

Abstract
Excessive alcohol consumption is a risk factor associated with colorectal cancer; however, some studies have reported that moderate alcohol consumption may not contribute additional risk for developing colorectal cancer while others suggest that moderate alcohol consumption provides a protective effect that reduces colorectal cancer risk. The purpose of this study was to determine the effects of moderate voluntary alcohol (20% ethanol) intake on alternate days for 3 months in outbred Wistar rats on risk factors associated with colorectal cancer development. Colonic gene expression of cyclooxygenase-2, RelA, 8-oxoguanine DNA glycosylase 1, superoxide dismutase, catalase, glutathione peroxidase, glutathione reductase, glutathione-S-transferase M1, and aldehyde dehydrogenase 2 were determined. Blood alcohol content, liver function enzyme activities, and 8-oxo-deoxyguanosine DNA adducts were also assessed. Alcohol-treated rats were found to have significantly lower 8-oxo-deoxyguanosine levels in blood, a marker of DNA damage. Alanine aminotransferase and lactate dehydrogenase were both significantly lower in the alcohol group. Moderate alcohol significantly decreased cyclooxygenase-2 gene expression, an inflammatory marker associated with colorectal cancer risk. The alcohol group had significantly increased glutathione-S-transferase M1 expression, an antioxidant enzyme that helps detoxify carcinogens, such as acetaldehyde, and significantly increased aldehyde dehydrogenase 2 expression, which allows for greater acetaldehyde clearance. Increased expression of glutathione-S-transferase M1 and aldehyde dehydrogenase 2 likely contributed to reduce mucosal damage that is caused by acetaldehyde accumulation. These results indicate that moderate alcohol may reduce the risk for colorectal cancer development, which was evidenced by reduced inflammation activity and lower DNA damage after alcohol exposure.
Keywords: colorectal cancer, alcohol, moderate, inflammation, DNA damage
Introduction
Colorectal cancer (CRC) is ranked third in the leading cancer types for estimated new cases and deaths for both genders in the United States (Siegel, Miller, & Jemal, 2015). The American Cancer Society estimates 69,090 and 63,610 new cases and 26,100 and 23,600 deaths in 2015 for men and women, respectively (Siegel et al., 2015). For most individuals, the development of CRC is sporadic, and risk factors include consuming a high-fat diet, low fiber intake, sedentary lifestyle, obesity, diabetes mellitus, smoking, and excessive alcohol intake (Cunningham et al., 2010).
Prior epidemiological studies have investigated the dose-response relationship between alcohol consumption and CRC risk. Recently, a meta-analysis found that alcohol consumption was positively associated with CRC risk with estimated relative risks of 1.03 (95% confidence interval [CI] 0.92–1.20) for 10 g/day, 1.08 (95% CI 1.02–1.19) for 25 g/day, 1.14 (95% CI 1.07–1.21) for 50 g/day, and 1.43 (95% CI 1.25–1.64) for 100 g/day alcohol consumption (Wang, Duan, Yang, & Lin, 2015). However, the authors acknowledged that the observed dose-response relationship could be prone to confounding due to the meta-regression analysis used, and that the results were exploratory in nature (Wang et al., 2015). Conversely, a recent review concluded that increased risk was significant only for individuals consuming above a threshold of 30 g of alcohol per day, which is equivalent to two standard alcoholic beverages (Klarich, Brasser, & Hong, 2015). Another study found significant reduced risk of CRC in a Mediterranean population that consumed moderate amounts of alcohol, between 12 and 35 g of alcohol per day (Kontou et al., 2012). However, other dietary factors may have influenced these results, indicating that moderate alcohol consumption provided a protective effect that reduced CRC development.
Uncertainty remains regarding the role of moderate alcohol consumption on risk for CRC. Highly controlled investigations that aim to determine risk factors that may influence the development of cancer are difficult to conduct in human subjects (Lu et al., 2014). Animal studies are critical to better understand the underlying mechanisms mediating effects of a given substance on cancer risk, as the animal studies allow for controlled examination of normal physiological states that may become altered from increased inflammation and DNA damage that precede cancer development (Lu et al., 2014).
Numerous preclinical studies have been used to investigate the relationship between alcohol consumption and CRC risk. Various approaches have been used in animal models, including administration of a pro-carcinogenic agent to attempt to induce CRC development, as well as the use of genetically modified strains of mice that are prone to developing intestinal tumors (Hamilton, Sohn, & Fiala, 1987; Hayashi et al., 2007; Kushida et al., 2009; Pérez-Holanda, Rodrigo, Viñas-Salas, & Piñol-Felis, 2005; Perse & Cerar, 2007; Roy et al., 2002; Wimberly et al., 2013). Results from these studies vary considerably and many limitations have been reported with their use (Perse & Cerar, 2007). For instance, Perse and Cerar (2007) advise against the use of 1,2-dimethylhydrazine and azoxymethane for animal models of CRC when administering alcohol, due to variation among studies in the timing of carcinogen administration and the dose of alcohol administered. Additionally, many studies have administered an excessive dose of alcohol to reflect the effect of chronic alcohol consumption in humans (Choi et al., 1999; Simanowski et al., 1994; Wimberly et al., 2013). However, the current literature assessing the effects of moderate alcohol on CRC development in rodent models is limited. Additional controlled preclinical studies are needed that investigate the influence of moderate alcohol exposure on physiologic changes related to CRC development and to characterize biomarkers that may be mechanistically involved in disease risk.
