT and LATS1/2 dKO cells were injected into opposite flanks in the same mouse (right panel). “WT [with LATS1/2 dKO(#1)]” (blue line) indicates WT tumor growth, and “LATS1/2 dKO(#1) [with WT]” (yellow line) indicates LATS1/2 dKO tumor growth, in the co-injected mice. Data are presented as means ± SEM; n = 8 tumors for WT or LATS1/2 dKO group, n = 6 tumors for each co-injected group. The p value was determined using two-way ANOVA test, comparing the “WT [with LATS1/2 dKO(#1)]” group to the WT group. ***p < 0.001.
(B and C) In (B), C57BL/6 mice were immunized intradermally at the base of the tail with equal numbers of irradiated WT or LATS1/2 dKO B16-OVA cells (or PBS control). 12 days after immunization, mice were challenged with WT B16-OVA melanoma, and tumor growth was monitored after the indicated times. Data are presented as means ± SEM; n = 8 tumors for each group. (C) Kaplan-Meier tumor-free survival curves for mice immunized and challenged as in (B) are shown (n = 12 mice for each group). The survival curve of C57BL/6 mice challenged with WT B16-OVA melanoma without vaccination in Figure 2C is also shown in light gray for reference. A schematic representation of vaccination experiment with irradiated-tumor cells is shown in the lower panel. The p value was determined using a two-way ANOVA test (B) or a log-rank test (C), comparing the WT-immunized group [WT / WT] to the LATS1/2 dKO-immunized group [LATS1/2 dKO(#1) / WT]. ***p < 0.001.
(D) C3H/HeOu mice were first injected with non-irradiated LATS1/2 dKO SCC7 cells. 60 days after the initial injection, mice designated tumor-free were re-challenged with WT SCC7 cells, and tumor growth was monitored [LATS1/2 dKO(#1) / WT]. The tumor growth curve of WT SCC7 injected into naive C3H/HeOu mice in Figure 2D is also shown in light gray for reference (WT). Data are presented as means ± SEM; n = 8 tumors for each group. ***p < 0.001, two-way ANOVA test. A schematic representation of the rechallenge experiment is shown in the lower panel.
(E) WT or LATS1/2 dKO B16-OVA cells were transplanted into Rag-1 knockout (KO) mice that lack mature B and T lymphocytes. Tumor growth was monitored after the indicated times. Data are presented as means ± SEM; n = 8 tumors for each group. ns, not significant (p > 0.05, two-way ANOVA test).
(F) Kaplan-Meier tumor-free survival curves for mice transplanted as in (E) are shown (n = 10 mice for each group). ns, not significant (p > 0.05, log-rank test).
(G) WT and LATS1/2 dKO B16-OVA melanoma cells were injected into opposite flanks in the same Rag-1 KO mouse. Data are presented as means ± SEM; n = 6 tumors for each group. The tumor growth curves shown in (E) are shown in a lighter color for reference. The p value was determined using a two-way ANOVA test, comparing the “WT [with LATS1/2 dKO(#1)]” group to the WT group. ns, not significant (p > 0.05).
Next, we tested whether adaptive immunity is required for tumor suppression by LATS1/2 deletion. We subcutaneously transplanted WT or LATS1/2 dKO B16-OVA cells into RAG-1 (recombination activating gene 1) knockout (KO) mice that are immune-compromised due to the lack of mature T and B cells. LATS1/2 dKO B16-OVA tumor cells grew similarly to WT cells (Figure 4E) and showed comparable mortality (Figure 4F) in the absence of an adaptive immune system. Consistently, co-injection of LATS1/2 dKO B16-OVA cells failed to inhibit the corresponding LATS1/2 WT tumor growth in RAG-1 KO mice (Figure 4G). Based on the aforementioned data, we conclude that LATS1/2 deletion in tumor cells enhances immunogenicity and provokes an adaptive immune response to eliminate tumor cells.
YAP or TAZ Overexpression in Tumor Cells Suppresses Tumor Growth In Vivo
YAP and TAZ are the most characterized downstream effectors of the Hippo pathway. LATS1/2 cells directly phosphorylate YAP/TAZ on multiple serine residues, leading to cytoplasmic retention, degradation, and thereby inactivation of YAP/TAZ. Because we observed high YAP/TAZ activation in LATS1/2 dKO B16-OVA tumors in vivo (Figures S4A and S4B), we examined whether YAP/TAZ hyperactivation phenocopies the effect of LATS1/2 deletion in tumor growth. To this end, we generated B16-OVA cells stably overexpressing YAP(5SA) or TAZ(4SA). YAP(5SA) and TAZ(4SA) are active mutants of YAP/TAZ, with the LATS1/2 phosphorylation sites mutated to alanine, thereby unresponsive to inhibition by LATS1/2. Notably, we observed a mutual inhibition between YAP and TAZ protein abundance in YAP(5SA)- or TAZ(4SA)-overexpressing B16-OVA cells (Figure S4C), consistent with the previously described negative-feedback response (Moroishi et al., 2015b). YAP(5SA)- or TAZ(4SA)-overexpressing B16-OVA cells showed increased anchorage-independent growth potential in comparison to the control cells in vitro (Figure S4D), while their tumor growth in vivo was significantly delayed (Figure S4E). We next investigated whether the effect of YAP(5SA) requires its transcriptional activity. YAP mainly binds to the TEA domain (TEAD) family of transcription factors (TEAD1–4) to induce gene expression, and Ser94 in YAP is required for TEAD binding (Zhao et al., 2008). As expected, mutating Ser94 abolished the ability of YAP(5SA) to induce target gene transcription (Figures S4F and S4G). Importantly, the TEAD-binding-defective YAP(5SA/S94A) was unable to suppress B16-OVA tumor growth (Figure S4H), suggesting that tumor suppression by YAP requires TEAD-dependent transcription. Together, these observations indicate that hyperactivation of YAP and TAZ significantly, though may not entirely, contributes to the in vivo tumor growth suppression by LATS1/2 deletion through a mechanism requiring TEAD-mediated transcription.
Extracellular Vesicles Released from LATS1/2-Null Tumor Cells Stimulate Immune Responses
We next explored how LATS1/2 deficiency in tumors stimulates host anti-tumor immune responses. Because we observed a preeminent CD8+ T cell cross-priming in mice injected with LATS1/2 dKO B16-OVA cells (Figure 3F), we hypothesized that LATS1/2 dKO B16-OVA cells stimulate cross-presentation by antigen-presenting cells. To test this, we examined the effects of LATS1/2 dKO B16-OVA cells on MHC (major histocompatibil ity complex) class I-restricted cross-presentation using bone-marrow-derived dendritic cells (BMDCs) as antigen-presenting cells. We found that pre-treatment of BMDCs with conditioned medium from LATS1/2 dKO B16-OVA cells significantly augmented antigen cross-presentation in comparison to WT conditioned medium (Figure 5A), and consistent with this, LATS1/2 dKO conditioned medium enhanced BMDC activation, as assessed by interleukin (IL)-12 production (Figure 5B). These results imply that factors released from LATS1/2 dKO B16-OVA cells activate BMDCs and thereby enhance antigen cross-presentation. Recent studies have revealed the emerging roles of extracellular vesicles (EVs) in immune regulation, both in an immunosuppressive and in an immunostimulatory manner (Robbins and Morelli, 2014). Therefore, we investigated whether EVs secreted from LATS1/2 dKO B16-OVA cells are capable of stimulating immune responses. We isolated EVs from culture supernatants of WT or LATS1/2 dKO B16-OVA melanoma cells by ultracentrifugation (Figure S5A) and found that EVs from LATS1/2 dKO B16-OVA cells were more potent than EVs from WT B16-OVA cells in activating BMDCs, as assessed by IL-12 production in vitro (Figure 5B). To discriminate EVs from extracellular non-membranous particles that may be enriched by ultracentrifugation, we treated the culture supernatants with detergent (Triton X-100) prior to EV purification. The detergent-treated EV pellets failed to activate BMDCs (Figure S5B). More importantly, LATS1/2 dKO EVs improved the tumor vaccine efficacy of irradiated WT B16-OVA cells and conferred a strong immunity against tumor challenge in vivo (Figure 5C). Thus, our results show that EVs released from LATS1/2-deficient tumor cells induce immune responses and are sufficient to render LATS1/2-adequate tumor cells highly immunogenic.
Figure 5. EVs Released from LATS1/2-Null Tumor Cells Stimulate Host Immune Responses.
