

SUMMARY
We present a systems strategy that facilitated the development of a molecular signature for glioblastoma (GBM), composed of 33 cell-surface transmembrane proteins. This molecular signature, GBMSig was developed through the integration of cell-surface proteomics and transcriptomics from patient tumors in the REMBRANDT (n=228) and TCGA datasets (n=547) and can separate GBM patients from controls with an MCC value of 0.87 in a lock-down-test. Functionally, 17/33 GBMSig proteins are associated with TGFβ signaling pathways, including: CD47, SLC16A1, HMOX1 and MRC2. Knockdown of these genes impaired GBM invasion, reflecting their role in disease-perturbed changes in GBM. ELISA assays for a subset of GBMSig (CD44, VCAM1, HMOX1, and BIGH3) on 84 plasma specimens from multiple clinical sites revealed a high degree of separation of GBM patients from healthy controls (AUC 0.98 in ROC). Additionally, a classifier based on these four proteins differentiated the blood of pre- and post-tumor resections, demonstrating potential clinical value as biomarkers.
eTOC Blurb
Multidimensional analysis of GBM cell-surface proteins reveals a disrupted membrane-signaling network that can be identified from the blood of GBM patients, a subset of which can distinguish between normal and diseased individuals.
INTRODUCTION
A systems approach that integrates multi-omic measurements offers an avenue for better understanding the emergent properties and complexities of a disease process. Considering the recent advancements in omics technologies and machine learning, the power of a systems approach, in contrast to the single parameter atomistic approach, can enable the development of molecular signatures for complex diseases such as cancer (Sung et al., 2012). However, such an approach that integrates data types across multiple sources also needs empirical validation since the separation of true disease signal from noise that arises out of variability in omics platforms-both biological and technical is essential. Here we have attempted to develop such a molecular signature for glioblastoma (GBM) through the integration of high-resolution proteomics and transcriptomics supported by end-to-end experimental validation.
Despite significant improvements in treatment and survival outcomes for other cancers, the median survival rate for GBM with treatment is still only 15 months—a figure that has been largely unchanged for decades (Demuth T, 2004; Mrugala, 2013; Delyon et al., 2015; Grabowski and Sehouli, 2015; Jorgensen and Knudtson, 2015; Limani et al., 2015; Milroy, 2015; Rollig et al., 2015). MRI scans are used to diagnose or evaluate tumor progression, but these studies are often difficult to interpret due to variability in the appearance of the tumor and include a degree of subjectivity (Thompson et al., 2011). The field of neurooncology would benefit from a blood-based molecular signature of GBM that could complement MRI scans and existing genomic tests (Hegi and Stupp, 2013; Kurscheid et al., 2015; Murat et al., 2008; Stupp et al., 2006).
Most attempts at developing robust biomarkers have failed to make it to the clinic (Omenn et al., 2012; Sung et al., 2012), and there is a process of validation that must be followed to generate a robust molecular signature appropriate for clinical use. That is not what we will present herein – that will be a subsequent downstream evaluation. Rather, here we have focused on the development of a molecular signature, GBMSig, which defines the set of transmembrane proteins whose transcript concentrations are perturbed in GBMs compared to healthy control tissues and on identifying the extent to which some of these have been released into the blood and can be observed by targeted mass spectrometry.
Cell-surface transmembrane proteins occupy a strategic location between the cell and its microenvironment, and can propagate signals from both exofacial and cytoplasmic ends of the membrane (Chen et al., 2008; De Marco et al., 2013; Kandouz, 2012; Murai and Pasquale, 2010; Pasquale, 2010). Since aberrant expression of these proteins on the cell-surface is known to disrupt normal cell activities and influence neoplastic transformation (Okumura et al., 2004; Teh and Chen, 2012), we hypothesized that integration of transcriptomic and proteomic expression data for these proteins would enrich for putative targets that could be the basis for a molecular GBM signature with a higher probability of being mechanistically linked to the underlying pathology. Cell-surface transmembrane proteins are often cleaved and shed into the blood in pathological conditions, making them ideal targets for diagnostic blood markers (Li et al., 2013; Shao et al., 2012; Varady et al., 2013).
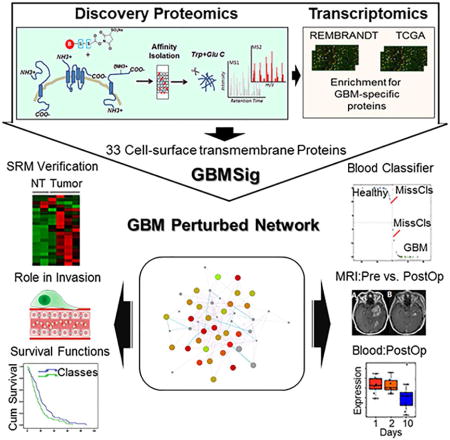
Cell-surface-transmembrane proteins tend to be low-abundant in the blood and are the proverbial needles in the haystack, presenting significant challenges in their detection even after depletion of highly abundant proteins. Reproducible quantification of these low abundant proteins can only be achieved after they have been identified. To first identify candidate tumor-derived, cell-surface transmembrane proteins, we performed comparative cell-surface proteomics analyses of four relevant GBM cell lines: CD133+ cancer stem cells, healthy neural stem cells, as well as the GBM cell lines U87MG and T98. Considering the possibility that the protein expression profile could be altered for a variety of reasons, including in-vitro culturing (Ertel et al., 2006; Vogel et al., 2005), we integrated cell-surface proteomics data with primary GBM tissue transcriptomic compendia of the REMBRANDT and TCGA. This integrative approach (Figure 1) helped us to verify mRNA expression of corresponding proteins identified through shotgun proteomics and also to develop a robust, GBM-specific membrane signature (GBMSig) composed of 33 proteins that reflects the biology of GBM with potential for tissue and blood-based diagnosis.
Figure 1.
Description of the rationale and accompanying filtering steps applied to the initial list of proteins identified through sulfo-NHS-SS-biotin tagging of intact cells from four cell lines: U87MG, T98, CD133+ cancer stem cells and neural stem cells. From the 1480 candidate proteins, 274 contained transmembrane domains. Corresponding probes were found for 202 targets in the REMBRANDT data set containing 228 GBM and 16 non-tumor brain tissue specimens. Genes found to be commonly expressed in oligodendrogliomas and astrocytomas were then removed to enrich for GBM-specific targets. This resulted in 33 targets identified as GBMSig. SRM mass spectrometry assays were developed for each of these targets. Twenty-one of the 33 GBMSig proteins could be detected across 4 GBM and 2 non-tumor brain specimens. Fourteen of the 33 proteins were also detected in the blood (following immunodepletion of the top 14 abundant blood proteins). Each of the 33 targets was evaluated as a classifier on 547 GBM and 10 control samples from TCGA. Four targets were selected based on the robustness as a classifier, ability to be detected in the blood and availability of a commercial ELISA assay.
RESULTS
Compositional analysis of GBM cell-surface proteome by shotgun proteomics
There is significant heterogeneity across tumors of individuals, even within each of the four molecular subtypes. To capture some of this heterogeneity, we performed cell-surface proteomics on four cell lines including two GBM cell lines UMG87 and T98, a GBM cancer stem cell line (grown at the source, Celprogen, that ensured CD133 expression) and a healthy neural stem cell line (positive for putative stem cell markers tub iii, oct-4, sox-2, and nestin from Millipore). Cell-surface proteins were labeled and captured with membrane-impermeable sulfo-NHS-SS-biotin from intact cells (Figure S1).
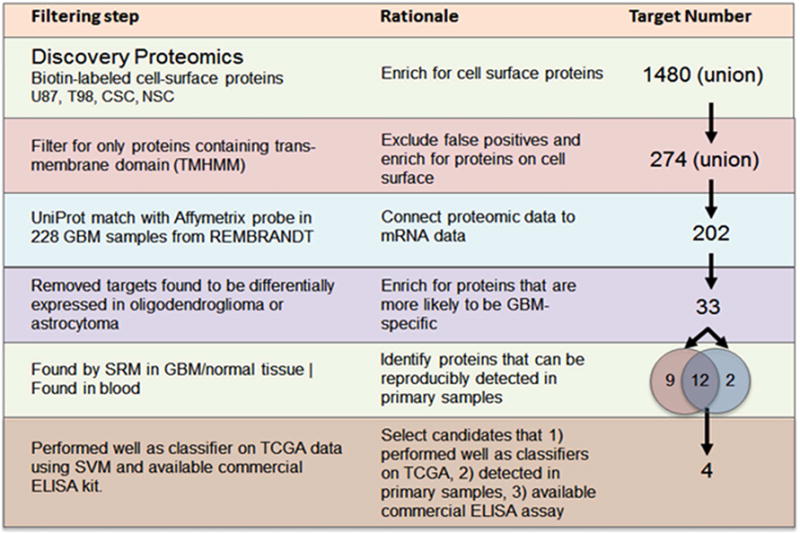
Captured cell-surface proteins were subjected to high-resolution mass spectrometry in triplicate (technical replicates) and the proteins were identified using the Global Proteome Machine [(the GPM) (http://www.theGPM.org)] with minimum log expectation scores of <10−3 (Craig and Beavis, 2003, 2004; Fenyo et al., 2007; Ghosh et al., 2010). We identified a total of 868, 813, 541, and 564 non-redundant proteins from U87MG, T98, neural stem cell, and cancer stem cell populations, respectively (Figure 2A–D and Table S1). While our experimental approach was designed to enrich for cell-surface proteins, we also employed the transmembrane prediction algorithm TMHMM (Krogh et al., 2001; Moller et al., 2001) to identify those cell-surface proteins with transmembrane domains from the total identified proteins. Although this filtering step was rather strict and likely eliminated several true positives, we were left with 157, 154, 98, and 80 cell-surface transmembrane proteins in U87MG, T98, neural stem cell, and cancer stem cell lines, respectively. Overall 274 different cell-surface transmembrane proteins were identified from all four cell lines. Among cell-surface transmembrane proteins identified, we found 53 cluster-of-differentiation (CD) markers, which in general offer an immunological basis for separating different cell types (Beare et al., 2008; Erber, 1990).
Figure 2.
Compositional analysis of cell-surface proteins (CSPs) from three independent runs by high resolution mass spectrometry in (A) U87MG, (B) T98, (C) cancer stem cell (Celprogen), and (D) neural stem cell (Millipore). Proteins with log(e)=−3 score (GPM) were considered valid. Numerical data represents proteins identified in each isolates. E) Functional analyses of cell-surface proteins with transmembrane domains identified in the study highlight the enrichment of those biological processes that are known to be associated with cell-surface activities. Fold enrichment is presented as a ratio of number of cell-surface transmembrane proteins identified for a given biological process relative to whole genome annotations with indicated p values. F) Cartoon diagram (KEGG) showing the identification (highlighted in orange) of those proteins associated with cell migration and invasion.
We also identified 98 multi-transmembrane domain containing cell-surface proteins, which are underrepresented in whole-cell proteomic datasets because of their hydrophobicity and limited cellular abundance. As would be expected, functional classification of these proteins highlighted the enrichment of those biological processes that are known to be associated with cell-surface activities such as cell adhesion and migration, transport, and bi-directional signaling (Figure 2E–F). We also observed a difference in enrichment of immune regulatory processes among all three distinct cancer cell lines (U87MG, T98, and cancer stem cell line) compared to the healthy neural stem cells—reflective of the functional differences between these cell types (Table S1). Our cell-surface proteome analysis identified transmembrane proteins expressed among various GBM cell lines related to different aspects of GBM biology. This approach provided us with a list of proteomic targets that, upon further selection based on RNA expression (described below), were good candidates for detection and measurement by quantitative selected reaction monitoring (SRM) mass spectrometry. The XML data files are available for viewing at http://human.thegpm.org/tandem/thegpm_upview.html. Unique Global Proteome Machine IDs for proteins identified in each cell line are provided in the key resources table of STAR Method section of this paper.