The aim of the current study was to investigate the effects of moderate voluntary alcohol consumption in outbred Wistar rats on several risk factors associated with CRC development. These included examining interactions between alcohol and cyclooxygenase-2 (COX-2), RelA, CD68, myeloperoxidase (MPO), the repair of 8-oxo-deoxyguanosine (8-oxo-dG) by 8-oxoguanine DNA glycosylase 1 (OGG-1), the antioxidant activities of superoxide dismutase (SOD), catalase (CAT), glutathione peroxidase (GPx), glutathione reductase (GR), and glutathione-S-transferase M1 (GSTM1), and expression of aldehyde dehydrogenase 2 (ALDH2). Additionally, liver function enzyme tests including alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), lactate dehydrogenase (LDH), creatine kinase (CK), and gamma-glutamyltransferase (γ-GT) were measured under non-intoxicated conditions (i.e., nonsignificant blood alcohol content). The goal was to provide a more thorough understanding of the effects of moderate alcohol consumption on measures related to CRC development and to identify potential mechanisms mediating disease risk.
Materials and Methods
Animals and diets
This study was approved by the Institutional Animal Care and Use Committee at San Diego State University and was in accordance with National Institutes of Health guidelines. Naïve adult male outbred Wistar rats (n = 24; 503.0 ± 10.9 g, Harlan Laboratories, Placentia, CA, USA) at 109 days of age were randomly assigned to two groups (n = 12/group). The experimental group was fed a regular chow diet (LabDiet, St. Louis, MO, USA), was given water ad libitum, and was provided access to a 20% ethanol solution (Pharmco-Aaper, Brookfield, CT, USA) on alternate days for 13 weeks (details below; Simms et al., 2008). In the control diet, rats were fed the same chow and drank exclusively water. Both chow diets contained 28.5% of energy as protein, 13.5% of energy as fat, and 58% of energy as carbohydrates. All rats were fed ad libitum throughout the experiment. During the course of experimental procedures, animals were housed individually in standard tub cages in a vivarium that maintained a 12:12-h light/dark cycle and an ambient temperature of approximately 23 °C.
Ethanol exposure
Rats were exposed for 13 weeks to either a 20% ethanol intermittent-access drinking paradigm (n = 12) or were given access only to water (n = 12). This alcohol exposure paradigm has previously been shown to result in blood alcohol concentrations (BACs) averaging ~30– 50 mg/dL in outbred Wistar rats when measured 30–120 min into a standard drinking session (Cippitelli et al., 2012; George et al., 2012; Simms et al., 2008). The first seven daily sessions served to acclimate the animals to the testing procedures and all rats received free access to food and water only. Following the acclimation period, ethanol-exposed rats began 22-h intake sessions involving voluntary access to a 20% (v/v) ethanol solution vs. water, alternating with 22-h abstinence periods involving voluntary access to water only (45 ethanol-drinking sessions total, over 13 weeks). The positions of ethanol and water bottles were rotated each ethanol session to control for position preferences. The control group was given voluntary access to water only during the entire duration of the chronic exposure period. All fluids were weighed to the nearest gram and replaced daily, and body weights were measured every 48 h. One day following their final experimental session, rats were sacrificed via carbon dioxide asphyxiation and tissues were collected for subsequent analysis.
Sample collection
Blood was collected after euthanasia and was centrifuged at 1,200 × g for 15 min at 4 °C; serum was stored at −80 °C. The colorectum was excised, opened longitudinally, and the feces pushed off gently. It was then washed with phosphate buffered saline (PBS) and the mucosa was scraped off gently with glass microscope slides. Colonic mucosal scrapings were frozen and stored at −80 °C.
Blood alcohol content
Blood alcohol content following sacrifice was assessed in serum using the Ethanol Assay Kit (Sigma-Aldrich, St. Louis, MO, USA). The protocol indirectly determined ethanol concentration by a coupled enzyme reaction, which results in a colorimetric (570 nm) product, proportional to the ethanol present.
Liver enzyme tests
Liver enzyme concentrations, including ALT and AST, were measured in serum using ALT/GPT (UV-Rate) (Stanbio, Boerne, TX, USA) and AST/GOT (UV-Rate) (Stanbio, Boerne, TX, USA), respectively. The Alkaline Phosphatase LiquiColor® Test (Stanbio, Boerne, TX, USA) was used to measure ALP, and γ-GT LiquiColor® Test (Stanbio, Boerne, TX, USA) was used to measure γ-GT in the serum. Lactate dehydrogenase activity was determined using the LDH (UV-Rate) (Stanbio, Boerne, TX, USA), and CK was measured using the CK Liqui-UV® Test (Creatine Kinase/Rate) (Stanbio, Boerne, TX, USA) in serum.
RNA extraction and gene expression
To isolate purified RNA from the colon mucosa, the samples were first homogenized with Trizol (Invitrogen, Carlsbad, CA, USA). Next, chloroform was added to the samples, followed by centrifugation to separate the solution into an upper aqueous phase containing RNA. Isopropanol was then added and tubes were centrifuged. The RNA was then washed with ethanol to remove contaminants and the purified total RNA was then eluted in RNase-free water. cDNA was synthesized by SuperScript III Reverse Transcriptase (Invitrogen) with oligo(dT)12–18primers.
