

Abstract
In an effort to discover potent camptothecin-derived antitumor agents, novel camptothecin analogues with sulfonylpiperazinyl motifs at position-7 were designed and synthesized. They were evaluated for in vitro cytotoxicity with the sulforhodamine-B (SRB) method in five types of human tumor cell lines, A-549, MDA-MB-231, KB, KB-VIN and MCF-7. With IC50 values in the low μM to nM level, most of the new analogues showed greater cytotoxicity activity than the reference compounds irinotecan and topotecan. Furthermore, compounds 12l (IC50, 1.2 nM) and 12k (IC50, 20.2 nM) displayed the highest cytotoxicity against the multidrug-resistant (MDR) KB-VIN cell line and merit further development as preclinical drug candidates for treating cancer, including MDR phenotype. Our study suggested that integration of sulfonylpiperazinyl motifs into position-7 of camptothecin is an effective strategy for discovering new potent cytotoxic camptothecin derivatives.
Keywords: Camptothecin, Cytotoxic activity, Sulfonylpiperazine, Synthesis
Graphical Abstract
Camptothecin (CPT, 1), a natural quinoline alkaloid isolated from the Chinese tree Camptotheca acuminata, has shown significant antitumor activity against various cancers via inhibition of topoisomerase I.1–3 Extensive structural modifications on the C-7-, 9- or 10-position of 1 led to the successful identification and development of the antitumor drugs topotecan (2) and irinotecan (3), as well as several other analogs that are currently in clinical trials. Particularly, the introduction of lipophilic substituents at the 7-position provides favorable molecular interactions and improved pharmacological features that could have potential therapeutic advantages. Through C-7 modification, some analogues were found to exhibit superior pharmacological properties to 1,4–15 and several newer-generation clinical candidates, including belotecan (CKD-602, 4), gimatecan (5), BNP-1350 (6), lurtotecan (7), sinotecan (8) and DB-67 (9) emerged as alternatives to overcome the drawbacks of 1.16–21 Overall, the excellent activity profiles of these agents, including improved water solubility, cytotoxic activity, drug resistance profiles, and antitumor spectra, suggested this compound class could be optimized through rational C-7 modification. A binding model of 1 with biological macromolecules also further demonstrated that the C-7 molecular area could accommodate considerable structural diversity.22,23
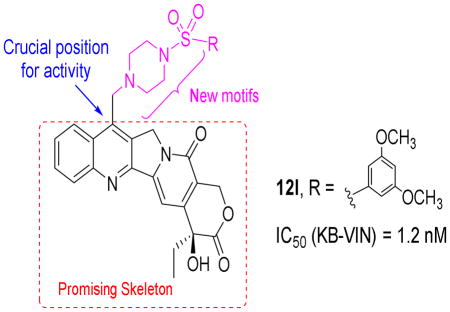
In our continuing studies on the chemistry of 1, we recently reported a series of 7-(N-substituted-methyl)-camptothecin derivatives that displayed potent cytotoxic activity with significantly different drug-resistance profiles from those of 1.24 Some compounds exhibited promising cytotoxicity against the KB-VIN drug resistant tumor cell line, while irinotecan lost activity completely. The encouraging preliminary results have prompted us to extend our investigation by synthesizing a novel series of 7-(N-[(substituted-sulfonyl)piperazinyl]-methyl)-CPT derivatives. Pharmacophore merging is an efficient well-known strategy that has been applied widely in drug design and discovery. Merging different pharmacophores, which might have different mechanisms of action and targets, into one molecule may lead to a new agent with enhanced efficacy and the ability to conquer resistance to the parent drug.25 Furthermore, a piperazine group is commonly found in various drugs26 and its introduction can contribute to improved drug-like properties, such as bioavailability and metabolism. Also, the introduction of a bioactive sulfonyl group into a heterocyclic skeleton results in significant changes in the bioactivity of the compounds.27 Thus, sulfonyl and piperazinyl groups may be useful structural motifs for optimization of the scaffold of various bioactive molecules. Given these considerations, in this paper, we proposed to incorporate the two functional fragments, sulfonyl and piperazine, into 1 at the C-7 position and synthesized a novel series of 1-derivatives.
