

Abstract
Activation of the renin angiotensin system resulting in stimulation of angiotensin-II (AngII) type I receptor (AT1R) is an important factor in the development of liver fibrosis. Here, we investigated the role of Janus kinase 2 (JAK2) as a newly described intra-cellular effector of AT1R in mediating liver fibrosis. Fibrotic liver samples from rodents and humans were compared to respective controls. Transcription, protein expression, activation, and localization of JAK2 and downstream effectors were analyzed by realtime polymerase chain reaction, western blotting, immunohistochemistry, and confocal microscopy. Experimental fibrosis was induced by bile duct ligation (BDL), CCl4 intoxication, thioacetamide intoxication or continuous AngII infusion. JAK2 was inhibited by AG490. In vitro experiments were performed with primary rodent hepatic stellate cells (HSCs), Kupffer cells (KCs), and hepatocytes as well as primary human and human-derived LX2 cells. JAK2 expression and activity were increased in experimental rodent and human liver fibrosis, specifically in myofibroblastic HSCs. AT1R stimulation in wild-type animals led to activation of HSCs and fibrosis in vivo through phosphorylation of JAK2 and subsequent RhoA/Rho-kinase activation. These effects were prevented in AT1R–/– mice. Pharmacological inhibition of JAK2 attenuated liver fibrosis in rodent fibrosis models. In vitro, JAK2 and downstream effectors showed increased expression and activation in activated HSCs, when compared to quiescent HSCs, KCs, and hepatocytes isolated from rodents. In primary human and LX2 cells, AG490 blocked AngII-induced profibrotic gene expression. Overexpression of JAK2 led to increased profibrotic gene expression in LX2 cells, which was blocked by AG490.
Conclusion
Our study substantiates the important cell-intrinsic role of JAK2 in HSCs for development of liver fibrosis. Inhibition of JAK2 might therefore offer a promising therapy for liver fibrosis.
Chronic liver diseases represent a major global health problem with annually approximately 800,000 deaths worldwide.1,2 Persistent liver injury induces hepatic fibrosis defined as excessive hepatic production and deposition of extracellular matrix (ECM) by myofibroblasts.3 In liver fibrosis, the main source of these myofibroblasts are activated hepatic stellate cells (HSCs) located in the space of Disse.3 These myofibroblastic HSCs express alpha-smooth muscle actin (α-SMA) as a marker of their activation.3-5 Stimulation of the angiotensin-II (AngII) type 1 receptor (AT1R) by local or systemic activation of the renin angiotensin system (RAS) plays a crucial role in HSC activation and fibrogenesis.6-13 Subsequent accumulation of ECM in the liver disturbs the intrahepatic angioarchitecture and therefore leads to further complications.14 Activated HSCs play a pivotal role in the progression of fibrosis to decompensated cirrhosis with high morbidity and mortality.14,15
AT1R is coupled to heterotrimeric G proteins (Gaq/11 and Ga12/13), allowing stimulation and activation of several signal pathways involved in cell contraction and ECM production.16 One of these pathways is the RhoA/Rho-kinase pathway, which is crucially involved in fibrosis and portal hypertension as previously shown by our group.17-20 Recently, a link between AT1 receptor and RhoA/Rho-kinase pathway was established in smooth muscle cells showing the involvement of the tyrosine kinase, Janus kinase 2 (JAK2).21 AT1R stimulation activates JAK2, which, in turn, induces Arhgef1, the nucleotide exchange factor responsible for activation of RhoA, which subsequently activates Rho-kinase.21
JAK2 is involved in the intracellular signaling of many other receptors, for example, for hormones and cytokines toward transcription regulators of the signal transducers and activators of transcription (STAT) family. In HSCs, JAK2 acts through the STAT pathway or independent of it.22,23 The role of these JAK2-induced pathways in hepatocytes has been investigated for hepatic steatosis, ischemia-reperfusion (I/R) injury and cancer, whereas the role of JAK2 in fibrosis, especially mediated by AT1R stimulation, has not been investigated to date.
The present study showed that AT1 receptor-mediated JAK2 activation induces liver fibrosis. Consequently, inhibition of JAK2 blunts fibrosis. This activation of the JAK2/Rho-kinase pathway—dependent on stimulation of AT1R and leading to activation of HSC—was shown in different animal models and cell culture experiments. Furthermore, we confirmed the up-regulation of JAK2/Rho-kinase expression in human fibrosis.
Materials and Methods
Animals
We used 190 Sprague-Dawley wild-type (WT) rats and 155 mice (95 C57BL/6J WT and 60 AT1aR–/– mice) for our experiments. AT1aR-deficient mice were kindly provided by Nikos Werner (Department of Internal Medicine II, University of Bonn, Bonn, Germany). The responsible committee for animal studies in North Rhine-Westphalia approved the study (LANUV 8.87-50.10.31.08.28).
Cholestatic Model of Fibrosis
Bile duct ligation (BDL) was performed in rats with an initial body weight of 180-200 g, as described previously.17,18 Experiments were carried out 2 weeks after BDL in 19 rats, whereas 28 sham-operated rats served as controls, respectively. Ten rats undergoing BDL for 2 weeks received AG490 (1 mg/kg/day, intraperitoneally [IP]) on the last 7 days before sacrifice. BDL and sham operation was performed in 5 20-25 g WT mice, which were sacrificed after 2 weeks.
Toxic Models of Fibrosis
Twenty-three rats with an initial body weight of 100-120 g underwent twice-weekly inhalation of 1 L/min CCl4 for 14-16 weeks until ascites were present, as described previously.17,18 Twenty-one age-matched control rats did not receive CCl4. Periodic CCl4 inhalation of 2 L/min was performed in 25 mice (15 WT and 10 AT1aR–/–) for 4 weeks, as described previously,24 whereas 38 mice served as controls (11 WT and 27 AT1aR–/–). Additionally, 24 rats with an initial body weight of 200-250 g underwent weekly thioacetamide (TAA) administration with adjusted dosing in their drinking water for 18 weeks, as described previously.25
AngII-Mediated Fibrosis
For continuous release of AngII, osmotic pumps (2ML2 for rats, 2002 for mice, Alzet; Charles River Laboratories, Sulzfeld, Germany) were subcutaneously implanted in vivo in 18 rats, 15 AT1aR–/–, and 19 WT littermates, as described previously.26 Each pump released 0.7 mg/kg/day of AngII in rats as well as mice for 14 days, which has been shown to induce hepatic fibrosis.9 Pumps releasing saline were implanted in 5 rats and 24 mice as controls. Additionally, 9 of the rats received AG490 (1 mg/kg/day, IP) for the last 7 days before sacrifice.
Human Liver Samples
The human ethics committee of the University of Bonn (202/01) approved the use of human liver samples, obtained during liver transplantation from patients with alcohol-induced cirrhosis (n=16). Liver samples from patients without cirrhosis undergoing liver resection served as controls (n=10). None of the patients or donors received catecholamines, angiotensin converting enzyme (ACE) inhibitors, or angiotensin receptor antagonists before transplantation. Samples were snap-frozen after excision.
Western Blotting
Snap-frozen cells and liver samples were processed as previously described using sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels and nitrocellulose membranes.17,18 Ponceau S staining assured equal protein loading. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) or β-actin served as endogenous controls. Membranes were incubated with the respective primary antibody (Ab; Supporting Table 1) and corresponding secondary peroxidase-coupled Ab (Santa-Cruz-Biotechnology, Santa Cruz, CA). After enhanced chemiluminescence (ECL; Amersham, Bucks, UK), digital detection was evaluated using Chemi-Smart (PeqLab Biotechnologies GmbH, Erlangen, Germany).
