

Abstract
The concept of the oligodendrocyte lineage as simply a source of myelinating cells in the vertebrate CNS is undergoing radical revision. Elucidation of the origins of oligodendrocytes in the CNS has led to identification of important signaling pathways, the timing and mechanism of lineage commitments and overlapping as well as redundant functionality among oligodendrocytes. The realization that a significant proportion of the oligodendrocyte lineage cells remain in a proliferative and immature state suggests they have roles other than as a reservoir of myelinating cells. While early studies were focused on understanding the development of oligodendrocytes, more recent work has begun to define the role of oligodendrocyte lineage cells in CNS functionality and the identification of new avenues for neural repair. A relatively unexplored aspect of the oligodendrocyte lineage is their contribution either directly or indirectly to the pathology of neurodegenerative diseases such as ALS and Alzheimer’s disease. Here we briefly consider the potential role of oligodendrocyte lineage cells as mediators of neural repair and neurodegeneration in the vertebrate CNS.
This article is part of the Special Issue entitled ‘Oligodendrocytes in Health and Disease’.
Keywords: Oligodendrocytes, Myelination, Neurodegeneration, Multiple sclerosis, ALS, Alzheimer’s disease
1. Introduction
Oligodendrocytes, the myelinating cells of the vertebrate CNS are generated from oligodendrocyte precursor cells (OPCs) that arise in discrete locations of the early neural tube (Rowitch and Kriegstein, 2010). The best understood role for oligodendrocytes is the generation of myelin sheaths around target axons that results in enhanced rates of axonal conduction through the establishment of saltatory conduction (Raine, 1984). The commitment of OPCs from multipotent neural stem cells (NSCs) has been extensively studied and several general themes have emerged. In caudal regions of the neural tube the earliest OPCs arise in the ventral midline as a result of inductive cues from adjacent tissues including the notochord and floor plate (Miller, 1996). Later in development a second source of OPCs arises in dorsal spinal cord that generates a second wave of myelinating oligodendrocytes (Cai et al., 2005). Whether the two populations of oligodendrocytes have similar properties or whether there is selective functionality between ventral and dorsal derived spinal cord OPCs is not well defined.
In more rostral regions of the CNS Cre-lox fate mapping experiments have demonstrated multiple waves of OPC generation beginning at an embryonic age in a ventral to dorsal progression and populating the entire telencephalon. The earliest cortical oligodendrocytes arise from Nkx2.1 expressing precursors in the ventricular zone of the medial ganglionic eminence (MGE) and the anterior entopeduncular area (AEP) starting at E12.5, migrating to the entire dorsal and ventral telencephalon to populate the cerebral cortex by E16. The second and third waves of OPCs arise from Gsh2-expressing precursors in the lateral and caudal ganglionic eminences (LGE and CGE) at E16 and Emx1-expressing precursors in the postnatal dorsal cortex. Paradoxically, most of the early born OPCs disappear after birth, such that majority of the oligodendrocytes in the adult dorsal cortex are derived from later arising Emx1-expressing cells (Kessaris et al., 2006).
During development not all OPCs that are generated differentiate into myelinating oligodendrocytes. Some OPCs that appear to commit to the myelination pathways but fail to associate with a target axon undergo programed cell death or apoptosis (Trapp et al., 1997), while other OPCs remain undifferentiated and form a significant population of adult OPCs, whose functions are not well defined.
The generation of OPCs from NSCs continues into adulthood and, in some experimental models, NSCs generate OPCs that contribute to neural repair. Two primary sources of NSCs have been defined: the subventricular zone (SVZ) and the subgranular zone (SGZ) of the hippocampus. In these regions anatomically distinct populations of stem cells retain the capacity to generate neurons, astrocyte and oligodendrocytes. The discovery of the existence of stem cells in animals and humans has led to speculation about the therapeutic potential of these cells. Mammalian adult NSCs support neurogenesis and gliogenesis within restricted areas of the CNS throughout adulthood. These cells or their progeny can undergo extensive expansion upon isolation and appropriate stimulation, and possess the capacity to generate all the major classes neural cells which can integrate into, and repair, the tissue of origin (Nunes et al., 2003). Lineage plasticity can be modulated in-vitro by exposure to growth factors that will direct them towards either neuronal or glial lineages suitable for regenerative transplantation in selected CNS diseases. This potential has lead to the utilization of NSCs or their progeny as cell replacement therapies in a number of diseases such as Parkinson’s disease and more recently in CNS demyelinating disease including multiple sclerosis.
2. Multiple sclerosis
Multiple sclerosis (MS) is one of the most commonly acquired neurological disease of young adults, affecting nearly 2.5 million people worldwide. The initial trigger for disease onset is not clearly characterized, however at diagnosis MS is characterized by an autoimmune attack targeted largely at myelin and oligodendrocytes (Noseworthy et al., 2000). Both T and B cells contribute to the immunological response and the majority of current therapies for MS are directed toward inhibiting the entry of lymphocytes and other peripheral immune cells into the CNS (Hauser et al., 1986; McFarland and Martin, 2007). The consequence of such an autoimmune insult is local demyelination and subsequent axonal loss that is the pathological hallmark of MS (De Stefano et al., 1998). Early in the disease, myelin loss is followed by remyelination- an endogenously regulated process orchestrated by the generation of new mature oligodendrocytes where new myelin sheathes are restored to demyelinated axons to recover saltatory conduction, axonal integrity and functional deficits (Keirstead and Blakemore, 1999) (Franklin and Ffrench-Constant, 2008). Although in experimental models, remyelination occurs with great efficiency, in MS, it is insufficient to repair severe demyelination events such as the ones occurring in later progressive phases of the disease, resulting in a worsening prognosis. An understanding of the reasons for remyelination failure is key to devising effective therapeutic strategies to enhance it. There are potentially two major sources of remyelinating cells in MS and experimental animal models: first, the parenchymal or adult OPCs which can be recruited to differentiate into mature oligodendrocytes and second, the endogenous NSCs which can be stimulated to regenerate lesions.
2.1. Remyelination by adult OPCs
Adult OPCs were first characterized in rat optic nerve (Ffrench-Constant and Raff, 1986) and have subsequently been shown to constitute 5–8% of the total cell population in the adult CNS (Horner et al., 2000; Dawson et al., 2003) where they can be identified through expression of markers such as PDGFR, NG2 etc. (Stallcup and Beasley, 1987; Nishiyama et al., 1996). In the pathological/demyelinating brain several cellular factors (Olig1/2, retinoid receptors, Nkx2.2, Sox17 etc.) are transiently upregulated in endogenous adult OPCs to give them an “activated OPC” phenotype. The first evidence that adult OPCs are remyelinating cells came from retroviral labeling of dividing cells in the adult white matter (Gensert and Goldman, 1997). In addition, cells with transitional expression of OPC and oligodendroglial markers were found at the onset of remyelination (Fancy et al., 2004); and transplanted OPCs remyelinate lesions with great efficacy (Windrem et al., 2004). Subsequently a number of fate mapping studies using transgenic animals have also demonstrated that adult OPCs generate remyelinating oligodendrocytes (Tripathi et al., 2010; Zawadzka et al., 2010). If adult OPCs are abundant, effective remyelinating cells present throughout the CNS, it is surprising that remyelination fails. Remyelination may fail due to local unavailability of precursor cells, failure in OPC recruitment or a failure in OPC differentiation and maturation. One of the surprising features of a demyelinating lesion and lack of myelin repair is that it is not simply caused by an exhaustion of the source of OPCs; in fact repeated focal demyelination events do not appear to deplete local OPCs or prevent subsequent remyelination (Wolswijk, 1998; Penderis et al., 2003). Instead, in more chronic lesions remyelination may be impaired is the OPC recruitment phase, which involves proliferation, migration and repopulation of lesions. Activated microglia and astrocytes in the setting of demyelination are the source of mitogens/growth factors such as FGF-2 and PDGF-2A that induce rapid proliferative responses in OPCs (Armstrong et al., 2002; Woodruff et al., 2004) this OPC response is regulated by P27-Kip1 (Crockett et al., 2005) and Cdk (Caillava and Baron-Van Evercooren, 2012). In chronic MS lesions, changes in the extracellular matrix composition as well as the formation of an astroglial scar (Franklin and Ffrench-Constant, 2008) might further contribute to the reduction in cell proliferation but perhaps more importantly inhibit the migration and accumulation of OPCs in the lesion (Franklin, 2002). The recruitment of OPCs into areas of demyelination is clearly an important aspect of repair since manipulations in the expression profiles of FGF2, guidance cues such as semaphorins 3A and 3F and signaling molecules such as CXCL12-CXCR4 (Williams et al., 2007; Carbajal et al., 2010; Clemente et al., 2011, 2013) all contribute to regulating the migration of activated OPCs and repopulation of MS lesions.