The synthetic route to target compounds 12a-p is outlined in Scheme 1. Firstly, 7-hydroxymethylcamptothecin (10) was obtained by Minisci free radical reaction of 1 with hydrogen peroxide and ferrous sulfate in an aqueous methanol-sulfuric acid solution in 80% yield28. Subsequently, precursor 10 was converted into the key intermediate 7-bromomethylcamptothecin (11) in 66% yield by heating in hydrobromic acid.29 Intermediate 11 was coupled with various substituted sulfonylpiperazines in dry DMF to produce the target compounds 12a-p in 19–45% yields.30
Scheme 1.

General synthetic procedure for target compounds 12a-p.
The new analogs 12a-p were evaluated for in vitro cytotoxicity against five human cancer cell lines (A-549, MDA-MB-231, KB, KB-VIN and MCF-7) using a sulforhodamine B colorimetric assay.31,32 Compounds 1–3 were used as reference compounds. The results of the cytotoxicity studies are shown in Table 1.
Table 1.
In vitro cytotoxicity data for 12a-p against five tumor cell lines.
| Entry | IC50 (nM) | ||||
|---|---|---|---|---|---|
|
| |||||
| A-549 | MDA-MB-231 | KB | KB-VIN | MCF-7 | |
| 12a | 7.22±1.77 | 336±15.4 | 78.6±7.4 | 141±38.8 | 71.5±17.2 |
| 12b | 4.01±0.43 | 134±13.7 | 8.5±3.0 | 203±52.5 | 74.9±10.1 |
| 12c | 0.25±0.02 | 46.4±4.1 | 2.0±0.2 | 70.4±0.4 | 4.9±0.6 |
| 12d | 0.89±0.12 | 84.2±17.6 | 84.5±50.8 | 34.6±11.2 | 18.2±5.4 |
| 12e | 0.65±0.18 | 105±16.8 | 10.9±4.1 | 126±14.2 | 61.8±0.7 |
| 12f | 4.37±0.54 | 122±17.1 | 15.9±7.2 | 81.4±7.1 | 67.6±5.7 |
| 12g | 27.4±8.53 | 552±41.3 | 195±32.4 | 416±150.0 | 362±86.4 |
| 12h | 29.7±5.82 | 350±20.6 | 55.5±8.9 | 481±64.4 | 96.2±2.2 |
| 12i | 5.36±0.17 | 124±23.3 | 20.7±5.1 | 142±30.3 | 70.9±14.1 |
| 12j | 0.50±0.19 | 62.8±5.2 | 76.3±34.4 | 25.7±2.6 | 37.6±4.4 |
| 12k | 6.92±0.78 | 75.4±10.3 | 96.9±47.9 | 20.2±1.0 | 34.5±7.6 |
| 12l | 1.42±0.19 | 6.4±0.2 | 2.9±1.1 | 1.2±0.1 | 34.0±4.2 |
| 12m | 13.0±4.10 | 113±11.8 | 44.2±8.6 | 165±29.8 | 81.3±17.0 |
| 12n | 0.70±0.03 | 82.6±21.6 | 21.0±5.6 | 266±104.9 | 56.3±14.4 |
| 12o | 21.9±10.90 | 106±14.3 | 67.8±17.2 | 327±135.3 | 61.0±7.2 |
| 12p | 0.21±0.29 | 30.5±10.6 | 71.3±0.7 | 57.5±8.0 | 8.0±2.6 |
| 1 | 8.63±0.66 | 307±118.0 | 50.0±9.7 | 15.7±1.1 | 87.5±6.4 |
| 2 | 8,300±280 | 15,600±302 | 7,990±312 | >20,000 | 11,300±249 |
| 3 | 112±28.44 | 773±36.0 | 102±8.1 | 287±25.3 | 666±26.7 |
As shown in Table 1, all of the new compounds exhibited more potent in vitro cytotoxic activity against five tested tumor cell lines than 2, a clinically used 1-derived chemotherapeutic drug, and most of the new derivatives showed comparable or superior potency to 3. The IC50 values of the new derivatives spanned a broad range from 0.21 to 481 nM, equivalent or superior compared with those of the parent compound 1. Remarkably, all of compounds were more potent than 2 (IC50 >20,000 nM) against the MDR KB-VIN cell line, with 12l (IC50 1.2 nM) and 12k (IC50 20.2 nM) showing the greatest cytotoxicity against this cell line. All of target compounds also showed increased cytotoxic potency against the triple-negative breast cancer (MDA-MB-231) cell line compared with 2 or 3. This result indicated that the introduction of a sulfonyl-piperazinyl group at C-7 might combat the tumor MDR phenotype caused by P-glycoprotein overexpression.
