

Abstract
Genetic factors play an important role in determining risk and resistance to increased breast cancer. Recent technological advances have made it possible to analyze hundreds of thousands of single nucleotide polymorphisms in large-scale association studies in humans and have resulted in identification of alleles in over 20 genes that influence breast cancer risk. Despite these advances, the challenge remains in identifying what the functional polymorphisms are that confer the increased risk, and how these genetic variants interact with each other and with environmental factors. In rodents, the incidence of mammary tumors varies among strains, such that they can provide alternate ideas for candidate pathways involved in humans. Mapping studies in animals have unearthed numerous loci for breast cancer susceptibility that have been validated in human populations. In a reciprocal manner, knockin and knockout mice have been used to validate the tumorigenicity of risk alleles found in population studies. Rodent studies also underscore the complexity of interactions among alleles. The fact that genes affecting risk and resistance to mammary tumors in rodents depend greatly upon the carcinogenic challenge emphasizes the importance of gene x environment interactions. The challenge to rodent geneticists now is to capitalize on the ability to control the genetics and environment in rodent models of tumorigenesis to better understand the biology of breast cancer development, to identify those polymorphisms most relevant to human susceptibility and to identify compensatory pathways that can be targeted for improved prevention in women at highest risk of developing breast cancer.
Keywords: Breast cancer, Animal models, Genetic mapping, Modifier genes, p53
Unveiling the Roots of Breast Cancer
Breast cancer remains the most common cancer in women occurring more than 2.5-fold more frequently than any other malignancy among women in the United States [1], and the rapid rise in breast cancer among developing nations is a grave trend as healthcare resources are already limiting in these regions [2]. Environmental factors are undoubtedly important in determining breast cancer risk, with the most reproducible factors identified in epidemio-logic studies being early menarche and late menopause, which likely reflects the important role of ovarian hormones in promoting breast cancer [3–5]. However, the study of twins has suggested that as much as 27% of sporadic breast cancer risk can be attributed to genetic factors [6]. Results from genome-wide association studies (GWAS) support the significant role of common, low-penetrance risk alleles in determining breast cancer risk. Even in cases of hereditary breast cancer, less than one third of these breast cancers can be explained by known moderate-high penetrance alleles in genes such as BRCA1, BRCA2, PTEN and TP53. Therefore polygenic models are being developed to explain the remaining familial risk. These models assume common, low penetrance risk alleles exist in numerous genes. Although the risk alleles are common, a relatively small proportion of carriers develop disease because the increase in risk is small, or the carriers must also be exposed to specific environmental agents to result in breast cancer, or the carriers also possess other compensatory alleles that decrease risk [7, 8].
The study of the high penetrance breast cancer alleles has exposed a consistent theme implicating double strand break DNA repair as a fundamental pathway protecting the breast epithelium from carcinogenesis. This view is affirmed by the observation that direct DNA damage, especially double strand breaks resulting from ionizing radiation, is the best documented carcinogenic exposure associated with breast cancer in women [9, 10]. However, it is also clear that compensatory pathways exist that can overcome deficiencies in these fundamental pathways. Initial estimates of risk associated with BRCA1/2 were subject to ascertainment biases due to use of probands with very frequent breast cancers. It is now clear that the risk associated with BRCA1/2 mutations is highly variable and is likely to be due to differences in genetic background among individuals [11, 12].
While our understanding of breast cancer genetics has developed extensively in recent years, and advances in genomics have rapidly expanded the number of breast cancer risk alleles identified in humans, the field is now confronted by several daunting challenges. First, the GWAS studies identify genomic regions linked to risk, but defining the identity of the underlying genes has been problematic. Second, it is unlikely that all of the inherited risk alleles can be detected using association strategies in humans as the statistical power to detect infrequent low-moderate risk alleles remains insufficient, even with very large studies. Further, GWAS studies will have genomic regions of limited marker density where alleles may be missed. Third, it is becoming clear that there are significant interactions between risk alleles and between risk alleles and environmental factors that are challenging to define in heterogeneous human populations. Lastly, but most importantly, is the challenge to adequately translate the knowledge of breast cancer risk alleles into actions for the benefit of patients. A comparison of the approaches of GWAS in humans and mapping of quantitative trait loci in mice has recently been described in detail [13]. In this review, we highlight some examples where rodent models can contribute to overcoming these challenges to the breast cancer genetics community (Fig. 1).
Figure 1.