Western blotting analysis was quantified by densitometry of all experiments (means ± standard error of the mean [SEM]) with values of controls set to 100 densitometric units (representative western blottings are shown in the Supporting Figures). Expression of phosphorylated JAK2 (pJAK2) at Tyr1007/1008 served as a marker of JAK2 activation, and Rho-kinase activity was analyzed as phosphorylation of its substrate moesin at Thr558 detected by phospho- and site-specific Abs.
Quantitative Real-Time Polymerase Chain Reaction
RNA isolation, reverse transcription, and detection by real-time polymerase chain reaction (RT-PCR) were performed as described previously.17,18 Assays provided by Applied Biosystems (Foster City, CA) are listed in Supporting Table 2. 18S ribosomal RNA served as an endogenous control. The results of HSC and liver samples were expressed as 2−ΔΔCt, and the data are reported as relative gene expression compared to the control group.
Hepatic Hydroxyproline Content
Hepatic hydroxyproline content was determined photometrically, as described previously.19
Sirius Red Staining
For the detection of collagen fibers, paraffin-embedded liver sections were stained with Sirius red using standard methods described previously.19
Immunohistochemical Staining for JAK2, pJAK2, and α-SMA
Staining for JAK2, pJAK2, and α-SMA was performed in cryosections from liver tissue (3 and 7 μm). The detailed method is described in the Supporting Information. The amount of staining was evaluated by computational analysis (Histoquant; 3DHistech, Budapest, Hungary), as described previously.27,28
Quantification (% of stained area) of immunohisto-chemical (IHC) staining is expressed as mean ± SEM of all experiments. For representative sections, please see the Supporting Figures.
Coimmunofluoresence Stainings of pJAK2 and α-SMA
Colocalization of pJAK2 and α-SMA was analyzed by immunofluorescent staining of 7-μm cryo-sections, as described in the Supporting Information.
Isolation of Primary HSCs
Rat and mouse HSCs were isolated as described previously.18,29 Briefly, primary HSCs were isolated in a two-step pronase-colla-genase perfusion from livers of healthy rats (n=36), as well as WT (n = 8) or AT1aR–/– mice (n = 8), and fractionated by density-gradient centrifugation. Viability and purity were systematically over 95%. Cells were seeded on uncoated plastic culture dishes. Experiments were performed 7 days after isolation or after the first passage (10 days) when HSCs were fully activated.
Primary human HSCs were obtained from ScienCell (San Diego, CA) and were cultured and harvested as described previously.30-32
Isolation of Hepatocytes
Primary hepatocytes were isolated from male WT rats (n=5) or WT mice (n=8), as described previously.29
Isolation of Kupffer Cells
Primary Kupffer cells (KCs) were isolated from WT mice (n=10), as described previously.29
Incubation with AngII, AG490, and Losartan
LX2 cells were provided by Vijay H. Shah (Mayo Clinic, Rochester, NY), originally established by Scott Friedman. AngII (10 μM) and/or AG490 (1.5, 5, and 25 μM) and/or Losartan (10 μM) was added to the culture medium of these cells, as indicated, for 3 days, or cells remained untreated.
Transfection With JAK2 Plasmid
Twenty-four hours before transfection, 6 × 105 LX2 cells were incubated with transfection media (Dulbecco's modified Eagle's medium [DMEM] with 10% fetal calf serum [FCS] without penicillin/streptomycin). The JAK2 plasmid and the respective empty plasmid control were isolated according to manufacturer instructions (Nucle-oBond Xtra Maxi kit; Machery, Nagel, Germany). Plasmid (15 μL) and 37.5 μL of lipofectamine were incubated for 20 minutes with a total volume of 3.6 μL of media. This plasmid/lipofectamine mix was added drop-wise to cells after removal of media. After 3-4 hours, cells were again incubated with media, containing 10% FCS, and harvested after 3 days. Efficacy of transfection was tested by RT-PCR and western blotting.
Transduction With AdJAK2
The recombinant replication-deficient adenoviral vector, AdJAK2, was generated using the adenoviral backbone vector, pAdEasy-1, containing the sequence of human adenovirus 5 with deletion of E1- and E3-genes with the transfer vector, pShuttleCMV (Stratagene, La Jolla, CA). JAK2 transgene was obtained from the plasmid, pUNO1-mJAK2a (Invivogen, Toulouse, France).
LX2 cells (6 × 10) were cultured in 10-cm plates. After 24 hours, cells were transduced with AdJAK2 and AdLacZ as a control at a transfection multiplicity of infection of 250 in DMEM supplemented with 2% FCS and 1% penicillin/streptomycin for 2 hours. Cells were washed twice with media, and AdJAK2-transduced cells were treated with 10% FCS and 1% penicillin/streptomycin alone, or additionally with 5 μM of AngII, or with 5 μM of AngII and AG490. AdLacZ-transduced cells were prepared in the same way as AdJAK2-transduced cells. Efficacy of transfection was tested by RT-PCR and western blotting.
Detection of Reactive Oxygen Species
Serum-starved LX2 cells (1.8 × 104 cells) plated in six-well plates were loaded with the reagent, 2′,7′-dichloro-fluorescein diacetate (DCFDA), a fluorogenic dye that measures hydroxyl, peroxyl, and other reactive oxygen species (ROS) activity within the cell (DCFDA-Cellular Reactive Oxygen Species Detection Assay Kit, catalog no. ab113851; Abcam, Cambridge, MA). After 20 minutes at 37° C incubation with AngII (10−5 M) with or without AG490 (5 μM), cells were washed and measured for the indicated time in a multiwell fluorescence plate reader using excitation and emission filters of 485 and 535 nm, respectively.
Apoptosis and Cycle Analysis
Analysis of apoptosis (Annexin V Apoptosis Detection Kit; BD Biosciences, Heidelberg, Germany) and cell-cycle analysis was performed as previously described.33
Statistical Analysis
Data are presented as mean- ± SEM. The Student t test was used for comparison, where appropriate. Mann-Whitney's U test or analysis of variance were used for comparison between groups (minimum n = 5/group). P values <0.05 were considered statistically significant.
Results
Increased Expression and Phosphorylation of JAK2 in Experimental and Human Liver Fibrosis is Found Predominantly in Activated HSCs and Myofi-broblasts
In human and experimental fibrosis, hepatic expression of the components of RAS was increased (Fig. 1A), suggesting that RAS is activated in liver fibrosis and cirrhosis. Accordingly, hepatic expression of AT1R and its downstream effectors (JAK2, Arhgef1, RhoA, and Rho-kinase) were increased in human liver fibrosis (Fig. 1B,D and Supporting Fig. S1A). We also observed that activation of JAK2, analyzed as JAK2 phosphorylation at Tyr1007/1008 (pJAK2),21 and of Rho-kinase, analyzed by the phosphorylation of its substrate, moesin, at Thr558, were increased in human cirrhosis, when compared to non-fibrotic controls (Fig. 1B and Supporting Fig. S1A). This was confirmed by IHC staining. Similarly, using two different models of fibrosis in mice and three different models in rats, we found that AT1R-mediated JAK2 phosphorylation at Tyr1007/100821 was increased in fibrotic livers, when compared to control livers (Fig. 2A,B and Supporting Fig. S1B,C). Furthermore, the JAK2 downstream effector, Arhgef1, its target, RhoA, and the downstream effector, Rho-kinase, were strongly expressed and activated in liver fibrosis (Fig. 2A,B).