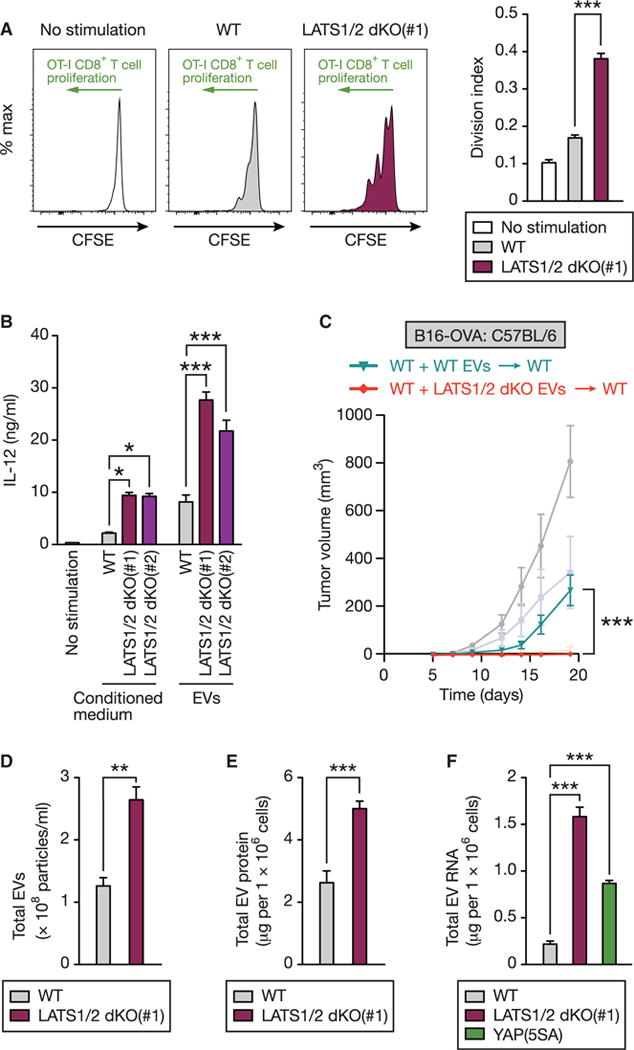
(A) Bone marrow-derived dendritic cells (BMDCs) were pretreated with conditioned medium from WT or LATS1/2 dKO B16-OVA melanoma cells (or control medium) and pulsed with OVA protein. BMDCs were then subjected to an in vitro cross-presentation assay using CFSE-labeled CD8+ T cells isolated from OVA-specific T-cell-receptor transgenic OT-I mice. OT-I CD8+ T cells proliferate when stimulated with OVA antigen via cross-presentation by BMDCs, resulting in dilution of CFSE content. Representative histograms of the gated CD8+ T cells are shown in the left panel. The division index was calculated, and data are presented as means ± SEM of three independent experiments in the right panel. ***p < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test.
(B) BMDCs were stimulated with conditioned medium or EVs from WT or LATS1/2 dKO B16-OVA cell culture supernatants, and then IL-12 levels in the culture supernatants were determined by ELISA. Data are presented as means ± SEM; n = 3 independent experiments for conditioned medium stimulation, n = 4 independent experiments for EVs stimulation. *p < 0.05; ***p < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test.
(C) C57BL/6 mice were inoculated with irradiated WT B16-OVA cells at the base of the tail, and EVs freshly isolated from culture supernatants of equal numbers of WT or LATS1/2 dKO B16-OVA cells were injected every 3 days (days 0, 3, 6, and 9). At day 12, mice were challenged with WT B16-OVA melanoma and tumor growth was monitored. Data are presented as means ± SEM; n = 8 tumors for each group. The tumor growth curves shown in Figure 4B are presented in a lighter color for reference. The p value was determined using a two-way ANOVA test, comparing WT EV-immunized group (WT + WT EVs / WT) to LATS1/2 dKO EV-immunized group (WT + LATS1/2 dKO EVs / WT). ***p < 0.001.
(D) EVs isolated from culture supernatants of equal numbers of WT or LATS1/2 dKO B16-OVA cells were subjected to nanoparticle tracking analysis (NanoSight) to quantify the number and size distribution. The numbers of particles are presented as means ± SEM of three independent experiments. **p < 0.01, unpaired t test.
(E) Protein concentrations of EVs isolated as in (D) were determined. Data are presented as means ± SEM of six independent experiments. ***p < 0.001, unpaired t test.
(F) EVs were isolated from culture supernatants of equal numbers of WT, LATS1/2 dKO, or YAP(5SA)-overexpressing B16-OVA cells, and RNA concentrations were determined by Agilent TapeStation. Data are presented as means ± SEM of three independent experiments. ***p < 0.001, one-way ANOVA test followed by Tukey’s multiple comparison test. See also Figures S4, S5, and S6.
LATS1/2-Deficient Tumor Cells Secrete Nucleic-Acid-Rich EVs
To elucidate the mechanistic basis for immunostimulatory effects of LATS1/2 dKO EVs, we characterized the nature of EVs released from WT or LATS1/2 dKO B16-OVA cells. We found that LATS1/2 dKO B16-OVA cells produced slightly more EVs compared with WT cells, as assessed by nanoparticle tracking analysis (Figures 5D and S6A) as well as by protein quantification (Figure 5E). We then analyzed the proteome of EVs using quantitative mass spectrometry. We identified a total of 1,772 proteins in EVs, which showed enrichment of previously reported exosomal and microvesicle cargo proteins (Figure S6B), supporting the quality of our EV purification. Most of the protein expression was almost identical between WT and LATS1/2 dKO EVs, but a subset of proteins were highly elevated in LATS1/2 dKO EVs (Figure S6C; Table S1). Among the top increased proteins in LATS1/2 dKO EVs were those involved in RNA and nucleic acid binding (Figure S6D). These observations prompted us to hypothesize that LATS1/2-null tumor EVs contain higher amounts of nucleic acids, which are previously reported contents of EVs (Yáñez-Mó et al., 2015) and are also well-known immunostimulators (Junt and Barchet, 2015). Because RNA is the most abundant nucleic acid in EVs (Robbins and Morelli, 2014), we characterized total RNA isolated from EVs. We found that RNA contents in LATS1/2 dKO or YAP(5SA)-over-expressing EVs were dramatically increased in comparison to WT EVs (Figures 5F and S6E). The RNAs in EVs were sensitive to single-strand-specific ribonuclease treatment (Figure S6F). Taken together, our observations suggest a model in which LATS1/2-deficient tumor cells secrete higher amounts of nucleic-acid-rich EVs that may contribute to the potent immunostimulatory effects.
EVs from LATS1/2 dKO Tumor Cells Stimulate the Toll-like Receptors-Type I Interferon Pathway
To test our hypothesis that nucleic-acid-rich EVs released from LATS1/2-null tumors stimulate host anti-tumor immunity, we examined whether alterations in the host nucleic-acid-sensing pathways impair the tumor-protective effects of LATS1/2 deletion in vivo. Both microbial (non-self) and self nucleic acids can be recognized by distinct families of pattern recognition receptors, including endosomal Toll-like receptors (TLRs) and cytosolic non-TLR sensors (Figure S7A). Activation of these pathways results in the production of inflammatory cytokines as well as type I IFN, which stimulates innate and adaptive immunity (Junt and Barchet, 2015). We subcutaneously transplanted WT or LATS1/2 dKO B16-OVA cells into C57BL/6 mice deficient in the following key molecules in the endogenous nucleic-acid-sensing pathways: MYD88 (myeloid differentiation primary response 88) and TRIF (TIR-domain-containing adaptor-inducing interferon-β, also known as TICAM1), two adaptor proteins required for TLR signaling; STING (stimulator of interferon genes, also known as TMEM173), an adaptor protein required for the cGAS (cyclic GMP-AMP synthase, also known as MB21D1) cytoplasmic DNA-sensing pathway; and caspase-1 (also known as CASP1), an effector protein involved in IL-1β maturation under the AIM2 (absent in melanoma 2) cytoplasmic DNA-sensing pathway. We found that deletion of MYD88 largely (Figures 6A and S7B), and TRIF deficiency considerably (Figures 6B and S7C), attenuated the tumor-suppressive effects of LATS1/2 deletion, as assessed by tumor mortality. In contrast, deletion of STING (Figure 6C) or caspase-1 (Figure 6D) in recipient mice had no effect on tumor protection by LATS1/2 deficiency, suggesting that the TLRs-MYD88/TRIF nucleic-acid-sensing pathway, not the cytoplasmic DNA-sensing pathway, is required for immunostimulatory effects of LATS1/2 deletion.
Figure 6. LATS1/2-Deleted Tumor EVs Stimulate Anti-tumor Immunity via the TLRs-Type I IFN Pathway.