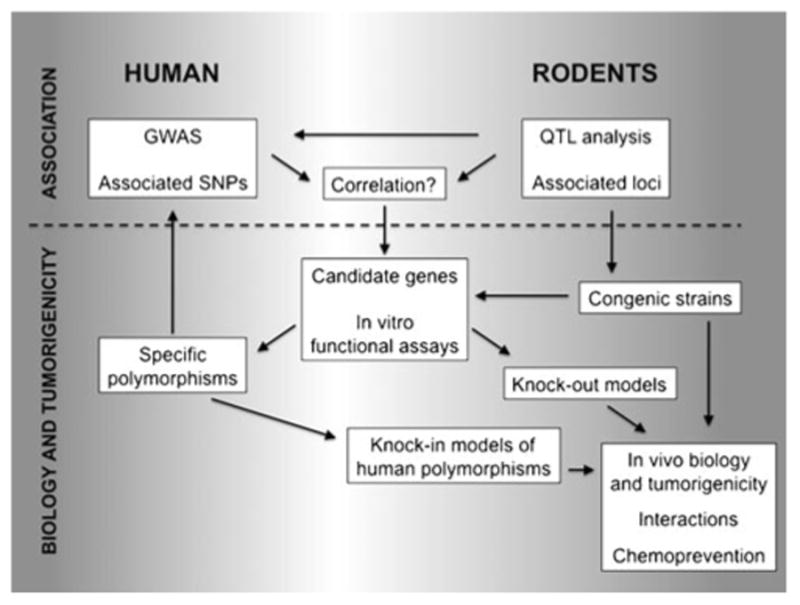
Strategies for unveiling and understanding genetic breast cancer susceptibility factors. Association studies in humans or rodents can identify genomic regions that confer risk. Typically, candidate genes containing polymorphisms are then selected for specific in vitro biological analysis to demonstrate potential functional significance based on our current understanding of cancer biology. Information from rodent models can feed into the candidate gene selection process. In addition, rodent models with defined genetics—either congenic strains possessing specific susceptibility/resistance loci, gene knockouts, or humanized gene knockins of specific polymorphisms—can be generated for in vivo biological testing of the tumorigenicity of risk alleles, and for the analysis of gene-gene or gene-environment interactions. Further, genetic risk pathways identified in rodents may provide targets for chemopreventive strategies and agents that can then be tested in these defined animal models
Rodent Models for Discovery of Hereditary Breast Cancer Alleles
Initial knockout studies in mice of breast cancer-associated genes (eg. Trp53, Brca1, Brca2, Atm) did not result in a high incidence of mammary tumors. Therefore, it was unclear if the function of these genes or if the process of carcinogenesis differed in mice, bringing into question the utility and relevance of mice to the study of breast cancer genetics. However, it is now recognized that this situation highlights the complexities of genetic backgrounds and genetic interactions, and the importance they play in the determination of risk, even in the context of highly penetrant alleles.
Modifier Loci in the Trp53+/− Mouse Model of Li-Fraumeni Syndrome
Li-Fraumeni Syndrome (LFS) is characterized by the occurrence of various cancers at an early age, including breast cancer, soft tissue sarcomas, osteosarcomas, and adrenocortical carcinoma [14]. Approximately half of LFS families carry germline mutations in the tumor suppressor gene TP53, which confer a 50% risk of developing breast cancer by 50 years of age [15] with 80% of women between 16 and 45 years of age developing breast cancer and lifetime incidence approaching 100% [16]. Mice heterozygous for the p53 gene (designated Trp53 in mice) are a model for LFS and succumb to a range of tumor types. While breast cancer is the most common tumor in women bearing heterozygous mutations in the TP53 gene, there was a near complete absence of mammary tumors in Trp53+/− mice in the C57BL/6 strain [17, 18]. However, tumor phenotypes associated with loss of Trp53 differed among strains of mice [19, 20]. This is highlighted by the prevalence of mammary tumors in BALB/c mice heterozygous for the p53 gene, with 55% of females developing spontaneous mammary tumors at 8–14 months of age [21], whereas 129/Sv, C57BL/6 and DBA2 mice are nearly completely resistant to mammary tumors [19–22]. Of note, while the risk of mammary tumors varied among strains, tumors in other tissues (e.g. lymphomas) had a similar prevalence in BALB/c-Trp53+/− and C57/BL6J-Trp53+/− mice. Thus the C57BL/6 J strain possesses alleles that can compensate for deficiencies in a major tumor suppressor pathway such as p53 in a mammary gland specific manner, demonstrating that in mice, as with women, the genetic background in which a mutation occurs can greatly impact on the outcome.
Genetic mapping strategies using deficiency in the p53 tumor suppressor gene have proven successful in localizing a number of natural genetic variants that contribute to development of spontaneous mammary tumors. Genetic linkage analysis using crosses of BALB/c and C57BL/6 mice that are Trp53+/− have identified at least 3 different modifiers. Using a backcross approach, a major recessive modifier locus (designated SuprMam1 for Suppressor of Mammary tumors) was identified on distal chromosome 7 with BALB/c alleles associated with ∼2-fold increase in relative risk [23]. In an independent study, a dominant-acting locus (designated Mtsm1) on proximal chromosome 7 (which does not overlap SuprMam1) and a recessive-acting modifier on chromosome X were identified [24].
Within the SuprMam1 locus, the tumor suppressor gene Dmbt1 (Deleted in malignant brain tumors 1) was investigated further as it showed reduced expression in the BALB/c-Trp53+/− mammary tissue (but not in small intestine) compared to the C57BL/6-Trp53+/− mice, consistent with a mammary-specific tumor suppressor role. Reduced expression levels in normal human breast tissue were also associated with the occurrence of sporadic breast cancer in women [23]. Further, a promoter polymorphism that reduces expression of DMBT1 was recently shown to be associated with increased risk of breast cancer (odds ratio = 1.66) among women >60 years old [25]. These studies demonstrate the potential use of Trp53+/− mice as a sensitized background to screen for low-penetrance modifiers of sporadic breast cancer risk in women.