Remyelination may also fail due to dysregulation of the signaling environment needed for differentiation and maturation of newly recruited OPCs in the lesion. It has been proposed that the differentiation of oligodendroglial lineage cells in demyelinated lesions may be inhibited by the lack of clearance and accumulation of myelin debris and myelin fragments (Kotter et al., 2006; Baer et al., 2009) since in vitro studies demonstrate that OPCs fail to differentiate when grown on a myelin substrate (Robinson and Miller, 1999). Several signaling pathways have been implicated in the inhibition of OPC differentiation. During development the Notch-Jagged inhibits OPC differentiation (Blaschuk and ffrench-Constant, 1998) however, both Notch and Jagged are expressed in lesions that undergo complete remyelination (Seifert et al., 2007) and targeted deletion of Notch1 following demyelination does not specifically increase remyelination (Stidworthy et al., 2004). These observations suggest that the canonical Notch pathway has a relatively minor role in remyelination and that noncanonical Notch signaling in OPCs may be primarily involved in the induction of OPC differentiation in development and MS lesions (Brosnan and John, 2009). More recently considerable evidence suggests that a dysregulation of the Wnt-B-catenin signaling may be a major regulator of developmental myelination and myelin repair both in animal models and in human MS lesions (Fancy et al., 2009). Signaling through the glycosaminoglycan hyaluronan, an inhibitor of OPC differentiation that accumulates in MS lesions may also contribute to remyelination failure (Back et al., 2005; Sloane et al., 2010). Similarly, axonally-expressed neuregulin prevents the differentiation of immature oligodendrocytes (Vartanian et al., 1999), however it is also needed for the terminal differentiation of oligodendrocytes and the absence of neuregulin expression in active MS lesions has been cited as a reason for the eventual failure of remyelination (Flores et al., 2000). The identification of multiple molecular pathways that appear to inhibit or block the differentiation of OPCs and myelination has led to the development of multiple interventions to promote repair. For example, a NRG-controlled switch has been shown to enhance myelination of axons and it has been suggested that therapeutic manipulations involving enhancement of NRG and NMDA dependent remyelination may promote myelin repair (Lundgaard et al., 2013). Demyelinated axons increase the expression of PSA-NCAM that inhibits remyelination (Charles et al., 2002) and removal of PSA by endosialidases has been shown to enhance remyelination (Charles et al., 2000). Similarly, pharmacological activation of retinoid receptor signaling (Huang et al., 2011), Notch/Jagged signaling (Blanchard et al., 2013), Cdk5 (Luo et al., 2014) and Protein kinase C (Baer et al., 2009) signaling may all be advantageous for repair of MS lesions. Clearly, multiple environmental signals have an impact on the outcome of remyelination working through different pathways. Why so many different environmental stimuli influence myelination is not clear but it likely reflect the multiple different steps that are required to transition a proliferating OPC to a fully myelinating oligodendrocyte. What is more surprising is that a single critical rate-limiting step in this process has yet to be clearly identified.
2.2. Remyelination by endogenous NSCs
Besides activation of parenchymal OPCs, new remyelinating oligodendrocytes can be generated by the stimulation of SVZ-derived stem cells. In neonatal animals, retroviral lineage studies show that SVZ NSCs can give rise to neurons, astrocytes and oligodendrocytes and NG2 cells in the corpus callosum and neocortex (Levison and Goldman, 1993) while the adult SVZ continues to generate olfactory bulb neurons that migrate through the RMS (Pencea et al., 2001). Neurospheres generated from adult SVZ cells have been shown to give rise to oligodendrocytes (Morshead et al., 1994) and GFAP expressing SVZ type-B cells generate transit-amplifying type-C cells that express NG2, Olig2, Mash1, Dlx2 and EGFR (Aguirre et al., 2004). In contrast to type-B cells, SVZ OPCs are an actively dividing population and their proliferation is increased after demyelinating insult in the corpus callosum (Menn et al., 2006; Aguirre et al., 2007; Nait-Oumesmar et al., 2007). Electrophysiological studies demonstrate that OPCs express glutamate receptors (Karadottir and Attwell, 2007) and form novel neuron-OPC synapses in the cerebellar cortex and white matter (Lin et al., 2005) (Karadottir et al., 2005), cortex (Chittajallu et al., 2004), corpus callosum (Kukley et al., 2007; Ziskin et al., 2007) and hippocampus (Bergles et al., 2000; Jabs et al., 2005). The formation of such synapses potentially enables OPCs to monitor and respond to the neuronal instruction and maybe even regulate neuronal activity (Gallo et al., 2008). Conversely, synaptic input has been shown to regulate OPC proliferation (Gallo et al., 1996), differentiation (Kukley et al., 2010) and myelination (Wake et al., 2011; Lundgaard et al., 2013; Gibson et al., 2014). Recent studies have also demonstrated the consequences of physical activity on myelination (Simon et al., 2011). For example, physical exercise increases OPC production and oligodendrogliogenesis in the corpus callosum of mice trained on a running wheel with irregularly spaced rungs (McKenzie et al., 2014). One interpretation of these data is that the increased activity associated with this learning paradigm stimulates the generation of new oligodendrocytes. In the context of remyelination, it has also been proposed that after focal demyelination in the corpus callosum adult born NG2+ OPCs migrating from the SVZ form functional glutamatergic synapses onto demyelinated axons (Etxeberria et al., 2010). Furthermore, a study by Gautier et al (Gautier et al., 2015). shows that demyelinated axons generate de-novo synapses with OPCs, and neuronal activity regulates remyelination, by synaptic release of glutamate, thereby decreasing the proliferation rate of OPCs and prompting their differentiation into new remyelinating oligodendrocytes. Analysis of MS lesions showed enhanced numbers of PSA-NCAM+ progenitors in the proximal SVZ, suggestive of increased oligodendrogliogenesis (Nait-Oumesmar et al., 2007). In-vivo and in-vitro experiments reveal a battery of factors acting on OPCs derived from the adult SVZ that influence their proliferation, migration and differentiation properties (Michailidou et al., 2014). For example it has been shown that in an acute model of demyelination, heparin-binding epidermal growth factor (HB-EGF) and CNTF controls increased proliferation and migration of SVZ neural progenitors towards the demyelinated corpus callosum (Cantarella et al., 2008; Vernerey et al., 2013). Recently it has been shown that a distinct subset of mouse NSCs characterized by the expression of Gli1- a transcriptional effector of the sonic hedgehog (shh) pathway are recruited from the SVZ to populate demyelinated lesions. Enhanced oligodendrocyte differentiation corresponded to a loss of Gli-1 expression, suggesting that inhibition of Shh signaling may promote remyelination (Samanta et al., 2015). In experimental models of demyelination, several molecules such as netrin-1, Chondrin and Noggin have been identified as key modulators of progenitor cell migration, proliferation and lineage commitment (Cate et al., 2010; Jablonska et al., 2010; Cayre et al., 2013) and these can be therapeutically manipulated to aid regeneration and repair. Various cytokines, growth factors, pro and anti-inflammatory molecules constitute the cellular environment of the demyelinated lesion, where they most likely regulate progenitor cell fate plasticity and lineage commitment during repair, thus the main focus of future studies will be to define a fine balance of signaling pathways conducive to repair.
Both parenchymal and SVZ-derived OPCs contribute to remyelination, however, their relative contributions to the generation of remyelinating oligodendrocytes have not yet been quantified. OPCs are a functionally heterogeneous cell pool with not all cells generating extensive myelin sheaths (Leong et al., 2014). It is therefore tempting to speculate if tempo-spatially distinct OPC populations are differentially responsive to demyelinating insults?
