

SUMMARY
Androgen receptor (AR) is critical for the progression of prostate cancer to castration resistant (CRPC) state. AR antagonists are ineffective due to their inability to repress the expression of AR or its splice variant, AR-V7. Here, we report that the tyrosine kinase ACK1 (TNK2) phosphorylates histone H4 at tyrosine 88 upstream of the AR transcription start site. WDR5/MLL2 complex reads the H4-Y88-phosphorylation marks and deposits the transcriptionally activating H3K4-trimethyl marks promoting AR transcription. Reversal of the pY88-H4 epigenetic marks by the ACK1 inhibitor (R)-9bMS sensitized naive and enzalutamide-resistant prostate cancer cells and reduced AR and AR-V7 levels to mitigate CRPC tumor growth. Thus, a feed-forward ACK1/pY88-H4/WDR5/MLL2/AR epigenetic circuit drives CRPC and is necessary for maintenance of the malignant state.
Keywords: AR, AR-V7, ACK1, TNK2, Castration resistance, Prostate Cancer, Small molecule inhibitor, Enzalutamide, Histone, Epigenetics, Tyrosine phosphorylation
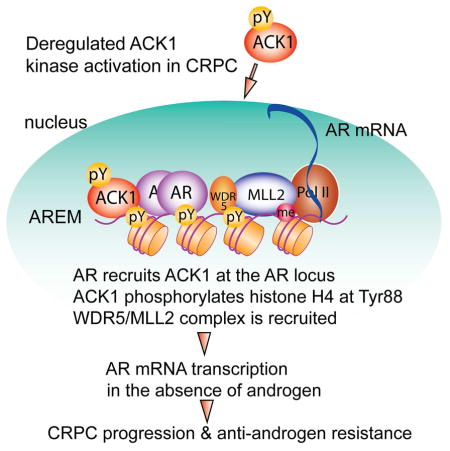
Graphical abstract
Mahajan et al. report that ACK1 phosphorylates histone H4 at Y88 upstream of the AR transcription start site, leading to the WDR5/MLL2 complex-mediated increase of AR transcription. Inhibition of ACK1 reverses the pY88-H4 marks and reduces AR and AR-V7 levels to mitigate castration resistant prostate tumor growth.
INTRODUCTION
Androgen receptor (AR) plays a paramount role in the onset and progression of prostate cancer (PC) (Burnstein, 2005; Lonergan and Tindall, 2011; Watson et al., 2015). This very facet underlies androgen deprivation therapy (ADT), a preferred treatment to negate AR transcriptional activity. While ADT provides immediate palliative benefits, it is ineffective long term, as the recalcitrant disease recurs within 2–3 years and progresses to a lethal stage, referred to as the metastatic Castration Resistant Prostate Cancer (mCRPC). The AR gene (AR) is amplified or mutated in >50% of mCRPCs (Grasso et al., 2012; Robinson et al., 2015), suggesting that PC cells may have reinvigorated AR transcription as a response to the loss of existing AR activity by ADT. Consequently, resistance to ADT has become one of the most vexing problems in PC therapy.
CRPC cells rely on AR for their growth despite androgen-depletion; not surprisingly, AR has been the epicenter of targeted therapies. Enzalutamide, a second generation AR antagonist, although efficiently antagonized AR transcriptional activity by overcoming its nuclear translocation (Tran et al., 2009), the overall survival advantage was found to be ~6 months, and most patients relapsed within 2 years (Bennett and Ingason, 2014). Interestingly, these relapsed patients exhibit renewed AR regulated genes expression by multiple mechanisms, suggesting that CRPCs overcome enzalutamide blockade (Arora et al., 2013; Balbas et al., 2013; Joseph et al., 2013; Korpal et al., 2013). The AR splice variant-7 (AR-V7) is a truncated form of AR that lacks the C terminal ligand-binding domain and remains constitutively active as a transcription factor (Dehm et al., 2008; Guo et al., 2009; Hu et al., 2009; Lu et al., 2015). Recent studies suggest that AR-V7 may be a clinically relevant mechanism of resistance to enzalutamide and the androgen-synthesis inhibitor abiraterone in CRPC patients (Antonarakis et al., 2014). The relative short-term efficacy of enzalutamide and abiraterone reveals two major caveats for tackling this complex disease; first, not all CRPCs are the same and second, other signaling events may be driving the disease. Moreover, because CRPCs display de novo or intrinsic ability to increase AR levels, inhibition of AR protein activity is not enough. To achieve complete remission, ablation of AR appears to be the key. However, targeted inhibition of transcription of AR and AR-V7 with small molecule inhibitors has not yet been accomplished.