The complexity of the SuprMam1 locus, however requires further investigation, as finer mapping now suggests that the two apparent peaks may indeed represent distinct genes (Griner, Blackburn, Jerry; unpublished data). Further, the genetic polymorphism underlying the expression difference between BALB/c and C57BL/6 mice is yet to be identified. While the relevance of the Trp53+/−model to humans is evident in its recapitulation of LFS, the long latency of the spontaneous mammary tumors (8–14 months) makes extensive phenotyping of additional backcrosses or congenic strains to further define the identified loci prohibitive. Unambiguous resequencing of the Dmbt1 gene is hindered by the repetitive nature of elements within the gene itself, and resequencing up to 3 kb upstream has failed to find any polymorphisms between BALB/c and C57BL/6 (Blackburn, unpublished data), leaving the potential for the variant altering gene expression to be distant from the gene itself. Furthermore, in the publicly available Cancer Genetics Markers of Susceptibility (CGEMS) GWAS data, no single nucleotide polymorphisms (SNPs) in or near DMBT1 were included, highlighting the limitation of GWAS studies to identify alleles which could be significant modifiers of risk if they are located within regions of limited marker coverage.
Genetic Complexities Unveiled in Rat Models of Mammary Carcinogensis
The incidence of spontaneous and induced mammary tumors also varies substantially among strains of rats (reviewed in ref 26). Dimethyl-α-benzanthracene (DMBA) is a potent inducer of mammary tumors in rodents and has been used extensively to identify loci contributing to mammary tumors by genetic linkage mapping. While DMBA is not encountered normally, it is representative of polyaliphatic hydrocarbons which are common environmental contaminants and have been associated with increased risk of breast cancer in some epidemiologic studies. As mammary tumors develop rapidly (3–5 months) after exposure to DMBA, genetic linkage studies have yielded a trove of genes that has been reviewed extensively [26]. Crosses between strains of rats that are susceptible (Wistar-Furth) or resistant (Copenhagen and Wistar-Kyoto) identified eight different loci (Mcs1-8).
The potential genetic complexity of a locus discovered by association or mapping studies is illustrated by the Mcs loci for susceptibility to DMBA induced mammary tumors. Finer mapping using congenic rat strains revealed that Mcs1 and Mcs5 are each composed of at least 3 independent alleles [27]. In the case of Mcs5, the 3 sub-alleles had similar absolute relative risk contributions, yet sub-alleles -a and -c contribute to resistance while sub-allele -b confers increased sensitivity [28]. The fact that Mcs5 displayed one of the strongest associations with mammary tumor susceptibility indicates that these alleles are interacting in more than an additive fashion, or detection of this locus would have been difficult [27]. While genetic models of human breast cancer most often assume independence among the risk alleles, these rodent models clearly demonstrate that complex interactions among risk alleles can occur, and that this can augment or compensate for the effects of other loci.
Further characterization of the Mcs5a locus demonstrates another benefit of the positional cloning approach. Ultrafine mapping using congenic strains of Mcs5a found it to contain at least two interacting elements: Mcs5a1 and Mcs5a2. These two elements need to be present on the same chromosome (i.e. cis-acting) for the resistance phenotype to be observed [29], and interestingly are both localized to non-coding DNA [27]. Emerging data from the Gould lab suggest that Mcs5a1 and Mcs5a2 interact physically, presumably by CTCF-mediated looping, to create a structural element affecting long-range gene expression [27]. Importantly, the relevance of these loci to human disease has been affirmed by association studies of the human orthologs with breast cancer in two case–control studies. MCS5A1 and MCS5A2 were independently associated with breast cancer risk, with odds ratios of 1.19 and 0.77 and minor allele frequencies of 0.26 and 0.12 for MCS5A1 and MCS5A2 respectively, classifying them as high population frequency, low penetrance modifier alleles [29]. This represents a novel genetic element determining breast cancer risk discovered through the use of rodent models, unbiased by our functional understanding of genetic elements in the region.
Modifying the Phenotype of Breast Cancer
Compared to the human population, the relative uniformity of tumors obtained in rodent models has allowed the mapping of loci involved in determining the aggressiveness and metastatic behavior in addition to the frequency and latency of mammary tumors [30]. The polyoma virus middle T antigen (PyMT) is a potent oncogene that strongly induces signaling through Src and Ras family PI3 kinases. Though polyoma viruses have not been implicated in human breast cancers, increased Ras signaling is commonly observed in breast cancers such as those overexpressing HER2. When PyMT is expressed in the mammary gland under the mouse mammary tumor virus promoter (MMTV-PyMT), tumors develop rapidly (1–2 months) and recapitulate the stages of progression observed in breast cancers [31]. A large-scale screen of the effects of MMTV-PyMT in 27 different strains of mice found that resistance to this powerful oncogenic stimulus was observed in seven strains [32]. Genetic mapping studies identified six loci affecting latency, tumor burden or metastasis. Of these loci, Mtes1, associated with metastasis efficiency, contained the candidate gene Sipa1 [33], and subsequent studies of polymorphisms in this gene have been associated with metastasis-free survival [34, 35]. Similar studies in rats have identified two further loci affecting aggressiveness [26, 36]. Thus germline polymorphisms alter the likelihood of a tumor to metastasize. Screening for a panel of these alleles may be useful in guiding treatment decisions for patients, and may improve the accuracy of predictions based on gene expression patterns in tumors [30].