Differentially expressed cell-surface transmembrane proteins in GBM
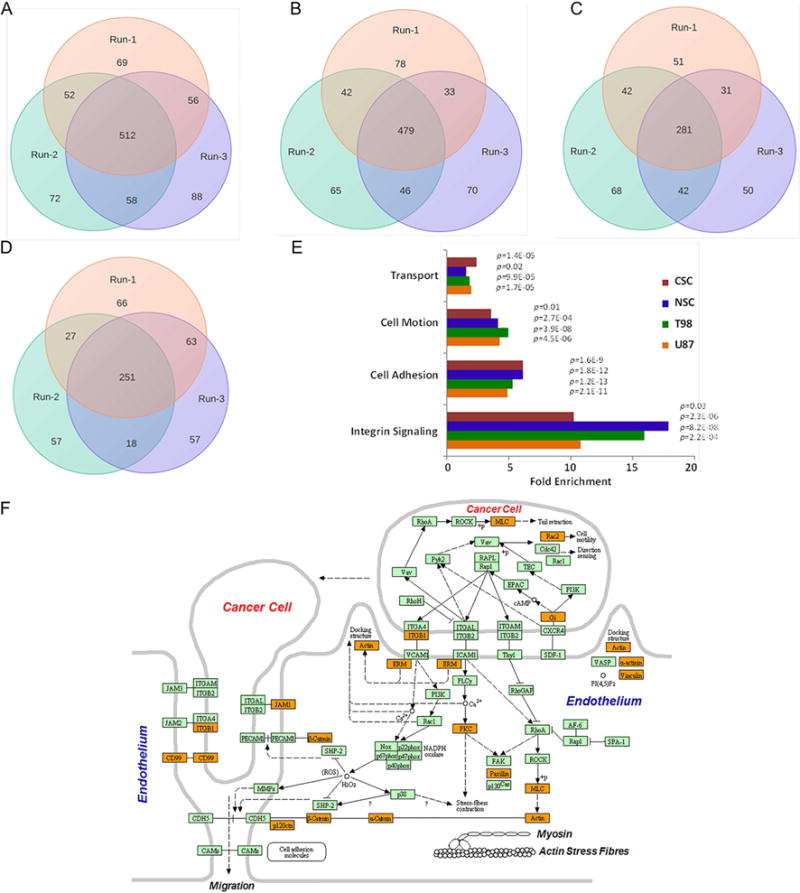
For a candidate list of differentially expressed transcripts between tumor and non-tumor regions of the brain, we utilized microarray data from the REMBRANDT tissue source (http://rembrandt.nci.nih.gov). Out of 274 cell-surface transmembrane proteins identified from the proteomics study, we found expression data for 202 (532 independent probes) corresponding transcripts in REMBRANDT. Expression levels in the tumor transcriptomes were, on average, much more abundant than those from their normal counterparts, possibly as an artifact of the small number of available normal control samples, but also likely due to a real difference in transcript expression. Because of the small number of control samples, we used a conservative cutoff of two-fold average expression change with FDR<0.05. Among the resulting 202 selected transcripts, we observed 155 of them were upregulated and 47 were down-regulated (Figure 3A). To identify GBM-specific cell-surface transmembrane protein expression changes, we filtered out transcripts found to be differentially expressed in non-GBM brain diseases such as astrocytoma (N=148 tumors) and oligodendroma (N=67 tumors) relative to the same REMBRANDT control samples. This filtering approach further reduced the number of candidate cell-surface transmembrane proteins from 202 to 33 (Figure 3A–F and Figure S2).
Figure 3.
Cell-surface proteins with transmembrane domains identified from shotgun proteomics were evaluated for their differential expressions using transcriptome compendiums of REMBRANDT and TCGA. A) The differential expression of 202 cell-surface transmembrane proteins in GBM tissues (N=228) relative to non-tumor brain specimens (N=16) of REMBRANDT transcriptome compendium. Expression values for these transcripts were log2 transformed, and a minimum of two-fold average expression change (FDR<0.05) between tumor and non-tumor brain tissues was used as cut-off for significance. Clustering reflects the directionality of cell-surface transmembrane transcript expression among GBM and controls. Non-tumor brain specimens are highlighted in yellow at the bottom. B) majority of the cell-surface proteins with transmembrane domains mapped to REMBRANDT transcripts were discarded due to common expressions in non-GBM diseases such as astrocytoma (N=148) and oligodendroma (N=67 tumors). Shown are 33 cell-surface transmembrane proteins that were subsequently tested for the development of GBMSig classifier. Each column of the heatmap is presented as the average log2 [tumor/non-tumor] ratios. CNT is non-tumor brain, AST is astrocytoma, and OLI is oligodendroma. C) Principal component analysis (PCA) of REMBRANDT GBM transcriptome arrays with GBMSig (n=33). Red dots represent non-tumor isolates and grey ones are GBM. Two principal components can explain 43% of the variability. D) Principal component analysis (PCA) of independent TCGA GBM transcriptome datasets composed of 547 GBM specimens and 10 non-tumor brain controls with GBMSig (n=33). Two principal components can explain 48% of the variability. Red dots represent non-tumor isolates and grey ones are GBM. E) ROC analysis of REMBRANDT tissue arrays for individual GBMSig proteins. Specificity (%) and Sensitivity (%) values of individual GBMSig were plotted on X and Y axis respectively. Standard error of AUC was calculated using the method described by DeLong et al. Orange color represents significance level P (Area=0.5) <0.0001 while gray color represents P>0.01.Detailed analysis is provided in table S2. F) ROC analysis of independent TCGA tissue arrays (GBM=547 subjects, NonTumor=10 subjects) for individual GBMSig proteins. Specificity (%) and Sensitivity (%) values of individual GBMSig were plotted on X and Y axis respectively. Standard error of AUC was calculated using the approach described by DeLong. The analysis revealed high degree of specificities and sensitivities in discriminating GBM populations from controls with significance level P (Area=0.5) <0.0001. Detailed analysis is provided in Table S2.
To increase confidence that these 33 candidate proteins could represent real differences between case and control samples, we tested their performance as a GBM signature (designated as GBMSig) in an independent dataset: TCGA (The Cancer Genome Atlas, 2008) (N=547 GBM tumor samples and 10 healthy brain tissues). We designed a Support Vector Machine (SVM) classifier (C. Cortes and Vapnik, 1995) with parameters tuned in 10-fold cross validation on the REMBRADT training set (Figure S2A). We then classified the TCGA test set with a lock-down test (Figure S2B). We obtained a Mathew’s correlation coefficient (MCC) of 0.87, exhibiting 99% positive predictive value and 89% negative predictive value for our classifier. Of note, while the high predictive values for both REMBRANDT and TCGA should be evaluated in the light of the disproportionately small control samples, the purpose of this step was to evaluate the robustness of the targets following integration of the proteomic and transcriptomic datasets and thus this transcriptomic step was a means for helping to select the protein signature (GBMSig) presented herein and not an end in and of itself.
Principal component analysis (PCA) of the GBMSig genes and individual specificities and sensitivities from ROC analyses of training and validation set (TCGA) are presented in Figures 3C–3F and Table S2 respectively. Taken together, these results support the predictive power of the 33 protein GBMSig classifier and suggest that the integration of both proteomic and transcriptomic data and stringent filtering have enriched for candidate proteins of interest that might be quantitatively different in their expression between GBM and control patients.
Quantitative SRM assays for GBMSig
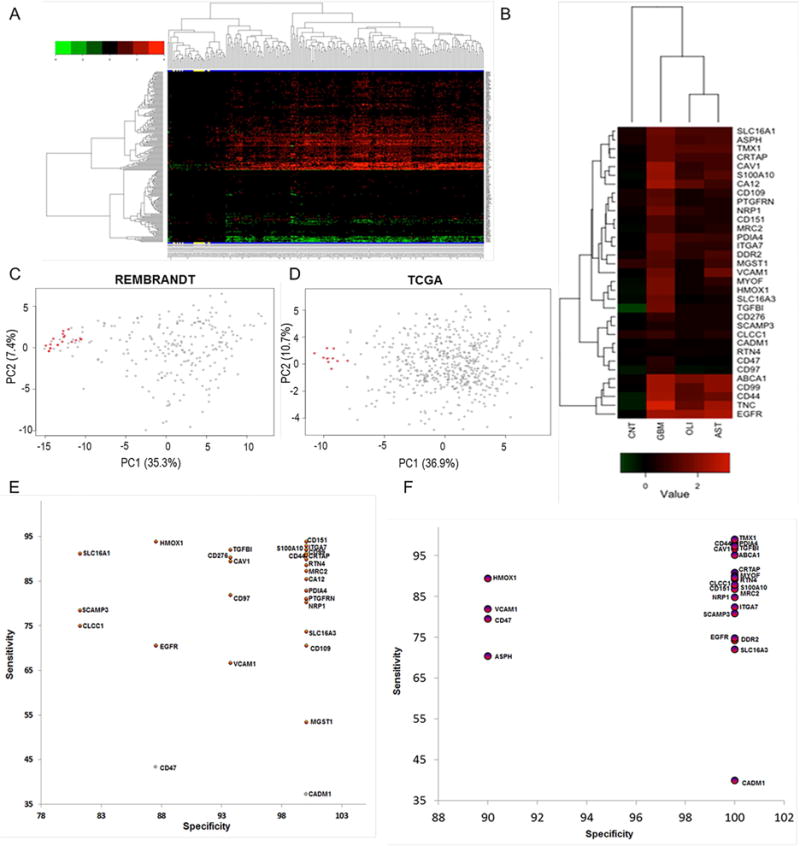
As an initial step to evaluating a subset of GBMSig as biomarkers in the blood, we developed quantitative SRM assays for all 33 proteins (Li et al., 2013; Picotti and Aebersold, 2012;Bereman et al., 2012). Our earlier cell-surface protein profiling of GBM cell lines provided data for selecting mass spectrometry compatible peptides with appropriate mass to charge ratios (m/z). Seventy cell-surface protein peptide representatives from 33 GBMSig proteins were generated for SRM assay development—approximately 2 for each protein. Synthetic peptides labeled (13C15N) C-terminally with either lysine (K) or arginine (R) act as surrogates of endogenous peptides. These peptides were subjected to collision energy (CE) optimization (Maclean et al., 2010) to maximize the release of trapped energy from each peptide bond. Three parental (Q1) charges (+2, +3, and +4) and two daughter (Q3) ion charges (+1 and +2) of peptides were tested in all feasible combinations for assay optimization; the Q1/Q3 transition-CE combination that demonstrated the highest SRM peak-intensity and was minimally affected by interfering ions was selected for assay validation (Table S3). In the final SRM assay, the best performing peptide with a minimum of three transitions was used for quantitation. Retention time of each surrogate peptide was determined empirically, which helped to develop dynamic-SRM assays (d-SRM). This targeted approach improved the sensitivity and specificity, enabling the reproducible measurement of low abundant GBMSig proteins in complex bio-specimens. Four resected GBM tumors and two non-tumor brain tissue samples were homogenized, enzymatically digested, clarified using C18, and spiked with synthetic C-terminally labeled (13C15N K/R) peptides for subsequent SRM mass spectrometry analysis (three technical replicates). Of the 33 GBMSig proteins assayed we were able to reliably detect 21 across all samples. Twelve of the 21 GBMSig proteins were overexpressed in all four GBM tissues relative to non-tumor brain tissues. The list of these proteins including nine proteins with p<0.05 are presented in Table S4. Protein expression data and PCA analyses are represented in Figure 4A–C. Although a majority of GBMSig proteins (12 of 21) revealed differential expression between tumor and non-tumor regions of the brain, intratumor heterogeneities in GBMSig expression as investigated through tumor subtyping (Phillips et al., 2006) underscored the nature of combinatorial perturbation of membrane networks across different clinical specimens (Figure 4D, Table-S4–5).