Resistance to ADT is closely associated with abnormal tyrosine kinase signaling; non-receptor tyrosine kinases (NRTKs) such as ACK1 and SRC are known to interact with AR in an androgen-independent manner to promote CRPC xenograft growth (Guo et al., 2006; Mahajan and Mahajan, 2010; Mahajan et al., 2007). ACK1 is a structurally unique NRTK upregulated in ~25% of prostate adenocarcinomas (Mahajan et al., 2010b; Mahajan and Mahajan, 2015; Taylor et al., 2010). Importantly, 10 out of 13 CRPCs exhibited 5- to >100-fold ACK1 overexpression (van der Horst et al., 2005). Further, LNCaP cells that are poorly tumorigenic in castrated mice formed robust CRPC tumors following expression of activated ACK1 (Mahajan et al., 2005). Moreover, the expression of activated ACK1 correlates positively with the progression of disease to CRPC stage and PC patients whose tumors display moderate to strong staining of activated ACK1 have poor prognosis (Mahajan et al., 2010a). Combined, these studies have established a crucial role for ACK1 in prostate cancer pathogenesis. In this study, we investigated whether ACK1 tyrosine kinase promotes chromatin alterations to drive CRPC progression.
RESULTS
Identification of Tyr88-phosphorylated histone H4 in human CRPCs
Epigenetic alterations have emerged to be an underlying mechanism in CRPC pathogenesis (Grasso et al., 2012). To examine a potential role for an epigenetic alteration/s in CRPCs, histones were purified from 5 freshly frozen human CRPCs and subjected to mass spectrometry–based identification of post-translational modifications. This unbiased approach led to the identification of phosphorylation of tyrosine 88 in histone H4 in 3 out of 5 CRPC biospecimens (Figure S1A–B). The Y88-phosphorylation of H4 in a human CRPC sample was also assessed by immunoblotting; as compared to a normal prostate sample, robust H4 Y88-phosphorylation was detected in the CRPC sample (Figure S1C). Notably, Tyr88 in histone H4 is evolutionarily conserved suggesting an important physiological function (Figure S1D). As the functional role of Tyr88-phosphorylated H4 (pY88-H4) is unknown, we generated a high affinity monoclonal antibody against pY88-H4. The pY88-H4 antibody specifically recognized the Tyr88-phosphorylated H4 peptide but failed to recognize the unphosphorylated peptide and the phosphopeptide competed with pY88-H4 antibody for binding, dampening the signal (Figure S2A). Moreover, pY88-H4 antibody was screened for cross-reactivity against 59 acetylation, methylation, phosphorylation, and citrullination modifications of histones using the Histone Peptide Array, as described in an earlier publication (Mahajan et al., 2012b). The pY88-H4 antibody did not cross-react with any of these histone modifications whereas the H3K4me3 antibody revealed the expected pattern of hybridization (Figure S2B). Phosphorylation of H4 at Tyr88 was also detected by direct immunoblotting (Figure S2C) as well as by immunofluorescence; pY88-H4 staining was predominantly seen in the nuclei of the ACK1 expressing cells (Figure S2D).
