

SUMMARY
Conformational dynamics plays a fundamental role in molecular recognition and activity in enzymes. The ubiquitin-conjugating enzyme (E2) Ube2g2 functions with the ubiquitin-ligase (E3) gp78 to assemble poly-ubiquitin chains on target substrates. Two domains in gp78, RING and G2BR, bind to two distant regions of Ube2g2, and activate it for Ubiquitin (Ub) transfer. G2BR increases the affinity between the RING and Ube2g2 by fifty-fold, while the RING catalyzes the transfer of Ub from the Ube2g2~Ub conjugate. How G2BR and RING activate Ube2g2 is unclear. In this work, conformational dynamics in Ube2g2 revealed a clear correlation of binding G2BR and RING with the sequential progression towards Ub transfer. The interrelationship of the existence and exchange between ground and excited states leads to a dynamic energy landscape model, in which redistribution of populations contributes to allostery and activation. These findings provide insight into gp78’s modulation of conformational exchange in Ube2g2 to stimulate ubiquitination.
Keywords: Ubiquitin-Conjugating Enzyme, Ube2g2, gp78, CPMG Dynamics, Energy Landscape, Allostery
eTOC
The ubiquitin conjugating enzyme Ube2g2 goes through a sequence of allosteric binding to its cognate ubiquitin ligase gp78, catalysis, and release from gp78 during the ubiquitination reaction. Chakrabarti et. al. reports that gp78 significantly modulates Ube2g2 dynamics and its energy landscape to drive the sequence of binding, catalysis and release.
INTRODUCTION
Ubiquitination is an important cellular mechanism that is involved in almost every aspect of cell signaling, including the endoplasmic reticulum (ER) associated degradation (ERAD). The ERAD pathway prevents accumulation of misfolded or unassembled proteins in the cell (Komander and Rape, 2012). The process of substrate ubiquitination starts by activation of ubiquitin (Ub) by the ubiquitin-activating enzyme (E1) in an ATP-dependent reaction. In the next step, the Ub is conjugated to an active site cysteine of another class of enzymes known as the ubiquitin-conjugating enzymes (E2). Finally, the Ubiquitin-ligase (E3) enzymes transfer Ub from the E2~Ub conjugate to the substrate or at the growing end of a poly-ubiquitin chain on the substrate(Komander and Rape, 2012). Ube2g2 is the cognate E2 of the first human identified ERAD E3 gp78 (Das et al., 2009). Ube2g2 functions along with gp78 to form K48 linkage specific ubiquitin chains that target unfolded proteins for proteasomal degradation (Das et al., 2009; Komander and Rape, 2012). gp78 activates Ube2g2 by interacting via two domains: RING (really interesting new gene, residues A327 to I384) and G2BR (Ube2g2 binding region, residues S574 to K600) (Chen et al., 2006).
The minimum, common catalytic unit of E2 enzymes is known as the UBC fold. A long loop is present between the fourth β-strand (β4) and second α-helix (α2) of the UBC fold, which we will refer to as the β4α2 loop. This loop contains the active site cysteine; C89 in the case of Ube2g2. Three mammalian E2s (Ube2g2, Ube2g1 and Cdc34) have an acidic long extension in the middle of the β4α2 loop (aa 96-108), which is also known as β4α2LL (Petroski and Deshaies, 2005). Adjacent to the β4α2LL is the α2α3 loop and these two loops together control access to the active site. These loops are observed in multiple conformations in the structure of free Ube2g2 (Figure 1A). Interestingly, they form a single conformation when G2BR binds Ube2g2. The β4α2LL and α2α3 loops come together and form a closed conformation that controls access to the active site (Figure 1B). When RING binds to the Ube2g2:G2BR binary complex, the electron density corresponding to the β4α2LL was unobserved in the Ube2g2:RING-G2BR structure (Figure 1C), suggesting dynamic mobility of this loop.
Figure 1. Conformational changes and binding sites in Ube2g2 upon interaction with gp78.

(A) Ube2g2 NMR structures (20 conformers in green, PDB: 2KLY) are superimposed with three structures from the crystal asymmetrical unit of Ube2g2 (yellow, PDB: 2CYX (Arai et al., 2006)). Binary Ube2g2:G2BR complex (B) (PDB: 3H8K) and ternary Ube2g2:RING-G2BR complex (C) (PDB: 4LAD), where Ube2g2 is green and G2BR and RING are light blue. The missing electron density of the loop in (C) is indicated by a dashed line. (D) The regions critical for binding and activity in Ube2g2 are depicted as: ‘backside’ G2BR binding site in orange, RING binding site in blue, and Ub binding in dark red. Regions around the active site are colored in yellow. The active site cysteine side-chain is shown in orange in (A–D).
The G2BR binds to the “backside” β sheet of Ube2g2 with high affinity (Kd= 21nM). Although the G2BR and RING bind at distinct regions in Ube2g2, the G2BR interaction has a positive allosteric effect on the binding of RING to Ube2g2. RING binds Ube2g2 with 144 μM affinity, but it binds to Ube2g2:G2BR with 3 μM affinity (Das et al., 2009). Interestingly, RING has a negative allosteric effect at the G2BR binding site, i.e. the G2BR binding to Ube2g2 becomes weaker when RING binds to Ube2g2, wherein multiple contacts between Ube2g2 and the N terminus of G2BR are disrupted. This is reflected in a lower binding, Kd of 38 nM between Ube2g2 and RING-G2BR compared to Ube2g2:G2BR (Kd = 21nM). The negative allosteric effect was found to be essential for the processivity of gp78 machinery through multiple ubiquitination cycles (Das et al., 2013).
Solution NMR studies of the picosecond-nanosecond (ps-ns) dynamics have been studied in four E2s: Ubc13, UbcH5b, UbcH5c and Ube2g2 (Houben et al., 2004; Ju et al., 2010; Pruneda et al., 2011; Rout et al., 2014; Soss et al., 2013). In UbcH5b and Ubc13, the β4α2 loop lacks the acidic extension found in Ube2g2 and Cdc34. Nevertheless, the ps-ns dynamics of the shorter β4α2 loop were shown to have an effect on the catalytic activity of Ubc13 (Rout et al., 2014). Although individual residues in the UbcH5b β4α2 loop were found to be crucial for its catalytic activity, the possible role of dynamics has not been revealed as yet (Houben et al., 2004). The longer β4α2 loop in Ube2g2 was previously found to be dynamic in the ps-ns timescale (Ju et al., 2010). The role of microsecond-millisecond (μs-ms) dynamics in controlling biological activity has been demonstrated in multiple systems (Boehr et al., 2006; Chakrabarti et al., 2016; Henzler-Wildman et al., 2007; Mulder et al., 2001a; Smith et al., 2016) Tzeng and Kalodimos, 2009). The μs-ms dynamics of E2 enzymes, either in the free form or in complex with partners, have not been explored. Here we report a study of the μs-ms timescale conformational dynamics (Palmer, 2004) in Ube2g2 in the freeform and in complex with two gp78 domains. The experimental data, in combination with all-atom molecular dynamics (MD) simulations, provides a coarse-grained picture of Ube2g2’s energy landscape during the initial phases of the ubiquitination reaction. Previous research has shown that Ube2g2 goes through a sequence of allosteric binding to gp78, transfer of Ub, and release from gp78 during the ubiquitination reaction. The allosteric effects were shown to be equivalent for both Ube2g2 and Ube2g2~Ub, using chemical shift perturbations (Das et al., 2013). The present study reveals that Ube2g2 dynamics is significantly modulated along this pathway, and the population distribution in the dynamic energy landscape drives the sequence of allosteric binding, catalysis and release. In addition, we observed (μs-ms) dynamics in UbcH5b indicating that such modulations of the intrinsic dynamics may be a general event during ubiquitination by other E2:E3 pairs.
RESULTS
G2BR attenuates dynamics in Ube2g2 across the μs-ms timescale
The dynamics in free Ube2g2 were studied by Tolman and co-workers using heteronuclear steady-state 15N-NOE (het-NOE) and residual dipolar coupling (RDC) experiments (Ju et al., 2010). While 15N het-NOE is a measure of ps-ns timescale dynamics, RDCs reflect motion on slower timescales. Ube2g2 was reported to be dynamic in the “backside” β-sheet, the extended region of β4α2 loop, and the α2α3 loop (Figure 1A). The 15N het-NOE for the 15N-Ube2g2:G2BR complex allowed comparison of the dynamics in the G2BR-bound form to that in free Ube2g2, providing information about the relative flexibility of Ube2g2 in the free and bound states. The majority of Ube2g2 residues were rigid on this time scale and had het-NOE values of 0.8 ± 0.05 in both the free and the G2BR-bound form (Figure 2A). However, some distinct regions showed an increase in their het-NOE values when G2BR is bound. Four regions or clusters in Ube2g2 have been shown to interact with gp78 domains and whose interaction is involved in catalysis (Figure 1D) (Das et al., 2009). Residues in cluster 1 are at the backside of Ube2g2 that bind the G2BR domain, including β-strands β1, β2, β3 and the C-terminal end of helix α4. Residues in cluster 2 form the binding site for the RING domain, including the N-terminus of helix α1, the N-terminal and C-terminal regions of β4α2. The active site cysteine (C89) and surrounding region (β4α2 loop and the α2α3 loop) form cluster 3. Residues in cluster 4 interact with the conjugated Ub and include helix α2. The 15N het-NOEs of clusters 1 and 3 were low in free Ube2g2 (Figure 2A). Interestingly, upon binding G2BR their het-NOE values increased (Figure 2A).
Figure 2. G2BR attenuates dynamics in Ube2g2.

(A) Comparison of the 15N Hetero-nuclear NOE for Ube2g2 and Ube2g2:G2BR at 25°C. The secondary structure and clusters of Ube2g2 are shown across the top. The extended loop, β4α2LL, is highlighted by red bars. Error bars reflect propagation of signal-to-noise error. (B) Comparison of Cα RMSF values (mean ± SEM, n = 3) observed in Ube2g2 and Ube2g2:G2BR from three 100ns MD simulations. (C) Residual dipolar couplings (RDCs) in Ube2g2 (Ju et al., 2010) and Ube2g2:G2BR plotted against predicted RDC values based on the respective structures. Residues in the dynamic loop are plotted in red, while the rest are plotted in black. See also Figure S4 and Table S1.