Gene expressions for COX-2, RelA, CD68, MPO, OGG-1, SOD, CAT, GPx, GR, GSTM1, and ALDH2 were analyzed using quantitative real time PCR (ViiA7, Applied Biosystems, Foster City, CA, USA). All real-time PCR reactions were performed in duplicate, and the results were analyzed using comparative method after target gene expression had been normalized to r18S expression. Every set of PCR contained a minus RT as a negative control.
DNA damage by 8-oxo-dG
DNA damage was assessed by measuring 8-oxo-dG in serum samples using the HT 8-oxo-dG ELISA kit II (Trevigen, Gaithersburg, MD, USA). The manufacturer’s protocol was followed. Serum samples were added to wells pre-coated with 8-OH-dG. Then anti-8-OH-dG monoclonal mouse antibody was added, followed by the addition of a horseradish peroxidase conjugated secondary antibody. The wells were washed four times in PBS with 0.1% Tween 20, and were incubated with TACS-Sapphire, a colorimetric substrate that turns blue in the presence of horseradish peroxidase. The color change reaction was stopped with the addition of 0.2-M HCl, and detection was performed using a spectrophotometer with absorbance read at 450 nm.
Statistical analysis
Ethanol intake in ethanol-exposed rats was calculated for each session during the 13-week chronic exposure phase to determine levels and patterns of ethanol consumption across time. Individual session data were averaged in 3-session blocks prior to analysis (15 blocks total). Ethanol intake (g/kg) and ethanol and water intake (mL/kg) for ethanol-exposed subjects were analyzed using repeated-measures analysis of variance (ANOVA) with block and solution as within-subject factors, followed by Newman-Keuls test where appropriate (α = 0.05). The significance of the compared mean values for physiological measures was determined using 2-tailed Student’s t test with a p value of <0.05 considered significant (SPSS, Version 21.0, Armonk, NY, USA). Results are reported as means ± standard error (SE). The correlations between variables were tested using Spearman rho and were performed on the measured values from individual rats in the experimental alcohol group for comparison and analysis.
Results
Ethanol intake and body weight gain
The intermittent-access 20% ethanol drinking paradigm resulted in increased g/kg intake of ethanol across the 13-week exposure phase (effect of session block: F(14,154) = 11.13, p < 0.001; Newman-Keuls test: block 1 <4–15; blocks 2–3 < 6–15; blocks 4–5 <14–15; block 6 <14; p’s < 0.05; Fig. 1A). Mean ethanol intake increased from 1.33 ± 0.38 g/kg/22 h during the first three sessions to 4.88 ± 0.62 g/kg/22 h by the end of the chronic exposure phase, with mean overall alcohol consumption of 3.58 ± 0.60 g/kg/22 h. Analysis of mL/kg ethanol and water intake in ethanol-exposed subjects revealed a similar increase in ethanol intake, while water intake declined (main effect of solution: F(1,11) = 12.24, p < 0.01; main effect of block: F(14,154) = 23.41, p < 0.001; solution × block interaction: F(14,154) = 22.35, p < 0.001; Newman-Keuls test: ethanol intake: block 1 <6–15, blocks 2–3 <14–15; water intake: blocks 1–3 >4–15, blocks 4–5 >7–15, block 6 >10–15, block 7 >15, p’s < 0.05). mL/kg ethanol intake was significantly less than water intake for all blocks (Newman-Keuls test, p’s < 0.05; Fig. 1B). Ethanol-exposed rats did not have significantly different body weight gain at the end of the study as compared to controls (Alcohol: 102 ± 21.0 g; Control: 110 ± 23.9 g). Initial body weight and final body weight was not different between the two groups. Water intake on non-ethanol days was also not significantly different between groups (Alcohol: 83.16 ± 4.23 mL/kg; Control: 82.87 ± 3.67 mL/kg).
Fig. 1. Alcohol intake levels.
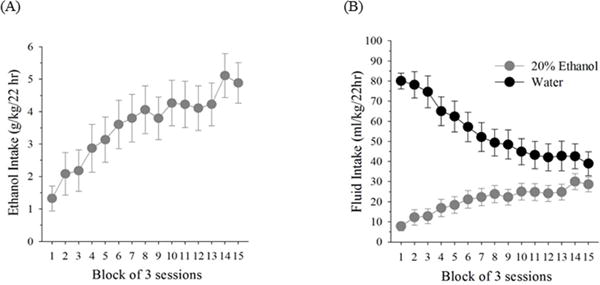
Ethanol intake (g/kg) (A) and 20% ethanol and water intake (mL/kg) (B) in male Wistar rats across all 15 session blocks of ethanol exposure (45 sessions total) in an intermittent-access 20% ethanol drinking paradigm. Individual session data were averaged in 3-session blocks prior to analysis. Values are means ± SEs, n = 12.
BAC and liver enzyme tests
The alcohol group and the controls did not have significantly different BAC measurements following sacrifice, nor did either group have significantly different measurements for their AST, ALP, CK, or γ-GT (Table 1). The ALT and LDH measurements in the alcohol group were significantly lower than the levels measured in the controls (p < 0.05; Table 1).
Table 1.