Figure 4. Proteomic verification of GBMSig expression in GBM tissues by SRM mass spectrometry.
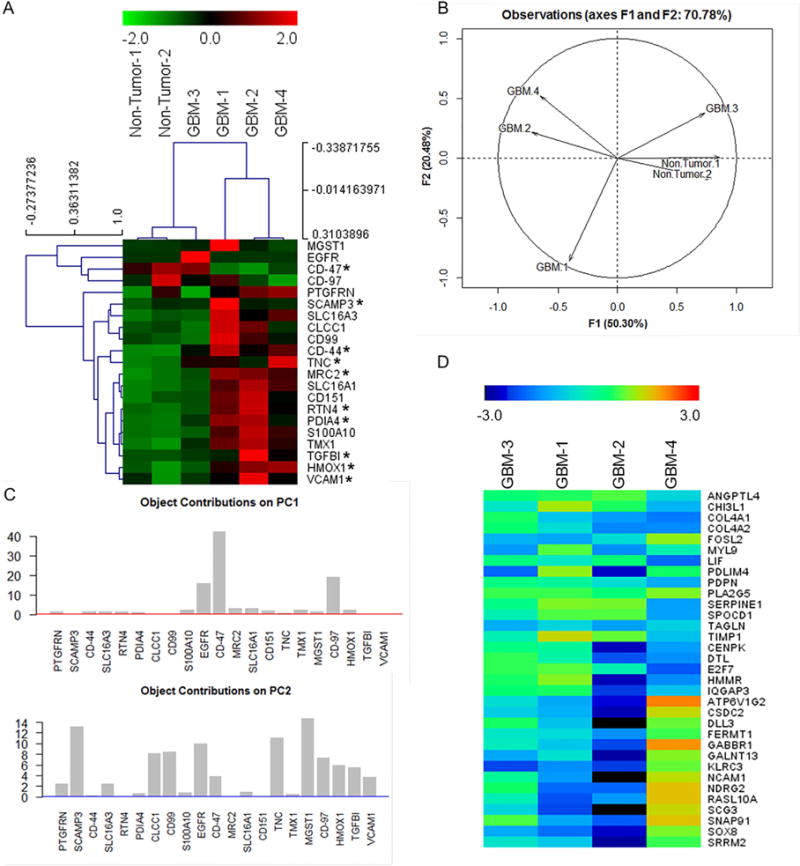
A) Equal quantities of tissue homogenates from tumor (n=4) and non-tumor isolates (n=2) were enzymatically digested, C18 clarified, and spiked with surrogate peptides labeled C-terminally with 13C15N K/R for SRM analysis. Ratios of endogenous and surrogate peptides were centroided and presented as Z-score in the heatmap. A subset of GBMSig (*) was also observed to be circulated in the blood plasma. Co-expression of several GBMSig proteins with BIGH3 (TGFBI) - a known TGFβ-inducible protein might be indicative of the presence of additional TGF-β responsive elements operating within GBMSig. Based on GBMSig expressions, GBM and non-tumor tissues can be arranged into groups as revealed through Spemann rank clustering. B) PCA analyses of GBMSig proteins as quantified by SRM mass spectrometry can distinguish GBM from non-tumor brain specimens with first two components explaining 70.78% of variability, highlighting the robustness of GBMSig in separating GBM from controls with high efficiency at both transcriptome and proteome levels. C) Contributions of each GBMSig protein onto respective principal components. Expected average contributions on PC1 and PC2 are denoted by a red and blue line respectively. D) Subtyping of GBM 1–4 tissues were performed using qPCR for 33 genes as described (Phillips et al., 2006). Accordingly, GBM-1 is assigned as prolifimes, GBM-2 as mesenchymal, GBM-3 as proliferative, and GBM-4 as proneuronal. This subtyping allowed us to explain the heterogeneities in GBMSig expression observed from proteomic analysis. Subtype expression data (qPCR) are provided in table S5.
Utility of GBMSig in the assessment of tumor progression and diagnosis through blood analysis
Cell-surface proteins are known to be secreted or shed into the blood stream by both healthy and tumor cells and the concentration changes in those proteins from tumor cells (as compared to normal cells) reflect the fact that their cognate networks have become perturbed (Li et al., 2013; Shao et al., 2012). As would be expected, given our initial decision to select for cell-surface proteins, we found twenty-one of 33 GBMSig proteins to possess N-terminal secretion signal sequences (SignalP4.1, www.cbs.dtu.dk). However, protein secretion in the blood is a complex process and can be controlled by a multitude of factors (Uhlen et al., 2015). Therefore, we sought to identify which GBMSig proteins could be identified in the blood of GBM patients through empirical means. In a pilot study, we evaluated four GBM plasmas (pre-operative blood collection) for circulating GBMSig proteins by SRM mass spectrometry (Table S6). Following immunodepletion of the 14 most abundant blood proteins, we detected 14 of 33 GBMSig proteins independently in triplicate SRM runs (Table S6). Four circulating GBMSig proteins (HMOX1, CD44, VCAM1, and BIGH3 (TGFBI)) were selected for further evaluation by ELISA assays for potential GBM diagnosis based on 1) detection by SRM in the blood of GBM patients; 2) high AUC values (>0.95) in ROC analyses of REMBRANDT and TCGA transcriptomic datasets; and 3) the availability of off-the shelf ELISA kits.
We assembled a collection of 84 plasma specimens from five different sources and subjected them to ELISA analyses for the four selected proteins. As we were unable to obtain GBM and normal plasma samples from the same source (and same collection procedure), we obtained each from multiple locations to help mitigate batch effects by performing analyses across data from multiple distinct sample sources, and different sources entirely in the training and test sets. All samples obtained were collected using the standard collection protocol for K2-EDTA (purple cap) blood. We subjected the samples to an independent training and validation format with each set being composed of age and gender matched GBM samples (21 training, 21 validation) and healthy samples (21 training, 21 validation) (Figures 5A–C and S3, Table S7). Following batch-normalization (standard score), the training set was modeled using linear discriminant analysis (LDA). LDA characterizes 2 classes as Gaussian densities of equal covariance using a linear combination of features. The performance of four GBMSig proteins in the identification of GBM class was assessed for validation set after locking down parameters. We observed 95.2% sensitivity and 95.2% specificity for the independent validation set (GBM=21, Healthy=21). ROC analyses of the training set exhibited an AUC of 0.99 while the validation set presented an AUC of 0.98 (Figure 5A–C), highlighting significant differences in the abundance of these proteins in GBM patients versus normal controls. Power analysis of ELISA results also indicated good agreement between the effect size (>|0.6|) and the sampling method (power>0.8) (Figure S3E).
Figure 5.
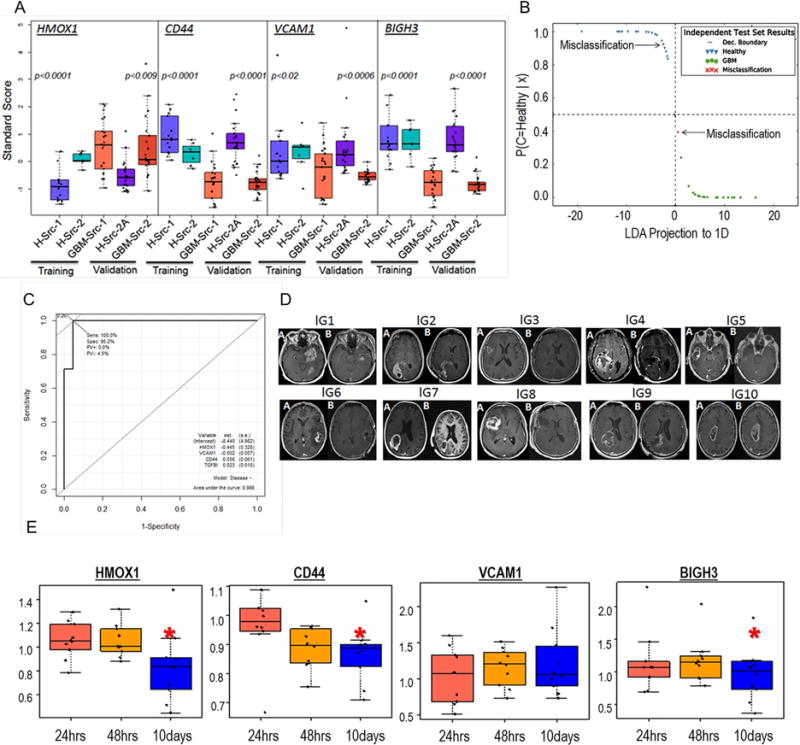
Blood Diagnostic potentials of four GBMSig proteins. A) four GBMSig proteins viz. CD44, VCAM1, HMOX1, and BIGH3 (TGFBI) were evaluated by ELISA assays using 84 plasma samples obtained from 3 different locations. Despite different sites of collection, both training set and validation set revealed statistically significant differences between GBM (GBM-Src-1 and GBM-Src-2) and healthy controls(H-Src-1,H-Src-2, and H-Src-2A). p values are two tailed and welch corrected. B) ELISA results from training set were modelled using Linear Discriminant Analysis (LDA). The training set created a classifier with a scaling factor of (0.72, −1.1, −.05. −.68) for HMOX1, BIGH3 (TGFBI), VCAM1 and CD44 respectively. The decision boundary coefficients are at (−1.83, 1.83) with an intercept of −2.38. Performance of these four GBMSig proteins was assessed for an independent validation set (GBM=21, Healthy=21). We observed 95.23% sensitivity and 95.2% specificity with 95.2% accuracy for the independent validation set. Dec. Boundary is Decision Boundary. C) Shown here is the ROC analysis of validation set that exhibited an AUC of 0.98, highlighting robust discerning ability of GBMSig proteins as attractive candidates. D) MRI images showing the changes in tumor volume before (A) and after resection (B) for ten GBM patients recruited prospectively for the blood analysis. E) Boxplot showing the changes in the plasma values for 4 GBMSig proteins at 24hrs, 48hrs, and 10days (~) post-resection as measured through ELISA assays. Data were normalized to preoperative condition for individual patient. Y-axis represents GBMSig values in ng/unit of total protein. Black dots represent each patient and ‘*’ indicates p<0.05.
We then examined the changes in blood concentrations of the same four proteins (HMOX1, CD44, VCAM1, and BIGH3) for ten GBM patients prior to and after tumor resection. Blood samples were collected preoperatively and postoperatively at 24 hrs, 48 hrs, and ~10 days post-surgery (first post-operative visit). From ELISA analysis, we observed significant changes (p<0.05; ROC AUC of 0.83) in the blood concentrations of three proteins (HMOX1, CD44, and BIGH3), possibly reflecting the pathophysiological changes related to tumor resection (Figure 5D–E) within ten days of surgery. PCA analysis also revealed a separation of 52.1% on PC1 and 27% on PC2 for changes in the blood concentrations of HMOX1, CD44, and BIGH3 between 24hrs and 10days post-resection (Figure S4A–B, Table S8). However, since there was no endpoint to this study, it was not possible to relate the changes in the blood concentrations of these GBMSig proteins to the overall survival (OS) or progression-free-survival (PFS) of patients, which could be evaluated in future by undertaking longer and more frequent post-operative follow-ups.