Similar information about dynamics can also be obtained from all-atom molecular dynamics (MD) simulations (Figure 2B). A MD simulation with a well calibrated force-field can provide detailed information about the different conformational states and their populations, if the states are sufficiently sampled within the simulation time. Three independent 100 nanosecond simulations were run on the free Ube2g2 and the Ube2g2:G2BR binary complex (Table S1 available online). The averaged Cαroot-mean-square-fluctuations (RMSF) observed in Ube2g2 in the free and binary complex forms are shown in Figure 2B. Overall, there is a general agreement between the heteronuclear NOEs and the MD simulations. The regions of free Ube2g2 with low het-NOE (< 0.7; Figure 2A) have high RMSF (>1 Å; Figure 2B) in the MD trajectory. Several regions in Ube2g2 show a significant reduction in RMSF values upon binding to G2BR. Regions in cluster 1 have lower RMSF, consistent with contacts between this region and G2BR. Surprisingly, the extended acidic region of β4α2 in cluster 3, which is far from the G2BR interface, also exhibits significantly reduced fluctuations (Figure 2B). MD simulations are reasonable approximations of the conformational fluctuations in macromolecules; however, they cannot be exact representations due to uncertainties in force-fields, solvent approximation, finite size effects and limited time sampling. However, good correlations have been reported between the dynamics revealed by MD trajectories and NMR relaxation behavior (Gill et al., 2016; Robustelli et al., 2013). In the present case, we observe different behavior between het-NOE and the MD simulations in the β3 region (residues 50-65) (Figure 2A versus 2B), which may be due to a difference in the timescale of the MD (up to 0.1 μs) and the motions affecting the het-NOE (ps-ns).
Residual dipolar couplings (RDCs) are sensitive to a wider range of motional timescales from ps to ms (Lange et al., 2008). Fluctuations in the angle between the HN-N amide bond and a molecule fixed coordinate system causes dynamic averaging of the RDC values, and any change in the flexibility upon binding G2BR can be assessed by comparing the RDC values in free Ube2g2 and Ube2g2:G2BR. Tolman and coworkers have reported that the dynamics of the β4α2 loop in free Ube2g2 leads to a poor correlation between the experimental RDCs and the back-calculated RDCs for the β4α2loop (Figure 2C) (Ju et al., 2010). RDCs of Ube2g2 were measured and fit to the experimental structure of Ube2g2:G2BR (Figure 2C). The fit shows that the experimental and back-calculated RDCs for the β4α2 loop residues in Ube2g2:G2BR agree well in contrast to the free Ube2g2 (Figure 2C). The RDCs in the β4α2 loop of free Ube2g2 are averaged by motion and have a lower magnitude compared to the core of the protein. In contrast, the magnitudes of RDCs for the β4α2 loop in Ube2g2:G2BR were similar to the core region of the protein, confirming the loss of mobility.
Observing μs-ms dynamics in Ube2g2
CPMG relaxation dispersion (CPMG-RD) experiments can provide more detailed information about the dynamics in the μs-ms timescale. This experiment is unique in providing thermodynamic (from population distribution, pB), kinetic (rate of exchange, kex) and structural information (chemical shifts of the invisible population via the differences in chemical shift, Δω). The 15N-(TROSY) CPMG-RD experiment (Loria et al., 1999a, b) was carried out on [2H, 15N]-labeled Ube2g2, using the C89K variant of Ube2g2, as described previously in our study of the Ube2g2-Ub (E2~Ub) conjugate (Das et al., 2013). The C89K-Ube2g2 had relaxation dispersion profiles very similar to wild-type Ube2g2 (Figure S1) and was subsequently used for all further experiments. The Rex values in Ube2g2 were undetectable at 25°C; however, they were found to increase with a decrease in temperature (Figure S2). Hence, all reported CPMG-RD experiments were performed at 1.5°C. This behavior indicates that the rates of conformational dynamics are faster at 25–37°C, consistent with recent observations made using adiabatic R1ρ and R2ρ experiments (Chao and Byrd, 2016). The strategy of using low temperature, while beneficial to bring the kinetics of exchange, kex, within the CPMG detection window, limits our ability to measure exchange in Ube2g2~Ub due to the increased transverse relaxation rate R2 of this species. However, we have previously established using chemical shift perturbations, which are exquisitely sensitive to chemical environments and the populations of exchanging species, that the allosteric perturbations in the binary and ternary complexes of Ube2g2 upon binding G2BR and RING domains are equivalent to the complexes formed with Ube2g2~Ub (Das et al., 2013). Therefore, we have investigated the dynamic exchange behavior of Ube2g2 as a surrogate for the population redistribution in Ube2g2~Ub upon binding G2BR and RING domains. The 15N relaxation dispersion data of Ube2g2 (Figure 3) indicated that 59 residues exhibit conformational exchange (Supplemental Data Set 1). They are distributed mainly in four regions/clusters in Ube2g2 (Figure 3). RD data were collected at magnetic field strengths corresponding to 850MHz and 700MHz and were fit simultaneously to a two-site exchange model using the Bloch-McConnell equation (Mcconnell, 1958) to determine kex, pB and ΔωN (STAR Methods, Supplemental Data Set 1, Table S2). Forty-five of the residues exhibiting exchange could be clustered based on similarity of exchange parameters kex and pB. If the χ2 value of residues fit globally does not increase by more than 100% of the χ2 value when fit individually, then the set of residues is considered to be experiencing a global exchange (Mulder et al., 2001a). Once the member residues of a cluster were identified, they were fit globally to determine the global kex and pB (for that cluster) and |ΔωN| values, suggesting that residues within each cluster are exchanging between conformations with an approximately common frequency (Figure 3, Table S2). Interestingly, the residues having similar dynamics are localized in the same four clusters identified above. Dispersion profiles from the residues in the different clusters were fit to the Carver-Richards equation (Carver and Richards, 1972) to determine kex for each cluster. The frequency of exchange averaged over the different clusters was kex ~ 3100 s−1 (Figure 3). The chemical shift difference between the two sites, ΔωN, can be obtained from fits of the CPMG data and reflects the different environment/conformation of the two sites. The independent estimation of population and chemical shift differences is most efficient when the conformational exchange is in the intermediate regime of the NMR timescale, which is defined by α ≤ 1.5, where α is the scaling factor described by Palmer and coworkers (Millet et al., 2000). In the case of fast exchange (α > 1.5) (Mulder et al., 2001a), the pB and ΔωN are highly correlated, and cannot be determined with high accuracy. However, for the case of global exchange, the global population derived from a fraction of dispersion profiles, which are in intermediate exchange, can be extrapolated to the residues which are in fast exchange (Mulder et al., 2001a). For Ube2g2, under our experimental conditions, a sufficient number of residues in clusters 1 and 2 satisfy the criteria to determine both pB and ΔωN. We are able to obtain ΔωN values for clusters 3 and 4 (Table S2), which are in fast exchange, but show a similar correlation in comparison with the ΔωN values calculated for clusters 1 and 2 (Figure S3B–D, vide infra). The sign of the ΔωN is obtained from a pair of HMQC and HSQC experiments (Skrynnikov et al., 2002) (Figure S3A), which enables determination of the change in chemical shift ΔδN and the determination of the chemical shift δNB of the minor population species. The likelihood that the minor species represents the closed or open conformation of Ube2g2 seen in the MD trajectories (vide infra) can be assessed by comparing the δNB shifts with the chemical shifts predicted from the conformational snapshots found in the MD trajectories using SHIFTX+ (Han et al., 2011) (Figure S3B–D). When the CPMG-RD experiments were repeated for the [2H, 15N]-Ube2g2:G2BR complex, most residues do not exhibit any conformational exchange, as evidenced by flat dispersion profiles (Figure 3).
Figure 3. Observed μs-ms dynamics in Ube2g2.

Ube2g2 is shown in green ribbon representation on the left (active site C89 shown in orange, ball-and-stick representation). Residues experiencing motion at the same timescale are clustered and in red, stick representation. CPMG relaxation dispersion profiles (squares) and fits (solid line) for a representative residue in each cluster are shown on the right. (A) Y13 for cluster 1, (B) W110 for cluster 2, (C) E104 for cluster 3 and (D) V123 for cluster 4. Dispersion profiles represent both free Ube2g2 (orange) and Ube2g2 in Ube2g2:G2BR (magenta). See also Figures S1, S2, Supplemental Data Set 1 and Table S2.
The presence of μs-ms dynamics in E2 enzymes is not unique to Ube2g2. When CPMG-RD experiments were carried out on UbcH5b at 850MHz, little or no exchange could be detected at 25°C. However, at 1.5°C, 42 resolved residues showed μs-ms dynamics distributed in three clusters (Figure 4). These data suggest that dynamics are present and, at physiological temperatures, occur at rates faster than can be readily sampled by CPMG. The trends and implications become accessible at lower temperatures. Interestingly, these clusters were at regions similar to Ube2g2, except that dynamics was absent in cluster 4, which is the interaction site of conjugated Ub.
Figure 4. Observed μs-ms dynamics in UbcH5b.

(A–D) Relaxation dispersion profiles (blue circles) and fits (solid line) are given as in Figure 3. Clustered residues are shown in red stick representation on the solution structure (yellow) of UbcH5b (PDB: 1W4U). The active site C85 is depicted as in Figure 3. Residues in cluster 4 (colored red) represent residues similar to members of cluster 4 in Ube2g2.
MD simulations indicate that RING binding revives dynamics in Ube2g2:G2BR
To observe the effect of the RING domain in Ube2g2:G2BR complex, we carried out all-atom MD simulations of the Ube2g2:RING-G2BR complex (PDB:4LAD). The crystal structure of the complex lacks electron density in the extended region of β4α2 loop, and the missing region was modeled using Rosetta (Raman et al., 2009; Song et al., 2013). Three lowest-energy Rosetta structures were used as the starting structure in the three independent 85ns trajectories, respectively. In the ternary Ube2g2:RING-G2BR complex, residues 27-32 in the ‘backside’ G2BR binding region (cluster 1) and the β4α2 extended region (cluster 3) showed different mobility compared to the binary Ube2g2:G2BR complex. Intriguingly, the Cα RMSF values of residues 27-32 were similar to free Ube2g2 (Figure 5A). The Cα RMSF values of the β4α2 extension were in between the free Ube2g2 and the binary Ube2g2:G2BR complex (Figure 5A).