To determine the tyrosine kinase modifying H4 at Y88, total histones were purified from activated ACK1 or SRC expressing cells, followed by mass spectrometry. The phosphorylation of H4 at Y88 was detected only in histones isolated from the activated ACK1 expressing cells (Figure S2E–G). To confirm the kinase responsible for the H4 phosphorylation, HEK293T cells were transfected with MYC-tagged H4, followed by pull down using MYC-beads and mass spectrometry. It revealed the presence of ACK1 which was phosphorylated at Tyr 827 (Figure S2H and I), suggesting that activated ACK1 is a major kinase that binds to H4. In addition to ACK1, many other well-known nuclear proteins e.g. PARP, RNA helicase DHX9, XRCC5, XRCC6, DAXX, MSH2, MRE11 and Histone acetyl transferase HAT1 were also identified in this proteomics approach (Table S1).
To further assess the role of ACK1 as a bona fide histone kinase in vitro and in vivo, we treated PC cells with an ACK1 small molecule inhibitor, (R)-9bMS (Figure 1A, S3A–C, Table S2). (R)-9bMS is a mesylate salt derivative of (R)-9b that we developed and characterized as an ACK1 inhibitor (IC50 of 48 nM) with considerable stability in human plasma and a long half-life (Lawrence et al., 2015). (R)-9bMS is significantly more soluble in aqueous media than the parent compound, thus making it an attractive candidate for in vivo studies.
Figure 1. Androgen-independent phosphorylation of histone H4 at tyrosine 88 by ACK1.

(A) Chemical structure of (R)-9b mesylate salt or (R)-9bMS.
(B) Purified histone H4 alone or incubated with equimolar of purified ACK1 in the absence or presence of 100 nM (R)-9bMS then subjected to immunoblotting using the indicated antibodie.
(C) In vitro kinase assay performed using purified ACK1 and indicated histones, followed by immunoblotting with indicated antibodie
(D) HEK293T cells co-transfected with ACK1 and MYC-tagged H4 or mutants, followed by immunoblottin.
(E) Serum starved LNCaP (left) and LAPC4 (right) cells were treated with (R)-9bMS overnight followed by stimulation with IGF for 20 min or PDGF for 60 min, respectively. Lysates were immunoprecipitated with pY88-H4 or ACK1 antibodies followed by immunoblotting with H4 or pTyr antibodies, respectivel
(F) WT C4-2B cells or two distinct clones of C4-2B cells lacking ACK1 generated using CRISPR-Cas9 gene editing system were serum starved and treated with IGF for 20 min. Lysates were immunoprecipitated with pY88-H4 antibody followed by immunoblotting with H4 antibody.
(G) Androgen-deprived LNCaP cells were electroporated with control or three distinct ACK1 siRNAs, followed by treatment with IGF for 20 min. Lysates were immunoprecipitated with pY88-H4 antibody followed by immunoblotting with H4 antibody. Lysates were also subjected to immunoblotting with ACK1 antibody.
(H, I) Serum and androgen starved C4-2B (H) and LNCaP (I) cells were treated with vehicle, (R)-9bMS (5 μM), enzalutamide (5 μM) or DHT (10 nM) overnight. Cells were then treated with IGF for 20 min, and lysates were immunoprecipitated with pY88-H4 or ACK1 antibodies followed by immunoblotting with H4 or pTyr antibodies, respectively.
See also Figures S1–S3, and Tables S1 and S2.
To uncover any other kinases that may be potentially targeted by (R)-9bMS, we performed specificity profiling wherein its activity against a large panel of kinases (n=369) at 250 nM was assessed. Besides ACK1, only 11 other kinases were inhibited in vitro to >90% (Table S2 and Figure S3C). Human Protein Atlas reveals that 8 of these 11 kinases, TNK1, LRRK2, JAK1-3, TYK2 and NUAK1 and NUAK2, are either not expressed or expressed at very low levels in prostate tissues and cancers. Remaining three kinases, FLT3, PKCmu and SIK2, have not yet been shown to be critical for CRPCs. Thus, while in vitro studies indicate other potential targets, at least in CRPCs, ACK1 appears to be the predominant target of (R)-9bMS.