Liver function enzyme activities in male Wistar rats that consumed water (control) or moderate voluntary alcohol (20% ethanol) on alternate days for 13 weeks a
| Control | Alcohol | |
|---|---|---|
| ALT, U/L | 51.02 ± 7.99 | 32.79 ± 6.61* |
| AST, U/L | 70.00 ± 9.91 | 65.49 ± 8.56 |
| ALP, U/L | 42.36 ± 2.48 | 44.61 ± 3.71 |
| LDH, U/L | 173.9 ± 28.6 | 112.3 ± 20.3* |
| CK, U/L | 550.3 ± 78.1 | 534.1 ± 79.9 |
| γ-GT, U/L | 19.05 ± 4.85 | 15.15 ± 3.04 |
ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase; γ-GT, gamma–glutamyltransferase; LDH, lactate dehydrogenase.
Values are means ± SEs, n = 12 per group.
p < 0.05 compared with the controls.
Gene expression
The measured gene expression for COX-2 was significantly lower for alcohol-treated rats as compared to the controls (p < 0.05; Fig. 2A). There was a trend for lower CD68 expression in the ethanol group (1.35 ± 0.28) compared to the control group (2.18 ± 0.47; p = 0.074). Additionally, the measured gene expressions for GSTM1 and ALDH2 were significantly higher for the experimental alcohol group as compared to controls (p < 0.05; Fig. 2B, C). The measured gene expression for RelA, MPO, OGG-1, SOD, CAT, GPx, and GR were not significantly different for alcohol-treated and control subjects.
Fig. 2. Colonic biomarker gene expression.
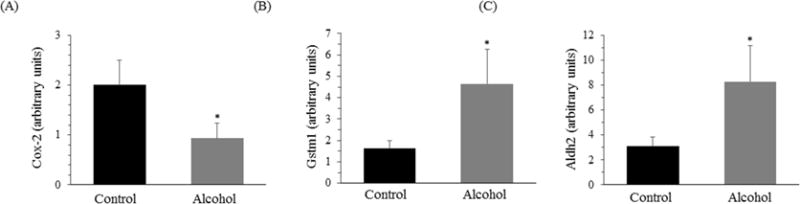
Gene expression measured from the colon mucosa in male Wistar rats that consumed water (control) or moderate voluntary alcohol (20% ethanol) on alternate days for 13 weeks. Values are means ± SEs, n = 12 per group. *p < 0.05 compared with the controls. ALDH2, aldehyde dehydrogenase 2; COX-2, cyclooxygenase-2; GSTM1, glutathione-S-transferase M1.
DNA damage by 8-oxo-dG
The DNA damage, expressed as 8-oxo-dG measured in serum, was significantly lower in the alcohol group, with an average of 4.54 ± 0.91 nM when compared to the control group that averaged 16.92 ± 4.57 nM (p < 0.05; Fig. 3).
Fig. 3. DNA damage measured in serum.
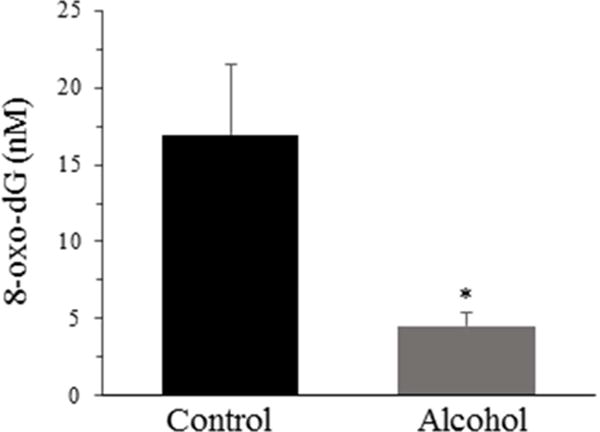
DNA damage expressed as 8-oxo-dG was measured in male Wistar rats that consumed water (control) or moderate voluntary alcohol (20% ethanol) on alternate days for 13 weeks. Values are means ± SEs, n = 12 per group. *p < 0.05 compared with the controls. 8-oxo-dG, 8-oxo-deoxyguanosine.
Correlations among measured values
Measured values from individual rats in the alcohol group were correlated and analyzed to determine statistical significance. Greater alcohol consumption was correlated with higher OGG-1 and ALDH2 gene expression (coefficient 0.745 and 0.612, respectively; p’s < 0.05; Table 2). Other significant positive correlations were found between 8-oxo-dG levels and liver enzyme levels for AST, ALT, and LDH (coefficient 0.577, 0.520, and 0.468, respectively; p’s < 0.05). 8-oxo-dG levels were moderately correlated with gene expression for COX-2, RelA, OGG-1, and GSTM1 (coefficient 0.597, 0.422, 0.404, and 0.381, respectively; p’s < 0.05; Table 2). ALDH2 gene expression was negatively correlated with ALT levels (coefficient −0.447) and positively correlated with OGG-1 gene expression (coefficient 0.629; p < 0.05; Table 2).
Table 2.
Correlations between measured values for individual male Wistar ratsa
| Correlation Coefficient (R) | p value | ||
|---|---|---|---|
| Alcohol intakeb | |||
| OGG-1 | 0.745 | 0.008 | |
| ALDH2 | 0.612 | 0.030 | |
|
| |||
| 8-oxo-dGc | |||
| AST | 0.577 | 0.003 | |
| ALT | 0.520 | 0.009 | |
| LDH | 0.468 | 0.021 | |
| COX-2 | 0.597 | 0.003 | |
| RelA | 0.422 | 0.050 | |
| OGG-1 | 0.404 | 0.028 | |
| GSTM1 | 0.381 | 0.033 | |
|
| |||
| ALDH2c | |||
| ALT | -0.447 | 0.042 | |
| OGG-1 | 0.629 | 0.002 | |
ALDH2, aldehyde dehydrogenase 2; ALT, alanine aminotransferase; AST, aspartate aminotransferase; COX-2, cyclooxygenase 2; GSTM1, glutathione-S-transferase M1; LDH, lactate dehydrogenase; OGG-1, 8-oxoguanine DNA glycosylase 1; 8-oxo-dG, 8-oxodeoxyguanosine.