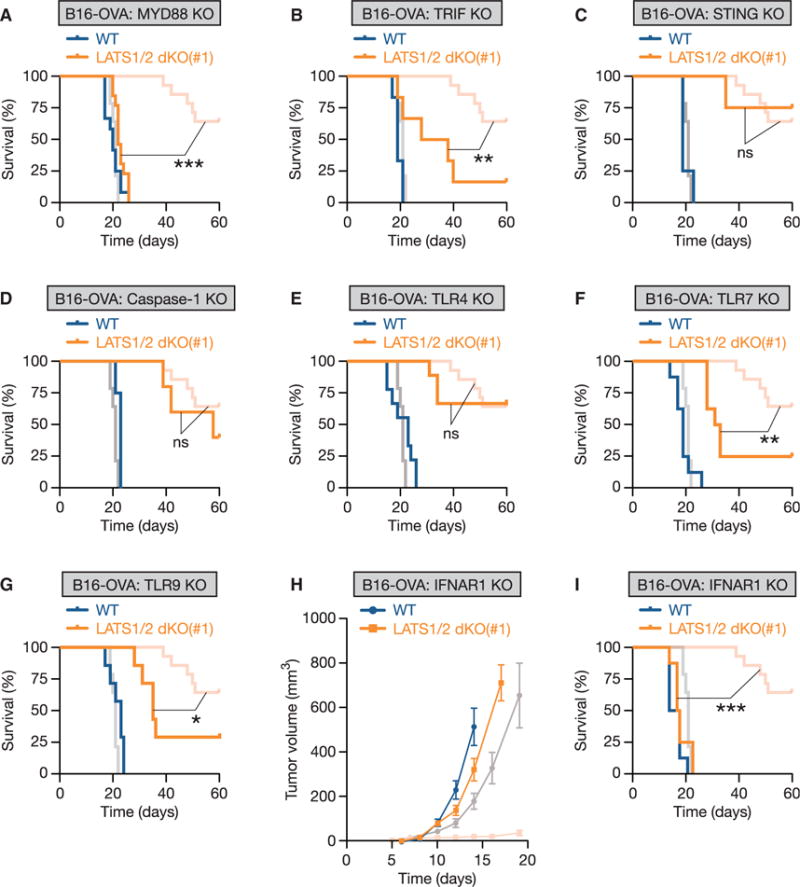
(A–G) WT or LATS1/2 dKO B16-OVA cells were transplanted into mice deficient in Myd88 (A; n = 12 mice for the WT group, and n = 13 mice for the LATS1/2 dKO group), Ticam1 (also known as TRIF) (B; n = 6 mice for each group), Tmem173 (also known as STING) (C; n = 4 mice for each group), Casp1 (also known as caspase-1) (D; n = 4 mice for the WT group, and n = 5 mice for the LATS1/2 dKO group), Tlr4 (E; n = 9 mice for each group), Tlr7 (F; n = 8 mice for each group), or Tlr9 (G; n = 7 mice for each group), and Kaplan-Meier tumor-free survival curves are shown. The survival curves of WT C57BL/6 mice injected with WT or LATS1/2 dKO B16-OVA in Figure 2C are shown in a lighter color for reference. The p value was determined using a log-rank test, comparing KO mice injected with LATS1/2 dKO B16-OVA cells (orange) to corresponding wild-type C57BL/6 mice injected with LATS1/2 dKO B16-OVA cells (light red). ns, not significant (p > 0.05); *p < 0.05; **p < 0.01; ***p < 0.001.
(H) WT or LATS1/2 dKO B16-OVA cells were injected into Ifnar1 KO mice that lack functional type I IFN receptor, and tumor growth was monitored after the indicated times. The tumor growth curves of WT or LATS1/2 dKO B16-OVA cells injected into wild-type C57BL/6 mice in Figure 2A are shown in a lighter color for reference. Data are presented as means ± SEM; n = 8 tumors for each group.
(I) Kaplan-Meier tumor-free survival curves for mice injected as in (H) are shown (n = 8 mice for each group). The survival curves of WT C57BL/6 mice injected with WT or LATS1/2 dKO B16-OVA in Figure 2C are shown in a lighter color for reference. The p value was determined using a log-rank test, comparing Ifnar1 KO mice injected with LATS1/2 dKO B16-OVA cells (orange) to corresponding WT C57BL/6 mice injected with LATS1/2 dKO B16-OVA cells (light red). ***p < 0.001. See also Figure S7.
Distinct types of TLRs utilize MYD88 or TRIF as adaptor proteins and specifically respond to a wide range of ligands on the cell surface, as well as in the endosome (Figure S7A). The endosomal TLRs are intrinsically capable of detecting nucleic acids. We further investigated which TLR is required for tumor suppression by LATS1/2 loss. Whereas LATS1/2 deletion in tumors still protected mice from tumor challenge in TLR4 (which senses bacterial lipopolysaccharides) KO mice (Figure 6E), deletion of TLR7 (which senses single-stranded RNA) (Figure 6F) or TLR9 (which senses double-stranded DNA) (Figure 6G) in recipient mice partially, but significantly, impaired tumor protection by LATS1/2 loss. Thus, our data suggest that multiple TLRs, and probably not a single TLR, cooperatively sense the nucleic-acid-rich EVs secreted from LATS1/2-null tumors and trigger immune responses through the MYD88/TRIF signaling pathway.
Activation of TLRs-MYD88/TRIF signaling results in pro-inflammatory cytokine production as well as type I IFN (in particular, IFNα and IFNβ) production, which stimulates anti-tumor immune responses (Figure S7A). Particularly, type I IFN plays a central role in anti-tumor immunity by promoting dendritic cell maturation, antigen cross-presentation, and CD8+ T cell clonal expansion (Fuertes et al., 2013). Therefore, we examined whether host type I IFN signaling is required for establishing host anti-tumor immunity induced by LATS1/2 dKO tumor cells. To test this, we subcutaneously transplanted WT or LATS1/2 dKO B16-OVA cells into IFNAR1 (interferon α and β receptor subunit 1) KO mice that are deficient in a functional type I IFN receptor. We found that loss of host type I IFN signaling largely obliterated the protective role of LATS1/2 deletion in tumor growth (Figure 6H) as well as tumor mortality (Figure 6I). Thus, collectively, our data provide in vivo evidence supporting a model in which EVs secreted from LATS1/2-deficient tumor cells stimulate the host TLRs-MYD88/TRIF nucleic-acid-sensing pathways to incite type I IFN signaling and establish robust anti-tumor immunity.
DISCUSSION
In this study, we demonstrate that LATS1/2 deletion unmasks a malignant cell’s immunogenic potential and restrains tumor growth due to the induction of anti-tumor immune responses. The effects of LATS1/2 deletion on tumor growth are striking insofar as LATS1/2 dKO completely abolishes the tumor growth potential of SCC7 and dramatically reduces tumor growth and the metastasis of B16 and 4T1 cells. LATS1/2-null B16 melanomas secrete nucleic-acid-rich EVs that stimulate the host TLRs-MYD88/TRIF-IFN pathways to induce anti-tumor immunity and the eventual elimination of tumor cells (Figure 7). LATS1/2 deletion similarly stimulates host immune responses in both SCC7 and 4T1 syngeneic models (Figures 4D, S3A, and S3B), though the involvement of EVs has only been examined in the B16 model in this study.
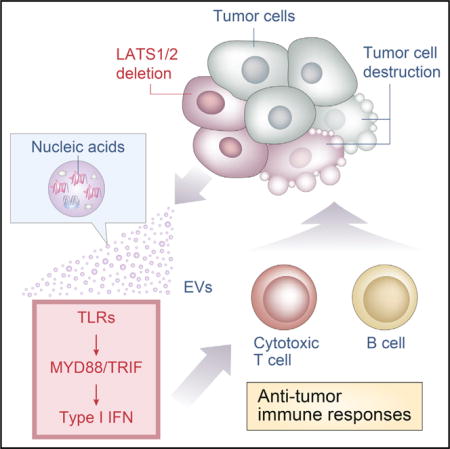
Figure 7. Proposed Model for the Regulation of Anti-tumor Immunity by the Hippo Pathway in Tumors.
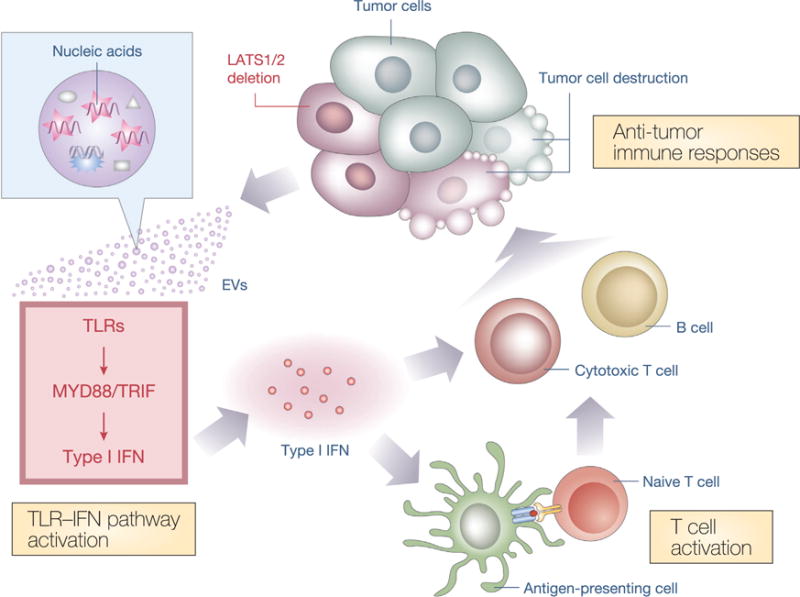
Poorly immunogenic tumor cells evade host immune defenses, despite expressing antigenic neoepitopes. LATS1/2 deletion in tumor cells stimulates nucleic-acid-rich EV secretion, which induces a type I IFN response via the TLRs-MYD88/TRIF pathway. Type I IFN stimulates multiple components of host immune responses, including cross-presentation of tumor-derived antigens by antigen-presenting cells and T cell activation. Activated T cells, in turn, facilitate tumor-specific responses of cytotoxic T cells and antibody production by B cells, promoting tumor destruction. Thus, loss of LATS1/2 in tumors leads to the rejection of both LATS1/2-deficient and LATS1/2-adequate tumor cells by enhancing host anti-tumor immune responses.