The IC50 values in Table 1 also revealed that the A-549 cell line was more sensitive than the other four cell lines to these compounds, which is consistent with the clinical behavior of other derivatives of 1.33 From Table 1, phenyl (12c), 4-methylphenyl (12d), 4-ethylphenyl (12e), 4-fluorophenyl (12j), 3,5-difluorophenyl (12n), and 2-thienyl (12p) R groups within the sulfonylpiperazinyl side chain led to significantly enhanced cytotoxicity, as these derivatives displayed IC50 values of less than 1 nM against the A-549 tumor cell line. In comparison, compounds without an aromatic ring directly attached to the sulfonyl group, including 12a (R = NMe2), 12b (R = n-Bu), and 12g (R = Bn) showed reduced cytotoxic activity. In addition, the identity of the aromatic group was important as a sharp drop in potency was also observed against most cell lines, when 2-naphthyl, a bicyclic aryl group, or 2-pyridinyl was introduced rather than phenyl, compare 12m (IC50 13.0–165 nM) and 12o (IC50 21.9–327 nM) to 12c (IC50 0.25–70.4 nM), respectively.
The data also indicated that the size or electronic density of the substituents at C-7 might greatly influence the potency of the new 1-derivatives. Compounds containing phenyl rings with simple electron-donating alkyl substituents, such as 12d (p-CH3) and 12e (p-CH2CH3), exhibited similar and significant cytotoxic activity against A549 cells (IC50 0.89 and 0.65 nM, respectively). Meanwhile, compound 12h with a strongly electron-withdrawing 4-nitro group on the phenyl ring was dramatically less potent (A549 IC50 29.7 nM). However, compound 12j with weakly electron-withdrawing p-fluoro substitution on the phenyl ring was highly active against the tested tumor cells (A549 IC50 0.50 nM) and much more potent than 12k with p-chloro substitution (A549 IC50 6.92 nM), indicating that the substituent’s size, in addition to electronic characteristics, could affect the activity. Compound 12l, with a 3,5-dimethoxyphenyl R group, exhibited much better activity than compound 12i, with a 4-methoxyphenyl R group, again indicating that steric characteristics are critical.
In summary, we have reported the synthesis and cytotoxicity evaluation of a series of new 7-(N-[(substituted-sulfonyl)-piperazinyl]-methyl)-CPT derivatives. Preliminary in vitro cytotoxicity evaluation demonstrated that some of the derivatives showed very potent cytotoxicity from 0.21 to 481 nM and were more potent than 2 and 3. Notably, all of the compounds were more potent than 2 (IC50 >20 μM) against the MDR KB-VIN cell line, with 12l (IC50 1.2 nM) and 12k (IC50 20.2 nM) showing the greatest cytotoxicity against this cell line. Furthermore, SAR study suggested that both aromatic and aliphatic substituents on the sulfonyl-piperazinyl side chain at C-7 can promote potency to some extent, while selected variations of these substituents can greatly affect the activity. These findings support our further optimization of 1 to develop potential anticancer drug candidates, particularly against the MDR phenotype. Continuing studies to substantiate and improve activity profiles are underway in our laboratories and will be reported in due course.
Supplementary Material
Fig. 1.