To assess the direct binding of ACK1 to histone H4, an in vitro kinase assay was performed with purified proteins. Purified recombinant human ACK1 protein was incubated with purified H4 followed by immunoblotting with pTyr antibody. H4 Tyr-phosphorylation was abrogated upon (R)-9bMS treatment (Figure 1B). Further, purified ACK1 was incubated with all the four core histones, followed by immunoblotting with pY88-H4 antibody. The presence of a phosphorylated H4 band exclusively when ACK1 was incubated with H4 indicates the specificity of pY88-H4 antibody (Figure 1C). To further validate the H4 Y88-phospho-antibody, point mutant constructs of FLAG-tagged H4 lacking either the Tyr88 site (Y88F-H4) or the Tyr72 site (Y72F-H4) were generated. ACK1 phosphorylated H4 and the Y72F-H4, but failed to phosphorylate the Y88F-H4 mutant (Figure 1D).
To evaluate the status of endogenous H4 phosphorylation upon ACK1 activation, serum and androgen starved LNCaP and LAPC4 cells were treated with insulin like growth factor (IGF) or platelet derived growth factor (PDGF). Growth factor stimulation led to robust ACK1 activation and H4 Y88-phosphorylation, both of which were suppressed by (R)-9bMS treatment (Figure 1E). To corroborate that ACK1 kinase is essential for histone H4 Y88-phosphorylation, ACK1 expression was knocked down by using siRNA or CRISPR-Cas9 gene editing system. ACK1 stimulation led to the phosphorylation of H4 in control cells and its knockdown resulted in a significant loss of H4 Y88-phosphorylation (Figure 1F, G and S3D). Moreover, to explore the sensitivity of endogenous H4 Y88-phosphorylation to enzalutamide and dihydrotestosterone (DHT), serum-deprived LNCaP-C4-2B (C4-2B here onwards) and LNCaP cells were treated with vehicle, (R)-9bMS, enzalutamide or DHT, followed by IGF treatment. The endogenous Y88-phosphorylation of H4 was eliminated upon treatment with (R)-9bMS, but was unaffected by DHT or enzalutamide treatments (Figure 1H and I).
ACK1 deposits the pY88-H4 marks upstream of the AR coding region
To decipher the pY88-H4 epigenetic footprint in CRPCs, chromatin prepared from vehicle or (R)-9bMS treated C4-2B cells was immunoprecipitated (ChIP) with pY88-H4 antibody followed by sequencing (ChIP-sequencing). The pY88-H4 marks were found to be deposited at ~370 distinct locations throughout the genome (Table S3). Closer examination of X chromosome revealed pY88-H4 deposition at 3 distinct locations upstream of the AR transcription start site; AREM1 (nt 66,669,600–66,670,200), AREM2 (nt 66,649,550–66,649,850) and AREM3 (nt 66,631,300–66,631,650) (Figure 2A and B). To validate ACK1 mediated deposition of the pY88-H4 marks upstream of AR, ChIP was performed followed by real time PCR using site specific primers. The pY88-H4 marks were specifically deposited at the three AREMs but not at a control site, however, upon (R)-9bMS treatment there was a significant loss of pY88-H4 at AREMs (Figure 2C–F), suggesting that the deposition of pY88-H4 marks is abolished by an ACK1 inhibitor.
Figure 2. Deposition of pY88-H4 epigenetic marks upstream of the AR gene.

(A) Chromatin was isolated from C4-2B cells and ChIP was performed using pY88-H4 antibody or IgG followed by sequencing. The peaks upstream of the AR coding region, designated as AREM1-3 are shown in graphical format. The numbers indicate nucleotide position at the midpoint of the peaks.
(B) Peak regions that were amplified using PCR primers are shown.
(C–F) C4-2B cells treated with vehicle or (R)-9bMS and ChIP was performed using pY88-H4 antibody or IgG, followed by qPCR using primers corresponding to AREM1(C), AREM2 (D), AREM3 (E) and control (gene desert on chromosome 12) sites (F) (n = 3, three replicates).
(G) C4-2B cells transfected with control or AR siRNAs and ChIP was performed using AR antibody followed by qPCR using primers corresponding to AREM1 (n = 2, three replicates).
(H) C4-2B cells transfected with control, ACK1, or AR siRNA and subjected to ChIP using pY88-H4 antibody or IgG followed by qPCR using primers corresponding to AREM2 (n = 2, three replicates).