Dual Functions of LATS1/2 in Cancer
It is generally accepted that the Hippo pathway is a tumor suppressor that inhibits proliferation and survival of normal cells, preventing tumorigenesis (Harvey et al., 2013; Moroishi et al., 2015a; Wang et al., 2014), yet a few studies did suggest an onco-genic role of the Hippo pathway in certain contexts (Barry et al., 2013; Cottini et al., 2014). We have analyzed human epidemiological data using the PrognoScan database (Mizuno et al., 2009) to find any correlation between LATS1/2 mRNA expression levels and patient outcome in different types of human cancer (Table S2). Among 107 epidemiological datasets available, 26 studies show significant (p < 0.05) correlation between LATS2 mRNA levels and patient outcome, which includes 17 studies showing better patient survival with low LATS2 expression. Moreover, 12 studies show significant correlation between LATS1 mRNA levels and patient outcome, which includes 5 studies showing better patient survival with low LATS1 expression. In addition, low YAP expression predicted worse patient survival in human colorectal cancer (Barry et al., 2013) and multiple myeloma (Cottini et al., 2014). Therefore, although YAP/TAZ hyperactivation is frequently observed in human cancers (Harvey et al., 2013; Moroishi et al., 2015a), the precise role of the Hippo pathway in human cancer might be context dependent. In this study, we show that deletion of LATS1/2 in tumor cells strongly suppresses tumor growth in vivo. On the surface, our data cannot be easily reconciled with the tumor suppressor model of LATS1/2 in the Hippo field.
We propose the following model: LATS1/2 suppress tumor initiation as well as inhibit immunogenicity. These two activities are important for the physiological role of LATS1/2 in maintaining tissue homeostasis. LATS1/2 normally provide growth-inhibitory signals to the cells; therefore, they function cell autonomously to limit tissue overgrowth. We also propose that LATS1/2 suppresses immunogenicity, serving as a built-in mechanism to prevent overgrowth of undesirable cells at the wrong places in the organism. For example, inactivation of LATS1/2 is needed to promote cell proliferation during wound healing and tissue regeneration. However, cells with impaired LATS1/2 activity may over-proliferate and migrate to the wrong place. Such undesirable cells should be eliminated to maintain tissue homeostasis and integrity. This can be achieved because inactivation of LATS1/2 in these cells can induce a strong immune response. Therefore, the immunosuppressive function of LATS1/2 is consistent with its physiological roles in tissue homeostasis.
In the established tumor cell lines of B16, SCC7, and 4T1, YAP and TAZ are not constitutively active. In fact, YAP and TAZ are readily regulated (in B16 and 4T1 cells) or even largely inactive (in SCC7 cells). Therefore, the tumorigenicity of these cancer cell lines is independent of the Hippo pathway. Nevertheless, deletion of LATS1/2 causes a moderate increase of anchorage-independent growth of these tumor cells in vitro, consistent with the growth inhibitory effect of LATS1/2. However, the enhanced immunogenicity unmasked by the LATS1/2 deletion in these cells can induce strong immune responses and overwhelm any growth advantage that might be gained due to LATS1/2 deletion, leading to strong inhibition of tumor growth in the immune-competent mice. The dual functions of LATS1/2 in suppressing cell growth and immunogenicity can explain previous observations along with our present data.
Hippo Pathway in Inflammation and Tumor Immunogenicity
Our results indicate that inactivation of the Hippo pathway in tumor cells induces host inflammatory responses. Interestingly, recent studies revealed that the Hippo pathway can respond to (Nowell et al., 2016; Taniguchi et al., 2015) and mediate (Liu et al., 2016) inflammatory signals. Our study, together with these recent findings, suggests a reciprocal interaction between the Hippo pathway and inflammatory responses. LATS1/2-deficient tumor-derived EVs contain higher amounts of nucleic acids, which stimulate the host TLRs-MYD88/TRIF nucleic-acid-sensing pathways, provoking a type I IFN response to establish robust anti-tumor immunity. Recent studies indicate that tumor cells themselves can produce type I IFN in response to chemotherapy, thus enhancing anti-tumor immune responses (Chiappinelli et al., 2015; Sistigu et al., 2014). Because WT and LATS1/2 dKO B16-OVA cells showed similar expression levels of type I IFN genes such as Ifna4 and Ifnb1 (Figure S7D), it is less likely that type I IFN secreted from LATS1/2-null tumor cells is the main mechanism conferring the anti-tumor immunity evoked by LATS1/2 deletion. Given that nucleic-acid-rich EVs from LATS1/2-deficient tumors can stimulate dendritic cells in vitro (Figure 5B), and that the host TLRs-MYD88/TRIF nucleic-acid-sensing pathways are required for immunostimulatory effects of LATS1/2 deletion in vivo (Figure 6), we speculate that host immune cells in the tumor microenvironment may be the major source of type I IFN (Fuertes et al., 2013).
A series of unbiased Hippo pathway interactome studies have linked endosomal compartments to the Hippo pathway (Moya and Halder, 2014). It is possible that the Hippo pathway may regulate endocytic trafficking and, therefore, regulate EV biogenesis. Little is known about the signaling mechanisms involved in EV biogenesis and incorporation of proteins or nucleic acids into EVs. Given the known effect of YAP on global microRNA (miRNA) biogenesis (Mori et al., 2014) and the functional importance of miRNA in EVs (Yáñez-Mó et al., 2015), one may speculate that the effect of YAP/TAZ on miRNA biogenesis may increase immunogenicity of LATS1/2-null cells. However, we show that TEAD-mediated transcription is required for tumor suppression by YAP (Figure S4H), whereas TEAD-dependent transcription is dispensable for YAP-influenced miRNA biogenesis (Mori et al., 2014). Moreover, LATS1/2 dKO cells do not increase miRNA contents in EVs (Figure S6E). Therefore, YAP/TAZ hyperactivation suppresses tumor growth in vivo via a transcription-dependent, but miRNA-biogenesis-independent, mechanism. The tumor growth suppression by YAP/TAZ overexpression (Figure S4E) is not as strong as that of LATS1/2 deletion (Figure 2A), suggesting that LATS1/2 may have additional targets to suppress immune responses. Recent studies revealed new LATS1/2 substrates in spindle orientation (Dewey et al., 2015; Keder et al., 2015). Because aneuploidy plays a role in tumor immunogenicity (Senovilla et al., 2012), these new LATS1/2 substrates in spindle regulation could contribute to immunosuppression. Further investigations delineating the mechanistic link between the Hippo pathway and EV biogenesis will have important implications in understanding both the basic biology of EVs and the inflammation provoked by alteration of the Hippo pathway.
Targeting the Hippo Pathway for Cancer Immunotherapy
Recent advances in cancer immunotherapy have provided new therapeutic approaches for cancer, and several immune checkpoint inhibitors indeed show impressive effects in the clinic (Sharma and Allison, 2015). However, individual immune response to cancer immunotherapy often relies on tumor immunogenicity that varies extensively between different cancer types and different individuals; therefore, immune checkpoint inhibitors may not work in cases where tumor immunogenicity is intrinsically limited (Pico de Coaña et al., 2015). Our study revealed that inactivation of LATS1/2 in tumor cells increases tumor immunogenicity and enhances tumor vaccine efficacy. Therefore, we speculate that inhibiting LATS1/2 may enhance anti-tumor immune response and, therefore, would be an attractive approach to treat cancer. Furthermore, LATS1/2 inhibition to improve immunogenicity of tumor cells may enhance immune checkpoint inhibitor efficacy. Thus, a combination of LATS1/2 inhibitors and immune checkpoint inhibitors would be a novel and exciting therapeutic approach for poorly immunogenic cancers, especially in cases where malignancy is driven by oncogenic alterations that leave the Hippo signaling pathway intact. It is noteworthy that germline or somatic mutations affecting the core components of the Hippo pathway are uncommon in human cancers (Harvey et al., 2013; Moroishi et al., 2015a). Therefore, inhibition of LATS1/2 may enhance tumor immunity in most cancer types. However, the caveat remains that the immune system of mice is considerably different from that of humans, and whether our findings in mice can directly be translated to humans remains to be determined. Moreover, the effect of LATS1/2 inhibition as an intervention for established tumors needs to be explored. Nevertheless, future studies expanding the therapeutic potentials of the Hippo pathway will have important clinical implications.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-YAP | Cell Signaling | Cat#: 14074 |