Gene-Gene and Gene-Environment Interactions in Rodents
Interactions Between Genetics and Radiation-Induction of Tumors
The sensitivity to mammary tumors induced by hormones or DMBA differs greatly among strains of rats, demonstrating the importance of interactions between genes and the carcinogenic stimulus. Ionizing radiation, which causes double strand DNA breaks, is a well documented carcinogenic exposure associated with breast cancer in women [9, 10, 37] to which rodents are also vulnerable [38–40]. BALB/c mice are uniquely sensitive to ionizing radiation and have high rates of radiation-induced apoptosis in thymus which are linked to modifier loci designated as Rapop1, 2 and 3 on mouse chromosomes 16, 9 and 3 respectively [41, 42]. However, these loci did not correspond with those mapped for mammary tumor sensitivity on the Trp53+/− background [23, 24], providing another example of both tissue and carcinogen specificity in the phenotype of modifier loci. Polymorphisms in the gene encoding the catalytic subunit of DNA Protein Kinase (Prkdc) are located within Rapop1 [43] and Atm is within Rapop2. Linkage of a known polymorphism in Prkdc to the high incidence of spontaneous mammary tumors in BALB/c-Trp53+/− mice was tested [20], however no association was found. Furthermore, DBA/2-Trp53+/− mice carry the BALB/c allele of Prkdc but are resistant to radiation-induced tumors while BALB/c-Trp53+/− mice are susceptible [22]. While these results do not exclude a role for Prkdc in mammary tumorigenesis, its role appears to be restricted to radiation-induced tumors.
Homozygous mutations in the ATM gene belie a rare, recessively-inherited neurological syndrome. Women who are heterozygous carriers are sensitive to ionizing radiation and have increased incidence of breast cancer. Despite substantial genetic and functional evidence supporting a role for ATM mutations in predisposing women to breast cancer, mice heterozygous for a null allele of Atm failed to develop mammary tumors [44]. However, when analyzed in combination with Trp53, the compound Atm+/−, Trp+/−heterozygotes developed spontaneous mammary tumors in 50% of females compared to 32% in the Atm+/+, Trp53+/−controls. Irradiation of the compound heterozygous mice decreased mammary tumor latency by ∼30 weeks and increased the number of tumors per mouse. Therefore, the mouse model recapitulates the features of radiation-sensitivity to mammary tumors conferred by heterozygosity for Atm; however, this was only evident in the background of genetic deficiencies in Trp5 3 [44]. Extending this result to humans, it suggests that ATM heterozygosity may only confer increases in breast cancer risk in those women in whom the p53 pathway is also compromised.
Interactions Between Hormones, Diet and Genes
In addition to carcinogen exposure, variations in hormone exposure (endogenous and exogenous) and diet are likely to contribute to cancer susceptibility in humans, and therefore to the confounding heterogeneity in population studies. Further, interactions between these environmental factors and genetics are clearly evident in rodent models. The ACI strain is susceptible to estrogen-induced mammary tumors (3–6 month latency) while Copenhagen and Brown-Norway rats are resistant. Crosses between these strains have yielded seven distinct loci controlling the latency and multiplicity of mammary tumors [45, 46]. There is very little overlap between the loci for estrogen stimulated mammary tumors and the DMBA-induced loci discussed earlier. These results emphasize the specificity of interactions between risk alleles and environmental exposures in driving tumorigenesis and determining the outcomes [26]. Interactions between genotype and a high-fat diet have recently been examined using the MMTV-PyMT mouse model of breast cancer. The study identified 13 loci modifying mammary cancer risk and pulmonary metastasis, the majority of loci demonstrating interactions with a very high-fat diet [47]. While the underlying genes and biology are still being investigated [48], these studies highlight the potential to unravel these complex interaction issues in the shorter latency animal models.
Validating Human Polymorphisms in Mice
The examples of SuprMam1, Mcs5 and Mtes1 demonstrate the validation of loci discovered in rodent models in human population studies. Although GWAS in humans have proven effective in identifying regions linked to breast cancer risk, and in vitro experiments can determine the functional effect of polymorphisms at the molecular level, it is ultimately necessary to functionally validate the impact of candidate risk alleles on tumor phenotypes (Fig. 1). Polymorphisms in CHEK2 were among the first moderately penetrant alleles to be associated with increased risk of breast cancer, but effects were variable among studies. To gain a better understanding of the impact of this allele, mice bearing a knockin of the protein-truncating polymorphism Chk2*1100delC were developed [49]. Homozygotes for this allele did produce more tumors in various tissues than wild-type mice, confirming that this allele can elevate cancer risk. This was especially so in female mice, indicating an interaction between hormones and this allele [50]. Administration of DMBA also showed a significant reduction in latency and increase in the incidence of mammary tumors in the homozygous Chk2*1100delC mice compared to the wild-type indicating an interaction between the carcinogen and the risk allele.