Figure 5. Comparison of dynamics for three states of Ube2g2 from MD simulations.

(A) Comparison of per-residue Cα RMSF profiles (mean + SEM, n = 3 trajectories) of Ube2g2 in Ube2g2, Ube2g2:G2BR and Ube2g2:RING-G2BR. Secondary structural elements, extended loop, and clusters are shown as in Figure 2. (B) Populations (fraction of frames) versus the distance between the centroid of the extended loop and C89-Cα are plotted based on trajectories for Ube2g2, Ube2g2:G2BR and Ube2g2:RING-G2BR. The conformation of the active site and the β4α2 loop corresponding to the maximum population is termed ‘open’ for Ube2g2, ‘closed’ for Ube2g2 and partially open ‘p-open’ for Ube2g2:RING-G2BR. (C) Trajectories for the C89Cα-Y103Cα distance in Ube2g2 in free Ube2g2 (orange), Ube2g2:G2BR (magenta) and two runs of Ube2g2:RING-G2BR (cyan and light-cyan). See also Figure S4 and Table S1.
The distance between the Cα-atom of residue C89 and the centroid of the extended β4α2 loop (residues between 97 and 106) was used as a reporter to classify the different conformations of the extended β4α2 loop. When the fractional occupancy of a given structure across the entire trajectory was plotted versus the distance, three predominant conformations of the β4α2 loop are observed in the different states of Ube2g2 (Figure 5B). These conformations are designated as open, partially open(p-open) and closed conformations. A similar reporter is the distance between C89-Cα and Y103-Cα in the individual trajectories (Figure 5C). The majority of structures in the free-Ube2g2 trajectory presented the β4α2 loop in the open conformation, where the C89-Cα atom – centroid distance is > 15Å. In a typical free-Ube2g2 trajectory, the distance between C89-Cα and Y103-Cα is ~20Å (Figure 5C). In the Ube2g2:G2BR complex, the β4α2 loop goes into a closed conformation, which is characterized by a shorter distance of ≤12Å between C89-Cα and the centroid. In a typical Ube2g2:G2BR trajectory, the distance between the C89-Cα and Y103-Cα is always <10Å (Figure 5C).
The β4α2 loop shows interesting dynamics in the Ube2g2:RING-G2BR ternary complex. In one trajectory, the β4α2 loop is in the p-open conformation, where the C89 Cα – Y103 Cα distance remains within 15 ± 2 Å for about 40 ns (Figure 5C). After 40ns, the β4α2 loop closes and remains closed during the rest of the MD trajectory. In two other trajectories, the β4α2 loop remains somewhere in between the open and the closed conformation (Figure 5C and Figure S4). Consequently, the distance C89 Cα – centroid of the β4α2 loop is ~ 15 Å (Figure 5B). Hence, the ternary complex exhibits the loop to be primarily in the p-open conformation, although there are excursions to the open and closed conformation for a considerable fraction of simulation time (Figure 5B and C).
Measurements of μs-ms dynamics in the Ube2g2:RING-G2BR complex
Binding of gp78 RING to the Ube2g2:G2BR complex triggers ubiquitin transfer when Ube2g2 is loaded with ubiquitin. To examine if RING binding impacts the μs-ms timescale dynamics in Ube2g2, CPMG-RD data were collected on [2H,15N]-labeled Ube2g2:RING-G2BR complex at 1.5°C, for two magnetic fields corresponding to 900 MHz and 700 MHz spectrometers. Indeed, the CPMG-RD data indicates that the residues in the four clusters regain significant dynamics in the presence of RING (Figure 6). This revival of dynamics is clear when Figure 6 is compared with the CPMG-RD profiles of the G2BR bound state (Figure 3). The data were simultaneously fit to the two-site exchange model to yield an average exchange rate of kex~3800 s−1 (Supplemental Data Set 2 and Table S3). The ΔωN of residues in clusters 3 and 4 do not correlate with the ΔδN corresponding to exchange between any two known/observable conformations. This is consistent with the MD simulations, which show that residues in clusters 3 and 4 are exchanging between the closed, open and p-open conformations with significant populations distributed across these conformations.
Figure 6. Observed μs-ms dynamics in Ube2g2 in the Ube2g2:RING-G2BR complex.

(A–D) The protein structure and residue coloring is as depicted in Figure 3. The relaxation dispersion profiles for representative residues in each cluster (cyan squares) and fit (solid line) of the data are given as in Figure 3. See also Supplemental Data Set 2 and Table S3.
Mutations in the β4α2 loop extension reduce the ubiquitination activity of Ube2g2
The dynamics data and previous structures of Ube2g2:G2BR and Ube2g2:RING-G2BR complexes indicate that the conformational dynamics of the β4α2 loop could be critical for ubiquitination. Mutations were made at the β4α2 extended region and tested for ubiquitination activity with gp78 as the E3. The mutant Ube2g2-Δ13 was designed by deleting the entire extended region (96-108) of the β4α2 loop, which retains the native fold (Figure S5). Ube2g2-Δ13 showed a drastic reduction in poly-ubiquitination activity compared to the wild-type (Figure 7A), consistent with loss of activity observed upon mutating the acidic residues (Kleiger et al., 2009) or the Ube2g2E108••R379RING salt bridge (Das et al., 2013) in the β4α2 loop. This data serves as a negative control for experiments focused on β4α2loop (M101-Y103) residues.
Figure 7. Functional role and dynamics of the gating loop.

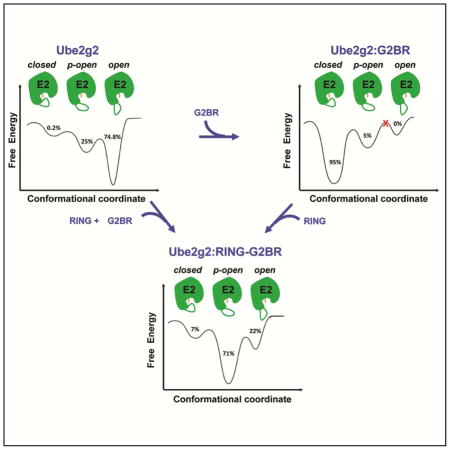
(A) Reduced catalytic efficiency observed as changes in auto-ubiquitination of gp78c for different β4α2LL loop mutants in Ube2g2 compared to wt-Ube2g2. The Ube2g2:G2BR complex is depicted as Ube2g2 (green), G2BR (blue), and residues C89 (active site), M101 and Y103 shown as orange sticks. The Δ13 segment is shown in yellow. (B) ITC measurement of the interaction between G2BR and Y103G-Ube2g2. Kd = 59 ± 6 nM, stoichiometry 1:0.95, ΔH = −39.5 ± 0.4 kCal mol−1, and ΔS = −99.3 ± 0.5 Cal mol−1 K−1. (C) Dynamic energy landscape model of Ube2g2 and the redistribution of population among conformations in different gp78 domain bound states. The different conformations of Ube2g2, marked as either open, partially-open (p-open) or closed, were obtained from MD simulations performed at 298 K (Figure 5B). The MD trajectories sampled the different conformers multiple times within the duration of the simulation. The populations were obtained by binning the frames from MD runs into either the open, p-open or closed conformations. Ube2g2 is shown in green with active site C89 indicated in orange and the β4α2LL loop drawn as a green curve. See also Figure S4.
In the G2BR:Ube2g2 complex, residues M101 and Y103, at the tip of the loop, make several contacts with the core region around the active site to stabilize the loop in the closed conformation (Figure 7A). This conformation facilitates formation of the critical Ube2g2E108••R379RING salt-bridge between the β4α2 loop and the RING. If the closed conformation of β4α2, or interactions which stabilize it, is perturbed by mutagenesis, then the G2BR-induced positive allosteric effects on the Ube2g2E108••R379RING salt-bridge at β4α2 is expected to diminish and reduce activity. In addition to G102 in wt-Ube2g2, M101 and Y103 were mutated to glycine to perturb the closed conformation and to enable maximum conformational flexibility of the loop. Y103G-Ube2g2 showed a significant decrease in activity compared to wild-type Ube2g2 (Figure 7A), although Y103G-Ube2g2 has similar binding affinities for G2BR as the wild-type (Figure 7B). The double mutant M101G, Y103G-Ube2g2 showed a further decrease in activity compared to the single mutant (Figure 7A). These data underscore the importance of the conformational dynamics in the extended region and its role in the poly-ubiquitination activity of the Ube2g2:gp78 machinery.
DISCUSSION
Ubiquitination is a multi-step reaction involving the three classes of enzymes (E1, E2 and E3), Ub and the substrate. Given the multiple events of binding, release, Ub conjugation and Ub transfer steps in the reaction, it is anticipated that the enzymes are dynamic and that conformational dynamics regulate populations of the respective effective species at each step along this process (Liu and Nussinov, 2013). Multiple structural snapshots of E1, in the presence and absence of Ubiquitin, indicate a high degree of conformational motion in E1 (Huang et al., 2007; Huang et al., 2005; Schulman and Harper, 2009; Souphron et al., 2008). Multiple molecular dynamics studies of SCF-E3s indicate the role of functional dynamics in the E3s (Liu and Nussinov, 2009, 2013). For E2s UbcH5c and Ubc13, the ps-ns dynamics of the β4α2 loop near the active site were correlated to its function (Pruneda et al., 2012; Pruneda et al., 2011; Rout et al., 2014). However, the dynamics in these enzymes upon interaction with their reaction partners has not been previously explored, and no studies have addressed the μs-ms timescale, which can be relevant to the recognition of binding partners and may correlate with the Ub transfer step. Using a combined approach of NMR experiments and molecular dynamics, we have explored the dynamics in the ubiquitin E2 conjugating enzyme Ube2g2 when it interacts with two domains of its cognate E3 ligase enzyme gp78, G2BR and RING. These findings have interesting parallels with studies of faster timescale (ps-ns) dynamics in the cases of Ub conjugated UbcH5b (Buetow et al., 2015) and UbcH5c (Pruneda et al., 2011; Soss et al., 2013), which show that modulation of dynamics in the E2~Ub conjugate is relevant for enzymatic activity.