A primary goal in promoting the proliferation, migration and differentiation of OPCs into oligodendrocytes is the formation of new myelin sheaths to reestablish saltatory conduction. Oligodendrocytes, however, have a more complex relationship with axons than simple myelination. For example, diseases thought to be primarily targeted to the oligodendrocyte lineage such as MS and leukodystrophies are characterized by widespread axonal damage indicating that oligodendrocytes play a critical role in maintaining axonal integrity (Nave and Trapp, 2008; Nave, 2010a). A seminal study where 2′, 3′-cyclic nucleotide 3′-phosphodiesterase (CNP) expression was disrupted using cre-lox strategy demonstrated a critical role for oligodendrocytes in the maintenance of axonal integrity (Lappe-Siefke et al., 2003). In CNP-null mice the ultrastructure and periodicity of myelin or myelin assembly remained relatively unaffected, but adult mice experienced severe neurodegeneration clearly demonstrating that the role of oligodendrocytes in myelin sheath maintenance was uncoupled from their role in supporting axonal integrity. These results imply that oligodendrocyte dysfunction alone is sufficient to cause secondary axonal degeneration and raises the possibility that oligodendrocytes may be a primary cellular target in neurodegenerative diseases. In support of this concept studies in which diphtheria toxin was used to disrupt MOG, PLP and MBP function also demonstrated severe myelin loss followed by acute axonal damage (Ghosh et al., 2011; Pohl et al., 2011; Oluich et al., 2012) consistent with the concept that in addition to promoting action potential propagation along axons, myelination and oligodendrocytes are important mediators of axonal viability (Fig. 1). While oligodendrocyte dysfunction or demyelination are obvious candidates for contributing to pathology in demyelinating diseases such as MS, increasing evidence suggests that oligodendrocyte dysfunction may be important in a number of other neurodegenerative diseases.
Fig. 1.
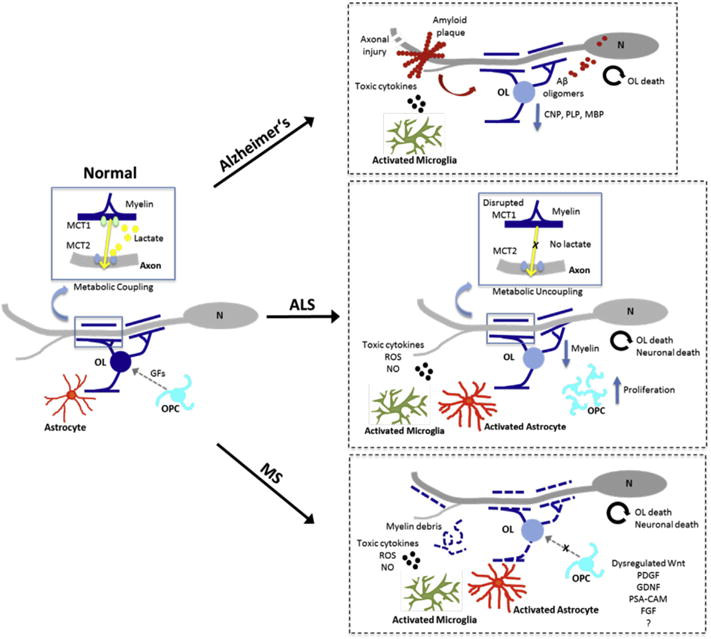
Potential mechanisms mediating oligodendrocyte neuron interactions that contribute to remyelination failure and neurodegeneration.
3. ALS
Amyotrophic lateral sclerosis (ALS) is a debilitating neurodegenerative disease of adulthood, characterized by death of upper and lower cortical and spinal motor neurons and a progressive loss of motor function (Pasinelli and Brown, 2006). Within the affected cells, disease pathology is characterized by abnormal accumulation of insoluble and misfolded proteins in degenerating motor neurons and the loss of motor neurons results in progressive paralysis that is typically fatal 3–5 years after disease onset. Among the most prevalent disease symptoms in ALS are extensive muscle weakness, atrophy and spasticity. The causes of sporadic ALS are not well understood, however 10% of cases are inherited, caused by mutations in a number of different genes including superoxide dismutase 1 (SOD1) and hexanucleotide repeats in C9orf72 (Chen et al., 2013). How mutations in these genes result in neurodegeneration is unclear and likely involves multiple pathways. SOD1 is a ubiquitously expressed enzyme that converts reactive superoxide to oxygen and hydrogen peroxide. Different gain of function mutations in the SOD1 gene result in toxicity and mutant SOD1 overexpression in mouse models recapitulates human ALS-like symptoms (Gurney et al., 1994). While it was originally assumed that ALS was a disease intrinsic to motor neurons, there is now compelling experimental evidence from animal models for a contribution from non-neuronal cells to disease progression. Expression of mutant SOD1 by microglia and astrocytes contributes to the loss of motor neurons (Boillee et al., 2006) although the precise mechanisms responsible are not well defined. Recent studies also suggest a contribution of oligodendrocyte lineage cells to ALS pathology. For example, the impaired function of gray matter oligodendrocytes in SOD1G93A transgenic animals increases the vulnerability of motor neurons to ALS-linked gene insults and accelerates disease progression (Kang et al., 2013). Morphological studies revealed alterations in myelin structure in mutant SOD1G93A transgenic animals that included abnormalities such as a loss of compact myelin, lamellae detachment, and a decrease in lipid content. Such changes are detectable in presymptomatic spinal cords and increasingly morphological and biochemical myelin degeneration is seen in symptomatic stages (Niebroj-Dobosz et al., 2007). The detection of myelin changes in presymptomatic spinal cord suggests they occur early in disease progression, but whether they precede neuronal loss or are a consequence of neuronal perturbation remains unclear.
Interactions between myelinating glia and axons are important for neuronal health. Post myelination, mature oligodendrocytes that are incapable of using the electron transport chain (ETC) to generate ATP, undergo a metabolic “developmental switch” and instead use aerobic glycolysis to generate lactate and pyruvate (Funfschilling et al., 2012); this lactate is in-turn rapidly utilized by myelinated axons in maintaining their structure and function. It has been shown that the lactate is transported exclusively via the monocarboxylate transporter-1 (MCT1) (Lee et al., 2012). Notably, oligodendrocytes express high levels of MCT1 compared with its expression in other CNS cell types such as astrocytes and neurons. In vitro disruption of MCT1 function suggested that it is important for neuronal survival while the viability of oligodendrocytes was not affected by MCT1 knockdown. Animals with a knockdown of endogenous MCT1 in all cells as well as oligodendrocyte-specific MCT1 knockouts developed axonal swellings and motor neuron loss in oligodendrocyte-associated axonal populations without any apparent disruptions in myelination or oligodendrocyte morphology. Immunohistochemical studies suggested that MCT1 expression is reduced in individuals with ALS and in animal models of ALS, consistent with the concept that aberrant lactate transport by oligodendrocytes in the CNS is a key feature of the disorder (Lee et al., 2012; Philips et al., 2013). The oligodendrocyte lineage responds to motor neuron degeneration in mouse models of ALS with an increase in the proliferation of NG2+ oligodendroglial precursors, which are prominent contributors to the extensive gliosis symptomatic of the disease. Fate mapping studies reveal that by the end-stage of the disease the proliferation of OPCs is 20-fold higher than in normal animals and these oligodendroglial precursors remain lineage restricted (Kang et al., 2010). It may be that newly generated oligodendrocytes remain in a differentiated but non-myelinating state in the disease and subsequently degenerate due to a disrupted signaling environment. This loss of oligodendrocyte support may contribute to further motor neuron death as oligodendrocytes are crucial for metabolic support of axons (Nave, 2010a). Astrocytes have also been implicated in ALS pathology and although OPCs appear to remain fate restricted in ALS, alterations in growth factor availability and inflammatory cytokines in degenerating regions might promote greater lineage plasticity as has been reported in-vitro where increased astrocyte differentiation of SOD1G93A glial restricted progenitors has been observed (Lepore et al., 2008). Additional analyses into the behaviors of oligodendroglial progenitors under different environmental contexts could yield further strategies to manipulate their proliferation rates and fate commitment in order to design better therapeutics for repair or disease progression in a number of different neurodegenerative diseases.