Fig. 1.
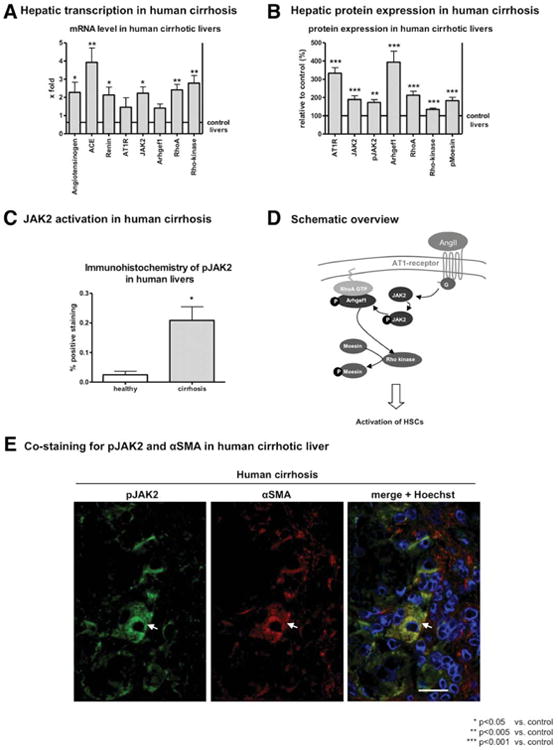
Expression and activity of AT1R and its downstream effectors in human fibrosis were up-regulated in activated HSCs. (A) mRNA levels of angiotensinogen, ACE, Renin, AT1R, JAK2, Arhgef1, RhoA, and Rho-kinase were analyzed by RT-PCR in liver samples of patients with cirrhosis and compared to noncirrhotic liver samples. (B) Protein expression of AT1R, JAK2, pJAK2, Arhgef1, RhoA, Rho-kinase, and pMoesin were analyzed in liver samples of patients with cirrhosis and compared to noncirrhotic liver samples. (C) Expression and distribution of pJAK2 were investigated by IHC (see representative section in Supporting Fig. S3A,B). (D) Scheme of the AT1R signaling pathway, elucidating the downstream effectors and their relation to each other. (E) Staining for pJAK2 in green, α-SMA in red and overlay with Hoechst33258 elucidate the localization of pJAK2 in α-SMA-positive cells in human liver fibrosis sections (see also Supporting Fig. S3C). Scale bar, 20 μm.
Fig. 2.
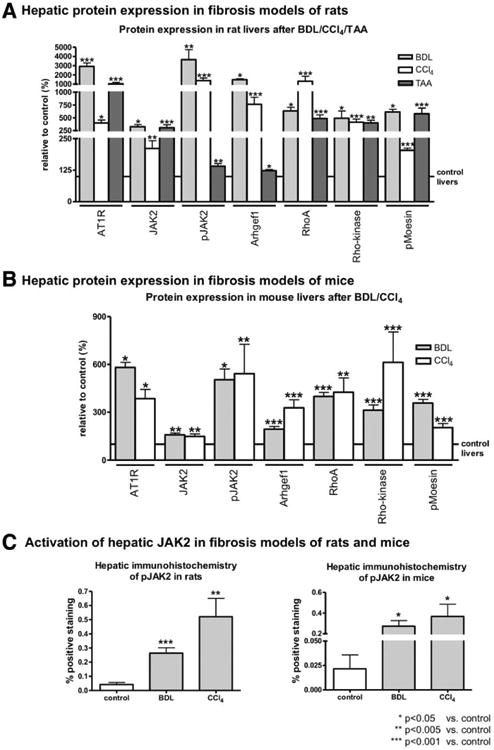
Expression and activity of AT1R and its downstream effectors in experimental fibrosis were up-regulated. (A) Protein expression of AT1R, JAK2, pJAK2, Arhgef1, RhoA, Rho-kinase, and pMoesin, analyzed by western blotting, was increased in liver samples from rats with BDL and CCl4-induced fibrosis, compared to their respective controls. (B) Significantly increased protein levels of AT1R, JAK2, pJAK2, Arhgef1, RhoA, Rho-kinase, and pMoesin were detected using western blotting in liver samples from mice after 2 weeks of BDL or 4 weeks of CCl4 treatment, compared to their respective controls. (C) Expression and distribution of pJAK2 were investigated by IHC for rats and mice (see representative section in Supporting Figs. S4 and S5).
Interestingly, although faint JAK2 staining was found in most liver cells, it predominated in fibrotic septa as well as in perivascular and -sinusoidal regions (Supporting Figs. S3A-S5A). In contrast, pJAK2 was only found in fibrotic septa and around vessels and small sinusoids (Figs. 1C and 2C and Supporting Figs. S3B-S5B). Immunofluorescence costaining of pJAK2 and α-SMA in fibrotic livers showed the expression of pJAK2 in α-SMA-positive cells in fibrotic septa as well as in perivascular and -sinusoidal areas (Fig. 1E and Supporting Fig. S3C). These data show, for the first time, that AT1R-dependent JAK2 phosphorylation at Tyr1007/1008 occurs in hepatic myofibroblasts, which derive mainly from activated HSCs.
Protein expression of JAK2-dependent STAT3/suppressor of cytokine signalling 3 signaling was also activated in different fibrosis models of mice and rats (Supporting Fig. S2A,B), suggesting an increased activity of JAK2.
AT1R Stimulation Induced Fibrosis Through the JAK2 Pathway In Vivo
We assessed the effect of JAK2 inhibition on hepatic fibrogenesis in rats and mice. AG490, injected daily for 1 week before killing of BDL rats (14 days of BDL in total) attenuated fibrogenesis, as assessed by hydroxyproline content and Sirius red staining. This treatment attenuated the activation of HSCs, as mirrored by α-SMA immunostaining as a result of inhibition of JAK2 phosphorylation at Tyr 1007/1008, as assessed by IHC (Fig. 3A,B and Supporting Fig. S6).
Fig. 3.
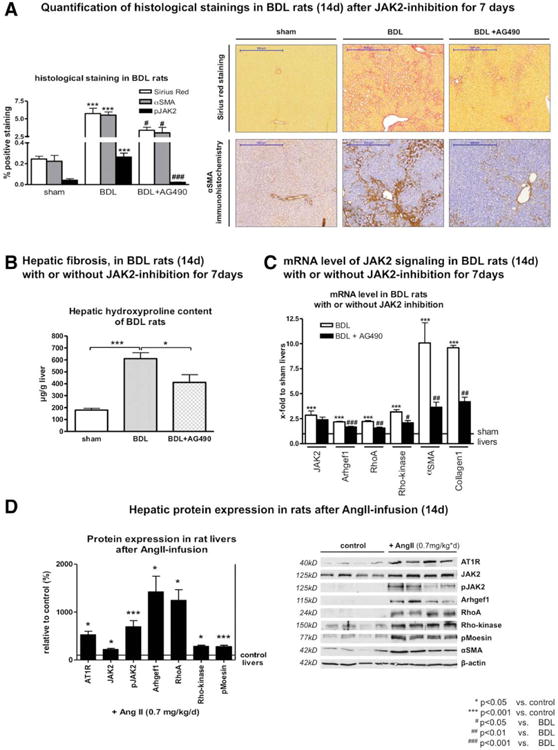
Stimulation of AT1R activated JAK2, but JAK2 inhibition attenuated hepatic fibrosis. (A) Quantification of Sirius red, α-SMA, and pJAK2 IHC staining is shown along with representative stained sections of rat liver after BDL for 14 days, with or without AG490 treatment (1 mg/kg/day) 7 days before sacrifice (see quantifications of Sirius red and α-SMA stainings, as well as representative sections of pJAK2 staining, in Supporting Fig. S6). (B) Less hepatic hydroxyproline content as a marker for fibrosis is shown. (C) mRNA levels of JAK2, Arhgef1, RhoA, Rho-kinase, α-SMA, and Col1 were analyzed by RT-PCR in liver samples of rats after BDL for 14 days, with or without AG490 treatment (1 mg/kg/day) 7 days before sacrifice. (D) Protein expression of AT1R, JAK2, pJAK2, Arhgef1, RhoA, Rho-kinase, pMoesin, and α-SMA was analyzed by western blotting in liver samples of rats receiving AngII infusion (0.7 mg/kg/day by osmotic pumps for 14 days) or solvent. Expression and activation of AT1R downstream effectors were increased after AT1 receptor stimulation.