Given the fundamental role of p53 in the decision to arrest and repair or undergo apoptosis in response to cellular stresses, it is not surprising that over 150 factors have been implicated in regulating or mediating the actions of p53 [51]. Thus, there is a vast potential of allelic polymorphisms that could impact on p53 function. In a search for such polymorphisms, a SNP in the intronic promoter of MDM2, a key p53 inactivating protein, was described which increased MDM2 expression and associated with decreased p53 function in cell lines [52]. Association of the G allele of SNP309 with earlier age of onset and increased tumor multiplicity in LFS patients was also demonstrated [52, 53]. However, subsequent studies of this SNP in more extensive populations has yielded conflicting reports [54]. To directly assess the impact of MDM2 SNP309 on cancer development, knockin mice bearing either the Mdm2SNP309G or Mdm2SNP309T alleles were characterized. Homozygosity for SNP309G resulted in higher Mdm2 levels, reduced p53 levels, and decreased apoptosis. Further, this was shown to result in a reduced latency of spontaneous tumor development, in both wild-type mice and mice bearing a mutant p53 (hotspot missense) allele, clearly demonstrating the ability of this allele to contribute to cancer susceptibility [55]. As these experiments were carried out on a C57BL/6 background, the incidence of mammary tumors was very low and so the impact of this SNP on breast cancer could not be assessed. Also of note was the lack of gender effects in the mouse, unlike in human cancers [54, 56]. Nonetheless, this demonstrates the ability of rodent models to clarify the tumorigenic potential of human SNPs where population studies continue to produce conflicting results.
Translating Genetic Knowledge into Better Health Outcomes
One of the most important challenges to the genetics community is to exploit our increasing knowledge of breast cancer genetics for the benefit of patients. The abundance of common low-penetrance modifiers of risk suggests that it may be very difficult to use this information for the prediction of an individual's risk in the absence of a high penetrance allele [57]. This is particularly the case when we concede that we are only scratching the surface when it comes to understanding the complex interactions that may be occurring between different alleles (known and unknown) or alleles and the environment. However, the ability to control the genetics, carcinogen and hormone exposures in animal models gives the opportunity to define the relative importance of these factors in a given genetic context, which can contribute to public health decisions and policy. For example, carrying alleles interacting with radiation to increase risk of mammary tumors would contraindicate mammographic screening, or women with risk alleles that interact with hormonal exposure such as MDM2-SNP309 may be advised differently about hormone replacement therapy options compared to non-carriers [54]. Treatment decisions may also be influenced. Where variants have been identified that can alter the efficiency of metastasis, patients may be screened for these germline variants, and this information used in conjunction with tumor expression phenotypes to guide treatment decisions of patients [30]. In the context of high risk alleles, our knowledge of modifier alleles could influence risk management strategies of patients. In the case of BRCA2 mutation carriers, analysis of seven risk-associated SNPs found that the 5% of BRCA2 carriers at highest risk (i.e., between 95th and 100th percentiles) had 80–96% chance of developing breast cancer by age 80, compared with 42–50% for the 5% of carriers at lowest risk [58]. This difference in risk may be sufficient to prompt a woman to undertake more severe preventive measures.
Even in cases where genetic testing can define a high risk individual, women may choose not to be tested as the interventions available to prevent the occurrence of breast cancer are limited and are not appealing (prophylactic mastectomy or oophorectomy) [59]. However, the genetic complexity unveiled in recent years demonstrates that there is a wealth of pathways that can compensate for high risk alleles. Thus while the majority of variants underlying risk of mammary tumors in rodents may not be expected to be present in humans, they may unearth compensatory pathways that are ideal targets for therapeutic interventions for chemoprevention or treatment of breast cancers arising from inherited risk alleles or spontaneous mutations. For example, the lack of mammary tumors in C57BL/6-Trp53+/− mice indicates that the C57BL/6 strain harbors alleles with sufficient power to overcome even a defect as fundamental as p53 deficiency to protect the mammary epithelium and prevent tumors. As the p53 tumor suppressor pathway is disrupted in over half of breast cancers, the C57BL/6-Trp53+/− compensatory pathways may have particularly broad relevance to human disease. Having appropriate pre-clinical models with various genetic and environmental influences that mimic human breast cancer development will be an essential step for the testing of agents targeting these pathways. Further, by exploiting this aspect of rodent models, we may be able to develop agents for preventing cancer in those individuals clearly at elevated risk from their family history, even in the absence of being able to define the causative genetic factors.
Acknowledgments
Financial and Material Support The research was supported by funds from Avon Foundation (DJJ), National Institutes of Health (R01-CA105452, R01-ES015739, DJJ), Department of Defense (W81XWH-10-1-0637, DJJ), National Health and Medical Research Council, Australia (ACB) and National Breast Cancer Foundation, Australia (ACB).
Abbreviations
- GWAS
Genome-wide association studies
- LFS
Li-Fraumeni Syndrome
- CGEMS
Cancer Genetics Markers of Susceptibility
- SNP
Single nucleotide polymorphism
- DMBA
Dimethyl-α-benzanthracene
- QTL
Quantitative trait locus
Footnotes
Conflict of Interest: The authors have no competing interests.
Permissions: None.