(I) Chromatin isolated from ACK1 deleted C4-2B cells were subjected to ChIP with pY88-H4 antibody or IgG, followed by qPCR using primers corresponding to AREM2 (n = 2, three replicates).
(J) Total RNA isolated from C4-2B cells lacking AREM1 or AREM2 was subjected to qRT-PCR with AR and actin primers (n = 3, two replicates).
Data are represented as mean ± SEM as in (C–J). **p<0.01, *p<0.05.
To assess whether the pY88-H4 mark deposition is critically dependent on AR and ACK1 recruitment, C4-2B cells were transfected with AR or ACK1 siRNAs followed by ChIP-qPCR. AR was recruited at AREM1 (Figure 2G). Further, ACK1 or AR silencing significantly reduced the pY88-H4 deposition at AREM1 and AREM2 (Figure 2H and S3E). In addition, deletion of ACK1 by CRISPR-Cas9 significantly compromised deposition of pY88-H4 marks (Figure 2I). Taken together, these data suggest that AR is needed to recruit ACK1 upstream of AR locus to deposit the pY88-H4 marks.
To examine a direct role of AREMs on AR mRNA expression, AREM1 and AREM2 were deleted using CRISPR-Cas9 gene editing method in C4-2B cells (Figure S3F). A significant reduction of AR mRNA was seen upon deletion of either AREM1 or AREM2 suggesting that these sites act as enhancers for AR (Figure 2J).
ACK1 kinase activity is required to sustain AR mRNA levels in an androgen deficient environment
Towards understanding the outcome of the androgen-independent AR/ACK1 cross talk, we chose an unbiased approach wherein androgen-deprived VCaP, LAPC4, or C4-2B cells were treated with dihydrotestosterone (DHT), enzalutamide, or (R)-9bMS. Unexpectedly, we discovered that treatment with (R)-9bMS resulted in a significant downregulation of AR protein levels (Figure 3A and B; Figure S3G and H). In contrast, the AR antagonist enzalutamide had a modest or no effect on AR expression. To confirm that the loss of AR expression is not due to the ‘off target effect’ of the ACK1 inhibitor, C4-2B and LNCaP cells were transfected with control and ACK1 siRNAs or ACK1 was deleted using CRISPR-Cas9 gene editing technique. Loss of ACK1 expression resulted in a significant loss of AR mRNA (Figure 3C and D) and protein levels (Figure 3E and F). Further, loss of AR expression was also reflected in a decrease in prostate specific antigen (PSA) levels (Figure 3G), suggesting that ACK1 is required for sustaining AR mRNA expression in an androgen-deficient environment.
Figure 3. ACK1 kinase activity is required to maintain androgen-independent AR mRNA and protein levels.

(A, B) Androgen starved VCaP (A) and C4-2B (B) cells were treated with vehicle, DHT (10 nM, 16 hr), (R)-9bMS (5 μM, 36 hr) and enzalutamide (5 μM, 36 hr) followed by immunoblotting. Relative levels of AR expression are shown below top panels.
(C) Androgen-deprived LNCaP (left) and C4-2B (right) cells were transfected with control or ACK1 siRNA. Total RNA was isolated followed by qRT-PCR with AR and actin primers (n = 3, three replicates).
(D) Total RNA isolated from WT C4-2B or C4-2B in which ACK1 was deleted using CRISPR-Cas9 gene editing system was subjected to qRT-PCR with AR and actin primers (n = 2, three replicates).
(E) C4-2B (left) and androgen starved LAPC4 (right) cells transfected with control or ACK1 siRNAs and lysates were immunoblotted for AR, actin or ACK1. Relative levels of AR expression are shown below top panels.
(F) ACK1 was deleted using CRISPR-Cas9 strategy in LAPC4 cells and the lysates were immunoblotted for AR, ACK1 or actin.
(G) Total RNAs isolated from WT C4-2B cells or C4-2B cells in which ACK1 was deleted using CRISPR-Cas9 were subjected to qRT-PCR with PSA and actin primers (n = 2, three replicates).