| Anti-pYAP (S127 in humans and S112 in mice) | Cell Signaling | Cat#: 4911 |
| Anti-YAP/TAZ | Cell Signaling | Cat#: 8418 |
| Anti-LATS1 | Cell Signaling | Cat#: 3477 |
| Anti-YAP/TAZ | Santa Cruz | Cat#: sc-101199; RRID: AB_1131430 |
| Anti-CD81 | Cell Signaling | Cat#: 10037 |
| Anti-ALIX | Cell Signaling | Cat#: 2171 |
| Anti-LATS2 (also weakly recognize LATS1) | Bethyl Laboratories | Cat#: A300-479A; RRID: AB_2133375 |
| Anti-FLOT1 | BD Biosciences | Cat#: 610821; RRID: AB_398140 |
| Anti-HSP90 | BD Biosciences | Cat#: 610418; RRID: AB_397798 |
| Anti-actin | Abcam | Cat#: ab3280; RRID: AB_303668 |
| Anti-ovalbumin | Abcam | Cat#: ab1221; RRID: AB_298931 |
| Anti-CD45 (PE-conjugated) | eBioscience | Cat#: 12-0451-82; RRID: AB_465668 |
| Anti-CD16/CD32 (Fc block) | BD Biosciences | Cat#: #553142 |
| Anti-CD45 (Fluorochrome-conjugated) | eBioscience | Clone 30F-11 |
| Anti-CD3e (Fluorochrome-conjugated) | eBioscience | Clone 145-2C11 |
| Anti-CD8a (Fluorochrome-conjugated) | BD Biosciences | Clone 53-6.7 |
| Anti-Granzyme B (Fluorochrome-conjugated) | BD Biosciences | Clone GB11 |
| Anti-IFNγ (Fluorochrome-conjugated) | eBioscience | Clone XMG1.2 |
| Anti-Mouse IL-12 p40/p70 (ELISA capture antibody) | BD Biosciences | Cat#: #551219; RRID: AB_394097 |
| Anti-Mouse IL-12 p40/p70 (ELISA detection antibody) | BD Biosciences | Cat#: #554476; RRID: AB_395419 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Kb-SIINFEKL-tetramer (PE-conjugated) | MBL International | Cat#: T03000 |
| Phos-tag reagent | Wako Chemicals | Cat#: 304-93521 |
| Ovalbumin recombinant protein | Worthington Biochemical | Cat#: LS003056 |
| SIINFEKL peptide | AnaSpec | Cat#: AS-60193-1 |
| Protein Transport Inhibitor | BD Biosciences | Cat#: 555029 |
| CFSE (carboxyfluorescein succinimidyl ester) | Thermo Fisher Scientific | Cat#: C34554 |
| Mouse GM-CSF Recombinant Protein | eBioscience | Cat#: 14-8331-80 |
| RNase A | Thermo Fisher Scientific | Cat#: EN0531 |
| Latrunculin B (LatB) | Abcam | Cat#: ab144291 |
| 2-deoxy-D-glucose (2-DG) | Sigma-Aldrich | Cat#: D8375 |
| Poly(I:C) | InvivoGen | Cat#: tlrl-pic |
| PolyJet | SignaGen Laboratories | Cat#: SL100688 |
| Critical Commercial Assays | ||
| RNeasy Plus Mini Kit | QIAGEN | Cat#: 74134 |
| iScript cDNA Synthesis Kit | BIO-RAD | Cat#: 1708890 |
| KAPA SYBR FAST qPCR Kit | Kapa Biosystems | Cat#: KK4605 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat#: 554714 |
| EasySep Mouse CD8a Positive Selection Kit | STEMCELL | Cat#: 18753 |
| Mouse IFN-gamma DuoSet ELISA | R&D Systems | Cat#: DY485-05 |
| TRIzol reagents | Thermo Fisher Scientific | Cat#: 15596026 |
| Experimental Models: Cell Lines | ||
| Mouse cell line: B16-F10 cells | ATCC | CRL-6475 |
| Mouse cell line: SCC7 cells | Laboratory of John Lee | N/A |
| Mouse cell line: 4T1 cells | ATCC | CRL-2539 |
| Mouse cell line: EL4 cells | ATCC | TIB-39 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6 | The Jackson Laboratory | JAX: 000664 |
| Mouse: C3H/HeOu | The Jackson Laboratory | JAX: 000635 |
| Mouse: BALB/c | The Jackson Laboratory | JAX: 000651 |
| Mouse: Myd88 KO | Laboratory of Shizuo Akira | N/A |
| Mouse: Tlr4 KO | Laboratory of Shizuo Akira | N/A |
| Mouse: Tlr7 KO | Laboratory of Shizuo Akira | N/A |
| Mouse: Tlr9 KO | Laboratory of Shizuo Akira | N/A |
| Mouse: Ticam1 (also known as TRIF) KO | Laboratory of Bruce Beutler | N/A |
| Mouse: Casp1 (also known as Caspase-1) KO | Laboratory of Richard A. Flavell | N/A |
| Mouse: Rag1 KO | The Jackson Laboratory | JAX: 002096 |
| Mouse: Tmem173 (also known as STING) KO | The Jackson Laboratory | JAX: 025805 |
| Mouse: OT-I transgenic | The Jackson Laboratory | JAX: 003831 |
| Mouse: Ifnar1 KO | B&K Universal | N/A |
| Recombinant DNA | ||
| Plasmid: PX459 (CRISPR) | Addgene | Plasmid #48139 |
| Plasmid: pBABE | Cell Biolabs | Cat#: RTV-001-puro |
| Plasmid: pBABE YAP(5SA) | This paper | N/A |
| Plasmid: pBABE YAP(5SA/S94A) | This paper | N/A |
| Plasmid: pBABE TAZ(4SA) | This paper | N/A |
| Sequence-Based Reagents | ||
| CRISPR targeting sequence: mouse Lats1: #1: AGACGTTCTGCTCCGAAATC | This paper | N/A |
| CRISPR targeting sequence: mouse Lats1: #2: ACGTTTCCATTGGCGAATGA | This paper | N/A |
| CRISPR targeting sequence: mouse Lats2: #1: GAGTGTCCAGCTTACAAGCG | This paper | N/A |
| CRISPR targeting sequence: mouse Lats2: #2: GCTGGGTGGTGCAAACTACG | This paper | N/A |
| qPCR primers for mouse Gapdh: 5′-GCCTGGAGAAACCTGCCAAGTATG-3′ and 5′-GAGTGGGAGTTGCTGTTGAAGTCG-3′ | This paper | N/A |
| qPCR primers for mouse Ctgf: 5′-AGCTGACCTGGAGGAAAACA-3′ and 5′-GACAGGCTTGGCGATTTTAG-3′ | This paper | N/A |
| qPCR primers for mouse Cyr61: 5′-GCTCAGTCAGAAGGCAGACC-3′ and 5′-GTTCTTGGGGACACAGAGGA-3′ | This paper | N/A |
| qPCR primers for mouse Amotl2: 5′-AGGAGAAGAGTTGCCCACCTATGAG-3′ and 5′-TCGAAGAGCTTCATCCTGTCGC-3′ | This paper | N/A |
| qPCR primers for mouse Ifna4: 5′-CCTGAGAAAGAAGAAACACAGCCTC-3′ and 5′-GCAAGTTGGTTGAGGAAGAGAGGG-3′ | This paper | N/A |
| qPCR primers for mouse Ifnb1: 5′-GAAGAGTTACACTGCCTTTGCCATC-3′ and 5′-AAACACTGTCTGCTGGTGGAGTTC-3′ | This paper | N/A |
| Software and Algorithms | ||
| Optimized CRISPR Design | Ran et al., 2013 | http://crispr.mit.edu |
| GraphPad Prism 5 | GraphPad Software | N/A |
| FlowJo | Treestar | N/A |
| Proteome Discoverer 1.4 | Thermo Fisher Scientific | Cat#: IQLAAEGABSFAKJMAUH |
| PANTHER | Mi et al., 2013 | http://geneontology.org |
| NetWalker | Komurov et al., 2012 | N/A |
| PrognoScan | Mizuno et al., 2009 | http://www.abren.net/PrognoScan/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Please direct any requests for further information or reagents to the Lead Contact, Kun-Liang Guan (kuguan@ucsd.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
C57BL/6, C3H/HeOu, or BALB/c mice were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Myd88, Tlr4, Tlr7, and Tlr9 KO mice were kind gifts from Dr. Shizuo Akira (Osaka University, Osaka, Japan). Ticam1 (also known as TRIF) KO mice were kindly provided by Dr. Bruce Beutler (University of Texas Southwestern Medical Center, Dallas, TX, USA). Casp1 (also known as Caspase-1) KO mice were kindly provided by Dr. Richard A. Flavell (Yale University School of Medicine, New Haven, CT, USA). Rag1 KO mice, Tmem173 (also known as STING) KO mice, and OT-I transgenic mice were purchased from The Jackson Laboratory. Ifnar1 KO mice were purchased from B&K Universal (East Yorkshire, United Kingdom). These mouse strains were backcrossed for 10 generations onto the C57BL/6 background at the University of California, San Diego. Mutant mice were bled by the University of California, San Diego Animal Care Program. 7–12 weeks old mice were used and all animal experiments were approved by the University of California, San Diego, Institutional Animal Care and Use Committee.
METHOD DETAILS
Cell culture and gene deletion by CRISPR/Cas9 system
All cell lines were cultured under an atmosphere of 5% CO2 at 37°C. B16-OVA cells (B16F10 cells expressing ovalbumin) were cultured in DMEM (GIBCO) supplemented with 10% fetal bovine serum (FBS, GIBCO), penicillin (100 U/ml), and streptomycin (100 mg/ml). SCC7, 4T1, EL4, bone marrow-derived dendritic cells (BMDCs), mouse primary lymph node cells and CD8+ T cells were cultured in RPMI 1640 (GIBCO) supplemented with 10% FBS (GIBCO), penicillin (100 U/ml), and streptomycin (100 mg/ml).