Transcription of the JAK2 downstream proteins, as well as HSC activation documented by elevated α-SMA levels and collagen production, was significantly up-regulated at the messenger RNA (mRNA) level after BDL. This effect was attenuated by JAK2 inhibition with AG490 (Fig. 3C).
To further underline the crucial role of AT1R, continuous stimulation of AT1R was performed using AngII infusion in rats and mice for 14 days. Indeed, this treatment induced expression and activation of JAK2 pathway components (Fig. 3D), resulting in mild fibrosis.
Interestingly, concomitant JAK2 inhibition during the last 7 days before sacrifice of rats receiving AngII infusion significantly decreased fibrosis, as assessed by hydroxyproline content and Sirius red staining, as well as inhibition of JAK2 phosphorylation at Tyr 1007/1008 and JAK2 downstream signaling (Fig. 4 and Supporting Fig. S7). These findings demonstrate that AngII injection induced fibrosis as a result of AT1R-mediated activation of JAK2 and its downstream effectors.
Fig. 4.
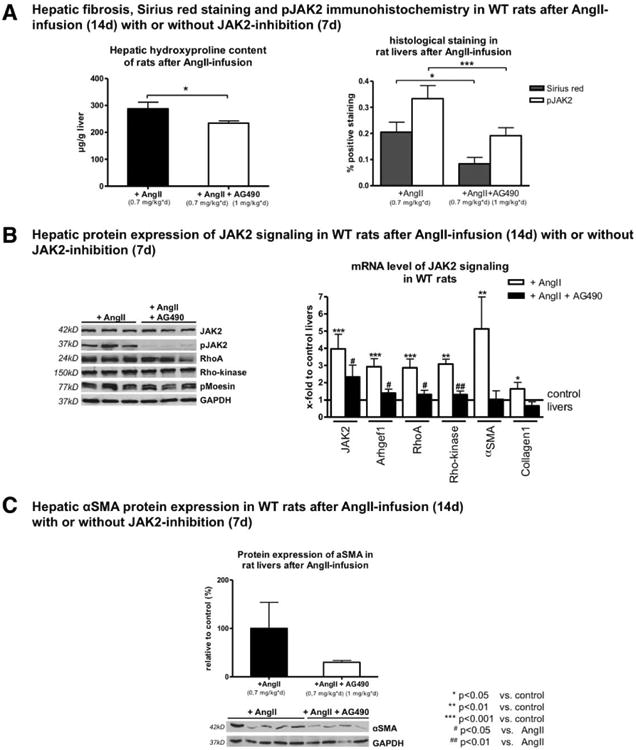
Stimulation of AT1R with AngII activated JAK2, but JAK2 inhibition attenuated hepatic fibrosis. (A) Hepatic hydroxyproline content and quantification of Sirius red and pJAK2 IHC staining are shown in rats after AngII infusion (0.7 mg/kg/day) for 14 days with or without additional AG490 (1 mg/kg/day) 7 days before sacrifice. JAK2 inhibition with AG490 blunted fibrosis development in these rats (for representative sections, see Supporting Fig. S7). (B) Protein levels of AT1R, JAK2, pJAK2, Arhgef1, RhoA, Rho-kinase, and pMoesin were analyzed using western blotting, and mRNA levels of JAK2, Arhgef1, RhoA, Rho-kinase, α-SMA, and Col1 were analyzed by RT-PCR in liver samples of rats after AngII infusion (0.7 mg/kg/day) for 14 days with or without additional AG490 (1 mg/kg/day) 7 days before sacrifice. (C) Hepatic protein levels of α-SMA are shown in rats after AngII infusion (0.7 mg/kg/day) for 14 days with or without additional AG490 (1 mg/kg/day) 7 days before sacrifice.
In these models, we could not detect any difference regarding thrombogenic or angiogenic effects of the JAK2 inhibitor, as assessed by histology.
Inhibition of JAK2 Pathway or Absence of AT1R With Consecutive Down-Regulation of JAK2 Pathway In Vivo Attenuated Fibrosis
To further assess the role of AT1R in vivo, we induced another form of chronic liver injury by means of CCl4 inhalation in AT1aR–/– mice and their respective WT littermates. (AT1aR is the subtype of AT1R in mice responsible for contraction of smooth muscle cells and important for development of liver fibrosis,10,16 further abbreviated as AT1R). Expression of JAK2 and its downstream effectors was significantly down-regulated in these mice (Fig. 5 and Supporting Fig. S8). This was associated with reduced hepatic fibrosis after challenge (Fig. 5 and Supporting Fig. S8). AngII induces fibrosis and activation of JAK2 and its downstream effectors in wt mice, but not in AT1R–/– mice. (Fig. 6A,B and Supporting Fig. S9).
Fig. 5.
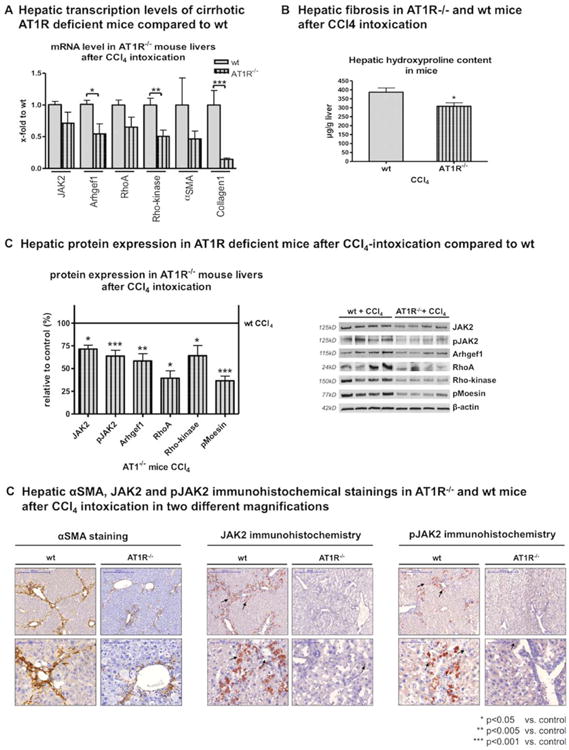
AT1R deficiency attenuated hepatic fibrosis. (A) Decreased hepatic transcription of JAK2, Arhgef1, RhoA, Rho-kinase, α-SMA, and Col1, (B) reduced hepatic hydroxyproline content, and (C) decreased protein expression of JAK2, pJAK2, Arhgef1, RhoA, Rho-kinase, and pMoesin in AT1R-deficient mice after CCl4-induced fibrosis, compared to CCl4-treated WT mice. (D) Representative sections of IHC stains for α-SMA, JAK2, and pJAK2 in AT1R-deficient mice versus WT mice after CCl4 treatment are shown (for quantification, see Supporting Fig. S8B).