(H, I) Total RNA isolated from androgen-deprived LAPC4 cells transfected with vector, FLAG-tagged H4 or Y88F mutant H4 expressing constructs was subjected to qRT-PCR with AR (H), and PSA (I) primers (n = 2, three replicates).
(J) Lysates from LAPC4 cells treated with vehicle, (R)-9bMS (5 μM, 24 hr) or MG132 (10 μM, 6 hr) or both were immunoblotted for AR and actin.
Data are represented as mean ± SEM as in (C, D and G–I). **p<0.01, *p<0.05.
See also Figure S3.
To examine whether the deposition of pY88-H4 marks upstream of AR is necessary for its transcriptional upregulation, total RNA was prepared from cells that were transfected with either the Y88F mutant of H4 or WT-H4. An overexpression of the Y88F-H4 mutant resulted in a significant decrease in both AR and PSA mRNA levels (Figures 3H and I). Taken together, these data illustrate the histone tyrosine kinase activity of ACK1 is required to promote AR expression in the androgen-deficient milieu of PC cells.
AR is a target of several E3 ubiquitin ligases which positively and negatively regulate its activity (Xu et al., 2009). To determine whether AR is targeted for proteosomal degradation following treatment with the ACK1 inhibitor, LAPC4 cells were treated with the proteasome inhibitor MG-132 in the presence or absence of (R)-9bMS. Treatment with MG132 did not reverse the loss of AR expression caused by ACK1 kinase inhibition (Figure 3J) opening a possibility that ACK1 regulates AR expression at the level of transcription.
(R)-9bMS attenuates AR and the splice variant-7 (AR-V7) transcription
To further interrogate whether the decrease in AR protein levels is a consequence of inhibition of AR transcription, prostate cancer cells grown in androgen-deprived media were treated with vehicle, (R)-9bMS, DHT or enzalutamide. Real time PCR revealed that AR mRNA expression was significantly suppressed in all (R)-9bMS treated PC cell lines (Figure 4A–D). A modest decrease in AR mRNA levels was observed in VCaP cells following both enzalutamide and DHT treatments (Figure 4A). In contrast, AR mRNA levels were not significantly altered in DHT or enzalutamide treated LNCaP and LAPC4 cells (Figure 4B, C). Notably, as an outcome of AR transcriptional suppression by (R)-9bMS, a significant reduction in the expression of the AR target gene PSA was observed (Figure 4E–H). As a control, LNCaP and LAPC4 cells treated with enzalutamide revealed a significant decrease in PSA expression without affecting AR expression. (Figure 4B, C, and F, G). Intriguingly, VCaP cells displayed resistance to enzalutamide and did not exhibit reduction in PSA mRNA expression (Figure 4E). Consistent with the mechanism of action of enzalutamide to inhibit AR nuclear translocation, PSA was downregulated without affecting AR mRNA levels (Figure 4F and G). This is in contrast to (R)-9bMS, which reduced PSA expression by suppressing AR transcription (compare Figure 4E–H).
Figure 4. Inhibition of ACK1 kinase activity suppresses AR transcription.

(A–D) Androgen-starved VCaP (A), LNCaP (B), LAPC4 (C) and C4-2B (D) cells were treated overnight with vehicle, (R)-9bMS at 3.5 μM (orange) or 7 μM (green) for 16 hr, enzalutamide (7 μM, 16 hr) and DHT (10 nM, 2 hr). Total RNA was isolated followed by qRT-PCR with AR and actin primers (n = 3, three replicates).
(E–H) Total RNA isolated from androgen-starved VCaP (E), LNCaP (F), LAPC4 (G) and C4-2B (H) cells treated as described above and subjected to qRT-PCR with PSA and actin primers (n = 3, three replicates).
(I) Total RNA isolated from vehicle or (R)-9bMS treated VCaP cells followed by qRT-PCR with AR-V7 and actin primers (n = 2, three replicates).
(J) The lysate prepared from vehicle or (R)-9bMS treated VCaP cells were subjected to immunoblotting with indicated antibodies.