Connections between the classifier and TGF-β responsiveness
Pathway analysis of GBMSig proteins using the KEGG database (Kanehisa and Goto, 2000) and GeneMANIA (www.genemania.org) reflected the role of GBMSig proteins in several established aspects of GBM biology, including: focal adhesion, ECM-receptor interaction, apoptosis, and the MAPK signaling pathway (Figures 2F and S2C). Additionally, co-expression analysis of GBM tissue transcriptomics and proteomics data (Figure 3A,4A,4C) indicated that a number of proteins within the GBMSig classifier co-expressed with TGFBI (BIGH3)—a known TGF-β-inducible protein (Lauden et al., 2014; Nummela et al., 2012). This observation, along with a known role for TGF-β in cancer and GBM (Pickup et al., 2013), highlighted possible TGF-β1 responsive network components operating within GBMSig classifier that could impact the modulation of other GBMSig proteins within the classifier. To determine whether GBMSig proteins could respond to TGF-β treatment and provide biological, mechanistic evidence for the classifier, we tested TGF-β1 responsiveness in the astrocytoma cell line U87MG through induction of C-terminal phosphorylation of SMAD2 (Figure 6A). Conversely, SMAD2 phosphorylation is diminished in the presence of TGF-β-inhibitor (SB 431542). Cell viability did not significantly change following treatment with TGF-β1 or its inhibitor relative to untreated cells (data not shown). Proteomic changes of GBMSig expressions following TGF-β/inhibitor treatment were evaluated by SRM assays. We observed 13 GBMSig proteins including TGFBI (BIGH3), which exhibited at least 1.5 fold higher expression following TGF-β treatment relative to cells treated with TGF-β-inhibitor alone (Figure 6B and Table-S10). As basal TGF- β expression could contribute to expression of our GBMSig proteins, we found that cells pretreated with TGF-β-inhibitor and subsequent TGF-β treatment modulated the expression of GBMSig proteins. There were four additional GBMSig proteins (CD47, MYOF, ABCA1, and CD44) that exhibited a positive enrichment (>1.2 fold over inhibitor treatment) for TGF-β treatment in comparison to TGF-β-inhibitor treatment alone. Details are presented in Table S10 and Figure S5. Changes in protein expression of SLC16A1, MRC2, SLC16A3, CD47, and CD97 after TGFβ treatment relative to inhibitor treatment were confirmed by alternate method flow cytometry (Figure 6C). Results were consistent with the changes in protein expression observed by SRM. These observations highlight the modular responsiveness of a subset of GBMSig classifier with TGF-β signaling that was previously undescribed (Figure 6D).
Figure 6. Association of GBMSig proteins with TGF-β1 signaling network.
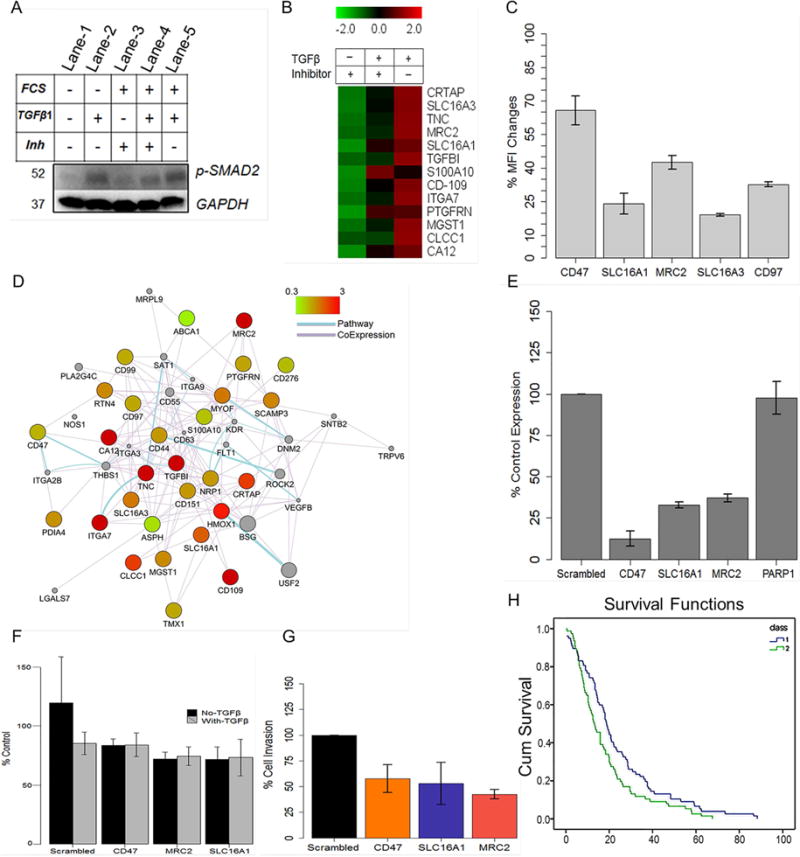
A) U87MG cells were treated with TGF-β1 or its inhibitor in presence or absence of FCS to evaluate i) endogenous c-terminally phosphorylated SMAD2 (lane-1), ii) the ability of TGF-β1 to phosphorylate SMAD2 (lane-2), iii) the ability of TGF-β1-inhibitor to inhibit SMAD2 phosphorylation when cells were grown in normal media (lane-3), iv) the ability of TGF-β1 to induce SMAD2 phosphorylation in cells grown earlier in presence of TGF-β1-inhibitor (lane-4), and v) the level of SMAD2 phosphorylation on prolong TGF-β1 exposure (50hrs) when cells were grown in normal growth media (lane-5). The results demonstrated i) the ability of TGF-β1-inhibitor in inhibiting c-terminal phosphorylation of SMAD2 (lane-3) similar to when cells were grown in serum-free media (lane-1&2) and ii) reversible nature of c-terminal phosphorylation inhibition of SMAD2 that could be reversed with TGF-β1 treatment (lane-3&4). GAPDH was used as loading controls. B) SRM analysis of TGF-β or its inhibitor treated U87MG cell lines revealed responsiveness of a subset of GBMSig proteins towards TGF-β signaling. Complete list of GBMSig proteins detected in various biospecimens and the responsiveness of these proteins towards TGF-β/Inhibitor is provided in the supplementary tables S9 and S10 respectively. Data from four replicates SRM analyses were centroided and presented as Z-score. C) flow cytometry analysis of a subset of GBMSig proteins following TGFβ/Inhibitor treatment. Percentage of changes in mean fluorescence intensity (MFI) was measured from treating U87MG cell lines with TGFβ relative to its inhibitor. Data represent means ± S.D. from four-replicate analyses. D) Network relationship (drawn using Cystoscape) between GBMSig proteins, which are presented as nodes. These nodes are connected through edges based on known pathways and co-expression. Color of the nodes is controlled by fold changes in expression of GBMSig proteins on TGFβ treatment. Grey colored nodes represent extended relationship with GBMSig proteins. E) qPCR analysis of siRNA mediated interference of a subset of TGFβ responsive GBMSig elements viz. MRC2, SLC16A1, and CD47 genes in U87MG cells. Results from three independent siRNA treatments (30hrs) were averaged (error bars represent S.D.) and presented as CT ratios normalized to HPRT housekeeping gene. PARP1 expression was used as non-targeted control. F) Calcein AM assay indicates no significant changes in cell growth and proliferation following siRNA mediated inhibition of SLC16A1, MRC2, and CD47 in U87 cell lines. Data represent means ± S.D. from five replicate analyses. G) siRNA treated U87 cells were allowed to migrate towards TGF-β1 gradient through basement membrane (Cell Biolabs Inc.). Invaded cells were analyzed through colorimetric assay. Results from three independent experiments were averaged and normalized to non-targeting siRNA pools (scrambled). As evident, loss of cell migration following siRNA mediated inhibition of SLC16A1 and MRC2 is similar to that of known invasive marker CD47. Data represent means ± S.D. from three replicate analyses. H) A panel of TGF-β responsive GBMSig (CA12, MRC2, TNC, CD44, SLC16A1, S100A10, ITGA7, CLCCI, and SLC16A3) highlights poor survival (p<0.003, log rank, Mantel-Cox) among GBM patients (class 2) when overexpressed relative to GBM patients where these genes were low expressed (class 1).
TGF-β1 responsive GBMSig subset and tumor invasion
As a known inducer of epithelial to mesenchymal transition (EMT), TGF-β1 plays an important role in the local metastasis of tumor cells (Picon et al., 1998, Hoelzinger et al., 2007). We determined if overexpression of TGFβ1-responsive transcripts from our GBMSig subset correlated with patient survival in the REMBRANDT data. Co-expression of these genes indeed correlated with poor patient survival (p<0.003, Log Rank, Mantel-Cox) (Figure 6G). This correlation could be the result of several factors related to tumor development concurrent with the hypothesis that invasion is, in part, attributable to the action of TGF-β1. To further investigate the role of our TGF-β targets in this context, we inhibited the expressions of SLC16A1, MRC2, and CD47 through siRNA silencing and quantified differences in U87MG cell invasion. We confirmed effective siRNA knockdown by qPCR (Figure 6E) and flow cytometry (Figure S5B), and observed no significant impact on cell viability following knockdown (Figure 6F). siRNA or non-targeting siRNA treated cells were seeded in transwell chambers, and the degree of cell invasion was evaluated as the percentage of cells invaded compared to non-targeting siRNA treated cells. The resultant cell invasion from three independent experiments is presented in Figure 6G. The silencing of SLC16A1 and MRC2 caused 52.88% ± 9.70SEM, and 42.26% ±2.19SEM reduced cell invasion respectively—similar to knockdown of the known invasion-mediating protein CD47 (57.74% ± 6.32SEM)-highlighting the role of these proteins in GBM invasion. We conclude that a subset of TGF-β responders play a crucial role in the migration and invasion of GBM cells, which in combination or alone may influence the clinical outcomes of GBM.
DISCUSSION
We formulated a systems strategy to develop GBMSig—a molecular signature for GBM composed of protein targets (table S9) originating from the cell-surface of diseased cells that provide excellent candidates that after shedding from the cell-surface can be detectable in the blood. We reasoned that cell-surface proteins would be the best candidates as they are most likely to be found in the blood due to their known role and function in disease associated cell-signaling processes (Li et al., 2013). The heterogeneity of GBM is well documented, and adds to the challenge of finding putative targets that can be reliably detected across heterogeneous tumors. For this reason, we wanted to enrich for putative targets that would likely be common to many GBM subtypes. Starting with four cell lines—two widely used cell lines of GBM, one established as having primary GBM cancer stem cell properties (e.g. expression of CD133), and a healthy neural stem cell line—we generated a list of putative targets by capturing and characterizing cell-surface proteins by mass spectrometry. Identification of cell-surface proteins by biotin labeling helped to enrich for proteins that are often difficult to detect because of their low abundance and hydrophobicity. By requiring all candidates to possess a known transmembrane domain, we likely eliminated many potential candidates. However, we feel our conservative approach greatly lessened the possibility of false positives. While reducing our target number from well over a thousand to 274, these targets were likely enriched for the properties most central to GBM blood biomarkers. We could detect that ten of the fourteen targets in the blood contained an N-terminal secretion signaling sequence (21 of 33 GBMSig targets) consistent with the properties our experimental design targeted.