LATS1/2-deficient cells were created through the CRISPR (clustered regularly interspaced short palindromic repeats)/Cas9 system (Ran et al., 2013). We use a transient CRISPR strategy for the deletion of LATS1/2 to avoid any potential unspecific effects mediated by stable Cas9/sgRNA genome integration. Cells were transiently transfected with a Cas9 and single-guide RNA (sgRNA) expression plasmid encoding puromycin resistance (PX459; Addgene plasmid #48139). The CRISPR-transfected cells will thus acquire transient resistance to puromycin. The guide sequences were designed using the Optimized CRISPR Design at http://crispr.mit.edu. The guide sequences used are 5′-AGACGTTCTGCTCCGAAATC-3′ or 5′-ACGTTTCCATTGGCGAATGA-3′ for mouse Lats1; 5′-GAGTGTCCAGCTTACAAGCG-3′ or 5′-GCTGGGTGGTGCAAACTACG-3′ for mouse Lats2. Following transfection and transient selection with puromycin for 3 days, cells were single-cell sorted by fluorescence-activated cell sorting (FACS) into 96-well plate without puromycin. Knockout clones were selected by immunoblot analysis for the lack of LATS1/2 proteins and YAP phosphorylation. We confirmed that LATS1/2 dKO cells were sensitive to puromycin after expansion, indicating a transient expression of CRISPR/Cas9 system in those cells. Two independent clones were analyzed as indicated and the parental LATS1/2 WT cells (not transfected with PX459) were used as control.
Retroviral infection
B16-OVA cells stably expressing empty vector, YAP(5SA), YAP(5SA/S94A), or TAZ(4SA) were generated by retroviral infection. 293 Phoenix retrovirus packaging cells were transfected with pBABE empty vector, pBABE YAP(5SA), pBABE YAP(5SA/S94A), or pBABE TAZ(4SA) constructs. Forty-eight hours after transfection, retroviral supernatant was supplemented with 5 μg/ml polybrene, filtered through a 0.45 μm filter, and used for infection. Forty-eight hours after infection, cells were selected with 4 μg/ml puromycin in culture medium.
Immunoblot analysis
Equal amount of protein samples were resolved in SDS-PAGE in reducing conditions unless otherwise mentioned in the Figure Legends. Antibodies to YAP (#14074), pYAP (S127 in humans and S112 in mice; #4911), YAP/TAZ (#8418), LATS1 (#3477), CD81 (#10037), and ALIX (#2171) were obtained from Cell Signaling; those to actin (#ab3280) and ovalbumin (OVA, #ab1221) were from Abcam; those to LATS2 (#A300-479A, also weakly recognize LATS1) were from Bethyl Laboratories; those to FLOT1 (#610821) and HSP90 (#610418) were from BD Biosciences. The phos-tag electrophoresis was performed as described previously (Moroishi et al., 2015b). YAP proteins can be separated into multiple bands in the presence of phos-tag depending on differential phosphorylation levels, with phosphorylated proteins migrating more slowly. Where indicated, cells were treated with serum starvation (DMEM or RPMI 1640 without other supplements), 1 μg/ml Latrunculin B (LatB), or 25 mM 2-deoxy-D-glucose (2-DG) for 1 hr before harvest.
Immunostaining of cells
Cells were treated with or without 1 μg/ml Latrunculin B (LatB) for 1 hr before harvest. Cells were then fixed for 10 min at room temperature with 4% paraformaldehyde in phosphate-buffered saline (PBS) and were permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature. Cells were then incubated consecutively with primary antibodies to YAP/TAZ (Santa Cruz, #sc-101199) (overnight at 4°C) and Alexa Fluor 488-labeled goat secondary antibodies (for 90 min at room temperature) in PBS containing 1% bovine serum albumin (BSA). Cells were covered with a drop of ProLong Gold antifade reagent with DAPI (Invitrogen) for observation. Cells in five randomly selected views (~100 cells) were selected for the quantification of YAP/TAZ localization.
Reverse transcription (RT) and real-time PCR analysis
Total RNA (500 ng) isolated from cells with the use of RNeasy Plus Mini Kit (QIAGEN) was reverse-transcribed to complementary DNA using iScript cDNA Synthesis Kit (Bio-Rad). Complementary DNA was then diluted and used for quantification by real-time PCR, which was performed using KAPA SYBR FAST qPCR Kit (Kapa Biosystems) and the 7300 real-time PCR system (Applied Bio-systems). The sequences of the PCR primers (forward and reverse, respectively) are 5′-GCCTGGAGAAACCTGCCAAGTATG-3′ and 5′-GAGTGGGAGTTGCTGTTGAAGTCG-3′ for mouse Gapdh; 5′-AGCTGACCTGGAGGAAAACA-3′ and 5′-GACAGGCTTGGCGATTTTAG-3′ for mouse Ctgf; 5′-GCTCAGTCAGAAGGCAGACC-3′ and 5′-GTTCTTGGGGACACAGAGGA-3′ for mouse Cyr61; 5′-AGGAGAAGAGTTGCCCACCTATGAG-3′ and 5′-TCGAAGAGCTTCATCCTGTCGC-3′ for mouse Amotl2; 5′-CCTGAGAAAGAAGAAACACAGCCTC-3′ and 5′-GCAAGTTGGTTGAGGAAGAGAGGG-3′ for mouse Ifna4; 5′-GAAGAGTTACACTGCCTTTGCCATC-3′ and 5′-AAACACTGTCTGCTGGTGGAGTTC-3′ for mouse Ifnb1. Reactions for Gapdh mRNA were performed concurrently on the same plate as those for the test mRNAs, and results were normalized by the corresponding amount of Gapdh mRNA.
Soft agar colony formation assay
Each 6-well plate was coated with 1.5 mL of bottom agar (DMEM or RPMI 1640 containing 10% FBS and 0.5% Difco agar noble). Cells (5 × 103 cells for B16-OVA and SCC7, 2.5 × 103 cells for 4T1) were suspended in 1.5 mL of top agar (DMEM or RPMI 1640 containing 10% FBS and 0.35% Difco agar noble) into each well. Cells were cultured for approximately two weeks and replaced with fresh medium every three days. Colonies were stained using 0.005% crystal violet in 5% methanol and quantified using ImageJ software.
Tumor transplantation and immunization
B16-OVA cells (2 × 105) were subcutaneously transplanted into both back flanks of C57BL/6 mice. Tumor height and width were measured with a caliper every 2–3 days to calculate tumor volume (= width2 × height × 0.523). Mice were sacrificed when tumors reached maximum allowed size (15 mm in diameter) or when signs of ulceration were evident. Likewise, 1 × 105 of SCC7 cells were subcutaneously transplanted into both back flanks of C3H/HeOu mice and 2.5 × 105 of 4T1 cells were transplanted into both mammary fat pads of BALB/c mice. For 4T1 lung metastasis assay, lungs were tracheally injected with India ink 28 days after transplantation, and then destained in Fekete’s solution to count tumor nodules.
For tumor vaccination experiments, C57BL/6 mice were immunized intradermally at the base of the tail with irradiated B16-OVA cells (100 Gy, 1 × 106) 12 days prior to challenge with B16-OVA cells (one time vaccination, without any adjuvant). For immunization with EVs, C57BL/6 mice were inoculated with irradiated B16-OVA cells (100 Gy, 1 × 106) at the base of the tail and EVs freshly isolated from culture supernatants of B16-OVA cells (6 × 106) were injected every 3 days (days 0, 3, 6, and 9) into the same place until challenged with B16-OVA cells at day 12.
Histopathology and immunostaining of tumors
Tumors were fixed with 4% paraformaldehyde in PBS, embedded in paraffin, sectioned with a microtome, and stained with hematoxylin-eosin according to standard procedures. Immunostaining of tumors was performed with frozen cryostat sections with PE-conjugated antibodies to CD45 (eBioscience, #12-0451-82).
Measurement of OVA specific antibodies
Serum anti-OVA IgG concentrations were measured by enzyme-linked immunosorbent assay (ELISA). Briefly, half area 96-well plates (Corning) were coated with 5 μg/ml OVA protein (Worthington Biochemical, #LS003056) in PBS overnight at 4°C. Plates were washed and then blocked for 3 hr at room temperature with blocking buffer [1% BSA (bovine serum albumin) in PBS], followed by wash and incubation with serum samples tested at a 1:100 to 1:125,600 dilutions in blocking buffer overnight at 4°C. Plates were then washed and incubated with HRP-conjugated detecting antibody in blocking buffer at room temperature for 2 hr. Plates were washed and incubated with TMB substrate (KPL, #95059-286), and then read at 450 nm and 650 nm after stopping the development with 1 M phosphoric acid. Each ELISA plate contained a titration of a previously quantified serum to generate a standard curve. Anti-OVA IgG concentrations were determined from the lowest dilution of serum samples within a standard curve and reported as U/ml.
Flow cytometry
Flow cytometry was performed using a BD LSRFortessa and results were analyzed using FlowJo software (Treestar). Cell suspensions were incubated in mouse Fc block (anti CD16/CD32; BD Biosciences, #553142) prior to staining. Fluorochrome conjugated anti-mouse CD45 (clone 30F-11), CD3e (clone 145-2C11), CD8a (clone 53-6.7), Granzyme B (clone GB11), and IFNγ (clone XMG1.2) antibodies were used following the manufacturers protocol. Kb-SIINFEKL-tetramer was used for identifying OVA-specific CD8+ T cells. Propidium iodide (PI) was used to stain dead cells.