Data calculated using Spearman rho.
Data from rats that consumed moderate voluntary alcohol (20% ethanol) on alternative days for 13 weeks.
Data from rats in control (water) or moderate voluntary alcohol (20% ethanol) on alternative days for 13 weeks.
Discussion
Ethanol Intake
The intermittent-access 20% ethanol-drinking paradigm resulted in gradually elevated levels of ethanol intake in outbred Wistar rats across the first 6 weeks of ethanol access, followed by stabilization and average daily ethanol intake values of ~4–5 g/kg during the remainder of the 13-week period of chronic exposure. These data are similar to previously reported intake levels (~5.8 g/kg/day maintenance of intake) by outbred Wistar rats in the 20% ethanol intermittent-access paradigm (Simms et al., 2008). Measurement of blood alcohol levels 30– 120 min into a standard drinking session, following stabilization of intake in this paradigm, have demonstrated moderate BACs in the Wistar strain, averaging ~30–50 mg/dL (Cippitelli et al., 2012; George et al., 2012; Simms et al., 2008). The latter BAC approximately corresponds to an acute 0.5-g/kg dose in human drinkers (Brasser, McCaul, & Houtsmuller, 2004), or a BAC that normally results from 1–2 standard drinks. Although the precise distribution of ethanol intake across daily 22-h intake sessions in the current study is not known, these data support that Wistar rats in this drinking paradigm maintain blood ethanol levels within a moderate range.
Liver Enzymes and BAC
The amount of alcohol voluntarily consumed by alcohol-treated rats in the present study produced no significant difference between groups for AST, ALP, γ-GT, and CK. By contrast, levels of ALT and LDH were significantly lower in the alcohol-treated group as compared to the controls. These results are consistent with another recent study demonstrating that low doses of alcohol administered in the drinking water to rats consuming a high-fat diet resulted in significantly lower serum levels of ALT and LDH when compared to controls (Osaki, Okazaki, Kimoto, Izu, & Kato, 2014). BAC at the time of sacrifice was not significantly different between groups (i.e., measures reflect sober levels).
Inflammatory Markers
The alcohol group showed significantly lower COX-2 expression when compared to controls, whereas RelA expression was not significantly different between groups. Increased attention has developed around COX-2 because its expression has been found primarily in pathological states related to inflammatory processes and cancer development (Doré, 2011). Other investigations have targeted gene regulatory factors that play a crucial role in controlling cell proliferation and apoptosis, including the transcription factor NF-κB (Yu et al., 2004). Various types of Rel proteins, like RelA, arrange as a dimer to form NF-κB (Yu et al., 2004), and dysregulation of the NF-κB pathway through constitutive activation has been shown to cause abnormal cell proliferation and apoptosis in those with CRC (Liu et al., 2014). Increased mRNA and protein expression of RelA has been reported in different CRC cell lines compared to controls (Liu et al, 2014). Another study used a transgenic mouse model to study the effects of overexpressing COX-2 in specific tissue, which resulted in an induced tumorigenic transformation in the affected tissue (Liu et al., 2001). Given the association between increased COX-2 and RelA expression and CRC (Greenhough et al., 2009; Liu et al., 2014), our results suggest that moderate alcohol levels may potentially provide a protective effect that reduces CRC risk, by lowering COX-2 expression, while no difference in RelA expression was found between groups.
DNA Damage by 8-oxo-dG and OGG-1 Expression
Alcohol-treated rats displayed significantly lower DNA damage, expressed as 8-oxo-dG measured in the serum, when compared to controls. Lower levels of 8-oxo-dG are associated with reduced oxidative stress, as reactive oxygen species (ROS) generation that is prone to attack DNA results in the formation of 8-oxo-dG. A study examining 8-OH-dG levels in human colorectal carcinoma patients found a positive correlation with the amount of 8-OH-dG damage and hOGG-1 expression (Kondo et al., 2000). Those with colorectal carcinomas produced greater levels of ROS, which accounted for higher levels of 8-OH-dG (Kondo et al., 2000). Given this association, our results suggest that the average level of alcohol consumed did not increase oxidative stress. It is possible that the lower 8-oxo-dG found in the alcohol-treated group may have reduced the induction of repair enzymes, which was evidenced by no significant difference in OGG-1 expression between the groups. It is important to note that there was a moderate correlation between 8-oxo-dG measured in the serum of the alcohol-exposed rats and the expression of OGG-1. Alcohol intake was also significantly correlated with OGG-1 expression, which suggests that the rats that consumed the highest levels of alcohol may have experienced greater oxidative stress. These correlations suggest that OGG-1 may be upregulated to repair damage resulting from higher intakes of alcohol.