The 15N-het NOE, RDC experiments and MD simulations unambiguously indicated that free Ube2g2 is dynamic and has motions in both the ps-ns and μs-ms timescale regimes. The CPMG-RD experiments observed μs-ms motions not detected by 15N-het NOE and RDC measurements, which is clustered in four distinct regions, each region exhibiting different rates of motion. There seem to be multiple conformations accessed by these clusters, which is clearly visible from MD simulations. Thus, the extracted 15N chemical shifts from fits of CPMG data to a two-site model show only a general agreement with the observed chemical shifts of the known states. The β4α2 loop region experiences extensive dynamics, including the acidic extension β4α2LL, which is exclusive to Ube2g2, Ube2g1 and Cdc34. Interestingly, the combination of NMR experiments and MD simulations indicated a drastic decrease in Ube2g2 dynamics (both ps-ns and μs-ms) upon interaction with the G2BR domain. The 15N-het-NOE values increased, RDC values fit well to the back-predicted values from the core of the protein, the Cα RMSF values decreased, and conformational exchange was not detected in the Ube2g2:G2BR complex. In addition to the G2BR binding region (cluster1), all other clusters experienced a dramatic loss of motion, including the β4α2extension. The correlated quenching implies a connectivity network between different regions of Ube2g2, which are distant in space, and could be essential to form the closed conformation of the β4α2loop and reorientation of E108 to promote formation of the critical salt bridge with R376 in RING (Das et al., 2013).
Similar to Ube2g2, we observe dynamics in the μs-ms timescale in free UbcH5b, which is a close homolog of UbcH5c (97% identity) (Kim et al., 2015). In previous studies, Pruneda et al. and Soss et al. observed that Ub in the UbcH5c~Ub conjugate is exchanging between multiple open conformations (Pruneda et al., 2011; Soss et al., 2013). Upon interaction with RING domain, the conjugates were found to be in a closed conformation where the L8-I44-V70 patch of Ub is packed against the helix α2of the E2 (Pruneda et al., 2012; Soss et al., 2013). In contrast, Ubc13~Ub and Ube2g2~Ub conjugates were found to have a substantial population in a closed conformation, even in the absence of RING domain (Das et al., 2013; Pruneda et al., 2011). Whereas these studies observed the segmental dynamics of Ub in the E2~Ub:E3 complex, the current study observes internal conformational dynamics of E2 that direct interactions in the E2:E3 complex. A comparison between UbcH5b and Ube2g2 shows that the μs-ms dynamics were simultaneously observed in all clusters of the E2s except cluster 4, which is the binding region for ligated Ub. Cluster 4 shows dynamics in Ube2g2 but not in UbcH5b. While Ube2g2 is known to preferably assemble K48-linked poly-ubiquitin chains, UbcH5b can promiscuously assemble different types of chains. Observation of the μs-ms dynamics in two different E2 enzymes with different preferences for lysine linkages indicates that the dynamics observed here could be common to several E2s.
Several non-RING domains of E3s or other co-factors (including Ub) bind to the backside of E2s (Metzger et al., 2014). MD simulations have shown that Ub backside binding reduces the ps-ns dynamics of helix α1 and the loop α1β1 in UbcH5b, facilitating the binding of RING and promoting ubiquitination (Buetow et al., 2015). The affinity between Ub and UbcH5b is weak and UbcH5b lacks the extended region of the β4α2 loop. G2BR has a high affinity for Ube2g2, modulates α1 and α1β1 dynamics, similar to Ubiquitin, but modulates μs-ms dynamics throughout Ube2g2, including the extended region of the β4α2 loop. It is interesting to note how the dynamics in two different E2s is fine-tuned by backside binding to promote function. It is reasonable to postulate that, for several E2s, backside binders such as E3-domains, accessory proteins or Ubiquitin play a vital role in modulating the functional dynamics in the E2s.
In the MD simulations, CαRMSF values of the β4α2LL loop increased in the Ube2g2:RING-G2BR ternary complex compared to the Ube2g2:G2BR binary complex (Figure 5A). Given the general agreement observed between 15N-hetNOE and RMSF values (Figure 2), it is expected that the RING binding would re-introduce motion in Ube2g2:G2BR in the ps-ns motion timescale. In addition, CPMG-RD experiments detected conformational exchange in all the four clusters of Ube2g2 in the ternary complex. Altogether, simulation and experiments indicated that interaction with RING domain revives the μs-ms dynamics in the Ube2g2. The revival is somewhat surprising in cluster 2, where RING binds; however, it suggests that flexibility nearby may be required to facilitate transfer, once the stable RING-bound conformation is established. For example, increased dynamics is observed in the nearby β4α2 loop (cluster 3), α2 helix (cluster 4) and the distant G2BR binding region (cluster 1). This motion suggests that the system is poised for the subsequent mechanistic transfer and release steps. We showed previously that binding of RING domain disrupts several Ube2g2:G2BR contacts in a negative-allosteric manner and the N-terminal region G2BR dissociates from Ube2g2 (Das et al., 2013). The change in dynamics observed in all the clusters is consistent with partial, or initiation of, G2BR dissociation, and foreshadows the release of gp78 from Ube2g2. The locking of dynamics facilitating RING binding has been reported in UbcH5b~Ub (Buetow et al., 2015), while restriction of dynamics for enzymatic activity has been seen in UbcH5c~Ub (Soss et al., 2013). The dynamics in both cases were in the faster ps-ns timescale. The appearance of functionally-relevant μs-ms dynamics playing mechanistic roles in catalysis and binding, upon binding cofactors or ligands, has been seen previously, e.g. the binding of metal atoms in the case of RNase H (Stafford et al., 2013) and the binding of DNA to the catabolite activator protein (Tzeng and Kalodimos, 2009), respectively. These are, to our knowledge, the first such observations in E2:E3 interactions.
The observed μs-ms dynamics of the β4α2 loop in Ube2g2 can be placed in the context of a dynamic energy landscape (Figure 7C) (Frauenfelder et al., 1991). The MD trajectories sampled the ‘open’, ‘p-open’ and ‘closed’ conformers multiple times within the duration of the simulation. The populations depicted in the energy landscape were obtained by binning the frames from MD runs into the relevant conformations. The loop is dynamic in free Ube2g2 and primarily in the open conformation (~ 75 %), with a minor 0.2% population in the closed conformation of the β4α2 loop. Intriguingly, upon G2BR binding, the loop primarily adopts the closed conformation (~ 95 %). In this state, only ~5% of the population has p-open conformation and the completely open conformation is never observed. Our previous study has indicated that the closed form is the result of an G2BR-induced positive allosteric effect, increasing binding to RING and enhanced ubiquitination (Das et al., 2013). Clearly, G2BR also decreases dynamics in Ube2g2 and keeps it in a poised, static mode for efficient RING binding. However, the G2BR-induced closed conformation around the active site is hardly conducive to attack by the substrate lysine and Ub transfer. The RING domain binds to Ube2g2 and catalytically activates it by releasing the β4α2 loop, enabling or facilitating the subsequent lysine attack. These events reintroduce dynamics into β4α2LL, and the region around the active site returns to a partially open conformation (~ 71 %). About 22% of the population is also observed in the complete open conformation. The partially and completely open conformations of β4α2 provide ample space for a substrate lysine side-chain to attack the thioester and transfer the Ub. In the ternary complex, the β4α2 loop accesses all three conformations. These studies of Ube2g2 dynamics did not involve the conjugated Ub. Previously, it was observed that conjugated Ub does not perturb the interactions between Ube2g2 and RING-G2BR (Das et al., 2013), indicating that it may not perturb the gp78 induced dynamics in Ube2g2. The implications of the dynamics studied here are corroborated by functional assays (vide infra), which involve Ub. The conjugated Ub may influence the dynamics in cluster 3 and 4. Future studies are required to elaborate how Ub will fine-tune the μs-ms dynamics in E2:E3 complexes.
The significance of interactions formed by the β4α2 loop was tested in mutational and functional experiments. Mutants were designed to interfere with the normal reaction coordinate of locking a conformation that favors RING binding and stabilizing contact interactions between (i) the extended loop and RING and (ii) the extended loop and the active site region. These mutations drastically reduced the ability to assemble poly-ubiquitin chains (Figure 7A), consistent with our observation that conformations of β4α2 are critical for the activity of Ube2g2. The sequential changes from dynamic to static and back to dynamic conformations for this loop (spanning the states of free, binary and ternary complexes) are tracked by the relaxation dispersion experiments and correlate extremely well with the positive- and negative-allosteric controls of the ubiquitination reaction for gp78 and Ube2g2. In addition, the loop could also play a role in the substrate attack and Ub transfer. In fact, the homologous extended acidic region in Cdc34 has been implicated to boost interaction with its cognate E3 SCF and deprotonate ionizable species at the active site or the acceptor lysine (Sandoval et al., 2015).
A strong correlation was observed between the conformational exchange revealed by CPMG experiments and MD simulations, where there is excellent qualitative agreement despite differences in the timescale of transitions up to an order of magnitude faster for MD simulations compared to CPMG experiments. This correlation has been observed previously between NMR parameters calculated from MD simulations and experimental values (Smith et al., 2016; Xue et al., 2012), and the difference in the quantitative timescale agreement has been suggested to reflect limitations in current force fields and the ability to accurately characterize barriers between conformational states. In conclusion, we have determined that E2 enzymes, particularly Ube2g2, exhibit multiple conformations, which are in exchange with one another on the μs-ms timescale, at different steps along the ubiquitination reaction pathway. The lower population, higher energy and invisible closed-state, is likely to be the conformation that interacts with the binding partner RING to move forward along the reaction coordinate. As the system moves along this reaction coordinate, the dynamics change to suit the next transition. For Ube2g2, the exchange between different functionally relevant conformations is selected by binding to the G2BR and RING domains of gp78. Our current work establishes the connection between the observed positive allosteric effect of binding of different domains of gp78 and the opening/closing of the β4α2 and α2α3 loops in the μs-ms timescale. E3 binding changes the energy landscape of Ube2g2 and redistributes the population. The redistribution of population underlies allosteric effects on binding and activation in E2 enzymes. The functional ubiquitination assays have underlined the importance of relevant μs-ms dynamics in the β4α2 loop and the higher energy conformations of Ube2g2. Given the presence of similar dynamics in UbcH5b, it raises the interesting possibility that this is a common mode of activation of other E2 enzymes. Detailed analysis of the E2:E3 molecular recognition dynamics is essential to understand the basics of ubiquitination.