Fig. 6.

AT1R deficiency attenuated hepatic fibrosis. AT1R-mediated activation of JAK2 signaling is predominant in primary HSCs and induced collagen production. (A) Hepatic hydroxyproline content, and protein expression of AT1R, JAK2, pJAK2, Arhgef1, RhoA, Rho-kinase, and pMoesin, were analyzed in liver samples of WT and AT1R-deficient mice after AngII infusion (0.7 mg/kg/day) or solvent infusion. Quantification of IHC staining for pJAK2 is also shown for these mice (for representative sections, see Supporting Fig. S9A). In AT1R-deficient mice, pJAK2 remained unchanged after AngII infusion and mice did not develop fibrosis. (B) Hepatic transcription levels of JAK2, Arhgef1, RhoA, Rho-kinase, α-SMA, and Col1 in liver samples of WT and AT1R-deficient mice after AngII infusion (0.7 mg/kg/day) or solvent infusion. (C) Protein expression of JAK2, pJAK2, Arhgef1, Rho-kinase, and pMoesin was analyzed by western blotting in activated (day 7) primary mouse HSCs, compared to quiescent (day 0) cells, hepatocytes, and KCs. Of note, expression of loading control proteins β-actin and GAPDH was higher in HSCs, compared to hepatocytes, despite the fact that the same amount of protein was used for each lane, whereas Ponceau S staining revealed equal loading. (D) Protein transcription level of JAK2, Arhgef1, RhoA, Rho-kinase, α-SMA, and Col1 in mouse primary HSCs, hepatocytes, and KCs. For cell culture experiments, minimum duplicates from a minimum of 3 different animals (primary cells) or samples (cell lines) were used.
Thus, JAK2 inhibition upon fibrosis induction blunted fibrosis. Similarly, AT1R-deficient animals showed attenuated fibrogenesis and lack of activation of JAK2 and its downstream effectors.
AT1R-Dependent Activation of JAK2 Pathway-Induced Activation of HSC and Collagen Production
In vitro experiments confirmed the results from IHC staining. The components of the JAK2 pathway (JAK2, pJAK2, Arhgef-1, Rho-kinase, and pMoesin) were highly expressed and activated in HSCs after their activation (day 7 in culture), when compared to hepatocytes and KCs (Fig. 6C,D), confirming that pJAK2 was localized in activated HSCs (as shown in Fig. 6C,D).
Activated HSCs showed increased expression and activation of components of the AT1R-mediated JAK2 pathway. To further decipher the role of JAK2, specifically for fibrogenesis, we overexpressed JAK2 in an immortalized human HSC line, namely, LX2 cells, using a JAK2 plasmid (Fig. 7A). We observed elevated transcription of its downstream effectors, RhoA and Rho-kinase, together with higher protein expression of α-SMA and collagen I (Col1) as markers for profibrotic activity of HSCs (Fig. 7B). Efficacy of JAK2 transfection is shown in Supporting Fig. S10.
Fig. 7.
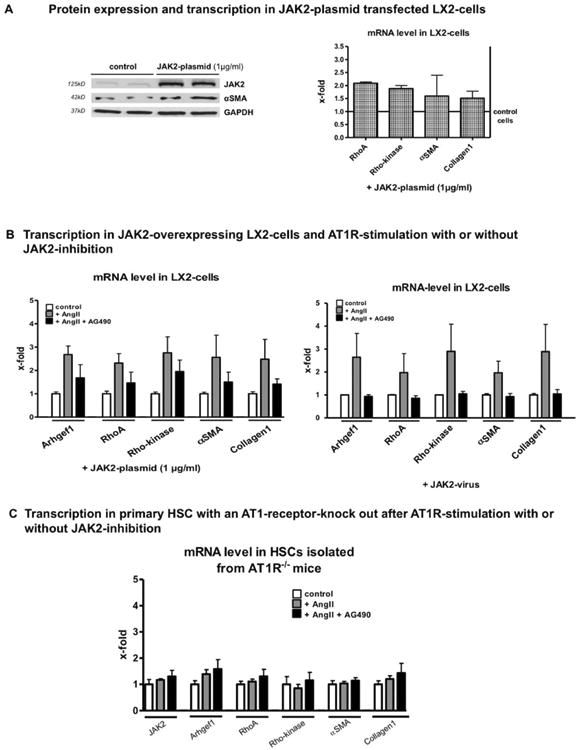
AT1R-mediated JAK2 stimulation activated and induced collagen production, which is attenuated by JAK2 inhibition. (A) LX2 cells transfected with a JAK2 plasmid (1 μg/mL) for 48 hours showed increased protein expression of JAK2 and α-SMA detected by western blotting, as well as increased transcription of JAK2, RhoA, Rho-kinase, α-SMA, and Col1 analyzed by RT-PCR. (B) Transfected JAK2-overexpressing LX2 cells incubated with AngII (10 μM) for 3 days showed further augmented mRNA levels of Arhgef1, RhoA, Rho-kinase, α-SMA, and Col1 measured by RT-PCR, but this was blunted after AG490 (5 μM) coincubation. LX2 cells transduced by a JAK2 adenovirus were incubated with AngII (10 μM) for 3 days and showed increased mRNA levels of Arhgef1, RhoA, Rho-kinase, α-SMA, and Col1 measured by RT-PCR, which was blunted after AG490 (5 μM) coincubation. (C) Primary HSCs isolated from AT1R–/– mice showed no effect of AngII or AngII and AG490. For cell culture experiments, minimum duplicates from a minimum of 3 different animals (primary cells) or samples (cell lines) were used.
Additional stimulation of these cells with AngII further increased the transcription of downstream effectors of JAK2 and their profibrotic activity (α-SMA and Col1; Fig. 7B). Again, the JAK2 inhibitor, AG490, blunted the AngII effect (Fig. 7B). In order to exclude a vector-specific effect, we repeated these experiments using adenoviral vectors containing JAK2 or LacZ (as a control) and obtained similar results (Fig. 7B). AT1R dependence was shown in further experiments using HSCs isolated from AT1R-deficient mice. In these mice, neither AngII nor AG490 elicited any significant effect on the transcription of AT1R/JAK2 pathway effectors (Fig. 7C). Efficacy of JAK2 transduction with the adenoviral vector is shown in Supporting Fig. S10.
In Vitro JAK2 Inhibition and Blocking Experiments
AT1R stimulation, assessed by incubation of human immortalized HSCs (LX2 cells) with AngII, induced JAK2 phosphorylation at Tyr 1007/1008 (Supporting Fig. S10). The pharmacologic inhibitor of JAK2, AG490, dose dependently blocked phosphorylation of JAK2 and expression of the downstream effectors, RhoA and Rho-kinase (Supporting Fig. S10). Also, the elevated Arhgef1 expression and Rho-kinase activation was blunted after AG490 coincubation (Fig. 8A). Experiments with human primary HSCs confirmed the findings in LX2 cells (Fig. 8B,C). AngII treatment of LX2 cells increased ROS, but after coincubation with AG490, ROS dropped to normal levels (Fig. 8E). Blocking experiments using AG490 (JAK2 inhibitor) or Losartan (AT1R blocker) demonstrated blunting of protein expression of downstream effectors of JAK2 (Arhgef1 and Rho-kinase) and α-SMA (activation marker) in primary HSCs similarly to LX2 cells (Fig. 8D). Interestingly, blockade of AT1R or inhibition of JAK2 had no effect on apoptosis or cell cycle (Supporting Fig. S10).