Other studies provide support that moderate alcohol consumption does not increase 8-oxo-dG formation (Petitpas et al., 2013) and may decrease its levels within a moderate intake range (Yoshida, Shioji, Kishida, & Ogawa, 2001). A prior human study of moderate alcohol consumers revealed that 8-OH-dG levels significantly decreased with the amount of alcohol consumed (Yoshida et al., 2001). The authors suggested that moderate alcohol consumption increased the levels of uric acid, which is considered an important antioxidant in plasma capable of inhibiting oxidative damage (Yoshida et al., 2001). Another study evaluated the effects of subchronic ethanol consumption in pigs and found no significant difference in the levels of 8-oxo-dG between groups (Petitpas et al., 2013). Our results are consistent with these studies, suggesting that the mean dose of alcohol consumed was not sufficient to induce greater ROS formation, and rather reduced 8-oxo-dG damage.
Enzymatic Antioxidant Activity
Related pathways that reduce DNA damage caused by ROS include the activities of the antioxidant defense system. Glutathione-S-transferase M1 is one of the antioxidant defense enzymes that responds to oxidative stress differently than the other measured antioxidant enzymes, due to its ability to conjugate a reduced glutathione to acetaldehyde (Gyamfi & Wan, 2006). Therefore, it is possible that the increased expression found for GSTM1 may have resulted from adaptive upregulation that was needed to facilitate clearance of acetaldehyde in the alcohol group.
Acetaldehyde Clearance by ALDH2
Gene expression for ALDH2 was found to be significantly higher in the alcohol group, an effect that would allow for improved clearance of acetaldehyde. The intermittent (alternate day) administration of alcohol may have enhanced the induction of ALDH2 in a positive manner. At higher alcohol intakes, ALDH activity has been shown to decrease significantly in rectal tissue, which may promote carcinogenesis through acetaldehyde accumulation that leads to mucosal damage (Pronko, Bardina, Satanovskaya, Kuzmich, & Zimatkin, 2002). A prior study comparing wild-type and ALDH2-deficient mice to study the effects of chronic ethanol administration found that plasma acetaldehyde was significantly higher for ALDH2 (+/−) compared to that of the wild-type (Chaudhry et al., 2015). These data support the conclusion that greater ALDH2 activity is a protective adaptation that reduces acetaldehyde accumulation and may reduce the risk for CRC. The expression of ALDH2 was also negatively correlated with ALT in the alcohol-treated rats, suggesting that enhanced acetaldehyde clearance by ALDH2 may be associated with reduced liver damage.
Further studies are needed that assess CRC development through aberrant crypt foci formation and polyp development to directly support that moderate alcohol exposure may be beneficial in preventing colon carcinogenesis. Additionally, future studies that systematically evaluate alcohol dose-related effects, including an experimental group administered a higher dose of alcohol, will allow for within-study comparison of the effects of moderate vs. excessive alcohol exposure on key measures of interest.
Conclusion
Taken together, the present findings of reduced COX-2 expression and 8-oxo-dG damage in alcohol-exposed rats indicate that moderate levels of alcohol consumption do not elevate risk factors for CRC development, but rather may provide beneficial effects through lower inflammation and DNA damage that reduce CRC risk. These data are consistent with the results of recent human studies that moderate alcohol consumption may provide a protective effect that reduces CRC risk (Kontou et al., 2012; Park et al., 2009; Zhao et al., 2012) and provide insight into relevant mechanisms. Importantly, the effects observed in the current study, as well as previously reported protective effects of ethanol exposure, have been observed in protocols yielding average blood alcohol levels in a low-to-moderate range (Collins et al., 2009; Godfrey et al., 2015; Gunzerath, Faden, Zakhari, & Warren, 2004; Standridge, Zylstra, & Adams, 2004). A significant body of literature supports detrimental health effects at higher levels of ethanol exposure (Standridge et al., 2004). The present data add to a growing number of studies showing that moderate ethanol intake has favorable effects on specific physiological indices of disease risk. These studies may contribute to a more thorough understanding of the underlying mechanisms mediating protective effects of moderate alcohol consumption.
Highlights.
Excessive alcohol intake has been linked to increased risk for colorectal cancer.
Effects of moderate alcohol on CRC risk are not well-characterized.
Moderate alcohol intake in Wistar rats reduced COX-2 expression and DNA damage.
Adaptive increases in GSTM1 and ALDH were found in alcohol-treated rats.
Potential protective mechanisms of moderate alcohol on disease risk are identified.
Acknowledgments
Funding
This work was funded by the National Institutes of Health [grant number AA023291]. Additional funds were received from the Kasch-Boyer Endowed Scholarship in Exercise and Nutritional Sciences from San Diego State University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
Authors have no conflict of interest.