Contributor Information
Anneke C. Blackburn, John Curtin School of Medical Research, Australian National University, Building 131, Garran Rd, Acton, Canberra, ACT 0200, Australia
D. Joseph Jerry, Pioneer Valley Life Sciences Institute, 3601 Main Street, Springfield, MA 01199, USA; Department of Veterinary & Animal Sciences, University of Massachusetts Amherst, Amherst, MA 01003, USA.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Althuis MD, Dozier JM, Anderson WF, Devesa SS, Brinton LA. Global trends in breast cancer incidence and mortality 1973–1997. Int J Epidemiol. 2005;34:405–12. doi: 10.1093/ije/dyh414. [DOI] [PubMed] [Google Scholar]
- 3.Singletary SE. Rating the risk factors for breast cancer. Ann Surg. 2003;237:474–82. doi: 10.1097/01.SLA.0000059969.64262.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunphy KA, Blackburn AC, Yan H, O'Connell LR, Jerry DJ. Estrogen and progesterone induce persistent increases in p53–dependent apoptosis and suppress mammary tumors in BALB/c–Trp53+/− mice. Breast Cancer Res. 2008;10:R43. doi: 10.1186/bcr2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jerry DJ, Dunphy KA, Hagen MJ. Estrogens, regulation of p53 and breast cancer risk: a balancing act. Cell Mol Life Sci. 2010;67:1017–23. doi: 10.1007/s00018-009-0244-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lichtenstein P, Holm NV, Verkasalo PK, et al. Environmental and heritable factors in the causation of cancer—analyses of cohorts of twins from Sweden, Denmark, and Finland. N Engl J Med. 2000;343:78–85. doi: 10.1056/NEJM200007133430201. [DOI] [PubMed] [Google Scholar]
- 7.Antoniou AC, Easton DF. Models of genetic susceptibility to breast cancer. Oncogene. 2006;25:5898–905. doi: 10.1038/sj.onc.1209879. [DOI] [PubMed] [Google Scholar]
- 8.Ponder BA. Cancer genetics. Nature. 2001;411:336–41. doi: 10.1038/35077207. [DOI] [PubMed] [Google Scholar]
- 9.Land CE. Studies of cancer and radiation dose among atomic bomb survivors. The example of breast cancer. JAMA. 1995;274:402–7. [PubMed] [Google Scholar]
- 10.Aisenberg AC, Finkelstein DM, Doppke KP, Koerner FC, Boivin JF, Willett CG. High risk of breast carcinoma after irradiation of young women with Hodgkin's disease. Cancer. 1997;79:1203–10. doi: 10.1002/(sici)1097-0142(19970315)79:6<1203::aid-cncr20>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Begg CB, Haile RW, Borg A, et al. Variation of breast cancer risk among BRCA1/2 carriers. JAMA. 2008;299:194–201. doi: 10.1001/jama.2007.55-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van der Kolk DM, de Bock GH, Leegte BK, et al. Penetrance of breast cancer, ovarian cancer and contralateral breast cancer in BRCA1 and BRCA2 families: high cancer incidence at older age. Breast Cancer Res Treat. 2010;124:643–51. doi: 10.1007/s10549-010-0805-3. [DOI] [PubMed] [Google Scholar]
- 13.Hunter KW, Crawford NP. The future of mouse QTL mapping to diagnose disease in mice in the age of whole–genome association studies. Annu Rev Genet. 2008;42:131–41. doi: 10.1146/annurev.genet.42.110807.091659. [DOI] [PubMed] [Google Scholar]
- 14.Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene. 2001;20:4621–8. doi: 10.1038/sj.onc.1204621. [DOI] [PubMed] [Google Scholar]
- 15.Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 1997;150:1–13. [PMC free article] [PubMed] [Google Scholar]
- 16.Chompret A, Brugieres L, Ronsin M, et al. P53 germline mutations in childhood cancers and cancer risk for carrier individuals. Br J Cancer. 2000;82:1932–7. doi: 10.1054/bjoc.2000.1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Donehower LA, Harvey M, Slagle BL, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumors. Nature. 1992;356:215–21. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 18.Jacks T, Remington L, Williams BO, et al. Tumor spectrum analysis in p53–mutant mice. Curr Biol. 1994;4:1–7. doi: 10.1016/s0960-9822(00)00002-6. [DOI] [PubMed] [Google Scholar]
- 19.Donehower LA, Harvey M, Vogel H, et al. Effects of genetic background on tumorigenesis in p53–deficient mice. Mol Carcinog. 1995;14:16–22. doi: 10.1002/mc.2940140105. [DOI] [PubMed] [Google Scholar]
- 20.Blackburn AC, Brown JS, Naber SP, Otis CN, Wood JT, Jerry DJ. BALB/c alleles for Prkdc and Cdkn2a interact to modify tumor susceptibility in Trp53+/− mice. Cancer Res. 2003;63:2364–8. [PubMed] [Google Scholar]
- 21.Kuperwasser C, Hurlbut GD, Kittrell FS, et al. Development of spontaneous mammary tumors in BALB/c p53 heterozygous mice: a model for Li–fraumeni syndrome. Am J Pathol. 2000;157:2151–9. doi: 10.1016/S0002-9440(10)64853-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Backlund MG, Trasti SL, Backlund DC, Cressman VL, Godfrey V, Koller BH. Impact of ionizing radiation and genetic background on mammary tumorigenesis in p53–deficient mice. Cancer Res. 2001;61:6577–82. [PubMed] [Google Scholar]