Fig. 8.
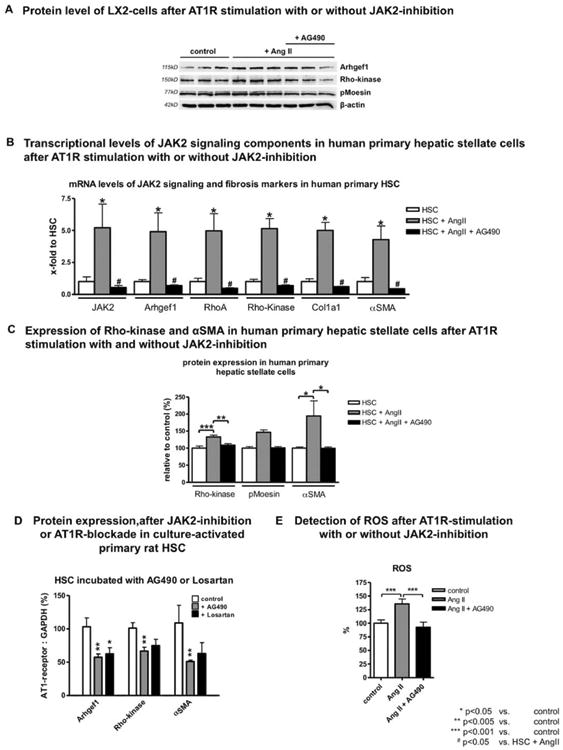
In vitro AT1R-mediated JAK2 activation and JAK2 inhibition in rodent and human HSCs. (A) Protein levels of human-derived LX2 cells after AngII treatment with or without JAK2 inhibition. The increased expression after AngII incubation of Arhgef1 and Rho-kinase as well as Rho-kinase activation, measured by phosphorylation of moesin, was blunted after coincubation with AG490. (B) mRNA levels of JAK2, Arhgef1, RhoA, Rho-kinase, Col1, and α-SMA and (C) protein levels of Rho-kinase, pMoesin, and α-SMA in human primary HSCs after AngII incubation, with or without JAK2 inhibition with AG490, compared to untreated control cells. (D) Primary HSCs from healthy rats were incubated with AG490 (1.5 μM) or AT1R blocker Losartan (10 μM) for 3 days, and expression of Arhgef1, Rho-kinase, and α-SMA was analyzed using western blotting. (E) ROS detection of LX2 cells incubated with AngII (10 μM) for 3 days with or without AG490 (5 μM), compared to untreated cells. For cell culture experiments, minimum duplicates from a minimum of 3 different animals (primary cells) or samples (cell lines) were used.
In summary, our in vitro data demonstrated that AT1R-dependent JAK2 induced activation and profibrotic activity of HSCs though its downstream effectors. Inhibition of JAK2 blunted AT1R-mediated activation, profibrotic properties, migration, and contraction of HSCs.
Discussion
Our in vivo and in vitro findings show the importance of AT1R-dependent JAK2 activation in hepatic fibrosis in mice, rats, and humans. These findings are uniformly supported by different approaches using several animal models of liver fibrosis as well as by pharmacological and genetic modulations. JAK2 expression and activation was found to occur mainly in activated HSCs, underlining the pivotal role of these cells for collagen production.
RAS is critically involved in the development of progressive fibrosis in different tissues (heart, kidney, and lung) as well as in the liver, mainly as a result of AT1R stimulation.6,34-36 In chronic liver injury, activation of RAS is a hallmark of progressive liver disease.2,7,14,15 RAS activation leads to sodium retention with formation of ascites in later stages of liver disease. But, it also leads to initiation, perpetuation, and augmentation of inflammatory and fibrogenic processes within the liver, where HSCs are key players.3
It has been shown only recently that AngII exerts its AT1R-dependent contractile effect in the vascular smooth muscle cells through phosphorylation of JAK2 and the nucleotide exchange factor for RhoA, Arhgef1, which then activates the RhoA cascade toward Rho-kinase, finally resulting in contraction of these cells.21 Importantly, JAK2-mediated signaling in fibrosis is caused by both increased JAK2 expression and increased JAK2 activation. In this landmark study, the researchers described the missing link between AT1R stimulation and the RhoA/Rho-kinase pathway in arterial smooth muscle cells as a potential mechanism for arterial hypertension.21 Here, we demonstrate that the same holds true for myofibroblastic-activated HSCs. We show that HSCs—but not hepatocytes or KCs— harbor an activated JAK2/RhoA/Rho-kinase signaling cascade that can be further stimulated by AngII and which is blocked or blunted by an AT1R blocker, AT1aR, deletion in mice (the subtype of AT1R in mice responsible for contraction of smooth muscle cells and important for development of liver fibrosis10,16) or JAK2 inhibition, respectively. This altered signaling clearly leads to functional changes with increased α-SMA and collagen expression of these cells. Of note, no effects on survival or cell-cycle progress were observed in these cells.
In line with these in vitro findings, we show that fibrosis in different animal models and in humans is associated with increased JAK2 expression and activation. Furthermore, fibrosis is attenuated by application of JAK2 inhibitors, whereas continuous exogenous AngII infusion9,37 increases collagen formation and HSC activation. In addition, we demonstrated in in vivo experiments that cells showing JAK2 phosphorylation were, in fact, activated myofibroblastic HSCs, because they were located in fibrotic septa and were positive for α-SMA, whereas other α-SMA-negative liver cells (e.g., hepatocytes and inflammatory cells) lacked evidence of JAK2 phosphorylation.
Activation of RAS is a uniform finding in advanced cirrhosis.14,15,38 One possible major reason for this could be a systemic hemodynamic derangement with splanchnic and peripheral vasodilation, leading to “underfilling” in the central intrathoracic compartment and reactive secretion of vasoconstrictors. However, the response from the peripheral and splanchnic vessels to AngII is inadequate, whereas the hepatic vascular bed overreacts. Although this has been shown repeatedly in human and animal models, it is only partially understood.14,15,38 It is questionable whether systemic RAS activation alone accounts for hepatic JAK2 activation, as shown here. Certainly, there must be a systemic effect because we were able to show that AngII infusion enhanced hepatic collagen formation and activated the AT1R/JAK2/Rho-kinase axis in the liver. However, there are probably also para-crine intrahepatic factors involved that lead to up-regulation of AT1R expression on activated HSCs and local formation of AngII with subsequent phosphorylation of JAK2, as shown for oxidative stress and the I kappa B kinase/RelA pathway.6,39 Furthermore, JAK2 may also be activated by cytokines or several concomitant pathological conditions in the liver.40-44
In our animal models, neither inflammatory infiltrates nor isolated KCs expressed pJAK2. Thombogenic and angiogenic properties of JAK2 were not observed in our models, as assessed by histology. We focused on the AT1R-dependent JAK2/Arhgef1/Rho-kinase pathway. Effects that might be driven by the JAK/STAT pathway, as shown for the transdifferentiation of HSC, I/R, ischemia/reperfusion liver injury, hepatocellular carcinoma, and steatohepatitis, could also play a role in liver cirrhosis.22,40,44 Indeed, these effects were present in our animal models as well.