To analyze intracellular cytokine expression, cells were re-stimulated ex vivo with 10 μg/ml SIINFEKL peptide (AnaSpec, #AS-60193-1) for 5 hr in the presence of protein transport inhibitor (BD biosciences, #555029) for the last 4 hr. Intracellular cytokine staining was then performed using Fixation/Permeabilization Solution Kit (BD Biosciences, #554714).
Ex vivo cytotoxicity assay
EL4 cells were pulsed with 8 μg/ml SIINFEKL peptide or irrelevant peptides for 2 hr at 37°C, and then labeled with 0.25 μM or 2.5 μM CFSE (carboxyfluorescein succinimidyl ester; Thermo Fisher Scientific, #C34554) for 10 min at 37°C, respectively. CFSElow (SIINFEKL loaded target) and CSFEhigh (irrelevant peptide control) EL4 cells were mixed at 1:1 ratio, and then co-cultured with CD8+ T cells isolated from splenocytes of C57BL/6 mice challenged (or not) with WT or LATS1/2 dKO B16-OVA cells at 8:1 effector to target cell ratio (E:T). CD8+ T cells were isolated using EasySep Mouse CD8a Positive Selection Kit (STEMCELL, #18753) from pooled splenocytes of 3–4 mice per group for each experiment. The frequencies of CFSElow and CSFEhigh EL4 cells in CFSE positive fraction were determined by flow cytometric analysis 18 hr after incubation and the percent of specific killing was calculated. Specific killing (%) = [1−”Sample ratio”/”Negative control ratio”] × 100; “Sample ratio” = [CFSElow(target)/CSFEhigh(irrelevant)] value of each samples co-cultured with CD8+ T cells; “Negative control ratio” = [CFSElow(target)/CSFEhigh(irrelevant)] value of EL4 cells not cultured with CD8+ T cells.
In vitro cross-presentation assay
For conditioned medium preparation, B16-OVA cells were seeded on culture plates and incubated in DMEM supplemented with 10% FBS for 24 hr at 37°C to allow cell attachment. The cells were then washed with PBS, and culture medium was switched to DMEM without serum. After incubation for 48 hr, conditioned medium was collected and centrifuged at 2,000 g for 10 min at 4°C to remove cell debris. The resulting supernatant was used for the experiment.
Naive CD8+ T cells were isolated from OVA-specific T cell receptor transgenic OT-I mice using EasySep Mouse CD8a Positive Selection Kit (STEMCELL) and labeled with 2 μM CFSE. Bone marrow derived dendritic cells (BMDCs) were generated by 6 days of GM-CSF (20 ng/ml; eBioscience, #14-8331-80) differentiation, and then incubated (or not) for 18 hr with conditioned medium (10% of the total volume) from WT or LATS1/2 dKO B16-OVA melanoma cells and pulsed with OVA protein (10 μg/ml) for the last 4 hr. The cells were washed and cultured with CFSE-labeled OT-I CD8+ T cells at 1:1 ratio for 3 days. OT-1 T cell proliferation was monitored by CFSE dilution using a flow cytometer and a division index was determined using FlowJo software (Treestar).
Cytokine ELISA
IFNγ or IL-12 levels in culture supernatants were determined by ELISA. For ex vivo IFNγ production from lymph node cells, draining lymph nodes (inguinal lymph nodes) were isolated from C57BL/6 mice challenged (or not) with B16-OVA cells and cultured with OVA protein (100 μg/ml) for 3 days. For IL-12 production from BMDCs, BMDCs were generated by 6 days of GM-CSF (20 ng/ml) differentiation and stimulated (or not) for 18 hr with conditioned medium (10% of the total volume) or EVs isolated from culture supernatants of equal numbers of WT or LATS1/2 dKO B16-OVA cells (EVs from 2 × 105 cells were used to stimulate 1 × 106 BMDCs). Both cultures were done in RPMI 1640 supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (100 mg/ml) under an atmosphere of 5% CO2 at 37°C, and then aliquots of cell culture supernatants were used for cytokine ELISA. For cell conditioned medium preparation, B16-OVA cells were seeded on culture plates and incubated in DMEM supplemented with 10% FBS for 24 hr at 37°C to allow cell attachment. The cells were then washed with PBS, and culture medium was switched to DMEM without serum. After incubation for 48 hr, conditioned medium was collected and centrifuged at 2,000 g for 10 min at 4°C to remove cell debris. The resulting supernatant was used for EV isolation, which is described in the “EV isolation and analysis” section.
IFNγ concentrations were determined using Mouse IFN-gamma DuoSet ELISA (R&D Systems, #DY485-05) according to a manufacturer’s protocol. For IL-12 ELISA, half area 96-well plates were coated with capture antibody (Purified Rat Anti-Mouse IL-12 p40/p70; BD Biosciences, #551219) in PBS overnight at 4°C. Plates were washed and then blocked for 3 hr at room temperature with blocking buffer (1% BSA in PBS), followed by wash and incubation with culture supernatants overnight at 4°C. Plates were then washed and incubated with biotinylated detection antibody (Biotin Rat Anti-Mouse IL-12 (p40/p70; BD Biosciences, #554476) in blocking buffer at room temperature for 1 hr, followed by wash and incubation with streptavidin–HRP conjugate for 20 min. Plates were washed and incubated with TMB substrate (KPL, #95059-286), and then read at 450 nm and 650 nm after stopping the development with 1 M phosphoric acid. Concentrations were determined by comparison to a standard curve.
EV isolation and analysis
B16-OVA cells were seeded in 150 mm culture plate and incubated in DMEM supplemented with 10% FBS for 24 hr at 37°C to allow cell attachment. The cells were then washed with PBS twice, and culture medium was switched to 35 mL of DMEM without serum. After incubation for 48 hr, conditioned medium was collected and centrifuged at 2,000 g for 10 min at 4°C to thoroughly remove cell debris. The resulting supernatant was then filtered through a 0.22 μm PVDF filter (Millipore, #SLGV033RB) to remove cell debris and microvesicles (for the detergent treatment experiment, the resulting flow-through was treated with 1% Triton X-100 for 10 min at 4°C prior to the ultracentrifugation). The flow-through was transferred into ultracentrifuge tubes (BECKMAN COULTER, #344058) and then ultracentrifuged in a Beckman SW32Ti rotor at 30,000 rpm for 90 min at 4°C. The resulting pellets were washed with 35 mL of PBS and then ultracentrifuged again at 30,000 rpm for 90 min at 4°C. The resulting EV pellets were re-suspended in PBS for experimental use. Protein concentrations of EVs were determined using Micro BCA Protein Assay Kit (Thermo, #23235). RNA in EVs was isolated using TRIzol reagents (Thermo, #15596026) according to the manufacturer’s protocol and concentrations were determined using Agilent 2200 TapeStation (Agilent Technologies). For ribonuclease treatment, total RNA isolated from EVs was digested for 30 min at 37°C with 100 μg/ml RNase A (Thermo, #EN0531) in a buffer comprising 10 mM Tris-HCl (pH 7.5), 5 mM EDTA, 300 mM NaCl. RNA was then resolved in agarose gels in non-denaturing conditions. Nanoparticle tracking analysis was performed using NanoSight NS300 system (Malvern Instruments, Ranch Cucamonga, CA, USA) on isolated EVs diluted 5,000-fold with PBS for analysis.
LC-MS/MS
EV samples were resolved in SDS-PAGE and the gels were cut into three regions, and then digested with trypsin. Extracted peptides were analyzed using a C18 column and an EASY-nLC-1000 (Thermo Scientific) coupled to a hybrid quadrupole-orbitrap Q-Exactive mass spectrometer (Thermo Scientific). A data-dependent, top 50 method was utilized for analysis. The resulting. RAW files were analyzed with Proteome Discoverer 1.4 and MASCOT. Results were filtered with 1% FDR at the protein level and exported to our in-house FileMakerPro database iSPEC and analyzed with Align!, which calculated intensity based absolute quantification (iBAQ) values (Schwanhäusser et al., 2011) that were used for subsequent analysis. The ratio of the iBAQ values for WT and LATS1/2 dKO EVs (DKO/WT ratio) in the individual experiments was calculated and scored according to the following criteria: score 2, > 5-fold; score 1, 5- to 2-fold; score 0, 2- to 0.5-fold; score 1, 0.5- to 0.2-fold; and score 2, < 0.2-fold. We then added each scores from the individual experiments and set the threshold as a score of > 3 for the top 100 most significantly increased proteins in LATS1/2 dKO EVs. Gene Ontology (GO) analysis was done using the PANTHER program (Mi et al., 2013). Heatmaps were generated using NetWalker (Komurov et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 5 software (GraphPad Software, Inc, La Jolla, CA, USA). Statistical parameters and methods are reported in the Figures and the Figure Legends. A value of p < 0.05 was considered statistically significant. Epidemiological data are obtained using the PrognoScan database (Mizuno et al., 2009). Association of gene expression with the survival of patients was evaluated using log-rank test and a value of p < 0.05 was considered statistically significant.
DATA AND SOFTWARE AVAILABILITY
Data Resources
Protein contents of the EVs comparing WT and LATS1/2 dKO B16-OVA cells: Table S1, related to Figure S6C. Patient epidemiological data obtained from the PrognoScan database: Table S2.
Supplementary Material
Highlights.