We next relied upon existing microarray expression data to further enrich for candidate targets and explored how well they might work as classifiers. With a number of regulatory steps occurring between transcription and translation, we used the transcript array data as a filter to identify targets with appreciable RNA expression. Because the REMBRANDT and TCGA data sets were created for the purpose of better understanding heterogeneity across GBM and other tumor types, their small control sample size is not ideal for identifying the genes differentially expressed between tumor and normal samples. As we did not rely on the microarray expression data for generation of targets, but rather as a filter for the elimination of targets, the small control sample size was less of an issue. We further filtered our candidate list by removing differentially expressed genes that were also found in astrocytomas (148 samples) and oligodendrogliomas (67 samples), thus enhancing the specificity for GBM. Additionally, this integrative analysis ensured that the classifier was not based on expression of proteins that are the product of artifactual changes such as those that arise from cell culturing. Utilization of laboratory grown cell lines for the generation of candidates is consistent with the prior reports (Geiger et al., 2010). As GBM tumors are typically more aggressive and metastatic than other brain tumors, this filtering step may have selected for those genes specific to those functions. Previous work by our group used microarray data alone to identify gene networks that differ across astrocytoma grades (Wang et al., 2013). This previous work did not identify the TGF-β network and did not utilize anything resembling the filtering and proteomic data integration presented here, highlighting the differences achieved through an integrative omics approach. This filtering step may have enabled the identification of several TGF-β network proteins. In doing so, we may also have enriched for targets more likely to be in the blood, as metastatic mechanisms require signaling external to the primary tumor. This is supported by our analysis of the survival data, where co-expression of TGF-β responsive GBMSig genes correlated with poorer outcome (p<0.003, Log Rank, Mantel-Cox) (Figure 6H).
Following this filtering, we utilized the REMBRANDT and TCGA data for evaluating the performance of putative targets as classifiers. While the sensitivity (99.8%) and specificity (80%) were encouraging, with an MCC value of 0.87, the small control sample size limits the ability to determine the robustness of any classifier based on this evidence alone. However, we performed end-to-end experimental verification of the classifier and demonstrated that an integrated-omics approach was capable of producing a list of candidates that performed well as classifiers across platforms (proteomics, transcriptomics, ELISA) and across experiments (REMBRANDT to TCGA).
With our GBMSig candidates that performed well as classifiers for both REMBRANDT and TCGA, we turned to identifying those candidates that could best be quantitatively and reproducibly detected in both tissue and blood. We were able to detect 14 of the 33 low-abundant proteins in the blood and 21 of the 33 in brain tissue using the recently developed and sensitive approach of SRM. Further efforts to detect all proteins would likely have been productive as absence of detection is just as likely to be due to technical as it is biological reasons. Rather than optimize detection of all GBMSig targets, we chose to evaluate targets that could be reliably detected via established ELISA assays—the gold standard of clinical detection.
The vast majority of published biomarker candidates have failed to make it to the clinic (Micheel CM, 2012). A contributing factor to this high failure rate is the lack of properly designed experiments that contain truly independent training and test sets. We were unable to secure GBM and normal blood samples collected and processed by the same source. To circumvent this limitation, we acquired samples from five different sources, distributing samples from different sources into separate training and test sets. This approach makes it improbable, though not impossible, that our high sensitivity and specificity are the result of a batch effect. We have previously shown that multiple sources can be beneficial for the purposes of robustness and have the ability to help mitigate batch effects (Ma et al., 2014; Sung J, 2013; Wang et al., 2013). Even when mixing and matching samples in different configurations, we found our test to be robust, with comparable sensitivity and specificity (data not shown).
Our integrated omics approach also produced a list of candidates that are related to known characteristics of GBM. Many of the resulting targets had connections to TGF-β. We investigated the role of SLC16A1 and MRC2 in TGF-β responsiveness, showing that knockdown of these transcripts greatly reduced invasion. Recently, we have demonstrated that HMOX1 expression is associated with GBM stemness and invasion (Ghosh et al., 2016). MRC2 was previously shown to correlate with TGF-β1 expression in hepatocellular carcinoma (Gai X, 2014). SLC16A1 has not previously been implicated in invasion nor linked with TGFβ1 signaling. We have provided experimental evidence in favor of modular roles for 17 GBMSig proteins (Table S10) in TGF-β signaling. How these proteins or other proteins in our GBMSig connected to TGFβ1 signaling function in relationship to each other is an outstanding question for future investigation.
Our ability to distinguish between blood from GBM patients and normal controls was robust, with high sensitivity (95.2%) and specificity (95.2%). This resulted in an AUC of 0.99 for the training set and 0.98 for the validation set (Figure 5A–C), with great agreement between the effect size (>|0.6|) and the sampling method (power>0.8) (Figure S3E). Although we were not able to secure the control and the GBM samples from the same source, a high AUC value was observed that was unexpected. It is possible that this high agreement could be due to the novelty of the multi-omics, systems approach we used herein. Further application and interpretation of the results in the context of other cancers and/or pathologies will be of interest moving forward. We attempted to demonstrate potential clinical utility in assaying changes in the blood concentrations of 3 of 4 GBMSig proteins following tumor resection (Figure 5D–E, S4A–B). Given the non-specificity of MRI imaging in evaluating the effectiveness of therapeutic options, performance of GBMSig in the longitudinal assessment of therapeutic changes might widen the scope for disease management in the future.
As our selection criteria for which targets to assay by ELISA were based on 1) SRM detection in the blood; 2) high AUC values (>0.95) in ROC analyses of transcriptomic data (Figure S2D–E); and 3) availability of commercial ELISA kits, we anticipate a canvasing of other targets for which reliable ELISA data could be generated would increase the options for development of a GBM-specific blood test—especially from the GBMSig extended candidate list. Targets with smaller variance or greater separation between case and control might also improve robustness or be the basis of distinguishing among diseases. Although our candidate selection process was designed to increase the likelihood of robust separation between GBM and lower grade brain tumors and healthy controls, performance of GBMSig in separating non-GBM cancer such as liver cancer is also encouraging (Figure S2G). However, it remains to be evaluated if other cancers or pathologies would exhibit similarities and/or differences in the pattern of GBMSig expression.
We have demonstrated the power of integrating large-scale transcriptomics data together with shotgun proteomics to identify proteins that can be quantitatively measured and used to distinguish between the blood of a GBM patient and a healthy individual. The TGF-β connection of several of our GBMSig targets lends itself to biological interpretation that is often lacking with statistically heavy approaches. Arguably, this is a product of how we integrated experimental proteomics data with carefully analyzed existing transcriptomics data. Translating what we have learned here to a clinical application requires significantly more work, but many of the principles and systems applications demonstrated here can enable future efforts. We have an excellent candidate list of GBM biomarkers with which to move forward into the validation stage. We believe this general approach will be applicable to generating diagnostic blood protein panels for virtually any disease where the phenotype distinctions between normal and disease are clear.
STAR*METHODS
Detailed methods are provided in the online version of this paper and include the followings:
Contact for Reagent and Resource Sharing
Further information and requests may be directed to Leroy Hood at the Institute for Systems Biology (lhood@systemsbiology.org).
Experimental Model and Subject Details
Human Studies
IRB committee approvals and informed consent were obtained from all patients. The inclusion criteria for GBM patients were: 1) diagnosed as GBM based on clinical assessment, 2) plasma samples (collected in K2-EDTA tubes) were obtained prior to surgical removal of tumor mass, 3) age was >15 yrs., 4) subject was not suffering from any other cancer, 5) GBM patients received standard care of treatment, 6) both male and female subjects were included, 7) plasma samples were archived, labeled and fresh frozen. Age and gender of the subjects are provided in table S7. Healthy controls were purchased from Bioreclamation (Healthy-source-1 and Healthy-source-2A), and Proteogenex (Healthy-source-2) with the following inclusion criteria 1) subjects were healthy with no known chronic diseases, 2) no previous history of cancer, 3) age and gender matched with that of GBM subjects, and 4) plasma samples were archived, labeled and fresh frozen. GBM plasmas for the training set and longitudinal set (collected pre-operatively and post-operatively at 24hrs, 48hrs, and ~10 days) were collected from Swedish Medical Center, Seattle, and the validation set were collected from California Pacific Medical Center Research Institute (CPMCRI), San Francisco. Equal number of GBM (21 subjects) and healthy (21 subjects) blood plasma specimens was selected for training data set. Power analysis (>0.8) justified the inclusion of similar number of GBM (21 subjects) and healthy (21 subjects) blood plasma samples for the validation set. For power analysis, please refer to python codes in Data S1 and the figure S3E.
Cell Culture
The human GBM cell lines U87MG and T98 obtained from ATCC were grown in DMEM high glucose culture medium supplemented with 10%FBS and Pen Strep. Neural stem cells (NSCs) from Millipore and cancer stem cells (CSCs) from Celprogen were grown according to suppliers’ specifications.
Methods Detail
Cell-Surface Labeling and Mass Spectrometry
EZ-Link-Sulfo-NHS-SS-biotin (Pierce) kit was used to surface label U87, T98, neural stem cell (Millipore) and cancer stem cell (Celprogen) according to manufacturer instruction. Biotinylated cell surface proteins were affinity-purified on neutravidin beads (supplied with the kit). After stripping off non-specifically bound proteins by several rounds of washing with the lysis buffer (supplied with the kit) followed by water wash (to remove excess reagents), labeled proteins were selectively eluted with DTT at elevated temperature to ensure higher recovery of bound proteins. Eluted proteins were concentrated onto 10kDa micro ultrafiltration unit and reduced in the same ultrafiltration unit with 10mM TCEP for 60 min at 37°C. Excess TCEP was neutralized by washing the membrane with equal volume of 100mM ammonium bicarbonate followed by alkylation with 55mM iodoacetamide for 1hr in dark. Excess alkylating agent was quenched with molar equivalent of TCEP by incubating at RT for 15min followed by washing the membrane several times with digestion buffer containing 50mM ammonium bicarbonate, 10% TFE and 1mM CaCl2. Proteins were digested on membrane serially with trypsin for 12hrs at 37°C with 1:25 enzyme-to-protein ratio and Glu C for 6 hrs with 1:50 enzyme-to-protein ratio at RT in dark, respectively. Enzymatic digestion was quenched by adding 20μl of 0.2%FA, and digested materials were collected by centrifugation. Peptides were lyophilized and re-dissolved in 1% ACN, 0.1% FA for mass spec analysis using a Thermo Electron Orbitrap mass spectrometer (LTQ ORBITRAP) equipped with an electrospray ionization source in line with an Agilent HP1100 liquid chromatography system. Peptide digests were enriched onto a 2cm pre-column packed in-house with 200Å Magic C18AQ resin and separated using a ProteoPep II 75μm i.d. × 10cm analytical column on 160 min ACN gradient as follows: 2%B (Buffer B-100%ACN/0.1%FA) for 5min, 2–10%B for 20min, 10–25%B for 65min, 25–40%B for 20min, 40–60%B for 15min, 60–100%B for 10min, and held at 100%B for 13 min followed by equilibration of column for 17min with buffer A (Buffer A-2%ACN/0.1%FA). Pump flow rate was maintained constantly at 0.350μl/min. Mass spectrometer was operated in positive data-dependent acquisition mode with 1 S MS scan (m/z 300–1800; 30,000 resolution) followed by 9 MS/MS events for peptides with charge states between +2 and +4. Dynamic exclusion was set for 60 sec. Each isolate was run three times. After data acquisition, Xcalibur (Thermo) raw data were converted to mzXML format using ReAdW profile and default parameters. Peptide assignments were done using the GPM (www.thegpm.org).