STAR * Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact: R. Andrew Byrd (byrdra@mail.nih.gov)
Site-directed mutagenesis
The C89K mutant of Ube2g2 was generated using a QuickChange Site-Directed Mutagenesis Kit.
Protein preparation
Ube2g2 was sub-cloned into the pET3a vector. UbcH5b in pET-DUET vector was a gift of Dr. A.M. Weissman. All three proteins, Ube2g2, C89K-Ube2g2 and UbcH5b were expressed and purified by the same protocol. The proteins were over-expressed in BL21 Star cells by induction with 1 mM IPTG at 37°C for 3 hours. Cells were lysed using a Microfluidizer M110S and separated into soluble and insoluble phase by centrifugation. The insoluble phase of cell lysate was solubilized with 4 M urea followed by a step-wise refolding procedure (4 M urea, 2 M urea and 0 M urea in 50 mM Tris HCl, pH 7.2, 2 mM DTT). These proteins were purified from refolded lysate using a Q Sepharose 26/10 column, followed by Superdex 75 column (Das et al., 2009).
The RING–G2BR represents a fusion protein containing the RING and the G2BR domains of gp78 connected by a 40-amino acid linker (Das et al., 2013). The RING–G2BR fusion construct was built into a pET3a vector with a nonspecific linker of 40-amino acids (sequence: GGGGGGGSSGSSGGSGGGSGSSSGGGGGSGGGSGGGGGGG). DNA fragments of RING (aa 313–393) and G2BR (aa 575–600) were PCR-amplified from gp78FL separately; the linker region was inserted by a two-step PCR using synthetic DNA oligos. The RING–G2BR protein was expressed in E. coli BL21 Star cells, grown at 37°C until the OD600 reaches 0.8–1.0. Thereafter, ZnSO4 and IPTG were added up to 50uM and 0.2mM, respectively, and the cell culture was grown overnight at room temperature. The cell pellet was lysed in 50mM Tris, pH 7.0, using Microfluidizer (Model M110S, Microfluidics, Westwood, MA). RING-G2BR protein was solubilized from the lysate pellet with 4M urea in Tris buffer (0.2M L-Arg, 0.5mM TCEP, 100uM ZnSO4, 50mM Tris pH 7.2), refolded and purified with Q Sepharose 26/10 column followed by Superdex 75 column in a similar fashion as described above for Ube2g2.
The G2BR(WSADERQRMLVQRKDELLQQARKRFLNK) peptide was custom synthesized by LifeTein, Somerset, NJ.
Sample preparation for 15N-relaxation dispersion experiments
The uniformly 15N, 2H-labeled Ube2g2 and UbcH5b were expressed in minimal media using 99% 2H2O (deuterium oxide from Cambridge Isotopes Limited) containing 15NH4Cl (Cambridge Isotopes Limited) and 1,2,3,4,5,6,6-D7 (98%) D-glucose (Cambridge Isotopes Limited). Both the Ube2g2 and UbcH5b were purified by refolding from the inclusion body pellet, thereby the labile amide deuterons were fully exchanged. The incorporation of 2H into non-exchangeable positions was generally 90–95%, based on mass spectroscopy. The NMR samples were prepared in 50 mM Tris, pH 7.0, 2 mM TCEP, 5% 2H2O buffer. The protein samples were concentrated using concentrators (Vivaspin from Sartorius Stedim). NMR samples for the 15N relaxation experiments were as follows:
0.7 mM wildtype Ube2g2 (15N, 2H labeled)
0.4 mM Ube2g2 (C89K, 15N, 2H labeled)
0.26 mM Ube2g2 (C89K, 15N, 2H labeled) + 0.3 mM G2BR (unlabeled)
0.4 mM Ube2g2 (C89K, 15N, 2H labeled) + 0.5 mM RING-G2BR (unlabeled).
0.8 mM UbcH5b (15N, 2H labeled)
CPMG 15N-relaxation dispersion experiments
15N R2 relaxation rates were measured in a relaxation-compensated Carr-Purcell-Meiboom-Gill (CPMG) experiment (Loria et al., 1999a,b; Mulder et al., 2001b; Tollinger et al., 2001). The pulse sequence for the CT CPMG 15N-relaxation experiment with TROSY implementation is modified to an interleaved pulse sequence with an RF heat compensation block (Loria et al., 1999a, b). All relaxation data were collected on Bruker Avance 850 MHz (room temperature TXI probe), Bruker Avance 900 MHz (cryoprobe TCI), and Bruker Avance 700 MHz (cryoprobe TCI) spectrometers running Topspin 3.0 (Bruker Biospin Corporation). The NMR samples were 250 μL in Shigemi tubes. The CPMG pulse train was applied during a constant-time delay of 50 ms (for Ube2g2, Ube2g2:G2BR and UbcH5b) with CPMG field strengths varied from 80 Hz to 1280 Hz and 30 ms for Ube2g2:RING-G2BR with CPMG field strengths ranging from 133 Hz to 1200 Hz. The probe temperature was calibrated using a methanol sample.
Reference spectra were collected without the CPMG delay period. The R2,eff was calculated as
Where νCPMG is the effective frequency of the CPMG field (νCPMG = 1/(4τ), where the time between the centers of consecutive 180° pulses is 2τ), T is the constant delay during which CPMG pulses were applied (50 ms or 30 ms), I0 is the intensity of the peak in reference experiment and I(νCPMG) is the intensity of the peak at that particular CPMG frequency. The constant time delay was chosen such that the intensity with maximum CPMG refocusing field has ~50% the intensity of the reference.
Ube2g2 CPMG-RD data were measured at temperatures of 1.5°C, 5°C, 10°C and 15°C and a magnetic field-strength of 850 MHz in order to measure the variation of Rex with temperature. Subsequently, all the Ube2g2 relaxation data for detailed analyses were measured at 1.5°C at 850 MHz (room temperature TXI probe) and at 700 MHz (cryoprobe TCI) for detailed analysis. Ube2g2 bound to G2BR was measured at 850 MHz (room temperature TXI probe). Ube2g2 bound to RING-G2BR was measured at 900 MHz (cryoprobe TCI) and in 700 MHz (cryoprobe TCI) spectrometers. Data on UbcH5b was measured at 850 MHz (room temperature TXI probe).
Relaxation dispersion data were extracted as peak intensities in the two-dimensional NMR spectra as a function of CPMG field strength and analyzed with the generalized Carver-Richards equation for two-site exchange (Carver and Richards, 1972; Korzhnev et al., 2004). The error-bars on individual data points reflect error-propagation of signal-to-noise ratio from duplicate measurement at one CPMG frequency. The kinetic parameters and their uncertainties were calculated using a Monte-Carlo approach by replacing the R2,eff value in the middle of the CPMG frequency range with 100 random values drawn from a normal distribution with the experimental S/N error as the standard deviation (σ) of the distribution (McElheny et al., 2005). The NMR data were processed using NMRpipe/NMRDraw (Delaglio et al., 1995), analyzed and intensity extracted using SPARKY (Goddard and Kneller). All the structures were visualized using Chimera (Pettersen et al., 2004) and PyMOL (Schrödinger, LLC.).
CPMG Data Fitting
The dispersion data from each residue were initially tested for significant amount of relaxation dispersion (> 2 s−1). The residues showing significant amount of relaxation dispersion were tested for statistical significance by fitting each to a horizontal line and to the two-site exchange expression vide infra. Residues for which the F test at the 99% confidence limit showed that the exchange is statistically significant were considered for analysis.
For each residue that displays statistically significant dispersion, the R2 relaxation data at two fields were fit simultaneously to the general Carver-Richards equation for exchange between two sites, A and B (Davis et al., 1994; Palmer et al., 2001):
where
τcp is the delay between CPMG 180° pulses, pA and pB are the populations in states A and B, R2A and R2B are the relaxation rates in sites A and B in absence of exchange (we made the assumption that R2A = R2B = R2(0)(McElheny et al., 2005)), kex is the rate of exchange, and Δω is the chemical shift difference between the two sites.
Initially, the data was fitted individually for each residue by non-linear least squares minimization using Levenberg-Marquardt algorithm implemented by Dmitry Korzhnev (CPMG_fit) (Korzhnev et al., 2004). Upon examination of individual fits it was clear that the exchanging residues were clustered in four clusters based on their relative populations and position on the protein structure. Finally, the residues within the same cluster were fitted globally, i.e., with the same global values of kex and pB, but the individual Δω and R2(0) values were optimized. The quality of fits was ascertained by measuring the ratio of χ2 values between individual fits and the global fits. The ratio of χ2 values between individual fits and global fits never exceeded by a factor of two.
The most significant structural difference between the closed, open and p-open conformations was observed in cluster 3. The β4α2 loop, α2α3 loop and the active site residues (cluster 3) populate the minor conformation to ~ 21 ± 3 % and the kex is 2640 + 270 s−1 (the dynamic parameters for the other clusters are included in Table S2). The kex value is close to the limit of the range of exchanges measurable using CPMG relaxation dispersion experiments employing CPMG refocusing pulse frequency up to ~ 1000 Hz (Kay, 2016).