LATS1/2 deletion in tumor cells inhibits tumor growth in vivo
Loss of LATS1/2 induces anti-tumor immune responses
Extracellular vesicles from LATS1/2-null tumors stimulate the host TLRs-IFN pathway
LATS1/2 deletion enhances tumor vaccine efficacy
Acknowledgments
We thank Kimberly C. Lin for critical reading of this manuscript; Shiyin Yao, Fumi Kaneko, Alast Ahmadiiveli, Tadashi Hosoya, Julissa Trahan, Baodong Qin, Le Yu, Min Luo, and Tuomas Vaisanen for technical assistance; Shizuo Akira, Bruce Beutler, Richard A. Flavell, Maripat Corr, Russell Vance, and Michael J. Bevan for mice; John Lee for SCC7 cells; Yang-Xin Fu, Wenxian Fu, Hyun Woo Park, Zhipeng Meng, David A. Cheresh, and Laetitia Seguin for discussion; and Emi Kosano for illustrating the mouse cartoon in Figure 4. This work was supported by grants from the NIH (EY22611, CA196878, and GM51586 to K.-L.G.; HHSN272201400051C to D.A.C.) and the Whitworth Immunotherapy Foundation (to D.A.C. and T.H.). K.-L.G. is a co-founder of Vivace Therapeutics.
Footnotes
AUTHOR CONTRIBUTIONS
T.M. and K.-L.G. designed the study, analyzed the data, and wrote the manuscript; T.H. and D.A.C. designed the animal experiments and provided technical and intellectual support; T.M. performed the experiments and analyzed the data with assistance from T.H., W.-W.P., Y.F., and M.V.H.; Y.F. performed nanoparticle tracking analysis of EVs; M.V.H. and J.Q. performed the mass spectrometry analysis of EVs. All authors discussed the results and commented on the manuscript.
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2016.11.005.
References
- Barry ER, Morikawa T, Butler BL, Shrestha K, de la Rosa R, Yan KS, Fuchs CS, Magness ST, Smits R, Ogino S, et al. Restriction of intestinal stem cell expansion and the regenerative response by YAP. Nature. 2013;493:106–110. doi: 10.1038/nature11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camargo FD, Gokhale S, Johnnidis JB, Fu D, Bell GW, Jaenisch R, Brummelkamp TR. YAP1 increases organ size and expands undifferentiated progenitor cells. Curr Biol. 2007;17:2054–2060. doi: 10.1016/j.cub.2007.10.039. [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2015;162:974–986. doi: 10.1016/j.cell.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri A, et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med. 2014;20:599–606. doi: 10.1038/nm.3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewey EB, Sanchez D, Johnston CA. Warts phosphorylates mud to promote pins-mediated mitotic spindle orientation in Drosophila, independent of Yorkie. Curr Biol. 2015;25:2751–2762. doi: 10.1016/j.cub.2015.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong J, Feldmann G, Huang J, Wu S, Zhang N, Comerford SA, Gayyed MF, Anders RA, Maitra A, Pan D. Elucidation of a universal size-control mechanism in Drosophila and mammals. Cell. 2007;130:1120–1133. doi: 10.1016/j.cell.2007.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff G. Experimental mouse tumour models: what can be learnt about human cancer immunology? Nat Rev Immunol. 2011;12:61–66. doi: 10.1038/nri3129. [DOI] [PubMed] [Google Scholar]
- Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol. 2013;34:67–73. doi: 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013;14:1014–1022. doi: 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hansen CG, Moroishi T, Guan KL. YAP and TAZ: a nexus for Hippo signaling and beyond. Trends Cell Biol. 2015;25:499–513. doi: 10.1016/j.tcb.2015.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer. 2013;13:246–257. doi: 10.1038/nrc3458. [DOI] [PubMed] [Google Scholar]
- Johnson R, Halder G. The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nat Rev Drug Discov. 2014;13:63–79. doi: 10.1038/nrd4161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junt T, Barchet W. Translating nucleic acid-sensing pathways into therapies. Nat Rev Immunol. 2015;15:529–544. doi: 10.1038/nri3875. [DOI] [PubMed] [Google Scholar]
- Keder A, Rives-Quinto N, Aerne BL, Franco M, Tapon N, Carmena A. The hippo pathway core cassette regulates asymmetric cell division. Curr Biol. 2015;25:2739–2750. doi: 10.1016/j.cub.2015.08.064. [DOI] [PubMed] [Google Scholar]
- Komurov K, Dursun S, Erdin S, Ram PT. NetWalker: a contextual network analysis tool for functional genomics. BMC Genomics. 2012;13:282. doi: 10.1186/1471-2164-13-282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei ZG, Ren XH, Wang SS, Liang XH, Tang YL. Immunocompromised and immunocompetent mouse models for head and neck squamous cell carcinoma. Onco Targets Ther. 2016;9:545–555. doi: 10.2147/OTT.S95633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Zheng Y, Yin F, Yu J, Silverman N, Pan D. Toll receptor-mediated Hippo signaling controls innate immunity in Drosophila. Cell. 2016;164:406–419. doi: 10.1016/j.cell.2015.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Mottier-Pavie V, Plouffe SW, Hansen CG, Hong AW, Park HW, Mo JS, Lu W, Lu S, et al. MAP4K family kinases act in parallel to MST1/2 to activate LATS1/2 in the Hippo pathway. Nat Commun. 2015;6:8357. doi: 10.1038/ncomms9357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Z, Moroishi T, Guan KL. Mechanisms of Hippo pathway regulation. Genes Dev. 2016;30:1–17. doi: 10.1101/gad.274027.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013;8:1551–1566. doi: 10.1038/nprot.2013.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Kitada K, Nakai K, Sarai A. PrognoScan: a new database for meta-analysis of the prognostic value of genes. BMC Med Genomics. 2009;2:18. doi: 10.1186/1755-8794-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori M, Triboulet R, Mohseni M, Schlegelmilch K, Shrestha K, Camargo FD, Gregory RI. Hippo signaling regulates microprocessor and links cell-density-dependent miRNA biogenesis to cancer. Cell. 2014;156:893–906. doi: 10.1016/j.cell.2013.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Hansen CG, Guan KL. The emerging roles of YAP and TAZ in cancer. Nat Rev Cancer. 2015a;15:73–79. doi: 10.1038/nrc3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moroishi T, Park HW, Qin B, Chen Q, Meng Z, Plouffe SW, Taniguchi K, Yu FX, Karin M, Pan D, Guan KL. A YAP/TAZ-induced feedback mechanism regulates Hippo pathway homeostasis. Genes Dev. 2015b;29:1271–1284. doi: 10.1101/gad.262816.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya IM, Halder G. Discovering the Hippo pathway protein-protein interactome. Cell Res. 2014;24:137–138. doi: 10.1038/cr.2014.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowell CS, Odermatt PD, Azzolin L, Hohnel S, Wagner EF, Fantner GE, Lutolf MP, Barrandon Y, Piccolo S, Radtke F. Chronic inflammation imposes aberrant cell fate in regenerating epithelia through me-chanotransduction. Nat Cell Biol. 2016;18:168–180. doi: 10.1038/ncb3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pico de Coaña Y, Choudhury A, Kiessling R. Checkpoint blockade for cancer therapy: revitalizing a suppressed immune system. Trends Mol Med. 2015;21:482–491. doi: 10.1016/j.molmed.2015.05.005. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robbins PD, Morelli AE. Regulation of immune responses by extracellular vesicles. Nat Rev Immunol. 2014;14:195–208. doi: 10.1038/nri3622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Senovilla L, Vitale I, Martins I, Tailler M, Pailleret C, Michaud M, Galluzzi L, Adjemian S, Kepp O, Niso-Santano M, et al. An immunosurveillance mechanism controls cancer cell ploidy. Science. 2012;337:1678–1684. doi: 10.1126/science.1224922. [DOI] [PubMed] [Google Scholar]
- Sharma P, Allison JP. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sistigu A, Yamazaki T, Vacchelli E, Chaba K, Enot DP, Adam J, Vitale I, Goubar A, Baracco EE, Remédios C, et al. Cancer cell-autonomous contribution of type I interferon signaling to the efficacy of chemotherapy. Nat Med. 2014;20:1301–1309. doi: 10.1038/nm.3708. [DOI] [PubMed] [Google Scholar]
- Taniguchi K, Wu LW, Grivennikov SI, de Jong PR, Lian I, Yu FX, Wang K, Ho SB, Boland BS, Chang JT, et al. A gp130-Src-YAP module links inflammation to epithelial regeneration. Nature. 2015;519:57–62. doi: 10.1038/nature14228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Du YC, Zhou XJ, Liu H, Tang SC. The dual functions of YAP-1 to promote and inhibit cell growth in human malignancy. Cancer Metastasis Rev. 2014;33:173–181. doi: 10.1007/s10555-013-9463-3. [DOI] [PubMed] [Google Scholar]
- Yáñez-Mó M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, Buzas K, Casal E, Cappello F, Carvalho J, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. 2015;4:27066. doi: 10.3402/jev.v4.27066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell. 2015;163:811–828. doi: 10.1016/j.cell.2015.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Wei X, Li W, Udan RS, Yang Q, Kim J, Xie J, Ikenoue T, Yu J, Li L, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes Dev. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang CY, Chinnaiyan AM, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes Dev. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, Bardeesy N. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.