Structures of 1-derivatives
Acknowledgments
This work was supported financially by the National Natural Science Foundation of China (31371975, 21672092); partial financial support was supplied by the Fundamental Research Funds for the Central Universities (lzujbky-2016-147). Support was also supplied by NIH grant CA177584 from the National Cancer Institute awarded to K.H. Lee. Thanks are also due to the support of Health and Welfare Surcharge of Tobacco Products, China Medical University Hospital Cancer Research Center of Excellence (MOHW103-TD-B-111-03, Taiwan).
Footnotes
Supplementary data, including analytical and spectroscopic data for all target compounds, associated with this article can be found, in the online version, at …….
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Li QY, Zu YG, Shi RZ, Yao LP. Curr Med Chem. 2006;13:2021. doi: 10.2174/092986706777585004. [DOI] [PubMed] [Google Scholar]
- 2.Burke TG. Ann N Y Acad Sci. 1996;803:29. doi: 10.1111/j.1749-6632.1996.tb26373.x. [DOI] [PubMed] [Google Scholar]
- 3.Liu YQ, Li WQ, Morris-Natschke SL, Qian KD, Yang L, Zhu GX, Wu XB, Chen AL, Zhang SY, Nan X, Lee KH. Med Res Rev. 2015;35:753. doi: 10.1002/med.21342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pisano C, De Cesare M, Beretta GL, Zuco V, Pratesi G, Penco S, Vesci L, Foderà R, Ferrara FF, Guglielmi MB, Carminati P, Dallavalle S, Morini G, Merlini L, Orlandi A, Zunino F. Mol Cancer Ther. 2008;7:2051. doi: 10.1158/1535-7163.MCT-08-0266. [DOI] [PubMed] [Google Scholar]
- 5.Dallavalle S, Delsoldato S, Ferrari A, Merlini L, Penco S, Carenini N, Perego P, De Cesare M, Pratesi G, Zunino F. J Med Chem. 2000;43:3963. doi: 10.1021/jm000944z. [DOI] [PubMed] [Google Scholar]
- 6.Dallavalle S, Ferrari A, Merlini L, Penco S, Carenini N, Perego P, De Cesare M, Pratesi G, Zunino F. Bioorg Med Chem Lett. 2001;11:291. doi: 10.1016/s0960-894x(00)00649-1. [DOI] [PubMed] [Google Scholar]
- 7.Wang L, Huang Y, Zhang J, Tong LJ, Chen Y, Lu W, Huang QQ. Bioorg Med Chem Lett. 2014;24:1597. doi: 10.1016/j.bmcl.2014.01.049. [DOI] [PubMed] [Google Scholar]
- 8.Xiao F, Xue YD, Luo Y, Zhang B, Lu W, Yang B. Chin Chem Lett. 2009;20:566. [Google Scholar]
- 9.Bom D, Curran DP, Zhang JS, Zimmer G, Bevins R, Kruszewski S, Howea JN, Bingcanga A, Latusc LJ, Burkea TGJ. Controlled Release. 2001;74:325. doi: 10.1016/s0168-3659(01)00343-1. [DOI] [PubMed] [Google Scholar]
- 10.Niizuma S, Tsukazaki M, Suda H, Murata T, Ohwada J, Ozawa S, Fukuda H, Murasaki C, Kohchi M, Morikami K, Yoshinari K, Endo M, Ura M, Tanimura H, Miyazaki Y, Takasuka T, Kawashima A, Nanba E, Nakano K, Ogawa K, Kobayashi K, Okabe H, Umeda I, Shimma N. Bioorg Med Chem Lett. 2009;19:2018. doi: 10.1016/j.bmcl.2009.02.031. [DOI] [PubMed] [Google Scholar]
- 11.Dallavalle S, Giannini G, Alloatti D, Casati A, Marastoni E, Musso L, Merlini L, Morini G, Penco S, Pisano C, Tinelli S, De Cesare M, Beretta GL, Zunino F. J Med Chem. 2006;49:5177. doi: 10.1021/jm060285b. [DOI] [PubMed] [Google Scholar]