Prediction of chemical shifts of closed, open and p-open conformations
The chemical shifts for the different states were calculated using SHIFTX+ (Han et al., 2011). The chemical shifts of the closed conformation of Ube2g2 were predicted using a conformation of Ube2g2 taken from the MD trajectory calculated using the crystal structure of Ube2g2:G2BR complex (PDB: 3H8K (Das et al., 2013)). The chemical shifts of the open conformation of Ube2g2 were predicted using a conformation taken from the MD trajectory calculated using the crystal structure of free Ube2g2 (PDB: 2CYX (Arai et al., 2006)). The chemical shifts of the p-open conformation of Ube2g2 were predicted using a conformation from the MD trajectory calculated using the crystal structure of Ube2g2:RING-G2BR (PDB: 4LAD (Das et al., 2013)).
Analyses of chemical shifts of the minor conformation from fits of CPMG profiles (ΔωNCPMG (ppm))
Comparison of the dynamic chemical shift differences (ΔωN) obtained from the fits of CPMG data (including the sign information from a pair of HMQC and HSQC experiments, Figure S3A) and differences of chemical shifts (ΔδN) of different conformations predicted by SHIFTX+ show correlation (Figure S3B–E). The reported RMS in SHIFTX+ predicted δ15N (2.3 ppm) is reflected in the error-bars along y-axis. The major state for clusters 2 and 3 in free Ube2g2 is the open conformation, and the minor state is the p-open conformation (Figures S3C and S3D). Although majority of residues are following the trend (blue points), there are outliers in these dynamic clusters (red points in Figures S3B–E). The major state for cluster 1 is the p-open conformation and the minor state is the open conformation (Figure S3B). In cluster 4 the sign information was not available for most of the residues, so only the magnitude of chemical shifts were analyzed showing that the residues are exchanging between open and closed conformations (Figure S3E). In the main text the closed, open and p-open conformations refer to the conformation of the β4α2 and α2α3 loops in cluster 3.
Determination of chemical shifts of the minor state
The direction of the shift in the15N dimension of the minor state compared to the major peak (the sign of ΔωN) was determined experimentally from a pair of HSQC and HMQC experiment (200 complex points in the 15N dimension) by noting that the HSQC peak is closer to the invisible minor conformation (Skrynnikov et al., 2002). This approach is valid for residues that satisfy the condition |ΔωN| < √3 kb (supplemental table S2). Measurements were made using a 2H, 15N labeled sample of Ube2g2. The correlation between ΔωNCPMG values obtained from relaxation dispersion experiments and ΔδN values obtained from the differences between the open, p-open and closed conformations were calculated using Origin (OriginLab, Northampton, MA).
Measurement of Residual Dipolar Couplings
Residual dipolar couplings (RDCs) were measured on the complex of Ube2g2 (2H, 15N labeled, 200 μM) saturated with G2BR (unlabeled, 400 μM) in 50 mM Tris, 2 mM TCEP, 100 mM NaCl, pH 7.2 and 10% 2H2O buffer containing 10 mg/mL bacteriophage Pf1 sample (ASLA Biotech) (Hansen et al., 1998) at 25°C in 850 MHz. The 1JNH coupling measurements were performed using IPAP-HSQC experiment (Ottiger et al., 1998) with one bond 15N-1H RDCs taken to be the difference between 1JNH measurements in isotropic and aligned samples. We could unambiguously measure RDCs for 60 residues distributed throughout Ube2g2. Back-calculation of RDCs were performed using MODULE-2 (Dosset et al., 2001), as described below.
Refinement of Ube2g2:G2BR structure with RDCs
The crystal structure of Ube2g2:G2BR (PDB: 3H8K) was subjected to refinement against the HN-N RDCs, allowing only minor torsional adjustments. We calculated 1H-1H distance restraints (NOESY like) from Ube2g2:G2BR crystal structure (PDB: 3H8K). These distance-restraints along with experimental RDC restraints were used for structure calculation using a typical annealing protocol in XPLOR-NIH (Schwieters et al., 2003). The resulting structure was very similar to the starting crystal structure, having Cα RMSF of 0.5 Å. We have validated the calculated structure in PDB validation tool and found the structure to be of acceptable stereo-chemical quality (Protein Data Bank Validation Server at http://deposit.pdb.org/validate/).
Fitting RDC data to the calculated structure
The Ube2g2:G2BR structure refined with Ube2g2:G2BR RDCs was used for analyzing the dynamics of the gating loop in Ube2g2:G2BR. The experimental Ube2g2:G2BR RDCs were fitted to the refined Ube2g2:G2BR structure in MODULE-2 (Dosset et al., 2001). The fit between experimental and back-calculated RDCs is excellent for all residues, including residues in the gating loop (in red, right in Figure 2B).
Measurement of 15N het-NOE of Ube2g2:G2BR
Steady state NOE values were measured using standard pulse sequence (Barbato et al., 1992; Kay et al., 1989) at 25°C at 700 MHz with a cryogenic probe. Het-NOE values were calculated as the ratio of peak intensities observed for experiments with and without 3 s of 1H pre-saturation during recycle delay of 5 s on 200 μM Ube2g2 (15N labeled) and 400 μM of G2BR mixture.
Molecular Dynamics simulations
Unbiased MD simulations of Ube2g2 (100 ns x 3), Ube2g2:G2BR (100 ns x 3) and Ube2g2:RING–G2BR (85 ns x 3) were run on GPU clusters using the NAMD package (Phillips et al., 2005). The protein and ions were described with the CHARMM36 force field (Huang and MacKerell, 2013; MacKerell et al., 2004), and water molecules with the TIP3P model. The proteins were solvated in a water box extending 12 Å from the outermost protein atom. The ionic strength of the solvating solution was 150 mM. The simulations were started from experimentally determined X-ray structures and were energy minimized. The energy-minimized structures were allowed to equilibrate for 5 ns before the production runs were started. A time step of 2 fs was used with the bonds involving hydrogen atoms being constrained using the SHAKE algorithm (Ryckaert et al., 1977). Electrostatic interactions were calculated using the PME method (Essmann et al., 1995), and the van der Waals interactions were truncated beyond 12 Å. Periodic boundary conditions were imposed in all directions. The temperature of the systems was controlled at 300 K using the Langevin dynamics and the pressure was kept at 1 atm using the Nose-Hoover Langevin piston method (Feller et al., 1995; Martyna et al., 1994). The MD data were analyzed and the figures were generated using Matlab (The Mathworks Inc.).
Isothermal Titration Calorimetry (ITC)
The ITC experiment was performed using a VP-ITC MicroCalorimeter from MicroCal, Northampton, US. Both proteins were dialyzed against identical buffer containing 50 mM Tris, pH 7.5, 1 mM TCEP. Forty μM G2BR in syringe was titrated into the cell containing 2.5 μM Y103G-Ube2g2 in 30 steps of 10 μL volume each (with 4 minutes delay between injections). The integrated heats of interaction for each step were corrected for the baseline using a blank (buffer into protein) experiment. The data were fit, excluding the first point, using a single site binding model using the data analysis template in Origin (OriginLab, Northampton, US) provided by MicroCal.
In-vitro Ubiquitination Assays
Auto-ubiquitination reactions using wt/mutants ofUbe2g2 and glutathione sepharose-bound glutathione S-transferase (GST) fusions of the entire wt-gp78 cytoplasmic tail (gp78C, aa: 309-643) were carried out as described previously (Lorick et al., 1999). GST-gp78C or its mutants were expressed in bacteria and bound to glutathione-Sepharose beads. One hundred microliter reactions containing 50 nM human E1 (Boston Biochem) and 1 μg ubiquitin (Sigma) in 1× ubiquitination buffer (50 mM Tris-HCl pH 7.4, 0.2 mM ATP, 0.5 mM MgCl2, 0.1 mM DTT, 1 mM phosphocreatine (Sigma)) were assembled on the beads and incubated for 90 minutes at 30°C. Following incubation, beads were washed in 1× TBS and bound material was eluted in SDS-reducing sample buffer. Reaction products were analyzed by SDS-PAGE and anti-ubiquitin (ThermoFisher) immunoblotting.