References
- Brasser SM, McCaul ME, Houtsmuller EJ. Alcohol effects during acamprosate treatment: a dose-response study in humans. Alcoholism: Clinical and Experimental Research. 2004;28:1074–1083. doi: 10.1097/01.alc.0000130802.07692.29. [DOI] [PubMed] [Google Scholar]
- Chaudhry KK, Samak G, Shukla PK, Mir H, Gangwar R, Manda B, et al. ALDH2 Deficiency Promotes Ethanol-Induced Gut Barrier Dysfunction and Fatty Liver in Mice. Alcoholism: Clinical and Experimental Research. 2015;39:1465–1475. doi: 10.1111/acer.12777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SW, Stickel F, Baik HW, Kim YI, Seitz HK, Mason JB. Chronic alcohol consumption induces genomic but not p53-specific DNA hypomethylation in rat colon. The Journal of Nutrition. 1999;129:1945–1950. doi: 10.1093/jn/129.11.1945. [DOI] [PubMed] [Google Scholar]
- Cippitelli A, Damadzic R, Singley E, Thorsell A, Ciccocioppo R, Eskay RL, et al. Pharmacological blockade of corticotropin-releasing hormone receptor 1 (CRH1R) reduces voluntary consumption of high alcohol concentrations in non-dependent Wistar rats. Pharmacology, Biochemistry, and Behavior. 2012;100:522–529. doi: 10.1016/j.pbb.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins MA, Neafsey EJ, Mukamal KJ, Gray MO, Parks DA, Das DK, et al. Alcohol in moderation, cardioprotection, and neuroprotection: epidemiological considerations and mechanistic studies. Alcoholism: Clinical and Experimental Research. 2009;33:206–219. doi: 10.1111/j.1530-0277.2008.00828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham D, Atkin W, Lenz HJ, Lynch HT, Minsky B, Nordlinger B, et al. Colorectal cancer. Lancet. 2010;375:1030–1047. doi: 10.1016/S0140-6736(10)60353-4. [DOI] [PubMed] [Google Scholar]
- Doré M. Cyclooxygenase-2 expression in animal cancers. Veterinary Pathology. 2011;48:254–265. doi: 10.1177/0300985810379434. [DOI] [PubMed] [Google Scholar]
- George O, Sanders C, Freiling J, Grigoryan E, Vu S, Allen CD, et al. Recruitment of medial prefrontal cortex neurons during alcohol withdrawal predicts cognitive impairment and excessive alcohol drinking. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:18156–18161. doi: 10.1073/pnas.1116523109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godfrey J, Jeanguenin L, Castro N, Olney JJ, Dudley J, Pipkin J, et al. Chronic Voluntary Ethanol Consumption Induces Favorable Ceramide Profiles in Selectively Bred Alcohol-Preferring (P) Rats. PLoS One. 2015;10:e0139012. doi: 10.1371/journal.pone.0139012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C, et al. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009;30:377–386. doi: 10.1093/carcin/bgp014. [DOI] [PubMed] [Google Scholar]
- Gunzerath L, Faden V, Zakhari S, Warren K. National Institute on Alcohol Abuse and Alcoholism report on moderate drinking. Alcoholism: Clinical and Experimental Research. 2004;28:829–847. doi: 10.1097/01.alc.0000128382.79375.b6. [DOI] [PubMed] [Google Scholar]
- Gyamfi MA, Wan YJ. The effect of ethanol, ethanol metabolizing enzyme inhibitors, and Vitamin E on regulating glutathione, glutathione S-transferase, and S-adenosylmethionine in mouse primary hepatocyte. Hepatology Research. 2006;35:53–61. doi: 10.1016/j.hepres.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Hamilton SR, Sohn OS, Fiala ES. Effects of timing and quantity of chronic dietary ethanol consumption on azoxymethane-induced colonic carcinogenesis and azoxymethane metabolism in Fischer 344 rats. Cancer Research. 1987;47:4305–4311. [PubMed] [Google Scholar]
- Hayashi N, Tsutsumi M, Fukura M, Yano H, Tsuchishima M, Takase S. Effect of chronic dietary ethanol consumption on colonic cancer in rats induced by 1,1-dimethylhydrazine. Alcoholism: Clinical and Experimental Research. 2007;31:S72–S76. doi: 10.1111/j.1530-0277.2006.00290.x. [DOI] [PubMed] [Google Scholar]
- Klarich DS, Brasser SM, Hong MY. Moderate Alcohol Consumption and Colorectal Cancer Risk. Alcoholism: Clinical and Experimental Research. 2015;39:1280–1291. doi: 10.1111/acer.12778. [DOI] [PubMed] [Google Scholar]
- Kondo S, Toyokuni S, Tanaka T, Hiai H, Onodera H, Kasai H, et al. Overexpression of the hOGG1 gene and high 8-hydroxy-2′-deoxyguanosine (8-OHdG) lyase activity in human colorectal carcinoma: regulation mechanism of the 8-OHdG level in DNA. Clinical Cancer Research. 2000;6:1394–1400. [PubMed] [Google Scholar]
- Kontou N, Psaltopoulou T, Soupos N, Polychronopoulos E, Xinopoulos D, Linos A, et al. Alcohol consumption and colorectal cancer in a Mediterranean population: a case-control study. Diseases of the Colon & Rectum. 2012;55:703–710. doi: 10.1097/DCR.0b013e31824e612a. [DOI] [PubMed] [Google Scholar]
- Kushida M, Wanibuchi H, Wei M, Kakehashi A, Ozaki K, Sukata T, et al. Ethanol Does Not Promote MeIQx-initiated Rat Colon Carcinogenesis Based on Evidence from Analysis of a Colon Cancer Surrogate Marker. Journal of Toxicologic Pathology. 2009;22:65–70. doi: 10.1293/tox.22.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CH, Chang SH, Narko K, Trifan OC, Wu MT, Smith E, et al. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. The Journal of Biological Chemistry. 2001;276:18563–18569. doi: 10.1074/jbc.M010787200. [DOI] [PubMed] [Google Scholar]