- 23.Blackburn AC, Hill LZ, Roberts AL, et al. Genetic mapping in mice identifies DMBT1 as a candidate modifier of mammary tumors and breast cancer risk. Am J Pathol. 2007;170:2030–41. doi: 10.2353/ajpath.2007.060512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Koch JG, Gu X, Han Y, et al. Mammary tumor modifiers in BALB/cJ mice heterozygous for p53. Mamm Genome. 2007;18:300–9. doi: 10.1007/s00335-007-9028-2. [DOI] [PubMed] [Google Scholar]
- 25.Tchatchou S, Riedel A, Lyer S, et al. Identification of a DMBT1 polymorphism associated with increased breast cancer risk and decreased promoter activity. Hum Mutat. 2009;31:60–6. doi: 10.1002/humu.21134. [DOI] [PubMed] [Google Scholar]
- 26.Szpirer C, Szpirer J. Mammary cancer susceptibility: human genes and rodent models. Mamm Genome. 2007;18:817–31. doi: 10.1007/s00335-007-9073-x. [DOI] [PubMed] [Google Scholar]
- 27.Gould MN. The utility of comparative genetics to inform breast cancer prevention strategies. Genetics. 2009;183:409–12. doi: 10.1534/genetics.109.108480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samuelson DJ, Aperavich BA, Haag JD, Gould MN. Fine mapping reveals multiple loci and a possible epistatic interaction within the mammary carcinoma susceptibility quantitative trait locus, Mcs5. Cancer Res. 2005;65:9637–42. doi: 10.1158/0008-5472.CAN-05-1498. [DOI] [PubMed] [Google Scholar]
- 29.Samuelson DJ, Hesselson SE, Aperavich BA, et al. Rat Mcs5a is a compound quantitative trait locus with orthologous human loci that associate with breast cancer risk. Proc Natl Acad Sci USA. 2007;104:6299–304. doi: 10.1073/pnas.0701687104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winter SF, Hunter KW. Mouse modifier genes in mammary tumorigenesis and metastasis. J Mammary Gland Biol Neoplasia. 2008;13:337–42. doi: 10.1007/s10911-008-9089-1. [DOI] [PubMed] [Google Scholar]
- 31.Lin EY, Jones JG, Li P, et al. Progression to malignancy in the polyoma middle T oncoprotein mouse breast cancer model provides a reliable model for human diseases. Am J Pathol. 2003;163:2113–26. doi: 10.1016/S0002-9440(10)63568-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lifsted T, Le VT, Williams M, et al. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int J Cancer. 1998;77:640–4. doi: 10.1002/(sici)1097-0215(19980812)77:4<640::aid-ijc26>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 33.Park YG, Zhao X, Lesueur F, Lowy DR, Lancaster M, Pharoah P, et al. Sipa1 is a candidate for underlying the metastasis efficiency modifier locus Mtes1. Nat Genet. 2005;37:1055–62. doi: 10.1038/ng1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crawford NP, Ziogas A, Peel DJ, Hess J, Anton-Culver H, Hunter KW. Germline polymorphisms in SIPA1 are associated with metastasis and other indicators of poor prognosis in breast cancer. Breast Cancer Res. 2006;8(2):R16. doi: 10.1186/bcr1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsieh SM, Look MP, Sieuwerts AM, Foekens JA, Hunter KW. Distinct inherited metastasis susceptibility exists for different breast cancer subtypes: a prognosis study. Breast Cancer Res. 2009;11(5):R75. doi: 10.1186/bcr2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Quan X, Laes JF, Stieber D, et al. Genetic identification of distinct loci controlling mammary tumor multiplicity, latency, and aggressiveness in the rat. Mamm Genome. 2006;17:310–21. doi: 10.1007/s00335-005-0125-9. [DOI] [PubMed] [Google Scholar]
- 37.Ronckers CM, Erdmann CA, Land CE. Radiation and breast cancer: a review of current evidence. Breast Cancer Res. 2005;7:21–32. doi: 10.1186/bcr970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Inano H, Suzuki K, Onoda M, Yamanouchi H. Susceptibility of fetal, virgin, pregnant and lactating rats for the induction of mammary tumors by gamma rays. Radiat Res. 1996;145:708–13. [PubMed] [Google Scholar]
- 39.Shellabarger CJ, Chmelevsky D, Kellerer AM. Induction of mammary neoplasms in the Sprague-Dawley rat by 430 keV neutrons and X-rays. J Natl Cancer Inst. 1980;64:821–33. [PubMed] [Google Scholar]
- 40.Ullrich RL. Tumor induction in BALB/c female mice after fission neutron or gamma irradiation. Radiat Res. 1983;93:506–15. [PubMed] [Google Scholar]
- 41.Mori N, Okumoto M, Hart AA, Demant P. Apoptosis susceptibility genes on mouse chromosome 9 (Rapop2) and chromosome 3 (Rapop3) Genomics. 1995;30:553–7. doi: 10.1006/geno.1995.1276. [DOI] [PubMed] [Google Scholar]