Brain Tissue Processing
Brain tissues were homogenized in tissue lysis buffer (TLB) composed of 100mM n-octyl-glucoside, 1% NP-40, 150mM NaCl, 1mM PMSF, 2mM sodium orthovanadate and 50mM sodium fluoride in 50mM TEAB, pH8.0. Tissue homogenate was clarified by centrifugation at 10,000×g for 10 min and the supernatant containing the proteins of interest was preserved at −80°C till further use. For SRM analysis, tissues were reduced, alkylated, and then digested with trypsin and Glu C o/n in dark. Digestion reaction was quenched with TFA, and peptides were lyophilized, C18 purified and solubilized in 1%ACN/0.1% FA for SRM analysis.
GBM Subtyping
Total RNA was isolated from tissue samples with Triazol, and then cleaned with RNeasy MinElute Cleanup Kit. 1 μg of total RNA was used to generate 100 μl of cDNA using the High Capacity cDNA Reverse Transcription Kit. Real-time PCR of MGMT was performed on the ABI PRISM 7900 HT detection system using 33 Taqman probes (Phillips et al., 2006) and Taqman reagents under default conditions: 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute with human beta-glucuronidase (hGUS) as endogenous control. All assays were done in triplicate. The expression level of each gene (Δct) for each tissue sample is calculated compared to the hGUS expression level using the formula 2−(Ct value of gene − Ct value of hGUS). qRT-PCR was performed for 4 GBM tissue samples (Table S5) for the 33 gene panel as described by Phillips et al. For each gene we obtained the average Δct (μ) and standard deviation (σ). For each tissue sample we calculate standard scores (z) for all 33 genes as follows: zg = Δctg − μ/σg, where g ∈ 33 gene panel. Three components: 1) Mesenchymal (M), 2) Proliferative (P) and 3) Proneuronal (N) expressions were calculated by taking the average of z scores of all genes belonging to the corresponding component.
M = μ(zg), where g ∈ component Mesenchymal
P = μ(zg), where g ∈ component Proliferative
N = μ(zg), where g ∈ component Proneuronal
Finally, the subtype is determined using the following reference range.
Mesenchymal M > P + 0.2, M > N + 0.2
Proliferative P > M + 0.2, P > N + 0.2
Proneuronal N > M + 0.2, N > P + 0.2
Prolifmes P > N + 0.2, M > N + 0.2, |P-M| < 0.2
Mesneuronal M > P + 0.2, N > P + 0.2, |N-M| < 0.2
Prolifneuronal P > M + 0.2, N > M + 0.2, |N-P| < 0.2
SRM Assay Development and Analysis
Agilent Triple Quadrupole equipped with ChipCube nanoelectrospray ionization source in line with 1200 nanoFlow HPLC system was employed for SRM assay development and subsequent analysis. Cell surface peptide library developed through prior high-resolution shotgun analysis (described earlier in shotgun mass spectrometry section) provided the foundation to synthesize C-terminally labeled (13C15N) surrogate peptides for 33 GBMSig. Collision energy (CE) values for each peptide bond were optimized using the Mass Hunter Optimizer for Peptides Software (Agilent Technologies). All combinations of +2, +3 and +4 of Q1 and +1 and +2 of Q3 ion pair (only y-series of ions) of each peptide were undertaken for CE optimization. The top 4–5 transition pairs (Q1/Q3) and corresponding CE values that ensured maximum signal intensity of SRM trace was employed for quantitation. To develop dynamic-SRM (d-SRM) assay, retention time of each peptide was determined a priori by spiking C-terminally labeled (13C15N) surrogate peptides in presence of corresponding biological isolates (cell, tissue or serum). The abundance of endogenous peptides was assessed from co-eluting surrogate peptides (13C15N) which ensured precise quantification, All SRM acquisition method was run with standard parameters viz. capillary voltage 1700–2100, a sheath gas flow of 11 L/min at a temperature of 380°C, a drying gas flow of 15 L/min at a temp of 150°C, nebulizer gas flow at 30psi, the fragmentor voltage at 380 V, the cell accelerator voltage at 7 V, an MS operating pressure of 5×105 Torr and Q1/Q3 set to operate in unit resolution.
All samples were loaded onto HPLC chip comprising of an enrichment column (160nL or 500nL) and a 150mm analytical C18 column. For plasma SRM analysis, 500nL enrichment column was used and peptides were eluted (with nanopump flow of 0.3μl/min) over step gradients as follows: 4%B for 2min, 25%B for 53min and maintained for 5min, 47%B for 12min and maintained for 10 min followed by column washing for 10 min at 0.5μl/min and equilibration for 11min at 0.3μl/min flow rate.
Plasma Depletion
All plasma samples were immunodepleted using a C10/10 column packed in-house with 6ml bulk Seppro IgY14 matrix capable of depleting top 14 abundant proteins from human plasma prior to SRM mass spectrometry. Flow through fraction was concentrated onto 10kDa micro ultrafiltration unit and reduced in the same ultrafiltration unit with 10mM TCEP for 60 min at 370C. Excess TCEP was neutralized by washing the membrane with equal volume of 100mM ammonium bicarbonate followed by alkylation with 55mM iodoacetamide for 1hr in dark. Excess alkylating agent was quenched with molar equivalent of TCEP and incubating at RT for 15min followed by washing the membrane several times with digestion buffer containing 50mM ammonium bicarbonate, 10% TFE and 1mM CaCl2. Proteins were digested on membrane serially with trypsin for 12hrs at 37°C with 1:25 enzyme-to-protein ratio and Glu C for 6 hrs with 1:50 enzyme-to-protein ratio at RT in dark respectively. Enzymatic digestion was quenched by adding 20μl of 0.2%FA and digested materials were collected by centrifugation. Peptides were lyophilized and re-dissolved in 1% ACN, 0.1% FA. After C18 clarification, each specimen was spiked with heavy (13C15N K/R) synthetic GBMSig peptides for SRM mass spectrometry analysis.
Flow Cytometry
The following antibodies were used in the study: SLC16A1, MRC2, SLC16A3, CD47, and CD97. Cells were harvested after washing with ice-cold washing buffer (PBS/0.1% sodium azide) and incubated with primary antibody in antibody incubation buffer (1% BSA in PBS/0.1% sodium azide) for 1hr on ice. After washing the cell pellets with washing buffer, cells were incubated with FITC conjugated anti-mouse or PE conjugated anti-rabbit for 30 min on ice at 1:100 or 1:200 dilutions (prepared in antibody incubation buffer) respectively. Unbound conjugates were removed by washing with the washing buffer. Flow cytometry analysis was performed on FACSCalibur (BD Biosciences). The mean fluorescence intensity (MFI) of FITC or PE-positive cells (three replicate runs) was measured in comparison to respective isotype controls and the data were analyzed using FlowJo.
Western Blotting
Total proteins from U87 cells treated with TGF-β or its inhibitor were lysed by sonication in TLB, composed of 100mM n-octyl-glucoside, 1% NP-40, 150mM NaCl, 1mM PMSF, 2mM sodium orthovanadate and 50mM sodium fluoride in 50mM TEAB, pH8.0. After protein estimation with Pierce 660, equal amount of protein (8μg) from each isolate was separated by electrophoresis on Mini-Protean TGX 4–15% precast gel (Biorad) and transferred to nitrocellulose membrane using a semi-dry blotting (Biorad) apparatus. The membrane was blocked in TBS containing 3% FBS for 30min/RT and incubated with anti-SMAD2 or C-terminally phosphorylated SMAD2 at 1:1,500 dilution or anti-GAPDH at 1:15,000 dilution O/N at 40C. After washing the membrane with TBS/0.1%T-20, membrane was incubated with appropriate HRP-conjugated secondary antibody. The Fisher SuperSignal West Femto Chemiluminescent Substrate was used for detection. The blot was visualized using Biorad gel documentation unit.
siRNA Treatment and qPCR Analysis
U87 cells (one million) maintained in DMEM high glucose culture medium supplemented with 10%FBS and penicillin-streptomycin were plated in a 10 cm dish. 24 hours after seeding, cells were washed with HBSS prior to reducing FBS to 0.2% in DMEM for siRNA treatment. ON-TARGETplus-SMARTpool siRNAs for SLC16A1, MRC2, CD47 and ON TARGETplus Non-targeting pool were used in the study. siRNAs were added at a 2:1 ratio of Dharmafect to siRNA for a final concentration of 40 nM per the manufacturer instructions. For Quantitative Real-time PCR (qRT-PCR) analysis of target gene inhibition, RNA was isolated per the manufacturer instructions using Qiagen RNeasy Kit. RNA was converted to cDNA using Applied Biosystems High Capacity cDNA Reverse Transcription Kit, per manufacturer instructions. qRT-PCR was performed on an ABI 7900 HT 384-well format using Power Syber Green. For the following genes, primers were designed using the online tools provided by Integrated DNA technologies (IDT): HPRT1 forward 5′-TGCTGAGGATTTGGAAAGGG, HPRT1 reverse 5′-ACAGAGGGCTACAATGTGATG, MRC2 forward 5′-ACCAGCAACATATCCAAGCC, MRC2 reverse 5′-GAGTTTCCCTGGATGGTGTAG, SLC16A1 forward 5′-GTGGCTTGATTGCAGCTTC, SLC16A1 reverse 5′-TGGTCGCCTCTTGTAGAAATAC, CD47 forward 5′-TGGTATGGATGAGAAAACAATTGC, CD47 reverse 5′-GTCACAATTAAACCAAGGCCAG. PCR pairs were designed to generate amplicons between 90–130 base-pairs and pre-evaluated for a dissociation curve.
Cell Invasion Assay
Quantitative cell invasion assay was performed for U87MG cells using the invasion kit according to manufacturer’s instructions. Percentage of siRNA treated cells invaded in respect to non-targeting RNA treated cells were assessed from three independent experiments with three replicates each time.
Quantification and Statistical Analysis
Data Normalization
All numerical raw data were normalized to standard score (Z-score) using the formula, is the raw value and μ is the mean of the population, σ is the standard deviation of the population.
SRM Quantification
SRM data were analyzed using Skyline (Bereman et al., 2012). SRM traces were integrated by default peak integration method and processed with Savitzky-Golay smoothing algorithm as described in Skyline. For precise identification and quantification of targets, each SRM trace was manually inspected for three- Q1/Q3 transition pairs with retention time determined empirically in prior runs. Peptide peak ratios (endogenous: surrogate) from three independent runs of each sample were averaged and expressed per μl of neat plasma or per μg of total protein as in the case of brain tissue homogenates. Final data were expressed as standard score (Z score).
Development of GBMSig
The human tissue expression array of GBM (N=228 subjects) and non-tumor (N=16 subjects) isolates in REMBRANDT [https://gdoc.georgetown.edu/gdoc/] were used as training datasets to distinguish differentially expressed cell-surface-transmembrane proteins as predicted using TMHMM algorithm (http://www.cbs.dtu.dk/services/TMHMM/). We employed R: Bioconductor (www.bioconductor.org) packages “affy” and “limma” for gene expression analysis. Cell surface proteins differentially expressed between GBM and non-tumor with log2-fold change >2X and FDR <0.05 was undertaken for further analysis. To identify GBM enriched expression, astrocytoma (N=148 subjects) and oligodendroma (N=67 subjects) tissue arrays in the same compendium were also assessed. Predictive accuracy of GBMSig was evaluated using independent transcriptome dataset in TCGA (547 GBM tumors and 10 non-tumor brain controls).