4. Alzheimer’s disease
Alzheimer’s disease (AD) is a progressive and irreversible neurodegenerative disorder characterized by neuronal loss in the hippocampus, entorhinal cortex and associated cortex, which leads to cognitive decline manifested as dementia (Schaeffer et al., 2009). A key feature in AD pathogenesis is the accumulation and extracellular deposition of “senile plaques” of amyloid-β peptide (Aβ1-42) derived from amyloid precursor protein (APP) (Geschwind, 2003). This is followed by formation of intraneuronal neurofibrillary tangles (NFT’s) of hyperphosphorylated microtubule-associated protein tau (Hardy and Higgins, 1992). AD has been classified as primarily a neuronal disorder, however it is not only neurons that are affected in AD, as many as 50% of all AD cases show loss of oligodendroglia lineage cells and other white matter components potentially due to glutamate excitotoxicity and oxidative stress (Brun and Englund, 1986; Mattson, 2008; Higgins et al., 2010). In the white matter of AD patients, concomitant with the increase in βamyloid plaques, there is a significant decrease in the myelin proteins MBP, PLP and CNP, indicative of white matter degeneration (Roher et al., 2002). It may be that Aβ1-42 is directly toxic to cells of the oligodendrocyte lineage since stereotaxic injection of Aβ1-42 into rat corpus callosum resulted in extensive white matter damage and oligodendrocyte death (Jantaratnotai et al., 2003). Evidence for a direct effect of Aβ1-42 on OPCs and oligodendrocytes comes from in vitro studies demonstrating apoptosis of undifferentiated and differentiated mouse oligodendrocyte precursors, as well as reduced NG2 expression (Desai et al., 2010; Nielsen et al., 2013). Furthermore, such toxic effects are not restricted to Ab1-42 since different Aβ fragments such as Aβ1-40 (Xu et al., 2001) and Aβ25-35 (Takao et al., 2004) have also been reported to be toxic. Such in vitro studies may reflect loss of oligodendrocyte lineage cells in the AD brain. Analysis of oligodendrocyte lineage cells in a mouse model with chronic plaque deposition (APPPS1 mice) and postmortem human AD cortex samples reveals that OPCs specifically react in these two conditions albeit with strikingly distinct outcomes: a higher number of Olig2+ were observed in APPPS1 mice, whereas OLIG2+ cells were decreased in number in human AD pathology (Behrendt et al., 2013). Interestingly, postmortem analysis of brain tissue from AD patients revealed decreased NG2 immunoreactivity and AD derived NG2+ OPCs displayed fewer, less branched, shorter processed with swollen cell bodies indicative of a reactive state. Likewise decreased levels of soluble NG2 found in the CSF from AD patients correlated with AD biomarkers such as Aβ1-42, T-tau and P-tau (Nielsen et al., 2013) suggesting that there is a loss of oligodendrocyte lineage cells in the setting of AD. Oligodendrocyte dysfunction is also a feature of brain aging and it has been hypothesized that AD pathology could potentially be related to protracted oligodendrocyte development in frontal lobe and medial temporal lobe- regions that are most vulnerable in AD. Indeed, late myelination in these regions is correlated with early AD pathology (Bartzokis, 2004). Major alterations in oligodendroglial populations in AD pathology suggest that targeting these cell types might provide novel therapeutic means for the prevention and treatment of AD.
5. Oligodendroglial pathways contributing to neurodegenerative diseases
The contribution of oligodendrocyte loss to the pathology of MS is potentially relatively easy to envision. In one commonly held concept immune mediate damage to myelin sheaths and mature oligodendrocytes results in localized demyelination that in turn exposes previously myelinated axons to a toxic environment resulting in axonal transection (Chang et al., 2008) and ultimately neuronal death. Other models in which the initial disease trigger is intrinsic to the oligodendrocyte lineage or primarily axonal may also account for some of the diverse pathology of MS.
In other neurodegenerative diseases such as ALS and AD the contribution of oligodendrocyte lineage cells to pathology is less well defined. It seems likely that perturbation of oligodendrocyte lineage cell viability will impact neuron health and survival in multiple ways. Clearly myelin sheaths themselves provide physical protection to long projection axons. Several independent lines of evidence support the notion that oligodendrocyte lineage cells provide metabolic support for adjacent axons (Nave, 2010a, 2010b) and in animal models of ALS, recent molecular studies implicate changes in the oligodendrocyte expression of the lactate transporter MCT1 as a contributor to neuronal loss (Lee et al., 2012). A more widespread mechanism of oligodendrocyte/OPC influence on neurodegenerative diseases may be associated with iron maintenance in the CNS. Oligodendrocytes are the major cellular source of iron in the CNS and express high levels of ferritin (Connor et al., 1990, 1995). Damage to oligodendrocytes may then release iron into the surrounding environment that can be taken up by macrophages and stimulate their cytotoxicity. Alternatively, elevated levels of iron have been implicated in elevation of reactive oxygen species and intracellular iron is capable of triggering a novel type of cell death termed ferroptosis (Dixon et al., 2012; Dixon and Stockwell, 2014) that is distinct from apoptosis or necrosis. Stimulation of neuronal death through such pathways could directly contribute to neurodegeneration secondary to oligodendrocyte loss. More specifically in AD, iron released from dying oligodendrocytes may promote the formation of AB oligomers and enhances Ab peptide toxicity as has been shown in other systems (Schubert and Chevion, 1995; Huang et al., 2004; Liu et al., 2011).
The interactions between cells of the oligodendrocyte lineage and their target neurons are complex and extend beyond the generation of myelin sheaths. Numerous insults can result in the death of oligodendroglial lineage cells and neuro-axonal degeneration through myelin breakdown, neuroinflammation, metabolic uncoupling, dysregulated signaling pathways and altered mitogenic environment.
In non-pathological conditions oligodendrocytes generated from OPCs myelinate their target axons and provide metabolic support to the axon mediated in part through lactate transport.
In Alzheimer’s disease (AD), amyloid β peptides derived from amyloid precursor protein (APP) by sequential protein cleavage, are deposited around neurons and form amyloid plaques, which contribute to disease pathogenesis. Aβ oligomers cause white matter damage including a reduction in myelin proteins and oligodendrocyte death. Loss of oligodendrocytes and their myelin leaves axons and neurons vulnerable to further insults.
Amyotrophic lateral sclerosis (ALS) is characterized by metabolic uncoupling and disrupted lactate transport between oligodendrocytes and neurons in the CNS, leading to oligodendrocyte loss and neuronal death. Extensive gliosis results in increased proliferation of OPCs but fails to provide support for motor neuron survival.
Demyelination in multiple sclerosis (MS) results from acute inflammation in which oligodendrocytes and myelin sheaths are damaged. This results in the generation of myelin debris that inhibit OPC differentiation. Absence of appropriate signaling environment further prevents the differentiation of OPCs to mature oligodendrocytes resulting in remyelination failure.
OL, oligodendrocyte; OPC, Oligodendrocyte precursor cell; N, Neuron; MCT1, monocarboxylate transporter 1; MCT2, monocarboxylate transporter 2; ROS, reactive oxygen species; NO, nitric oxide; GFs, growth factors.
In conclusion, our understanding of the role of oligodendrocyte lineage cells in development, maintenance, function and pathology of the CNS has expanded significantly over the last 3 decades. While many early studies focused on control of the development of oligodendrocytes, more recent work has been focused on how oligodendrocytes and their precursors contribute to CNS functionality including learning and memory as well as myelin repair. These advance have generated new potential therapeutic targets for CNS targeted myelin repair (Mi et al., 2007, 2009; Chong et al., 2012; Najm et al., 2015) that may prove important in both developmental and adult de/dysmyelinating conditions. How such insights will be translated for clinical benefit remains to be resolved. Two general approaches are the utilization of directed pharmacological intervention to stimulate the development or function of existing cells of the oligodendrocyte lineage to promote recovery (Deshmukh et al., 2013) or the generation of selectively engineered oligodendrocyte lineage cells for transplantation to replace defective host cells (Wang et al., 2013). In both approaches success will depend on a detailed understanding of the pathways controlling oligodendrocyte development and myelination. Elucidation of the role of oligodendrocyte lineage cells in neurodegenerative diseases has, with the exception of MS, moved more slowly in part because diseases such as ALS, AD and Parkinson’s disease have been considered to be intrinsic to the affected neuronal population. As the interdependency of oligodendrocytes and neurons becomes better defined at the molecular level, the contribution of oligodendrocyte dysfunction and the potential for oligodendrocyte-targeted therapies for neurodegenerative diseases will become increasingly evident.
Acknowledgments
This work was supported by NIH Grant # NS30800 to RHM.