In summary, in the present work, we showed that JAK2 expression and activation are increased in activated HSCs in fibrosis and inhibition of JAK2 decreased liver fibrosis. Recently, new JAK2 inhibitors have been released for human use in malignant disorders,45,46 which, based on these findings, might also be considered for treatment of fibrosis in a clinical setting. In this case, liver-specific targeting is warranted.
Supplementary Material
Acknowledgments
The authors thank G. Hack and S. Bellinghausen for their excellent technical assistance, as well as U.B. Kaupp and S. Dentler for their critical reading.
The study was supported by grants from Deutsche Forschungsgemeinschaft (SFB TRR57 projects 1, 10, 15, 18, and Q1).
Abbreviations
- Ab
antibody
- ACE
angiotensin converting enzyme
- AngII
angiotensin II
- AT1R
angiotensin-II type I receptor
- BDL
bile duct ligation
- Col1
collagen I
- DCFDA
dichlorofluorescein diacetate
- DMEM
Dulbecco's modified Eagle's medium
- ECL
enhanced chemiluminescence
- ECM
extracellular matrix
- FCS
fetal calf serum
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HSCs
hepatic stellate cells
- IHC
immunohistochemical
- IP
intraperitoneal
- I/R
ischemia-reperfusion
- JAK2
Janus kinase 2
- KCs
Kupffer cells
- mRNA
messenger RNA
- RAS
renin angiotensin system
- ROS
reactive oxygen species
- RT-PCR
realtime polymerase chain reaction
- pJAK2
phosphorylated JAK2
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- SEM
standard error of the mean
- α-SMA
alpha-smooth muscle actin
- STAT
signal transducers and activators of transcription
- TAA
thioacetamide
- WT
wild type
Footnotes
Potential conflict of interest: Nothing to report.
Supporting Information: Additional Supporting Information may be found in the online version of this article at the publisher's website.
References
- 1.WHO. The world health report 2002: reducing risks, promoting healthy life. Geneva: World Health Organisation; 2002. [DOI] [PubMed] [Google Scholar]
- 2.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655–1669. doi: 10.1053/j.gastro.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gressner AM, Weiskirchen R. Modern pathogenetic concepts of liver fibrosis suggest stellate cells and TGF-beta as major players and therapeutic targets. J Cell Mol Med. 2006;10:76–99. doi: 10.1111/j.1582-4934.2006.tb00292.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Parola M, Marra F, Pinzani M. Myofibroblast-like cells and liver fibro-genesis: Emerging concepts in a rapidly moving scenario. Mol Aspects Med. 2008;29:58–66. doi: 10.1016/j.mam.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 6.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, et al. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–1394. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bataller R, Gines P, Nicolas JM, Gorbig MN, Garcia-Ramallo E, Gasull X, et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 2000;118:1149–1156. doi: 10.1016/s0016-5085(00)70368-4. [DOI] [PubMed] [Google Scholar]
- 8.Croquet V, Moal F, Veal N, Wang J, Oberti F, Roux J, et al. Hemodynamic and antifibrotic effects of losartan in rats with liver fibrosis and/or portal hypertension. J Hepatol. 2002;37:773–780. doi: 10.1016/s0168-8278(02)00307-0. [DOI] [PubMed] [Google Scholar]
- 9.Bataller R, Gabele E, Parsons CJ, Morris T, Yang L, Schoonhoven R, et al. Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology. 2005;41:1046–1055. doi: 10.1002/hep.20665. [DOI] [PubMed] [Google Scholar]
- 10.Yang L, Bataller R, Dulyx J, Coffman TM, Gines P, Rippe RA, Brenner DA. Attenuated hepatic inflammation and fibrosis in angiotensin type 1a receptor deficient mice. J Hepatol. 2005;43:317–323. doi: 10.1016/j.jhep.2005.02.034. [DOI] [PubMed] [Google Scholar]
- 11.Sookoian S, Fernandez MA, Castano G. Effects of six months losartan administration on liver fibrosis in chronic hepatitis C patients: a pilot study. World J Gastroenterol. 2005;11:7560–7563. doi: 10.3748/wjg.v11.i48.7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Heller J, Shiozawa T, Trebicka J, Hennenberg M, Schepke M, Neef M, Sauerbruch T. Acute haemodynamic effects of losartan in anaesthetized cirrhotic rats. Eur J Clin Invest. 2003;33:1006–1012. doi: 10.1046/j.1365-2362.2003.01251.x. [DOI] [PubMed] [Google Scholar]
- 13.Heller J, Trebicka J, Shiozawa T, Schepke M, Neef M, Hennenberg M, Sauerbruch T. Vascular, hemodynamic and renal effects of low-dose losartan in rats with secondary biliary cirrhosis. Liver Int. 2005;25:657–666. doi: 10.1111/j.1478-3231.2005.01053.x. [DOI] [PubMed] [Google Scholar]
- 14.Bosch J, Garcia-Pagan JC. Complications of cirrhosis. I. Portal hypertension. J Hepatol. 2000;32:141–156. doi: 10.1016/s0168-8278(00)80422-5. [DOI] [PubMed] [Google Scholar]
- 15.Benvegnu L, Gios M, Boccato S, Alberti A. Natural history of compensated viral cirrhosis: a prospective study on the incidence and hierarchy of major complications. Gut. 2004;53:744–749. doi: 10.1136/gut.2003.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82–C97. doi: 10.1152/ajpcell.00287.2006. [DOI] [PubMed] [Google Scholar]
- 17.Trebicka J, Hennenberg M, Laleman W, Shelest N, Biecker E, Schepke M, et al. Atorvastatin lowers portal pressure in cirrhotic rats by inhibition of RhoA/Rho-kinase and activation of endothelial nitric oxide synthase. Hepatology. 2007;46:242–253. doi: 10.1002/hep.21673. [DOI] [PubMed] [Google Scholar]
- 18.Trebicka J, Hennenberg M, Schulze Probsting A, Laleman W, Klein S, Granzow M, et al. Role of beta3-adrenoceptors for intrahepatic resistance and portal hypertension in liver cirrhosis. Hepatology. 2009;50:1924–1935. doi: 10.1002/hep.23222. [DOI] [PubMed] [Google Scholar]
- 19.Trebicka J, Hennenberg M, Odenthal M, Shir K, Klein S, Granzow M, et al. Atorvastatin attenuates hepatic fibrosis in rats after bile duct ligation via decreased turnover of hepatic stellate cells. J Hepatol. 2010;53:702–712. doi: 10.1016/j.jhep.2010.04.025. [DOI] [PubMed] [Google Scholar]
- 20.Klein S, Van Beuge MM, Granzow M, Beljaars L, Schierwagen R, Kilic S, et al. HSC-specific inhibition of Rho-kinase reduces portal pressure in cirrhotic rats without major systemic effects. J Hepatol. 2012;57:1220–1227. doi: 10.1016/j.jhep.2012.07.033. [DOI] [PubMed] [Google Scholar]
- 21.Guilluy C, Bregeon J, Toumaniantz G, Rolli-Derkinderen M, Retailleau K, Loufrani L, et al. The Rho exchange factor Arhgef1 mediates the effects of angiotensin II on vascular tone and blood pressure. Nat Med. 2010;16:183–190. doi: 10.1038/nm.2079. [DOI] [PubMed] [Google Scholar]