DATA AND SOFTWARE AVAILABILITY
Data Resources
NMR data were acquired using Topspin software and pulse sequences in the Topspin libraries from Bruker Biospin, and NMR data were processed using NMRPipe. Modifications to pulse sequence are mentioned and cited. Copies of the modified pulse sequences are available from the Lead Contact. Data were analysed using SPARKY for peak picking, integration, and het-NOE. Analysis of peak intensities for determination of relaxation rates, exchange rates, populations and chemical shift differences were performed using Sparky, CPMG_fit and Matlab (sources listed in the Key Resources table). Chemical shift calculations based on protein structure were performed using SHIFTX+, and RDC data were analysed using XPLOR-NIH and MODULE-2. Structure refinements were done as described above using XPLOR-NIH and Rosetta. Molecular dynamics simulations were performed using NAMD and analyses partially assisted using VMD. Visualization and generation of molecular structure figures and images were created using Chimera and PyMOL. All software resources are listed in the Key Resources table, and the use of each package in the data analysis is described in the sub-headings of the Star Methods.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Ub | ThermoFisher | Cat# 710362 |
| Bacterial and Virus Strains | ||
| pET-DUET UbcH5b | Dr. Allan M. Weissman, NCI | N/A |
| pET3a | Novagen | Cat# 69418 |
| BL21 STAR (DE3) | ThermoFisher | Cat# C601003 |
| pET3a-hUbe2g2b | This paper | N/A |
| pET3a-hUbe2g2b-C89K | This paper | N/A |
| pET3a-hRING-G2BR | Das et al., 2013 | N/A |
| Pf1 phage particle suspension | ASLA Biotech | Cat# P-100-P |
| Chemicals, Peptides, and Recombinant Proteins | ||
| G2BR | LifeTein, Somerset,NJ | N/A |
| 15NH4Cl | CIL | Cat# NLM-467-PK |
| D-glucose (1,2,3,4,5,6,6-D7, 98%) | CIL | Cat# DLM-2062-PK |
| Deuterium oxide | CIL | Cat# DLM-6-1000 |
| hE1 Activating | Boston Biochem | Cat# E-305 |
| Ubiquitin | Sigma-Aldrich | Cat# U5382-1MG |
| Phosphocreatine | Sigma-Aldrich | CAS# 19333-65-4 |
| GST-gp78C | Dr. Allan M. Weissman, NCI | N/A |
| Critical Commercial Assays | ||
| QuickChange Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200519 |
| Deposited Data | ||
| CPMG relaxation dispersion profiles of Ube2g2 | Supplementary data sets 1 and 2 | Online with this publication |
| Software and Algorithms | ||
| Topspin | Bruker Corporation | https://www.bruker.com/products/mr/nmr/nmr-software/software/topspin |
| NMRPipe | Delaglio et al., 1995 | https://spin.niddk.nih.gov/NMRPipe/ |
| SPARKY | Goddard and Kneller | https://www.cgl.ucsf.edu/home/sparky/ |
| Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| PyMOL | Schrödinger | https://www.pymol.org/ |
| CPMG relaxation dispersion pulse program for Bruker | Loria et al., 1999a,b and this paper | Bruker library within Topspin software |
| CPMG fitting program CPMG_fit | Korzhnev et al., 2004 | http://www.nmr-relax.com |
| SHIFTX+ | Han et al., 2011 | http://shiftx.wishartlab.com/ |
| IPAP-HSQC pulse program for Bruker | Ottiger et al., 1998 | Bruker library within Topspin software |
| MODULE-2 | Dosset et al., 2001 | http://www.ibs.fr/research/scientific-output/software/module |
| XPLOR-NIH | Schwieters et al., 2003 | https://nmr.cit.nih.gov/xplor-nih/ |
| PDB validation server | Protein Data Bank | http://deposit.rcsb.org/validate/ |
| 15N-hetero NOE pulse program for Bruker | Barbato et al., 1992 and Kay et al., 1989 | Bruker library within Topspin software |
| Rosetta | Raman et al., 2009 and Song et al., 2013 | https://www.rosettacommons.org |
| NAMD | Phillips et al., 2005 | http://www.ks.uiuc.edu/Research/namd/ |
| VMD | Humphrey et. al., 1996 | https://www.ks.uiuc.edu/Research/vmd/ |
| Matlab | The Mathworks Inc. | https://www.mathworks.com/products/matlab.html |
| Origin | OriginLab | http://www.originlab.com/ |
| Other | ||
| Q Sepharose 26/10 column | GE Healthcare | Cat# 17-1066-01 |
| Superdex 75 column | GE healthcare | Cat# 28-9893-33 |
| 10 kDa m.w. cut-off Vivaspin 20 concentrator | Sartorius Stedim | Cat# VS2002 |
| Glutathione Sepharose beads | GE Healthcare | Cat# 17075601 |
| Cell disruptor – Microfluidizer M-110P | Microfluidics, Corp. | Cat# M-110S |
Supplementary Material
Highlights.
Two gp78 domains, G2BR and RING, bind Ube2g2 and work via allosteric mechanisms
Ube2g2 is dynamic in both the ps-ns and ms-ms timescale
G2BR domain binds and attenuates Ube2g2 dynamics to prime it for RING binding
RING domain revives dynamics around the active site to facilitate catalysis
gp78 modulates Ube2g2 energy landscape and drives efficient ubiquitination reaction
Acknowledgments
Jennifer Mariano & Allan Weissman (LPDS/NCI) and Vincenzo Venditti & Jinfa Ying (NIDDKD/NIH) are acknowledged for assistance with the ubiquitination assay and pulse-programs, respectively. This research was supported by the Intramural Research Program of the CCR, NCI, NIH, to RAB and the National Center for Biological Sciences, TIFR & Ramalingaswami fellowship from DBT-India to RD.
Footnotes
Supplemental information includes five figures and three tables and can be found with this article online at XXXXX.
AUTHOR CONTRIBUTIONS
Conceptualization, RD and RAB; Methodology, KSC, JL, and RD; Formal Analysis, KSC. and RD; Investigation, KSC, JL, and RD; Resources, RD and RAB.; Data Curation, KSC; Writing – Original Draft, KSC and RD; Writing – Review & Editing, RD and RAB; Visualization, KSC and RD; Supervision, RD and RAB; Project Administration, RD and RAB; Funding Acquisition, RAB.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Kalyan S. Chakrabarti, Email: kach@nmr.mpibpc.mpg.de.
Jess Li, Email: lije@mail.nih.gov.
Ranabir Das, Email: rana@ncbs.res.in.
R. Andrew Byrd, Email: byrdra@mail.nih.gov.
References
- Arai R, Yoshikawa S, Murayama K, Imai Y, Takahashi R, Shirouzu M, Yokoyama S. Structure of human ubiquitin-conjugating enzyme E2 G2 (UBE2G2/UBC7) Acta crystallogr Sect F, Struct Biol Cryst Commun. 2006;62:330–334. doi: 10.1107/S1744309106009006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbato G, Ikura M, Kay LE, Pastor RW, Bax A. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible. Biochemistry. 1992;31:5269–5278. doi: 10.1021/bi00138a005. [DOI] [PubMed] [Google Scholar]
- Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- Buetow L, Gabrielsen M, Anthony NG, Dou H, Patel A, Aitkenhead H, Sibbet GJ, Smith BO, Huang DT. Activation of a Primed RING E3-E2-Ubiquitin Complex by Non-Covalent Ubiquitin. Mol Cell. 2015;58:297–310. doi: 10.1016/j.molcel.2015.02.017. [DOI] [PubMed] [Google Scholar]
- Carver JP, Richards RE. General 2-Site Solution for Chemical Exchange Produced Dependence of T2 Upon Carr-Purcell Pulse Separation. J Magn Reson. 1972;6:89–105. [Google Scholar]
- Chakrabarti KS, Agafonov RV, Pontiggia F, Otten R, Higgins MK, Schertler GF, Oprian DD, Kern D. Conformational Selection in a Protein-Protein Interaction Revealed by Dynamic Pathway Analysis. Cell Rep. 2016;14:32–42. doi: 10.1016/j.celrep.2015.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao FA, Byrd RA. Geometric Approximation: A New Computational Approach To Characterize Protein Dynamics from NMR Adiabatic Relaxation Dispersion Experiments. J Am Chem Soc. 2016;138:7337–7345. doi: 10.1021/jacs.6b02786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Mariano J, Tsai YC, Chan AH, Cohen M, Weissman AM. The activity of a human endoplasmic reticulum-associated degradation E3, gp78, requires its Cue domain, RING finger, and an E2-binding site. Proc Natl Acad Sci USA. 2006;103:341–346. doi: 10.1073/pnas.0506618103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Liang YH, Mariano J, Li J, Huang T, King A, Tarasov SG, Weissman AM, Ji X, Byrd RA. Allosteric regulation of E2:E3 interactions promote a processive ubiquitination machine. EMBO J. 2013;32:2504–2516. doi: 10.1038/emboj.2013.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Mariano J, Tsai YC, Kalathur RC, Kostova Z, Li J, Tarasov SG, McFeeters RL, Altieri AS, Ji X, et al. Allosteric activation of E2-RING finger-mediated ubiquitylation by a structurally defined specific E2-binding region of gp78. Mol Cell. 2009;34:674–685. doi: 10.1016/j.molcel.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis DG, Perlman ME, London RE. Direct measurements of the dissociation-rate constant for inhibitor-enzyme complexes via the T1 rho and T2 (CPMG) methods. J Magn Reson Ser B. 1994;104:266–275. doi: 10.1006/jmrb.1994.1084. [DOI] [PubMed] [Google Scholar]
- Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- Dosset P, Hus JC, Marion D, Blackledge M. A novel interactive tool for rigid-body modeling of multi-domain macromolecules using residual dipolar couplings. J Biomol NMR. 2001;20:223–231. doi: 10.1023/a:1011206132740. [DOI] [PubMed] [Google Scholar]
- Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A Smooth Particle Mesh Ewald Method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- Feller SE, Zhang YH, Pastor RW, Brooks BR. Constant-Pressure Molecular-Dynamics Simulation - the Langevin Piston Method. J Chem Phys. 1995;103:4613–4621. [Google Scholar]
- Frauenfelder H, Sligar SG, Wolynes PG. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- Gill ML, Byrd RA, Palmer AG. Dynamics of GCN4 facilitate DNA interaction: a model-free analysis of an intrinsically disordered region. Phys Chem Chem Phys. 2016;18:5839–5849. doi: 10.1039/c5cp06197k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard TD, Kneller DG. SPARKY3. University of California; San Francisco: [Google Scholar]
- Han B, Liu Y, Ginzinger SW, Wishart DS. SHIFTX2: significantly improved protein chemical shift prediction. J Biomol NMR. 2011;50:43–57. doi: 10.1007/s10858-011-9478-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen MR, Mueller L, Pardi A. Tunable alignment of macromolecules by filamentous phage yields dipolar coupling interactions. Nat Struct Biol. 1998;5:1065–1074. doi: 10.1038/4176. [DOI] [PubMed] [Google Scholar]
- Henzler-Wildman KA, Thai V, Lei M, Ott M, Wolf-Watz M, Fenn T, Pozharski E, Wilson MA, Petsko GA, Karplus M, et al. Intrinsic motions along an enzymatic reaction trajectory. Nature. 2007;450:838–U813. doi: 10.1038/nature06410. [DOI] [PubMed] [Google Scholar]