- 12.Wang HK, Liu SY, Hwang KM, Taylorbv G, Lee KH. Bioorg Med Chem Lett. 1994;4:579. [Google Scholar]
- 13.Jew SS, Kim HJ, Kim MG, Roh EY, Cho YS, Kim JK, Cha KH, Lee KK, Han HJ, Choi JY, Lee H. Bioorg Med Chem Lett. 1996;6:845. [Google Scholar]
- 14.Liu YQ, Dai W, Wang CY, Morris-Natschke SL, Zhou XW, Yang L, Yang XM, Li WQ, Lee KH. Bioorg Med Chem Lett. 2012;22:7659. doi: 10.1016/j.bmcl.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 15.Bom D, Curran DP, Kruszewski S, Zimmer SG, Strode JT, Kohlhagen G, Du W, Chavan AJ, Fraley KA, Bingcang AL, Latus LJ, Pommier Y, Burke TG. J Med Chem. 2000;43:3970. doi: 10.1021/jm000144o. [DOI] [PubMed] [Google Scholar]
- 16.Dallavalle S, Ferrari A, Biasotti B, Merlini L, Penco S, Gallo G, Marzi M, Tinti MO, Martinelli R, Pisano C, Carminati P, Carenini N, Beretta G, Perego P, Cesare MD, Pratesi G, Zunino F. J Med Chem. 2001;44:3264. doi: 10.1021/jm0108092. [DOI] [PubMed] [Google Scholar]
- 17.Ahn SK, Choi NS, Jeong BS, Kim KK, Journ DJ, Kim JK. J Heterocycl Chem. 2000;37:1141. [Google Scholar]
- 18.Boven E, Van Hattum AH, Hoogsteen I, Schluper HM, Pinedo HM. Ann NY Acad Sci. 2000;922:175. doi: 10.1111/j.1749-6632.2000.tb07035.x. [DOI] [PubMed] [Google Scholar]
- 19.Yu Y, Zhan Y, Chen Z, Zhang Y, Zhong D. J Chromatog B. 2014;951:62. doi: 10.1016/j.jchromb.2014.01.017. [DOI] [PubMed] [Google Scholar]
- 20.Lerchen HG. Drugs of the Future. 2002;27:869. [Google Scholar]
- 21.Thomas CJ, Rahier NJ, Hecht SM. Bioorg Med Chem. 2004;12:1585. doi: 10.1016/j.bmc.2003.11.036. [DOI] [PubMed] [Google Scholar]
- 22.Fan J, Weinstein JN, Kohn KW, Shi LM, Pommier Y. J Med Chem. 1998;41:2216. doi: 10.1021/jm9605445. [DOI] [PubMed] [Google Scholar]
- 23.Staker BL, Hjerrild K, Feese MD, Behnke CA, Burgin AB, Stewart L. Proc Natl Acad Sci USA. 2002;99:15387. doi: 10.1073/pnas.242259599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao XB, Goto M, Song ZL, Morris-Natschke SL, Zhao Y, Wu D, Yang L, Li SG, Liu YQ, Zhu GX, Wu XB, Lee KH. Bioorg Med Chem Lett. 2014;24:3850. doi: 10.1016/j.bmcl.2014.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hopkins AL. Nat Chem Biol. 2008;4:682. doi: 10.1038/nchembio.118. [DOI] [PubMed] [Google Scholar]
- 26.Shaquiquzzaman M, Verma G, Marella A, Akhter M, Akhtar W, Khan MF, Tasneem S, Alam MM. Eur J Med Chem. 2015;102:487. doi: 10.1016/j.ejmech.2015.07.026. [DOI] [PubMed] [Google Scholar]
- 27.Casini A, Scozzafava A, Mastrolorenzo A, Supuran CT. Curr Cancer Drug Targ. 2002;2:55. doi: 10.2174/1568009023334060. [DOI] [PubMed] [Google Scholar]
- 28.Sawada S, Nokata K, Furuta T, Yokokura T, Miyasaka T. Chem Pharm Bull. 1991;39:2574. doi: 10.1248/cpb.39.2574. [DOI] [PubMed] [Google Scholar]