- 22.Lakner AM, Moore CC, Gulledge AA, Schrum LW. Daily genetic profiling indicates JAK/STAT signaling promotes early hepatic stellate cell transdifferentiation. World J Gastroenterol. 2010;16:5047–5056. doi: 10.3748/wjg.v16.i40.5047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Minicis S, Seki E, Oesterreicher C, Schnabl B, Schwabe RF, Brenner DA. Reduced nicotinamide adenine dinucleotide phosphate oxidase mediates fibrotic and inflammatory effects of leptin on hepatic stellate cells. Hepatology. 2008;48:2016–2026. doi: 10.1002/hep.22560. [DOI] [PubMed] [Google Scholar]
- 24.Domenicali M, Caraceni P, Giannone F, Baldassarre M, Lucchetti G, Quarta C, et al. A novel model of CCl4-induced cirrhosis with ascites in the mouse. J Hepatol. 2009;51:991–999. doi: 10.1016/j.jhep.2009.09.008. [DOI] [PubMed] [Google Scholar]
- 25.Laleman W, Vander Elst I, Zeegers M, Servaes R, Libbrecht L, Roskams T, et al. A stable model of cirrhotic portal hypertension in the rat: thioacetamide revisited. Eur J Clin Invest. 2006;36:242–249. doi: 10.1111/j.1365-2362.2006.01620.x. [DOI] [PubMed] [Google Scholar]
- 26.Rudolph V, Andrie RP, Rudolph TK, Friedrichs K, Klinke A, Hirsch-Hoffmann B, et al. Myeloperoxidase acts as a profibrotic mediator of atrial fibrillation. Nat Med. 2010;16:470–474. doi: 10.1038/nm.2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huss S, Schmitz J, Goltz D, Fischer HP, Buttner R, Weiskirchen R. Development and evaluation of an open source Delphi-based software for morphometric quantification of liver fibrosis. Fibrogenesis Tissue Repair. 2011;3:10. doi: 10.1186/1755-1536-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trebicka J, Racz I, Siegmund SV, Cara E, Granzow M, Schierwagen R, et al. Role of cannabinoid receptors in alcoholic hepatic injury: steatosis and fibrogenesis are increased in CB(2) receptor-deficient mice and decreased in CB(1) receptor knockouts. Liver Int. 2011;31:862–872. doi: 10.1111/j.1478-3231.2011.02496.x. [DOI] [PubMed] [Google Scholar]
- 29.Wojtalla A, Herweck F, Granzow M, Klein S, Trebicka J, Huss S, et al. The endocannabinoid N-arachidonoyl dopamine (NADA) selectively induces oxidative stress-mediated cell death in hepatic stellate cells, but not in hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2012;302:G873–G887. doi: 10.1152/ajpgi.00241.2011. [DOI] [PubMed] [Google Scholar]
- 30.Coenen M, Nischalke HD, Kramer B, Langhans B, Glassner A, Schulte D, et al. Hepatitis C virus core protein induces fibrogenic actions of hepatic stellate cells via toll-like receptor 2. Lab Invest. 2011;91:1375–1382. doi: 10.1038/labinvest.2011.78. [DOI] [PubMed] [Google Scholar]
- 31.Glassner A, Eisenhardt M, Kramer B, Korner C, Coenen M, Sauerbruch T, et al. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL-and NKG2D-dependent manner. Lab Invest. 2012;92:967–977. doi: 10.1038/labinvest.2012.54. [DOI] [PubMed] [Google Scholar]
- 32.Semela D, Das A, Langer D, Kang N, Leof E, Shah V. Platelet-derived growth factor signaling through ephrin-b2 regulates hepatic vascular structure and function. Gastroenterology. 2008;135:671–679. doi: 10.1053/j.gastro.2008.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein S, Klösel J, Schierwagen R, Körner C, Granzow M, Huss S, et al. Atorvastatin induces senescence in activated hepatic stellate cells and attenuates hepatic fibrosis in rats. Lab Invest. 2012;92:1440–1450. doi: 10.1038/labinvest.2012.106. [DOI] [PubMed] [Google Scholar]
- 34.Ma TK, Kam KK, Yan BP, Lam YY. Renin-angiotensin-aldosterone system blockade for cardiovascular diseases: current status. Br J Pharmacol. 2010;160:1273–1292. doi: 10.1111/j.1476-5381.2010.00750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wolf G. Novel aspects of the renin-angiotensin-aldosterone-system. Front Biosci. 2008;13:4993–5005. doi: 10.2741/3058. [DOI] [PubMed] [Google Scholar]
- 36.Uhal BD, Li X, Piasecki CC, Molina-Molina M. Angiotensin signalling in pulmonary fibrosis. Int J Biochem Cell Biol. 2012;44:465–468. doi: 10.1016/j.biocel.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moreno M, Ramalho LN, Sancho-Bru P, Ruiz-Ortega M, Ramalho F, Abraldes JG, et al. Atorvastatin attenuates angiotensin II-induced inflammatory actions in the liver. Am J Physiol Gastrointest Liver Physiol. 2009;296:G147–G156. doi: 10.1152/ajpgi.00462.2007. [DOI] [PubMed] [Google Scholar]
- 38.Hennenberg M, Trebicka J, Sauerbruch T, Heller J. Mechanisms of extrahepatic vasodilation in portal hypertension. Gut. 2008;57:1300–1314. doi: 10.1136/gut.2007.144584. [DOI] [PubMed] [Google Scholar]
- 39.Oakley F, Teoh V, Ching ASG, Bataller R, Colmenero J, Jonsson JR, et al. Angiotensin II activates I kappaB kinase phosphorylation of RelA at Ser 536 to promote myofibroblast survival and liver fibrosis. Gastro-enterology. 2009;136:2334–2344.e1. doi: 10.1053/j.gastro.2009.02.081. [DOI] [PubMed] [Google Scholar]
- 40.Shi SY, Martin RG, Duncan RE, Choi D, Lu SY, Schroer SA, et al. Hepatocyte-specific deletion of Janus kinase 2 (JAK2) protects against diet-induced steatohepatitis and glucose intolerance. J Biol Chem. 2012;287:10277–10288. doi: 10.1074/jbc.M111.317453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sos BC, Harris C, Nordstrom SM, Tran JL, Balazs M, Caplazi P, et al. Abrogation of growth hormone secretion rescues fatty liver in mice with hepatocyte-specific deletion of JAK2. J Clin Invest. 2011;121:1412–1423. doi: 10.1172/JCI42894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu FM, Li QL, Gao Q, Jiang JH, Zhu K, Huang XY, et al. IL-17 induces AKT-dependent IL-6/JAK2/STAT3 activation and tumor progression in hepatocellular carcinoma. Mol Cancer. 2011;10:150. doi: 10.1186/1476-4598-10-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu HC, Qin HY, He F, Wang L, Fu W, Liu D, et al. Canonical notch pathway protects hepatocytes from ischemia/reperfusion injury in mice by repressing reactive oxygen species production through JAK2/STAT3 signaling. Hepatology. 2011;54:979–988. doi: 10.1002/hep.24469. [DOI] [PubMed] [Google Scholar]
- 44.Freitas MC, Uchida Y, Zhao D, Ke B, Busuttil RW, Kupiec-Weglinski JW. Blockade of Janus kinase-2 signaling ameliorates mouse liver damage due to ischemia and reperfusion. Liver Transpl. 2010;16:600–610. doi: 10.1002/lt.22036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Verstovsek S, Kantarjian H, Mesa RA, Pardanani AD, Cortes-Franco J, Thomas DA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363:1117–1127. doi: 10.1056/NEJMoa1002028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lafave LM, Levine RL. JAK2 the future: therapeutic strategies for JAK-dependent malignancies. Trends Pharmacol Sci. 2012;33:574–582. doi: 10.1016/j.tips.2012.08.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.