References
- Aguirre A, Dupree JL, Mangin JM, Gallo V. A functional role for EGFR signaling in myelination and remyelination. Nat Neurosci. 2007;10:990–1002. doi: 10.1038/nn1938. [DOI] [PubMed] [Google Scholar]
- Aguirre AA, Chittajallu R, Belachew S, Gallo V. NG2-expressing cells in the subventricular zone are type C-like cells and contribute to interneuron generation in the postnatal hippocampus. J Cell Biol. 2004;165:575–589. doi: 10.1083/jcb.200311141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC. Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci. 2002;22:8574–8585. doi: 10.1523/JNEUROSCI.22-19-08574.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Tuohy TM, Chen H, Wallingford N, Craig A, Struve J, Luo NL, Banine F, Liu Y, Chang A, Trapp BD, Bebo BF, Jr, Rao MS, Sherman LS. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat Med. 2005;11:966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- Baer AS, Syed YA, Kang SU, Mitteregger D, Vig R, Ffrench-Constant C, Franklin RJ, Altmann F, Lubec G, Kotter MR. Myelin-mediated inhibition of oligodendrocyte precursor differentiation can be overcome by pharmacological modulation of Fyn-RhoA and protein kinase C signalling. Brain. 2009;132:465–481. doi: 10.1093/brain/awn334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25:49–62. 5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. author reply. [DOI] [PubMed] [Google Scholar]
- Behrendt G, Baer K, Buffo A, Curtis MA, Faull RL, Rees MI, Gotz M, Dimou L. Dynamic changes in myelin aberrations and oligodendrocyte generation in chronic amyloidosis in mice and men. Glia. 2013;61:273–286. doi: 10.1002/glia.22432. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–191. doi: 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- Blanchard B, Heurtaux T, Garcia C, Moll NM, Caillava C, Grandbarbe L, Klosptein A, Kerninon C, Frah M, Coowar D, Baron-Van Evercooren A, Morga E, Heuschling P, Nait Oumesmar B. Tocopherol derivative TFA-12 promotes myelin repair in experimental models of multiple sclerosis. J Neurosci. 2013;33:11633–11642. doi: 10.1523/JNEUROSCI.0774-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaschuk KL, ffrench-Constant C. Developmental neurobiology: notch is tops in the developing brain. Curr Biol. 1998;8:R334–R337. doi: 10.1016/s0960-9822(98)70215-5. [DOI] [PubMed] [Google Scholar]
- Boillee S, Vande Velde C, Cleveland DW. ALS: a disease of motor neurons and their nonneuronal neighbors. Neuron. 2006;52:39–59. doi: 10.1016/j.neuron.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Brosnan CF, John GR. Revisiting Notch in remyelination of multiple sclerosis lesions. J Clin Investig. 2009;119:10–13. doi: 10.1172/JCI37786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brun A, Englund E. A white matter disorder in dementia of the Alzheimer type: a pathoanatomical study. Ann Neurol. 1986;19:253–262. doi: 10.1002/ana.410190306. [DOI] [PubMed] [Google Scholar]
- Cai J, Qi Y, Hu X, Tan M, Liu Z, Zhang J, Li Q, Sander M, Qiu M. Generation of oligodendrocyte precursor cells from mouse dorsal spinal cord independent of Nkx6 regulation and Shh signaling. Neuron. 2005;45:41–53. doi: 10.1016/j.neuron.2004.12.028. [DOI] [PubMed] [Google Scholar]
- Caillava C, Baron-Van Evercooren A. Differential requirement of cyclin-dependent kinase 2 for oligodendrocyte progenitor cell proliferation and differentiation. Cell Div. 2012;7:14. doi: 10.1186/1747-1028-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarella C, Cayre M, Magalon K, Durbec P. Intranasal HB-EGF administration favors adult SVZ cell mobilization to demyelinated lesions in mouse corpus callosum. Dev Neurobiol. 2008;68:223–236. doi: 10.1002/dneu.20588. [DOI] [PubMed] [Google Scholar]
- Carbajal KS, Schaumburg C, Strieter R, Kane J, Lane TE. Migration of engrafted neural stem cells is mediated by CXCL12 signaling through CXCR4 in a viral model of multiple sclerosis. Proc Natl Acad Sci U S A. 2010;107:11068–11073. doi: 10.1073/pnas.1006375107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cate HS, Sabo JK, Merlo D, Kemper D, Aumann TD, Robinson J, Merson TD, Emery B, Perreau VM, Kilpatrick TJ. Modulation of bone morphogenic protein signalling alters numbers of astrocytes and oligodendroglia in the subventricular zone during cuprizone-induced demyelination. J Neurochem. 2010;115:11–22. doi: 10.1111/j.1471-4159.2010.06660.x. [DOI] [PubMed] [Google Scholar]
- Cayre M, Courtes S, Martineau F, Giordano M, Arnaud K, Zamaron A, Durbec P. Netrin 1 contributes to vascular remodeling in the subventricular zone and promotes progenitor emigration after demyelination. Development. 2013;140:3107–3117. doi: 10.1242/dev.092999. [DOI] [PubMed] [Google Scholar]
- Chang A, Smith MC, Yin X, Fox RJ, Staugaitis SM, Trapp BD. Neurogenesis in the chronic lesions of multiple sclerosis. Brain. 2008;131:2366–2375. doi: 10.1093/brain/awn157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Hernandez MP, Stankoff B, Aigrot MS, Colin C, Rougon G, Zalc B, Lubetzki C. Negative regulation of central nervous system myelination by polysialylated-neural cell adhesion molecule. Proc Natl Acad Sci U S A. 2000;97:7585–7590. doi: 10.1073/pnas.100076197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles P, Reynolds R, Seilhean D, Rougon G, Aigrot MS, Niezgoda A, Zalc B, Lubetzki C. Re-expression of PSA-NCAM by demyelinated axons: an inhibitor of remyelination in multiple sclerosis? Brain. 2002;125:1972–1979. doi: 10.1093/brain/awf216. [DOI] [PubMed] [Google Scholar]
- Chen S, Sayana P, Zhang X, Le W. Genetics of amyotrophic lateral sclerosis: an update. Mol Neurodegener. 2013;8:28. doi: 10.1186/1750-1326-8-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Aguirre A, Gallo V. NG2-positive cells in the mouse white and grey matter display distinct physiological properties. J Physiol. 2004;561:109–122. doi: 10.1113/jphysiol.2004.074252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong SY, Rosenberg SS, Fancy SP, Zhao C, Shen YA, Hahn AT, McGee AW, Xu X, Zheng B, Zhang LI, Rowitch DH, Franklin RJ, Lu QR, Chan JR. Neurite outgrowth inhibitor Nogo-A establishes spatial segregation and extent of oligodendrocyte myelination. Proc Natl Acad Sci U S A. 2012;109:1299–1304. doi: 10.1073/pnas.1113540109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente D, Ortega MC, Arenzana FJ, de Castro F. FGF-2 and Anosmin-1 are selectively expressed in different types of multiple sclerosis lesions. J Neurosci. 2011;31:14899–14909. doi: 10.1523/JNEUROSCI.1158-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemente D, Ortega MC, Melero-Jerez C, de Castro F. The effect of glia-glia interactions on oligodendrocyte precursor cell biology during development and in demyelinating diseases. Front Cell Neurosci. 2013;7:268. doi: 10.3389/fncel.2013.00268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor JR, Menzies SL, St Martin SM, Mufson EJ. Cellular distribution of transferrin, ferritin, and iron in normal and aged human brains. J Neurosci Res. 1990;27:595–611. doi: 10.1002/jnr.490270421. [DOI] [PubMed] [Google Scholar]
- Connor JR, Pavlick G, Karli D, Menzies SL, Palmer C. A histochemical study of iron-positive cells in the developing rat brain. J Comp Neurol. 1995;355:111–123. doi: 10.1002/cne.903550112. [DOI] [PubMed] [Google Scholar]
- Crockett DP, Burshteyn M, Garcia C, Muggironi M, Casaccia-Bonnefil P. Number of oligodendrocyte progenitors recruited to the lesioned spinal cord is modulated by the levels of the cell cycle regulatory protein p27Kip-1. Glia. 2005;49:301–308. doi: 10.1002/glia.20111. [DOI] [PubMed] [Google Scholar]