- Houben K, Dominguez C, van Schaik FM, Timmers HT, Bonvin AM, Boelens R. Solution structure of the ubiquitin-conjugating enzyme UbcH5B. J Mol Biol. 2004;344:513–526. doi: 10.1016/j.jmb.2004.09.054. [DOI] [PubMed] [Google Scholar]
- Huang DT, Hunt HW, Zhuang M, Ohi MD, Holton JM, Schulman BA. Basis for a ubiquitin-like protein thioester switch toggling E1-E2 affinity. Nature. 2007;445:394–398. doi: 10.1038/nature05490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang DT, Paydar A, Zhuang M, Waddell MB, Holton JM, Schulman BA. Structural basis for recruitment of Ubc12 by an E2 binding domain in NEDD8’s E1. Mol Cell. 2005;17:341–350. doi: 10.1016/j.molcel.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Huang J, MacKerell AD., Jr CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J Comput Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju T, Bocik W, Majumdar A, Tolman JR. Solution structure and dynamics of human ubiquitin conjugating enzyme Ube2g2. Proteins. 2010;78:1291–1301. doi: 10.1002/prot.22648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay LE. New Views of Functionally Dynamic Proteins by Solution NMR Spectroscopy. J Mol Biol. 2016;428:323–331. doi: 10.1016/j.jmb.2015.11.028. [DOI] [PubMed] [Google Scholar]
- Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- Kim JH, Choi JS, Kim S, Kim K, Myung PK, Park SG, Seo YS, Park BC. Synergistic effect of two E2 ubiquitin conjugating enzymes in SCF(hFBH1) catalyzed polyubiquitination. BMB Rep. 2015;48:25–29. doi: 10.5483/BMBRep.2015.48.1.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiger G, Hao B, Mohl DA, Deshaies RJ. The Acidic Tail of the Cdc34 Ubiquitin-conjugating Enzyme Functions in Both Binding to and Catalysis with Ubiquitin Ligase SCFCdc4. J Biol Chem. 2009;284:36012–36023. doi: 10.1074/jbc.M109.058529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, Rape M. The ubiquitin code. Annu Rev Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- Korzhnev DM, Salvatella X, Vendruscolo M, Di Nardo AA, Davidson AR, Dobson CM, Kay LE. Low-populated folding intermediates of Fyn SH3 characterized by relaxation dispersion NMR. Nature. 2004;430:586–590. doi: 10.1038/nature02655. [DOI] [PubMed] [Google Scholar]
- Lange OF, Lakomek NA, Fares C, Schroder GF, Walter KF, Becker S, Meiler J, Grubmuller H, Griesinger C, de Groot BL. Recognition dynamics up to microseconds revealed from an RDC-derived ubiquitin ensemble in solution. Science. 2008;320:1471–1475. doi: 10.1126/science.1157092. [DOI] [PubMed] [Google Scholar]
- Liu J, Nussinov R. The mechanism of ubiquitination in the cullin-RING E3 ligase machinery: conformational control of substrate orientation. PLoS Comput Biol. 2009;5:e1000527. doi: 10.1371/journal.pcbi.1000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Nussinov R. The role of allostery in the ubiquitin-proteasome system. Crit Rev Biochem Mol Biol. 2013;48:89–97. doi: 10.3109/10409238.2012.742856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loria JP, Rance M, Palmer AG. A relaxation-compensated Carr-Purcell-Meiboom-Gill sequence for characterizing chemical exchange by NMR spectroscopy. J Am Chem Soc. 1999a;121:2331–2332. [Google Scholar]
- Loria JP, Rance M, Palmer AG., 3rd A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J Biomol NMR. 1999b;15:151–155. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- Lorick KL, Jensen JP, Fang S, Ong AM, Hatakeyama S, Weissman AM. RING fingers mediate ubiquitin-conjugating enzyme (E2)-dependent ubiquitination. Proc Natl Acad Sci USA. 1999;96:11364–11369. doi: 10.1073/pnas.96.20.11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKerell AD, Jr, Feig M, Brooks CL., 3rd Improved treatment of the protein backbone in empirical force fields. J Am Chem Soc. 2004;126:698–699. doi: 10.1021/ja036959e. [DOI] [PubMed] [Google Scholar]
- Martyna GJ, Tobias DJ, Klein ML. Constant-pressure molecular-dynamics algorithms. J Chem Phys. 1994;101:4177–4189. [Google Scholar]
- Mcconnell HM. Reaction Rates by Nuclear Magnetic Resonance. J Chem Phys. 1958;28:430–431. [Google Scholar]
- McElheny D, Schnell JR, Lansing JC, Dyson HJ, Wright PE. Defining the role of active-site loop fluctuations in dihydrofolate reductase catalysis. Proc Natl Acad Sci USA. 2005;102:5032–5037. doi: 10.1073/pnas.0500699102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Pruneda JN, Klevit RE, Weissman AM. RING-type E3 ligases: master manipulators of E2 ubiquitin-conjugating enzymes and ubiquitination. Biochimica et biophysica acta. 2014;1843:47–60. doi: 10.1016/j.bbamcr.2013.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millet O, Loria JP, Kroenke CD, Pons M, Palmer AG. The static magnetic field dependence of chemical exchange linebroadening defines the NMR chemical shift time scale. J Am Chem Soc. 2000;122:2867–2877. [Google Scholar]
- Mulder FA, Mittermaier A, Hon B, Dahlquist FW, Kay LE. Studying excited states of proteins by NMR spectroscopy. Nat Struct Biol. 2001a;8:932–935. doi: 10.1038/nsb1101-932. [DOI] [PubMed] [Google Scholar]
- Mulder FA, Skrynnikov NR, Hon B, Dahlquist FW, Kay LE. Measurement of slow (micros-ms) time scale dynamics in protein side chains by (15)N relaxation dispersion NMR spectroscopy: application to Asn and Gln residues in a cavity mutant of T4 lysozyme. J Am Chem Soc. 2001b;123:967–975. doi: 10.1021/ja003447g. [DOI] [PubMed] [Google Scholar]
- Ottiger M, Delaglio F, Bax A. Measurement of J and dipolar couplings from simplified two-dimensional NMR spectra. J Magn Reson. 1998;131:373–378. doi: 10.1006/jmre.1998.1361. [DOI] [PubMed] [Google Scholar]
- Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- Palmer AG, 3rd, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Meth Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- Petroski MD, Deshaies RJ. Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell. 2005;123:1107–1120. doi: 10.1016/j.cell.2005.09.033. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda JN, Littlefield PJ, Soss SE, Nordquist KA, Chazin WJ, Brzovic PS, Klevit RE. Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol Cell. 2012;47:933–942. doi: 10.1016/j.molcel.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruneda JN, Stoll KE, Bolton LJ, Brzovic PS, Klevit RE. Ubiquitin in motion: structural studies of the ubiquitin-conjugating enzyme approximately ubiquitin conjugate. Biochemistry. 2011;50:1624–1633. doi: 10.1021/bi101913m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman S, Vernon R, Thompson J, Tyka M, Sadreyev R, Pei J, Kim D, Kellogg E, DiMaio F, Lange O, et al. Structure prediction for CASP8 with all-atom refinement using Rosetta. Proteins. 2009;77(Suppl 9):89–99. doi: 10.1002/prot.22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robustelli P, Trbovic N, Friesner RA, Palmer AG. Conformational Dynamics of the Partially Disordered Yeast Transcription Factor GCN4. J Chem Theory Comput. 2013;9:5190–5200. doi: 10.1021/ct400654r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rout MK, Hodge CD, Markin CJ, Xu X, Glover JN, Xiao W, Spyracopoulos L. Stochastic gate dynamics regulate the catalytic activity of ubiquitination enzymes. J Am Chem Soc. 2014;136:17446–17458. doi: 10.1021/ja505440b. [DOI] [PubMed] [Google Scholar]
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J Comput Phys. 1977;23:327–341. [Google Scholar]
- Sandoval D, Hill S, Ziemba A, Lewis S, Kuhlman B, Kleiger G. Ubiquitin-conjugating enzyme Cdc34 and ubiquitin ligase Skp1-cullin-F-box ligase (SCF) interact through multiple conformations. J Biol Chem. 2015;290:1106–1118. doi: 10.1074/jbc.M114.615559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulman BA, Harper JW. Ubiquitin-like protein activation by E1 enzymes: the apex for downstream signalling pathways. Nat Rev Mol Cell Biol. 2009;10:319–331. doi: 10.1038/nrm2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieters CD, Kuszewski JJ, Tjandra N, Clore GM. The Xplor-NIH NMR molecular structure determination package. J Magn Reson. 2003;160:65–73. doi: 10.1016/s1090-7807(02)00014-9. [DOI] [PubMed] [Google Scholar]
- Skrynnikov NR, Dahlquist FW, Kay LE. Reconstructing NMR spectra of “invisible” excited protein states using HSQC and HMQC experiments. J Am Chem Soc. 2002;124:12352–12360. doi: 10.1021/ja0207089. [DOI] [PubMed] [Google Scholar]
- Smith CA, Ban D, Pratihar S, Giller K, Paulat M, Becker S, Griesinger C, Lee D, de Groot BL. Allosteric switch regulates protein-protein binding through collective motion. Proc Natl Acad Sci USA. 2016;113:3269–3274. doi: 10.1073/pnas.1519609113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, DiMaio F, Wang RY, Kim D, Miles C, Brunette T, Thompson J, Baker D. High-resolution comparative modeling with RosettaCM. Structure. 2013;21:1735–1742. doi: 10.1016/j.str.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soss SE, Klevit RE, Chazin WJ. Activation of UbcH5c~Ub is the result of a shift in interdomain motions of the conjugate bound to U-Box E3 ligase E4B. Biochemistry. 2013;52:2991–2999. doi: 10.1021/bi3015949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souphron J, Waddell MB, Paydar A, Tokgoz-Gromley Z, Roussel MF, Schulman BA. Structural dissection of a gating mechanism preventing misactivation of ubiquitin by NEDD8’s E1. Biochemistry. 2008;47:8961–8969. doi: 10.1021/bi800604c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stafford KA, Ferrage F, Cho JH, Palmer AG. Side Chain Dynamics of Carboxyl and Carbonyl Groups in the Catalytic Function of Escherichia coli Ribonuclease H. J Am Chem Soc. 2013;135:18024–18027. doi: 10.1021/ja409479y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tollinger M, Skrynnikov NR, Mulder FA, Forman-Kay JD, Kay LE. Slow dynamics in folded and unfolded states of an SH3 domain. J Am Chem Soc. 2001;123:11341–11352. doi: 10.1021/ja011300z. [DOI] [PubMed] [Google Scholar]
- Tzeng SR, Kalodimos CG. Dynamic activation of an allosteric regulatory protein. Nature. 2009;462:368–372. doi: 10.1038/nature08560. [DOI] [PubMed] [Google Scholar]
- Xue Y, Ward JM, Yuwen T, Podkorytov IS, Skrynnikov NR. Microsecond time-scale conformational exchange in proteins: using long molecular dynamics trajectory to simulate NMR relaxation dispersion data. J Am Chem Soc. 2012;134:2555–2562. 3. doi: 10.1021/ja206442c. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.