- Liu S, Sun X, Wang M, Hou Y, Zhan Y, Jiang Y, et al. A microRNA 221- and 222-mediated feedback loop maintains constitutive activation of NFκB and STAT3 in colorectal cancer cells. Gastroenterology. 2014;147:847–859. doi: 10.1053/j.gastro.2014.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu L, Chan RL, Luo XM, Wu WK, Shin VY, Cho CH. Animal models of gastrointestinal inflammation and cancer. Life Sciences. 2014;108:1–6. doi: 10.1016/j.lfs.2014.04.036. [DOI] [PubMed] [Google Scholar]
- Osaki A, Okazaki Y, Kimoto A, Izu H, Kato N. Beneficial effect of a low dose of ethanol on liver function and serum urate in rats fed a high-fat diet. Journal of Nutritional Science and Vitaminology. 2014;60:408–412. doi: 10.3177/jnsv.60.408. [DOI] [PubMed] [Google Scholar]
- Park JY, Mitrou PN, Dahm CC, Luben RN, Wareham NJ, Khaw KT, et al. Baseline alcohol consumption, type of alcoholic beverage and risk of colorectal cancer in the European Prospective Investigation into Cancer and Nutrition-Norfolk study. Cancer Epidemiology. 2009;33:347–354. doi: 10.1016/j.canep.2009.10.015. [DOI] [PubMed] [Google Scholar]
- Pérez-Holanda S, Rodrigo L, Viñas-Salas J, Piñol-Felis C. Effect of ethanol consumption on colon cancer in an experimental model. Revista Espanola De Enfermedades Digestivas. 2005;97:87–96. doi: 10.4321/s1130-01082005000200003. [DOI] [PubMed] [Google Scholar]
- Perse M, Cerar A. Dimethylhydrazine model is not appropriate for evaluating effect of ethanol on colorectal cancer. Revista Espanola De Enfermedades Digestivas. 2007;99:463–466. doi: 10.4321/s1130-01082007000800007. [DOI] [PubMed] [Google Scholar]
- Petitpas F, Sichel F, Hébert B, Lagadu S, Beljean M, Pottier D, et al. Effects of alcohol consumption on biomarkers of oxidative damage to DNA and lipids in ethanol-fed pigs. Experimental and Toxicologic Pathology. 2013;65:263–269. doi: 10.1016/j.etp.2011.09.001. [DOI] [PubMed] [Google Scholar]
- Pronko P, Bardina L, Satanovskaya V, Kuzmich A, Zimatkin S. Effect of chronic alcohol consumption on the ethanol- and acetaldehyde-metabolizing systems in the rat gastrointestinal tract. Alcohol and Alcoholism. 2002;37:229–235. doi: 10.1093/alcalc/37.3.229. [DOI] [PubMed] [Google Scholar]
- Roy HK, Gulizia JM, Karolski WJ, Ratashak A, Sorrell MF, Tuma D. Ethanol promotes intestinal tumorigenesis in the MIN mouse. Multiple intestinal neoplasia. Cancer Epidemiology, Biomarkers & Prevention. 2002;11:1499–1502. [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA: A Cancer Journal for Clinicians. 2015;65:5–29. doi: 10.3322/caac.21254. [DOI] [PubMed] [Google Scholar]
- Simanowski UA, Suter P, Russell RM, Heller M, Waldherr R, Ward R, et al. Enhancement of ethanol induced rectal mucosal hyper regeneration with age in F344 rats. Gut. 1994;35:1102–1106. doi: 10.1136/gut.35.8.1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms JA, Steensland P, Medina B, Abernathy KE, Chandler LJ, Wise R, et al. Intermittent access to 20% ethanol induces high ethanol consumption in Long-Evans and Wistar rats. Alcoholism: Clinical and Experimental Research. 2008;32:1816–1823. doi: 10.1111/j.1530-0277.2008.00753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standridge JB, Zylstra RG, Adams SM. Alcohol consumption: an overview of benefits and risks. Southern Medical Journal. 2004;97:664–672. doi: 10.1097/00007611-200407000-00012. [DOI] [PubMed] [Google Scholar]
- Wang Y, Duan H, Yang H, Lin J. A pooled analysis of alcohol intake and colorectal cancer. International Journal of Clinical and Experimental Medicine. 2015;8:6878–6889. [PMC free article] [PubMed] [Google Scholar]
- Wimberly AL, Forsyth CB, Khan MW, Pemberton A, Khazaie K, Keshavarzian A. Ethanol-induced mast cell-mediated inflammation leads to increased susceptibility of intestinal tumorigenesis in the APC 468 min mouse model of colon cancer. Alcoholism: Clinical and Experimental Research. 2013;37:E199–E208. doi: 10.1111/j.1530-0277.2012.01894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida R, Shioji I, Kishida A, Ogawa Y. Moderate alcohol consumption reduces urinary 8-hydroxydeoxyguanosine by inducing of uric acid. Industrial Health. 2001;39:322–329. doi: 10.2486/indhealth.39.322. [DOI] [PubMed] [Google Scholar]
- Yu HG, Zhong X, Yang YN, Luo HS, Yu JP, Meier JJ, et al. Increased expression of nuclear factor-kappa B/RelA is correlated with tumor angiogenesis in human colorectal cancer. International Journal of Colorectal Disease. 2004;19:18–22. doi: 10.1007/s00384-003-0494-z. [DOI] [PubMed] [Google Scholar]
- Zhao JH, Zhu Y, Wang PP, West R, Buehler S, Sun ZY, et al. Interaction between alcohol drinking and obesity in relation to colorectal cancer risk: a case-control study in Newfoundland and Labrador, Canada. BMC Public Health. 2012;12:94. doi: 10.1186/1471-2458-12-94. [DOI] [PMC free article] [PubMed] [Google Scholar]