- Dawson MR, Polito A, Levine JM, Reynolds R. NG2-expressing glial progenitor cells: an abundant and widespread population of cycling cells in the adult rat CNS. Mol Cell Neurosci. 2003;24:476–488. doi: 10.1016/s1044-7431(03)00210-0. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Fu L, Narayanan S, Stanley J, Francis GS, Antel JP, Arnold DL. Axonal damage correlates with disability in patients with relapsing-remitting multiple sclerosis. Results of a longitudinal magnetic resonance spectroscopy study. Brain. 1998;121(8):1469–1477. doi: 10.1093/brain/121.8.1469. [DOI] [PubMed] [Google Scholar]
- Desai MK, Mastrangelo MA, Ryan DA, Sudol KL, Narrow WC, Bowers WJ. Early oligodendrocyte/myelin pathology in Alzheimer’s disease mice constitutes a novel therapeutic target. Am J Pathol. 2010;177:1422–1435. doi: 10.2353/ajpath.2010.100087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshmukh VA, Tardif V, Lyssiotis CA, Green CC, Kerman B, Kim HJ, Padmanabhan K, Swoboda JG, Ahmad I, Kondo T, Gage FH, Theofilopoulos AN, Lawson BR, Schultz PG, Lairson LL. A regenerative approach to the treatment of multiple sclerosis. Nature. 2013;502:327–332. doi: 10.1038/nature12647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon SJ, Stockwell BR. The role of iron and reactive oxygen species in cell death. Nat Chem Biol. 2014;10:9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B, 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etxeberria A, Mangin JM, Aguirre A, Gallo V. Adult-born SVZ progenitors receive transient synapses during remyelination in corpus callosum. Nat Neurosci. 2010;13:287–289. doi: 10.1038/nn.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fancy SP, Zhao C, Franklin RJ. Increased expression of Nkx2.2 and Olig2 identifies reactive oligodendrocyte progenitor cells responding to demyelination in the adult CNS. Mol Cell Neurosci. 2004;27:247–254. doi: 10.1016/j.mcn.2004.06.015. [DOI] [PubMed] [Google Scholar]
- Fancy SP, Baranzini SE, Zhao C, Yuk DI, Irvine KA, Kaing S, Sanai N, Franklin RJ, Rowitch DH. Dysregulation of the Wnt pathway inhibits timely myelination and remyelination in the mammalian CNS. Genes Dev. 2009;23:1571–1585. doi: 10.1101/gad.1806309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ffrench-Constant C, Raff MC. Proliferating bipotential glial progenitor cells in adult rat optic nerve. Nature. 1986;319:499–502. doi: 10.1038/319499a0. [DOI] [PubMed] [Google Scholar]
- Flores AI, Mallon BS, Matsui T, Ogawa W, Rosenzweig A, Okamoto T, Macklin WB. Akt-mediated survival of oligodendrocytes induced by neuregulins. J Neurosci. 2000;20:7622–7630. doi: 10.1523/JNEUROSCI.20-20-07622.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin RJ. Why does remyelination fail in multiple sclerosis? Nat Rev Neurosci. 2002;3:705–714. doi: 10.1038/nrn917. [DOI] [PubMed] [Google Scholar]
- Franklin RJ, Ffrench-Constant C. Remyelination in the CNS: from biology to therapy. Nat Rev Neurosci. 2008;9:839–855. doi: 10.1038/nrn2480. [DOI] [PubMed] [Google Scholar]
- Funfschilling U, Supplie LM, Mahad D, Boretius S, Saab AS, Edgar J, Brinkmann BG, Kassmann CM, Tzvetanova ID, Mobius W, Diaz F, Meijer D, Suter U, Hamprecht B, Sereda MW, Moraes CT, Frahm J, Goebbels S, Nave KA. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature. 2012;485:517–521. doi: 10.1038/nature11007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Mangin JM, Kukley M, Dietrich D. Synapses on NG2-expressing progenitors in the brain: multiple functions? J Physiol. 2008;586:3767–3781. doi: 10.1113/jphysiol.2008.158436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Zhou JM, McBain CJ, Wright P, Knutson PL, Armstrong RC. Oligodendrocyte progenitor cell proliferation and lineage progression are regulated by glutamate receptor-mediated K+ channel block. J Neurosci. 1996;16:2659–2670. doi: 10.1523/JNEUROSCI.16-08-02659.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard I, James F, Lao-Peregrin C, Reynolds R, Franklin RJ, Karadottir RT. Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun. 2015;6:8518. doi: 10.1038/ncomms9518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensert JM, Goldman JE. Endogenous progenitors remyelinate demyelinated axons in the adult CNS. Neuron. 1997;19:197–203. doi: 10.1016/s0896-6273(00)80359-1. [DOI] [PubMed] [Google Scholar]
- Geschwind DH. Tau phosphorylation, tangles, and neurodegeneration: the chicken or the egg? Neuron. 2003;40:457–460. doi: 10.1016/s0896-6273(03)00681-0. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Manrique-Hoyos N, Voigt A, Schulz JB, Kreutzfeldt M, Merkler D, Simons M. Targeted ablation of oligodendrocytes triggers axonal damage. PLoS One. 2011;6:e22735. doi: 10.1371/journal.pone.0022735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M. Neuronal activity promotes oligodendrogenesis and adaptive myelination in the mammalian brain. Science. 2014;344:1252304. doi: 10.1126/science.1252304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney ME, Pu H, Chiu AY, Dal Canto MC, Polchow CY, Alexander DD, Caliendo J, Hentati A, Kwon YW, Deng HX, et al. Motor neuron degeneration in mice that express a human Cu, Zn superoxide dismutase mutation. Science. 1994;264:1772–1775. doi: 10.1126/science.8209258. [DOI] [PubMed] [Google Scholar]
- Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- Hauser SL, Bhan AK, Gilles F, Kemp M, Kerr C, Weiner HL. Immunohistochemical analysis of the cellular infiltrate in multiple sclerosis lesions. Ann Neurol. 1986;19:578–587. doi: 10.1002/ana.410190610. [DOI] [PubMed] [Google Scholar]
- Higgins GC, Beart PM, Shin YS, Chen MJ, Cheung NS, Nagley P. Oxidative stress: emerging mitochondrial and cellular themes and variations in neuronal injury. J Alzheimers Dis. 2010;20(2):S453–S473. doi: 10.3233/JAD-2010-100321. [DOI] [PubMed] [Google Scholar]
- Horner PJ, Power AE, Kempermann G, Kuhn HG, Palmer TD, Winkler J, Thal LJ, Gage FH. Proliferation and differentiation of progenitor cells throughout the intact adult rat spinal cord. J Neurosci. 2000;20:2218–2228. doi: 10.1523/JNEUROSCI.20-06-02218.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JK, Jarjour AA, Nait Oumesmar B, Kerninon C, Williams A, Krezel W, Kagechika H, Bauer J, Zhao C, Baron-Van Evercooren A, Chambon P, Ffrench-Constant C, Franklin RJ. Retinoid X receptor gamma signaling accelerates CNS remyelination. Nat Neurosci. 2011;14:45–53. doi: 10.1038/nn.2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Atwood CS, Moir RD, Hartshorn MA, Tanzi RE, Bush AI. Trace metal contamination initiates the apparent auto-aggregation, amyloidosis, and oligomerization of Alzheimer’s Abeta peptides. J Biol Inorg Chem. 2004;9:954–960. doi: 10.1007/s00775-004-0602-8. [DOI] [PubMed] [Google Scholar]
- Jablonska B, Aguirre A, Raymond M, Szabo G, Kitabatake Y, Sailor KA, Ming GL, Song H, Gallo V. Chordin-induced lineage plasticity of adult SVZ neuroblasts after demyelination. Nat Neurosci. 2010;13:541–550. doi: 10.1038/nn.2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jabs R, Pivneva T, Huttmann K, Wyczynski A, Nolte C, Kettenmann H, Steinhauser C. Synaptic transmission onto hippocampal glial cells with hGFAP promoter activity. J Cell Sci. 2005;118:3791–3803. doi: 10.1242/jcs.02515. [DOI] [PubMed] [Google Scholar]
- Jantaratnotai N, Ryu JK, Kim SU, McLarnon JG. Amyloid beta peptide-induced corpus callosum damage and glial activation in vivo. Neuroreport. 2003;14:1429–1433. doi: 10.1097/00001756-200308060-00005. [DOI] [PubMed] [Google Scholar]
- Kang SH, Fukaya M, Yang JK, Rothstein JD, Bergles DE. NG2+ CNS glial progenitors remain committed to the oligodendrocyte lineage in postnatal life and following neurodegeneration. Neuron. 2010;68:668–681. doi: 10.1016/j.neuron.2010.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang SH, Li Y, Fukaya M, Lorenzini I, Cleveland DW, Ostrow LW, Rothstein JD, Bergles DE. Degeneration and impaired regeneration of gray matter oligodendrocytes in amyotrophic lateral sclerosis. Nat Neurosci. 2013;16:571–579. doi: 10.1038/nn.3357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadottir R, Attwell D. Neurotransmitter receptors in the life and death of oligodendrocytes. Neuroscience. 2007;145:1426–1438. doi: 10.1016/j.neuroscience.2006.08.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karadottir R, Cavelier P, Bergersen LH, Attwell D. NMDA receptors are expressed in oligodendrocytes and activated in ischaemia. Nature. 2005;438:1162–1166. doi: 10.1038/nature04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keirstead HS, Blakemore WF. The role of oligodendrocytes and oligodendrocyte progenitors in CNS remyelination. Adv Exp Med Biol. 1999;468:183–197. doi: 10.1007/978-1-4615-4685-6_15. [DOI] [PubMed] [Google Scholar]