Statistical Analysis and Modeling
True performance of GBMSig classifier was evaluated through SVM. For SVM modeling, tissue transcriptomics data in REMBRANDT were used as training set. Using R package CvTools, REMBRANDT data were splitted (K=10) into training and validation sets. To achieve, the highest level of accuracy, we performed autotuning of the hyperparameters and selected the best value for gamma and cost for subsequent modeling as follows:
| gamma | cost | error dispersion |
| 1.00E–06 | 0.1 | 0.072727 0.05749596 |
| 1.00E–05 | 0.1 | 0.072727 0.05749596 |
| 1.00E–04 | 0.1 | 0.072727 0.05749596 |
| 1.00E–03 | 0.1 | 0.072727 0.05749596 |
| 1.00E–02 | 0.1 | 0.072727 0.05749596 |
| 1.00E–01 | 0.1 | 0.072727 0.05749596 |
| 1.00E–06 | 1 | 0.072727 0.05749596 |
| 1.00E–05 | 1 | 0.072727 0.05749596 |
| 1.00E–04 | 1 | 0.072727 0.05749596 |
| 1.00E–03 | 1 | 0.072727 0.05749596 |
| 1.00E–02 | 1 | 0.072727 0.05749596 |
| 1.00E–01 | 1 | 0.022727 0.03214122 |
| 1.00E–06 | 10 | 0.072727 0.05749596 |
| 1.00E–05 | 10 | 0.072727 0.05749596 |
| 1.00E–04 | 10 | 0.072727 0.05749596 |
| 1.00E–03 | 10 | 0.068182 0.05769525 |
| 1.00E–02 | 10 | 0.013636 0.03067948 |
| 1.00E–01 | 10 | 0.009091 0.01916532 |
Best parameters: gamma=0.1 and cost=10.
SVM modeling was performed using the R package e1021. Minimization of the error function was calculated using the following formula: , constraints=yi(wT θ(xi)+b) ≥1− ζi and ζi ≥ 0, = 1, …. N;kernel θ=exp(−γ|xi−xj|; C=capacity constant, w= vector of coefficients, b= constant, ζi= parameters for handling inseparable data, y= class labels,xi=independent variables, and θ=kernel. To train the SVM model (C-classification) we used radial kernel, gamma=0.1, and cost=10 (as determined following autotuning of the hyperparameters). To assess the quality of the training results, accuracy of the model was calculated from 100fold cross validation, which revealed mean accuracy of 98.63%. Once the model was trained we evaluated the sensitivity, specificity, positive predictive value (PPV) and negative predictive value (NPV) of the classifier for independent validation set in TCGA: Sensitivity= TP/|P|; Specificity= TN/|N|; Precision= TP/TP+FP; PPV= TP/(TP+FP); NPV= TN/(TN+FN); where TP= True positive predicted by the model, |P|= Total Positive, TN= True negative predicted by the model, |N|= Total Negative, FP= False positive i.e. healthy instances predicted as GBM, FN= False negative i.e. true GBM instances predicted as healthy.
Power Analysis
To achieve empirical validity, power analysis was performed. Power analysis of ELISA data were performed using statsmodel library in python with alpha=0.05; effect size for ELISA data was calculated using Cohen’s d, where
For detailed analysis, please refer to the python code provided as Data S1.
Linear Discriminant Analysis (LDA)
A LDA method was developed to evaluate the performance of a subset of GBMSig (HMOX1, CD44, BIGH3, and VCAM1) proteins as blood classifier. ELISA Training data set (GBM plasmas= 21, and Healthy plasmas= 21) was modelled using LDA from the scikit-learn python package and the performance of the classifier were predicted for the validation set (GBM plasmas= 21, and Healthy plasmas= 21). For detailed analysis, please refer to the python codes provided as Data S1.
Receiver Operating Characteristic (ROC) Curve Analysis
Diagnostic performance and accuracy of the classifier in separating GBM and normal controls were assessed through ROC analysis as described in Medcalc. Sensitivity and specificity for a given GBMSig is calculated as: Sensitivity and specificity ; where a=True positive =(TP), b= False Negative (FN), c= False Positive (FP), and d= True Negative (TN).
Data and Software Availability
Python code used in the study is provided as Data S1.
Proteomics data for individual cell lines and replicate analyses are available from www.thegpm.org. Please refer to key resources table for unique GPM identification numbers that are required to access the data.
Supplementary Material
Table S1. Related to Figure 2. Proteins identified from cell surface proteomics study. Details of proteins identified from cell-surface proteomics study. All three replicate analyses of each cell line/primary (source) cells are presented as run1-3.
Table S3. Related to Figure 4 and Figure 6. SRM transition list. Details of Q1/Q3 SRM transition list used in the study are presented.
Table S8. Related to Figure 5 and Figure S4. ELISA data- Longitudinal. Changes in the level of four GBMSig proteins were investigated by commercial ELISA kits before and after tumor resection for prospectively collected blood plasmas from 10 patients.
Highlights.
Cell-surface proteomics of four cell lines relevant in glioblastoma.
Development of a 33-cell-surface-protein signature for glioblastoma.
Association of a subset of the signature with TGF-β signaling and cancer invasion.
Potential of a subset of GBMSig proteins as blood biomarkers.
Acknowledgments
We would like to sincerely acknowledge the life and contributions of Dr. Greg Foltz, who passed away during the preparation of this manuscript. Dr. Foltz was a passionate and dedicated neurosurgeon and was involved intricately with this project. We thank Drs. Sui Huang, Andrew Magis and James Eddy of ISB for critical readings and discussions, and Mr. Haidn E. Foster of SNI, for proof-reading the manuscript. We are grateful to the Proteomics facility at ISB where mass spectrometry analyses were completed. The work was supported by NIH/NCI NanoSystems Biology Cancer Center grant U54 CA151819A (LH), NIH Center for Systems Biology grant 2P50 GM076547 (LH, NDP), the Roy J. Carver Charitable Trust (NDP), and the Camille Dreyfus Teacher-Scholar Program (NDP).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
D.G. and L.H. designed the study, D.G.,C.C.F., and K.R performed experiments, G.F. and C.S.C performed craniotomy, and G.F.,C.S.C, and L.S. collected GBM plasma specimens, N.D.P.,D.G.,N.S.,J.C, and J.C.E. performed statistical analysis and computational modeling, D.G. and C.F. prepared the manuscript, all authors edited and revised the manuscript, and L.H. and N.P. supervised research.
References
- Aigner L, Bogdahn U. TGF-beta in neural stem cells and in tumors of the central nervous system. Cell and tissue research. 2008;331:225–241. doi: 10.1007/s00441-007-0466-7. [DOI] [PubMed] [Google Scholar]
- Audigier Y, Picault FX, Chaves-Almagro C, Masri B. G Protein-Coupled Receptors in cancer: biochemical interactions and drug design. Progress in molecular biology and translational science. 2013;115:143–173. doi: 10.1016/B978-0-12-394587-7.00004-X. [DOI] [PubMed] [Google Scholar]
- Beare A, Stockinger H, Zola H, Nicholson I. Monoclonal antibodies to human cell surface antigens. In: Coligan John E, et al., editors. Current protocols in immunology. 2008. p. 4A. Appendix 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bereman MS, MacLean B, Tomazela DM, Liebler DC, MacCoss MJ. The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics. 2012;12:1134–1141. doi: 10.1002/pmic.201200042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefoi H, Potti A, Delorenzi M, Mauriac L, Campone M, Tubiana-Hulin M, Petit T, Rouanet P, Jassem J, Blot E, et al. Validation of gene signatures that predict the response of breast cancer to neoadjuvant chemotherapy: a substudy of the EORTC 10994/BIG 00-01 clinical trial. The Lancet Oncology. 2007;8:1071–1078. doi: 10.1016/S1470-2045(07)70345-5. [DOI] [PubMed] [Google Scholar]
- Cortes C, Vapnik V. Support-Vector Networks. Machine Learning. 1995;20:273–297. [Google Scholar]
- Cancer Genome Atlas Research, N. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, Dai H, He YD, van’t Veer LJ, Bartelink H, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Fu AK, Ip NY. Bidirectional signaling of ErbB and Eph receptors at synapses. Neuron glia biology. 2008;4:211–221. doi: 10.1017/S1740925X09990287. [DOI] [PubMed] [Google Scholar]
- Craig R, Beavis RC. A method for reducing the time required to match protein sequences with tandem mass spectra. Rapid communications in mass spectrometry: RCM. 2003;17:2310–2316. doi: 10.1002/rcm.1198. [DOI] [PubMed] [Google Scholar]
- Craig R, Beavis RC. TANDEM: matching proteins with tandem mass spectra. Bioinformatics. 2004;20:1466–1467. doi: 10.1093/bioinformatics/bth092. [DOI] [PubMed] [Google Scholar]
- Dai L, Guinea MC, Slomiany MG, Bratoeva M, Grass GD, Tolliver LB, Maria BL, Toole BP. CD147-dependent heterogeneity in malignant and chemoresistant properties of cancer cells. The American journal of pathology. 2013;182:577–585. doi: 10.1016/j.ajpath.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delyon J, Maio M, Lebbe C. The ipilimumab lesson in melanoma: achieving long-term survival. Seminars in oncology. 2015;42:387–401. doi: 10.1053/j.seminoncol.2015.02.005. [DOI] [PubMed] [Google Scholar]
- De Marco P, Bartella V, Vivacqua A, Lappano R, Santolla MF, Morcavallo A, Pezzi V, Belfiore A, Maggiolini M. Insulin-like growth factor-I regulates GPER expression and function in cancer cells. Oncogene. 2013;32:678–688. doi: 10.1038/onc.2012.97. [DOI] [PubMed] [Google Scholar]
- Demuth T,BM. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol. 2004;70:217–228. doi: 10.1007/s11060-004-2751-6. [DOI] [PubMed] [Google Scholar]
- Erber WN. Human leucocyte differentiation antigens: review of the CD nomenclature. Pathology. 1990;22:61–69. doi: 10.3109/00313029009063782. [DOI] [PubMed] [Google Scholar]
- Ertel A, Verghese A, Byers SW, Ochs M, Tozeren A. Pathway-specific differences between tumor cell lines and normal and tumor tissue cells. Molecular cancer. 2006;5:55. doi: 10.1186/1476-4598-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenyo D, Phinney BS, Beavis RC. Determining the overall merit of protein identification data sets: rho-diagrams and rho-scores. Journal of proteome research. 2007;6:1997–2004. doi: 10.1021/pr070025y. [DOI] [PubMed] [Google Scholar]
- Gai X,TK, Lu Z, Zheng X. MRC2 expression correlates with TGFβ1 and survival in hepatocellular carcinoma. Int J Mol Sci. 2014;15:15011–15025. doi: 10.3390/ijms150915011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger T, Cox J, Ostasiewicz P, Wisniewski JR, Mann M. Super-SILAC mix for quantitative proteomics of human tumor tissue. Nature methods. 2010;7:383–385. doi: 10.1038/nmeth.1446. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Lippert D, Krokhin O, Cortens JP, Wilkins JA. Defining the membrane proteome of NK cells. Journal of mass spectrometry: JMS. 2010;45:1–25. doi: 10.1002/jms.1696. [DOI] [PubMed] [Google Scholar]
- Ghosh D, Ulasov IV, Chen L, Harkins LE, Wallenborg K, Hothi P, Rostad S, Hood L, Cobbs CS. TGFβ-Responsive HMOX1 Expression Is Associated with Stemness and Invasion in Glioblastoma Multiforme. Stem Cells. 2016 doi: 10.1002/stem.2411. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golestaneh N, B M. TGF-beta, neuronal stem cells and glioblastoma. Oncogene. 2005;24:5722–5730. doi: 10.1038/sj.onc.1208925. [DOI] [PubMed] [Google Scholar]