- 42.Mori N, Okumoto M, van der Valk MA, et al. Genetic dissection of susceptibility to radiation-induced apoptosis of thymocytes and mapping of Rapop1, a novel susceptibility gene. Genomics. 1995;25:609–14. doi: 10.1016/0888-7543(95)80001-3. [DOI] [PubMed] [Google Scholar]
- 43.Yu Y, Okayasu R, Weil MM, et al. Elevated breast cancer risk in irradiated BALB/c mice associates with unique functional polymorphism of the Prkdc (DNA-dependent protein kinase catalytic subunit) gene. Cancer Res. 2001;61:1820–4. [PubMed] [Google Scholar]
- 44.Umesako S, Fujisawa K, Iiga S, et al. Atm heterozygous deficiency enhances development of mammary carcinomas in p53 heterozygous knockout mice. Breast Cancer Res. 2005;7:R164–70. doi: 10.1186/bcr968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gould KA, Tochacek M, Schaffer BS, Reindl TM, Murrin CR, Lachel CM, et al. Genetic determination of susceptibility to estrogen-induced mammary cancer in the ACI rat: mapping of Emca1 and Emca2 to chromosomes 5 and 18. Genetics. 2004;168:2113–25. doi: 10.1534/genetics.104.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schaffer BS, Lachel CM, Pennington KL, Murrin CR, Strecker TE, Tochacek M, et al. Genetic bases of estrogen-induced tumorigenesis in the rat: mapping of loci controlling susceptibility to mammary cancer in a Brown Norway x ACI intercross. Cancer Res. 2006;66:7793–800. doi: 10.1158/0008-5472.CAN-06-0143. [DOI] [PubMed] [Google Scholar]
- 47.Gordon RR, Hunter KW, La Merrill M, Sørensen P, Threadgill DW, Pomp D. Genotype X diet interactions in mice predisposed to mammary cancer: II. Tumors and metastasis. Mamm Genome. 2008;19:179–89. doi: 10.1007/s00335-008-9096-y. [DOI] [PubMed] [Google Scholar]
- 48.Gordon RR, La Merrill M, Hunter KW, Sørensen P, Threadgill DW, Pomp D. Dietary fat-dependent transcriptional architecture and copy number alterations associated with modifiers of mammary cancer metastasis. Clin Exp Metastasis. 2010;27:279–93. doi: 10.1007/s10585-010-9326-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bahassi EM, Penner CG, Robbins SB, et al. The breast cancer susceptibility allele CHEK2*1100delC promotes genomic instability in a knock-in mouse model. Mutat Res. 2007;616:201–9. doi: 10.1016/j.mrfmmm.2006.11.025. [DOI] [PubMed] [Google Scholar]
- 50.Bahassi EM, Robbins SB, Yin M, et al. Mice with the CHEK2*1100delC SNP are predisposed to cancer with a strong gender bias. Proc Natl Acad Sci USA. 2009;106:17111–6. doi: 10.1073/pnas.0909237106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vousden KH, Prives C. Blinded by the light: the growing complexity of p53. Cell. 2009;137:413–31. doi: 10.1016/j.cell.2009.04.037. [DOI] [PubMed] [Google Scholar]
- 52.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell. 2004;119:591–602. doi: 10.1016/j.cell.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 53.Fang S, Krahe R, Lozano G, Han Y, Chen W, Post SM, et al. Effects of MDM2, MDM4 and TP53 codon 72 polymorphisms on cancer risk in a cohort study of carriers of TP53 germline mutations. PLoS ONE. 2010;5:e10813. doi: 10.1371/journal.pone.0010813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bond GL, Levine AJ. A single nucleotide polymorphism in the p53 pathway interacts with gender, environmental stresses and tumor genetics to influence cancer in humans. Oncogene. 2007;26:1317–23. doi: 10.1038/sj.onc.1210199. [DOI] [PubMed] [Google Scholar]
- 55.Post SM, Quintas-Cardama A, Pant V, et al. A high-frequency regulatory polymorphism in the p53 pathway accelerates tumor development. Cancer Cell. 2010;18:220–30. doi: 10.1016/j.ccr.2010.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bond GL, Hirshfield KM, Kirchhoff T, et al. MDM2 SNP309 accelerates tumor formation in a gender-specific and hormone-dependent manner. Cancer Res. 2006;66:5104–10. doi: 10.1158/0008-5472.CAN-06-0180. [DOI] [PubMed] [Google Scholar]
- 57.Varghese JS, Easton DF. Genome-wide association studies in common cancers–what have we learnt? Curr Opin Genet Dev. 2010;20:201–9. doi: 10.1016/j.gde.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 58.Antoniou AC, Beesley J, McGuffog L, Sinilnikova OM, Healey S, Neuhausen SL, et al. Common breast cancer susceptibility alleles and the risk of breast cancer for BRCA1 and BRCA2 mutation carriers: implications for risk prediction. Cancer Res. 2010;70:9742–54. doi: 10.1158/0008-5472.CAN-10-1907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Weitzel JN, Buys SS, Sherman WH, Daniels A, Ursin G, Daniels JR, et al. Reduced mammographic density with use of a gonadotropin-releasing hormone agonist-based chemoprevention regimen in BRCA1 carriers. Clin Cancer Res. 2007;13:654–8. doi: 10.1158/1078-0432.CCR-06-1902. [DOI] [PubMed] [Google Scholar]