- Kessaris N, Fogarty M, Iannarelli P, Grist M, Wegner M, Richardson WD. Competing waves of oligodendrocytes in the forebrain and postnatal elimination of an embryonic lineage. Nat Neurosci. 2006;9:173–179. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotter MR, Li WW, Zhao C, Franklin RJ. Myelin impairs CNS remyelination by inhibiting oligodendrocyte precursor cell differentiation. J Neurosci. 2006;26:328–332. doi: 10.1523/JNEUROSCI.2615-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- Kukley M, Nishiyama A, Dietrich D. The fate of synaptic input to NG2 glial cells: neurons specifically downregulate transmitter release onto differentiating oligodendroglial cells. J Neurosci. 2010;30:8320–8331. doi: 10.1523/JNEUROSCI.0854-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA. Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet. 2003;33:366–374. doi: 10.1038/ng1095. [DOI] [PubMed] [Google Scholar]
- Lee Y, Morrison BM, Li Y, Lengacher S, Farah MH, Hoffman PN, Liu Y, Tsingalia A, Jin L, Zhang PW, Pellerin L, Magistretti PJ, Rothstein JD. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature. 2012;487:443–448. doi: 10.1038/nature11314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leong SY, Rao VT, Bin JM, Gris P, Sangaralingam M, Kennedy TE, Antel JP. Heterogeneity of oligodendrocyte progenitor cells in adult human brain. Ann Clin Transl Neurol. 2014;1:272–283. doi: 10.1002/acn3.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepore AC, Rauck B, Dejea C, Pardo AC, Rao MS, Rothstein JD, Maragakis NJ. Focal transplantation-based astrocyte replacement is neuroprotective in a model of motor neuron disease. Nat Neurosci. 2008;11:1294–1301. doi: 10.1038/nn.2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levison SW, Goldman JE. Both oligodendrocytes and astrocytes develop from progenitors in the subventricular zone of postnatal rat forebrain. Neuron. 1993;10:201–212. doi: 10.1016/0896-6273(93)90311-e. [DOI] [PubMed] [Google Scholar]
- Lin SC, Huck JH, Roberts JD, Macklin WB, Somogyi P, Bergles DE. Climbing fiber innervation of NG2-expressing glia in the mammalian cerebellum. Neuron. 2005;46:773–785. doi: 10.1016/j.neuron.2005.04.025. [DOI] [PubMed] [Google Scholar]
- Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, Hider R, Marciniak SJ, Lomas DA, Crowther DC. Iron promotes the toxicity of amyloid beta peptide by impeding its ordered aggregation. J Biol Chem. 2011;286:4248–4256. doi: 10.1074/jbc.M110.158980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard I, Luzhynskaya A, Stockley JH, Wang Z, Evans KA, Swire M, Volbracht K, Gautier HO, Franklin RJ, Charles FC, Attwell D, Karadottir RT. Neuregulin and BDNF induce a switch to NMDA receptor-dependent myelination by oligodendrocytes. PLoS Biol. 2013;11:e1001743. doi: 10.1371/journal.pbio.1001743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo F, Burke K, Kantor C, Miller RH, Yang Y. Cyclin-dependent kinase 5 mediates adult OPC maturation and myelin repair through modulation of Akt and GsK-3beta signaling. J Neurosci. 2014;34:10415–10429. doi: 10.1523/JNEUROSCI.0710-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Glutamate and neurotrophic factors in neuronal plasticity and disease. Ann N Y Acad Sci. 2008;1144:97–112. doi: 10.1196/annals.1418.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- McKenzie IA, Ohayon D, Li H, de Faria JP, Emery B, Tohyama K, Richardson WD. Motor skill learning requires active central myelination. Science. 2014;346:318–322. doi: 10.1126/science.1254960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menn B, Garcia-Verdugo JM, Yaschine C, Gonzalez-Perez O, Rowitch D, Alvarez-Buylla A. Origin of oligodendrocytes in the subventricular zone of the adult brain. J Neurosci. 2006;26:7907–7918. doi: 10.1523/JNEUROSCI.1299-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi S, Hu B, Hahm K, Luo Y, Kam Hui ES, Yuan Q, Wong WM, Wang L, Su H, Chu TH, Guo J, Zhang W, So KF, Pepinsky B, Shao Z, Graff C, Garber E, Jung V, Wu EX, Wu W. LINGO-1 antagonist promotes spinal cord remyelination and axonal integrity in MOG-induced experimental autoimmune encephalomyelitis. Nat Med. 2007;13:1228–1233. doi: 10.1038/nm1664. [DOI] [PubMed] [Google Scholar]
- Mi S, et al. Promotion of central nervous system remyelination by induced differentiation of oligodendrocyte precursor cells. Ann Neurol. 2009;65:304–315. doi: 10.1002/ana.21581. [DOI] [PubMed] [Google Scholar]
- Michailidou I, de Vries HE, Hol EM, van Strien ME. Activation of endogenous neural stem cells for multiple sclerosis therapy. Front Neurosci. 2014;8:454. doi: 10.3389/fnins.2014.00454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller RH. Oligodendrocyte origins. Trends Neurosci. 1996;19:92–96. doi: 10.1016/s0166-2236(96)80036-1. [DOI] [PubMed] [Google Scholar]
- Morshead CM, Reynolds BA, Craig CG, McBurney MW, Staines WA, Morassutti D, Weiss S, van der Kooy D. Neural stem cells in the adult mammalian forebrain: a relatively quiescent subpopulation of subependymal cells. Neuron. 1994;13:1071–1082. doi: 10.1016/0896-6273(94)90046-9. [DOI] [PubMed] [Google Scholar]
- Nait-Oumesmar B, Picard-Riera N, Kerninon C, Decker L, Seilhean D, Hoglinger GU, Hirsch EC, Reynolds R, Baron-Van Evercooren A. Activation of the subventricular zone in multiple sclerosis: evidence for early glial progenitors. Proc Natl Acad Sci U S A. 2007;104:4694–4699. doi: 10.1073/pnas.0606835104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Najm FJ, et al. Drug-based modulation of endogenous stem cells promotes functional remyelination in vivo. Nature. 2015;522:216–220. doi: 10.1038/nature14335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave KA. Myelination and the trophic support of long axons. Nat Rev Neurosci. 2010a;11:275–283. doi: 10.1038/nrn2797. [DOI] [PubMed] [Google Scholar]
- Nave KA. Myelination and support of axonal integrity by glia. Nature. 2010b;468:244–252. doi: 10.1038/nature09614. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Niebroj-Dobosz I, Rafalowska J, Fidzianska A, Gadamski R, Grieb P. Myelin composition of spinal cord in a model of amyotrophic lateral sclerosis (ALS) in SOD1G93A transgenic rats. Folia Neuropathol. 2007;45:236–241. [PubMed] [Google Scholar]
- Nielsen HM, Ek D, Avdic U, Orbjorn C, Hansson O, Netherlands Brain B. Veerhuis R, Rozemuller AJ, Brun A, Minthon L, Wennstrom M. NG2 cells, a new trail for Alzheimer’s disease mechanisms? Acta Neuropathol Commun. 2013;1:7. doi: 10.1186/2051-5960-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiyama A, Lin XH, Giese N, Heldin CH, Stallcup WB. Co-localization of NG2 proteoglycan and PDGF alpha-receptor on O2A progenitor cells in the developing rat brain. J Neurosci Res. 1996;43:299–314. doi: 10.1002/(SICI)1097-4547(19960201)43:3<299::AID-JNR5>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- Noseworthy JH, Lucchinetti C, Rodriguez M, Weinshenker BG. Multiple sclerosis. N Engl J Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- Nunes MC, Roy NS, Keyoung HM, Goodman RR, McKhann G, 2nd, Jiang L, Kang J, Nedergaard M, Goldman SA. Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med. 2003;9:439–447. doi: 10.1038/nm837. [DOI] [PubMed] [Google Scholar]