- Grabowski JP, Sehouli J. Current management of ovarian cancer. Minerva medica. 2015;106:151–156. [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Hegi ME, Stupp R. Neuro-oncology: in search of molecular markers of glioma in elderly patients. Nature reviews Neurology. 2013;9:424–425. doi: 10.1038/nrneurol.2013.127. [DOI] [PubMed] [Google Scholar]
- Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. Journal of the National Cancer Institute. 2007;99:1583–1593. doi: 10.1093/jnci/djm187. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer metastasis reviews. 2006;25:435–457. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- Jorgensen B, Knudtson J. Stop cancer colon. Colorectal cancer screening–updated guidelines. South Dakota medicine: the journal of the South Dakota State Medical Association. 2015:82–87. Spec No. [PubMed] [Google Scholar]
- Kampen KR. Membrane proteins: the key players of a cancer cell. The Journal of membrane biology. 2011;242:69–74. doi: 10.1007/s00232-011-9381-7. [DOI] [PubMed] [Google Scholar]
- Kandouz M. The Eph/Ephrin family in cancer metastasis: communication at the service of invasion. Cancer metastasis reviews. 2012;31:353–373. doi: 10.1007/s10555-012-9352-1. [DOI] [PubMed] [Google Scholar]
- Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic acids research. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Lee JW. Membrane Proteins Involved in Epithelial-Mesenchymal Transition and Tumor Invasion: Studies on TMPRSS4 and TM4SF5. Genomics & informatics. 2014;12:12–20. doi: 10.5808/GI.2014.12.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. Journal of molecular biology. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- Kurscheid S, Bady P, Sciuscio D, Samarzija I, Shay T, Vassallo I, Criekinge WV, Daniel RT, van den Bent MJ, Marosi C, et al. Chromosome 7 gain and DNA hypermethylation at the HOXA10 locus are associated with expression of a stem cell related HOX-signature in glioblastoma. Genome biology. 2015;16:16. doi: 10.1186/s13059-015-0583-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauden L, Siewiera J, Boukouaci W, Ramgolam K, Mourah S, Lebbe C, Charron D, Aoudjit F, Jabrane-Ferrat N, Al-Daccak R. TGF-beta-induced (TGFBI) protein in melanoma: a signature of high metastatic potential. The Journal of investigative dermatology. 2014;134:1675–1685. doi: 10.1038/jid.2014.20. [DOI] [PubMed] [Google Scholar]
- Li XJ, Hayward C, Fong PY, Dominguez M, Hunsucker SW, Lee LW, McLean M, Law S, Butler H, Schirm M, et al. A blood-based proteomic classifier for the molecular characterization of pulmonary nodules. Science translational medicine. 2013;5:207ra142. doi: 10.1126/scitranslmed.3007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limani P, Samaras P, Lesurtel M, Graf R, DeOliveira ML, Petrowsky H, Clavien PA. [Pancreatic cancer- a curable disease] Praxis. 2015;104:453–460. doi: 10.1024/1661-8157/a001990. [DOI] [PubMed] [Google Scholar]
- Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. The New England journal of medicine. 2007;356:217–226. doi: 10.1056/NEJMoa063994. [DOI] [PubMed] [Google Scholar]
- Ma S, Sung J, Magis AT, Wang Y, Geman D, Price ND. Measuring the effect of inter-study variability on estimating prediction error. PloS one. 2014;9:e110840. doi: 10.1371/journal.pone.0110840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maclean B, Tomazela DM, Abbatiello SE, Zhang S, Whiteaker JR, Paulovich AG, Carr SA, Maccoss MJ. Effect of collision energy optimization on the measurement of peptides by selected reaction monitoring (SRM) mass spectrometry. Analytical chemistry. 2010;82:10116–10124. doi: 10.1021/ac102179j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micheel CM,NS, Omenn GS. Evolution of Translational Omics: Lessons Learned and the Path Forward. Washington (DC): National Academies Press (US); 2012. [PubMed] [Google Scholar]
- Milroy MJ. Breast cancer screening. South Dakota medicine: the journal of the South Dakota State Medical Association. 2015:69–73. Spec No. [PubMed] [Google Scholar]
- Moller S, Croning MD, Apweiler R. Evaluation of methods for the prediction of membrane spanning regions. Bioinformatics. 2001;17:646–653. doi: 10.1093/bioinformatics/17.7.646. [DOI] [PubMed] [Google Scholar]
- Mrugala MM. Advances and challenges in the treatment of glioblastoma: a clinician’s perspective. Discovery medicine. 2013;15:221–230. [PubMed] [Google Scholar]
- Murai KK, Pasquale EB. Restraining stem cell niche plasticity: a new talent of Eph receptors. Cell stem cell. 2010;7:647–648. doi: 10.1016/j.stem.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murat A, Migliavacca E, Gorlia T, Lambiv WL, Shay T, Hamou MF, de Tribolet N, Regli L, Wick W, Kouwenhoven MC, et al. Stem cell-related “self-renewal” signature and high epidermal growth factor receptor expression associated with resistance to concomitant chemoradiotherapy in glioblastoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2008;26:3015–3024. doi: 10.1200/JCO.2007.15.7164. [DOI] [PubMed] [Google Scholar]
- Nummela P, Lammi J, Soikkeli J, Saksela O, Laakkonen P, Holtta E. Transforming growth factor beta-induced (TGFBI) is an anti-adhesive protein regulating the invasive growth of melanoma cells. The American journal of pathology. 2012;180:1663–1674. doi: 10.1016/j.ajpath.2011.12.035. [DOI] [PubMed] [Google Scholar]
- Okumura S, Baba H, Kumada T, Nanmoku K, Nakajima H, Nakane Y, Hioki K, Ikenaka K. Cloning of a G-protein-coupled receptor that shows an activity to transform NIH3T3 cells and is expressed in gastric cancer cells. Cancer science. 2004;95:131–135. doi: 10.1111/j.1349-7006.2004.tb03193.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omenn, DeAngelis C, DeMets D, Fleming T, Geller G, Gray J, Hayes D, Henderson C, Kessler L, Lapidus S, et al. Evolution of Translational Omics: Lessons Learned and the Path Forward. Institute of Medicine Report; 2012. [PubMed] [Google Scholar]
- Pasquale EB. Eph receptors and ephrins in cancer: bidirectional signalling and beyond. Nat Rev Cancer. 2010;10:165–180. doi: 10.1038/nrc2806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen TN, Brunak S, von Heijne G, Nielsen H. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nature methods. 2011;8:785–786. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- Phillips HS, K S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Picon A, Gold LI, Wang J, Cohen A, Friedman E. A subset of metastatic human colon cancers expresses elevated levels of transforming growth factor beta1. Cancer epidemiology, biomarkers & prevention: a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology. 1998;7:497–504. [PubMed] [Google Scholar]
- Pickup M, Novitskiy S, Moses HL. The roles of TGFbeta in the tumour microenvironment. Nat Rev Cancer. 2013;13:788–799. doi: 10.1038/nrc3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picotti P, Aebersold R. Selected reaction monitoring-based proteomics: workflows, potential, pitfalls and future directions. Nature methods. 2012;9:555–566. doi: 10.1038/nmeth.2015. [DOI] [PubMed] [Google Scholar]
- Pietras A, Katz AM, Ekstrom EJ, Wee B, Halliday JJ, Pitter KL, Werbeck JL, Amankulor NM, Huse JT, Holland EC. Osteopontin-CD44 signaling in the glioma perivascular niche enhances cancer stem cell phenotypes and promotes aggressive tumor growth. Cell stem cell. 2014;14:357–369. doi: 10.1016/j.stem.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollig C, Knop S, Bornhauser M. Multiple myeloma. Lancet. 2015;385:2197–2208. doi: 10.1016/S0140-6736(14)60493-1. [DOI] [PubMed] [Google Scholar]
- Shao H, Chung J, Balaj L, Charest A, Bigner DD, Carter BS, Hochberg FH, Breakefield XO, Weissleder R, Lee H. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nature medicine. 2012;18:1835–1840. doi: 10.1038/nm.2994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R, Chinnaiyan AM, Ghosh D. Pathway analysis reveals functional convergence of gene expression profiles in breast cancer. BMC medical genomics. 2008;1:28. doi: 10.1186/1755-8794-1-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Bessarabova M, Dosymbekov D, Dezso Z, Nikolskaya T, Dudoladova M, Serebryiskaya T, Bugrim A, Guryanov A, Brennan RJ, et al. Functional analysis of multiple genomic signatures demonstrates that classification algorithms choose phenotype-related genes. The pharmacogenomics journal. 2010;10:310–323. doi: 10.1038/tpj.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stupp R, Hegi ME, van den Bent MJ, Mason WP, Weller M, Mirimanoff RO, Cairncross JG, et al. Changing paradigms–an update on the multidisciplinary management of malignant glioma. The oncologist. 2006;11:165–180. doi: 10.1634/theoncologist.11-2-165. [DOI] [PubMed] [Google Scholar]
- Sung J, Wang Y, Chandrasekaran S, Witten DM, Price ND. Molecular signatures from omics data: from chaos to consensus. Biotechnology journal. 2012;7:946–957. doi: 10.1002/biot.201100305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung J, K P, Ma S, Funk CC, Magis AT, Wang Y, Hood L, Geman D, Price ND. Multi-study integration of brain cancer transcriptomes reveals organ-level molecular signatures. PLoS Comput Biol. 2013;9 doi: 10.1371/journal.pcbi.1003148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England journal of medicine. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teh JL, Chen S. Glutamatergic signaling in cellular transformation. Pigment cell & melanoma research. 2012;25:331–342. doi: 10.1111/j.1755-148X.2012.00983.x. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- van ’t Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Varady G, Cserepes J, Nemeth A, Szabo E, Sarkadi B. Cell surface membrane proteins as personalized biomarkers: where we stand and where we are headed. Biomarkers in medicine. 2013;7:803–819. doi: 10.2217/bmm.13.90. [DOI] [PubMed] [Google Scholar]
- Vogel TW, Zhuang Z, Li J, Okamoto H, Furuta M, Lee YS, Zeng W, Oldfield EH, Vortmeyer AO, Weil RJ. Proteins and protein pattern differences between glioma cell lines and glioblastoma multiforme. Clinical cancer research: an official journal of the American Association for Cancer Research. 2005;11:3624–3632. doi: 10.1158/1078-0432.CCR-04-2115. [DOI] [PubMed] [Google Scholar]
- Wang C, Funk CC, Eddy JA, Price ND. Transcriptional analysis of aggressiveness and heterogeneity across grades of astrocytomas. PloS one. 2013;8:e76694. doi: 10.1371/journal.pone.0076694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung TL, Leung CS, Wong KK, Samimi G, Thompson MS, Liu J, Zaid TM, Ghosh S, Birrer MJ, Mok SC. TGF-beta modulates ovarian cancer invasion by upregulating CAF-derived versican in the tumor microenvironment. Cancer Res. 2013;73:5016–5028. doi: 10.1158/0008-5472.CAN-13-0023. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 2. Proteins identified from cell surface proteomics study. Details of proteins identified from cell-surface proteomics study. All three replicate analyses of each cell line/primary (source) cells are presented as run1-3.
Table S3. Related to Figure 4 and Figure 6. SRM transition list. Details of Q1/Q3 SRM transition list used in the study are presented.
Table S8. Related to Figure 5 and Figure S4. ELISA data- Longitudinal. Changes in the level of four GBMSig proteins were investigated by commercial ELISA kits before and after tumor resection for prospectively collected blood plasmas from 10 patients.