- 29.Synthesis of key intermediate 7-bromomethylcamptothecin (11). To a solution of 7-hydroxymethylcamptothecin (7) (300 mg, 0.80 mmol) in HBr (40%, 40 mL), 98% H2SO4 (0.1 mL) was added and the mixture was heated at reflux for 16 h. After completion of the reaction, the solvent was evaporated under vacuum and the residue recrystallized from MeOH to provide 7-bromomethylcamptothecin (8) as a light brown solid (234 mg, 66% yield). 1H NMR (400 MHz, DMS0-d6) δ: 0.87 (t, J=7.2Hz, 3H, 19-H), 1.84–1.90 (m, 2H, 18-H), 5.26 (s, 2H, -CH2-), 5.28 (s, 2H, 5-H), 5.45 (s, 2H, 17-H), 6.50 (s, 1H, 20-OH), 7.33 (s, 1H, 14-H), 7.76 (t, J=7.2Hz, 1H, 11-H), 7.89 (t, J=7.2Hz, 1H, 10-H), 8.21 (d, J=8.4Hz, 1H, 12-H), 8.42 (d, J=8.4Hz, 1H, 9-H); MS-ESI m/z: 441.4 [M+H]+.
- 30.General synthetic procedure for target compounds 12a–p. To a solution of 7-bromomethylcamptothecin (0.1 mmol) in dry DMF (10 mL), the appropriate sulfonylpiperazine (0.15 mmol) dissolved in DMF (3 mL) was added and the mixture was stirred at rt. After the reaction was completed, the mixture was evaporated to dryness and the residue was purified by chromatography on silica gel using CHCl3/MeOH as eluant to give target compounds 12a–p. Representative analytical and spectroscopic data of 7-(N-[(4-methylphenylsulfonyl)piperazinyl]-methyl)-(20S)-camptothecin (12a): Yield 28%; m.p. 245–247°C; 1H NMR (400MHz, DMS0-d6) δ: 0.87 (t, J=7.2Hz, 3H, 18-H), 1.84–1.89 (m, 2H, 19-H), 2.39 (s, 3H, -CH3), 2.59 (m, 4H, piperazine-H), 2.73–2.89 (m, 4H, piperazine-H), 4.11 (s, 2H, -CH2-), 5.28 (s, 2H, 5-H), 5.42 (s, 2H, 17-H), 6.54 (s, 1H, 20-OH), 7.32 (s, 1H, 14-H), 7.42 (d, J=8.0Hz, 2H, Ar-H), 7.58 (d, J=8.0Hz, 2H, Ar-H), 7.68 (t, J=8.0Hz, 1H, 10-H), 7.82 (t, J=7.6Hz, 1H, 11-H), 8.15 (d, J=8.4Hz, 1H, 12-H), 8.37 (d, J=8.4Hz, 1H, 9-H), MS-ESI m/z: 601.1 [M+H].
- 31.Skehan P, Storeng R, Scudiero D, Monks A, McMahon J, Vistica D, Warren JT, Bokesch H, Kenney S, Boyd MR. J Natl Cancer Inst. 1990;82:1107. doi: 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- 32.Cytotoxic activity was determined by the sulforhodamine B (SRB) colorimetric assay as previously described.31 In brief, the cells (3–5 × 103 cells/well) were seeded in 96-well plates filled with RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) containing various concentrations of samples, and incubated for 72 h. At the end of the exposure period, the attached cells were fixed with cold 50% trichloroacetic acid for 30 min followed by staining with 0.04% SRB (Sigma Chemical Co.) for 30 min. The bound SRB was solubilized in 10 mM Tris-base and the absorbance was measured at 515 nm on a Microplate Reader ELx800 (Bio-Tek Instruments, Winooski, VT) with a Gen5 software. All results were representative of three or more experiments and IC50 is expressed as the average with standard deviation (SD).
- 33.Cao Z, Harris N, Kozielski A, Vardeman D, Stehlin JS, Giovanella B. J Med Chem. 1998;41:31. doi: 10.1021/jm9607562. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.