- Oluich LJ, Stratton JA, Xing YL, Ng SW, Cate HS, Sah P, Windels F, Kilpatrick TJ, Merson TD. Targeted ablation of oligodendrocytes induces axonal pathology independent of overt demyelination. J Neurosci. 2012;32:8317–8330. doi: 10.1523/JNEUROSCI.1053-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nat Rev Neurosci. 2006;7:710–723. doi: 10.1038/nrn1971. [DOI] [PubMed] [Google Scholar]
- Pencea V, Bingaman KD, Freedman LJ, Luskin MB. Neurogenesis in the subventricular zone and rostral migratory stream of the neonatal and adult primate forebrain. Exp Neurol. 2001;172:1–16. doi: 10.1006/exnr.2001.7768. [DOI] [PubMed] [Google Scholar]
- Penderis J, Shields SA, Franklin RJ. Impaired remyelination and depletion of oligodendrocyte progenitors does not occur following repeated episodes of focal demyelination in the rat central nervous system. Brain. 2003;126:1382–1391. doi: 10.1093/brain/awg126. [DOI] [PubMed] [Google Scholar]
- Philips T, Bento-Abreu A, Nonneman A, Haeck W, Staats K, Geelen V, Hersmus N, Kusters B, Van Den Bosch L, Van Damme P, Richardson WD, Robberecht W. Oligodendrocyte dysfunction in the pathogenesis of amyotrophic lateral sclerosis. Brain. 2013;136:471–482. doi: 10.1093/brain/aws339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl HB, Porcheri C, Mueggler T, Bachmann LC, Martino G, Riethmacher D, Franklin RJ, Rudin M, Suter U. Genetically induced adult oligodendrocyte cell death is associated with poor myelin clearance, reduced remyelination, and axonal damage. J Neurosci. 2011;31:1069–1080. doi: 10.1523/JNEUROSCI.5035-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raine CS. Morphology of myelin and myelination. In: Morrell P, editor. Myelin. second. Springer; US: 1984. pp. 1–50. [Google Scholar]
- Robinson S, Miller RH. Contact with central nervous system myelin inhibits oligodendrocyte progenitor maturation. Dev Biol. 1999;216:359–368. doi: 10.1006/dbio.1999.9466. [DOI] [PubMed] [Google Scholar]
- Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, Anthony J, Watson D, Luehrs DC, Sue L, Walker D, Emmerling M, Goux W, Beach T. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–11090. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- Rowitch DH, Kriegstein AR. Developmental genetics of vertebrate glial-cell specification. Nature. 2010;468:214–222. doi: 10.1038/nature09611. [DOI] [PubMed] [Google Scholar]
- Samanta J, Grund EM, Silva HM, Lafaille JJ, Fishell G, Salzer JL. Inhibition of Gli1 mobilizes endogenous neural stem cells for remyelination. Nature. 2015;526:448–452. doi: 10.1038/nature14957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaeffer EL, Novaes BA, da Silva ER, Skaf HD, Mendes-Neto AG. Strategies to promote differentiation of newborn neurons into mature functional cells in Alzheimer brain. Prog Neuropsychopharmacol Biol Psychiatr. 2009;33:1087–1102. doi: 10.1016/j.pnpbp.2009.06.024. [DOI] [PubMed] [Google Scholar]
- Schubert D, Chevion M. The role of iron in beta amyloid toxicity. Biochem Biophys Res Commun. 1995;216:702–707. doi: 10.1006/bbrc.1995.2678. [DOI] [PubMed] [Google Scholar]
- Seifert T, Bauer J, Weissert R, Fazekas F, Storch MK. Notch1 and its ligand Jagged1 are present in remyelination in a T-cell- and antibody-mediated model of inflammatory demyelination. Acta Neuropathol. 2007;113:195–203. doi: 10.1007/s00401-006-0170-9. [DOI] [PubMed] [Google Scholar]
- Simon C, Gotz M, Dimou L. Progenitors in the adult cerebral cortex: cell cycle properties and regulation by physiological stimuli and injury. Glia. 2011;59:869–881. doi: 10.1002/glia.21156. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Batt C, Ma Y, Harris ZM, Trapp B, Vartanian T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc Natl Acad Sci U S A. 2010;107:11555–11560. doi: 10.1073/pnas.1006496107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stallcup WB, Beasley L. Bipotential glial precursor cells of the optic nerve express the NG2 proteoglycan. J Neurosci. 1987;7:2737–2744. doi: 10.1523/JNEUROSCI.07-09-02737.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stidworthy MF, Genoud S, Li WW, Leone DP, Mantei N, Suter U, Franklin RJ. Notch1 and Jagged1 are expressed after CNS demyelination, but are not a major rate-determining factor during remyelination. Brain. 2004;127:1928–1941. doi: 10.1093/brain/awh217. [DOI] [PubMed] [Google Scholar]
- Takao T, Flint N, Lee L, Ying X, Merrill J, Chandross KJ. 17beta-estradiol protects oligodendrocytes from cytotoxicity induced cell death. J Neurochem. 2004;89:660–673. doi: 10.1111/j.1471-4159.2004.02370.x. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Nishiyama A, Cheng D, Macklin W. Differentiation and death of premyelinating oligodendrocytes in developing rodent brain. J Cell Biol. 1997;137:459–468. doi: 10.1083/jcb.137.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi RB, Rivers LE, Young KM, Jamen F, Richardson WD. NG2 glia generate new oligodendrocytes but few astrocytes in a murine experimental autoimmune encephalomyelitis model of demyelinating disease. J Neurosci. 2010;30:16383–16390. doi: 10.1523/JNEUROSCI.3411-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartanian T, Fischbach G, Miller R. Failure of spinal cord oligodendrocyte development in mice lacking neuregulin. Proc Natl Acad Sci U S A. 1999;96:731–735. doi: 10.1073/pnas.96.2.731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernerey J, Macchi M, Magalon K, Cayre M, Durbec P. Ciliary neurotrophic factor controls progenitor migration during remyelination in the adult rodent brain. J Neurosci. 2013;33:3240–3250. doi: 10.1523/JNEUROSCI.2579-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Lee PR, Fields RD. Control of local protein synthesis and initial events in myelination by action potentials. Science. 2011;333:1647–1651. doi: 10.1126/science.1206998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Bates J, Li X, Schanz S, Chandler-Militello D, Levine C, Maherali N, Studer L, Hochedlinger K, Windrem M, Goldman SA. Human iPSC-derived oligodendrocyte progenitor cells can myelinate and rescue a mouse model of congenital hypomyelination. Cell Stem Cell. 2013;12:252–264. doi: 10.1016/j.stem.2012.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams A, Piaton G, Aigrot MS, Belhadi A, Theaudin M, Petermann F, Thomas JL, Zalc B, Lubetzki C. Semaphorin 3A and 3F: key players in myelin repair in multiple sclerosis? Brain. 2007;130:2554–2565. doi: 10.1093/brain/awm202. [DOI] [PubMed] [Google Scholar]
- Windrem MS, Nunes MC, Rashbaum WK, Schwartz TH, Goodman RA, McKhann G, 2nd, Roy NS, Goldman SA. Fetal and adult human oligodendrocyte progenitor cell isolates myelinate the congenitally dysmyelinated brain. Nat Med. 2004;10:93–97. doi: 10.1038/nm974. [DOI] [PubMed] [Google Scholar]
- Wolswijk G. Chronic stage multiple sclerosis lesions contain a relatively quiescent population of oligodendrocyte precursor cells. J Neurosci. 1998;18:601–609. doi: 10.1523/JNEUROSCI.18-02-00601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff RH, Fruttiger M, Richardson WD, Franklin RJ. Platelet-derived growth factor regulates oligodendrocyte progenitor numbers in adult CNS and their response following CNS demyelination. Mol Cell Neurosci. 2004;25:252–262. doi: 10.1016/j.mcn.2003.10.014. [DOI] [PubMed] [Google Scholar]
- Xu J, Chen S, Ahmed SH, Chen H, Ku G, Goldberg MP, Hsu CY. Amyloid-beta peptides are cytotoxic to oligodendrocytes. J Neurosci. 2001;21:RC118. doi: 10.1523/JNEUROSCI.21-01-j0001.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawadzka M, Rivers LE, Fancy SP, Zhao C, Tripathi R, Jamen F, Young K, Goncharevich A, Pohl H, Rizzi M, Rowitch DH, Kessaris N, Suter U, Richardson WD, Franklin RJ. CNS-resident glial progenitor/stem cells produce Schwann cells as well as oligodendrocytes during repair of CNS demyelination. Cell Stem Cell. 2010;6:578–590. doi: 10.1016/j.stem.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Rubio M, Fukaya M, Bergles DE. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]