(K) PC3 cells were transfected with MYC-tagged caACK, kdACK or vector, followed by immunoblotting.
(L, M) Total RNA was isolated from PC3 cells transfected as described in (K), followed by qRT-PCR with AR (L) and PSA (M) and actin primers (n = 2, three replicates).
(N) Chromatin isolated from PC3 cells transfected with full length ACK, kdACK or vector cells were subjected to ChIP with pY88-H4 antibody, followed by qPCR using primers corresponding to AREM1 (n = 2, three replicates).
Data are represented as mean ± SEM as in (A–I and L–N). **p<0.01, *p<0.05.
It has been reported that the expression of the full length AR is needed for the genesis of its variant, AR-V7 (Watson et al., 2010). We assessed the levels of AR-V7 mRNA and protein following treatment with (R)-9bMS. Both AR-V7 mRNA and protein levels were significantly reduced upon ACK1 inhibition (Figure 4I and J), suggesting that ACK1 regulates optimal AR and AR-V7 expression by a distinct epigenetic mechanism.
ACK1 kinase activity drives AR expression in an androgen-deficient environment
To validate an absolute requirement of ACK1 in sustaining AR transcription, we utilized the androgen-insensitive PC3 cells, which express undetectable levels of AR and PSA. Transfection of PC3 cells with constitutively active ACK1 (caACK), but not kinase dead ACK1 (kdACK) (Mahajan et al., 2007) lead to a detectable increase in AR protein levels (Figure 4K). This was due to a significant induction of AR (and PSA) mRNA expression (Figure 4L and M). Further, ChIP-qPCR analysis revealed the deposition of pY88-H4 epigenetic marks at the AREM1 in ACK1 expressing cells (Figure 4N). These data indicate that ACK1 kinase activity is required to maintain AR mRNA levels in prostate cancer cells.
Attenuation of global androgen-independent AR transcriptional program by (R)-9bMS
In order to determine whether AR transcriptional attenuation caused by ACK1 kinase inhibition resulted in changes in global gene expression, gene expression profiling was performed in VCaP cells treated with (R)-9bMS. Over- and under-expressed gene sets were generated by filtering to include data points that showed either over- or under-expression by at least two folds (log ratio with p3<30.001). A significant repression of AR expression was observed in (R)-9bMS treated VCaP cells (Figure 5A), indicating that AR transcription is critically dependent on ACK1 kinase activity. Further, microarray followed by Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005), revealed a significant decrease in DNA repair gene signature upon ACK1 inhibition (Figure 5B). In addition, inhibition of ACK1/AR signaling affected the E2F, MYC and estrogen-response regulated transcription programs (Figure 5C, D and E).
Figure 5. ACK1 regulates AR expression and DNA repair, E2F and MYC target gene signatures.

(A) Heatmap of microarray expression data of VCaP cells treated with vehicle or (R)-9bMS.
(B) GSEA of the DNA repair target gene signature in C4-2B (left), VCaP (middle) and LAPC4 (right) cells impacted by (R)-9bMS treatment.
(C) GSEA of the E2F target gene signature in C4-2B (left) and LAPC4 (right) cells impacted by (R)-9bMS treatment.
(D) GSEA of the Estrogen response gene signature in C4-2B (left) and VCaP (right) cells impacted by (R)-9bMS treatment.
(E) GSEA of the MYC target gene signature in C4-2B cells impacted by (R)-9bMS treatment.
(F) Heatmap of all the curated AR signatures in C4-2B, VCaP, LAPC4, PC3 and DU145 cells.
(G) Androgen-deprived LAPC4 (left), VCaP (middle) and LNCaP (right) cells were co-transfected with GFP and either FLAG-tagged AR or vector constructs, followed by treatment with vehicle or (R)-9bMS (3 μM, 96 hr). GFP positive cells were counted after 96 hr.
Data are represented as mean ± SEM (n = 3, three replicates). *p<0.05.
See also Figure S4.
To examine the differential sensitivity of PC cells to (R)-9bMS, total RNA was isolated from five PC cell lines that were either untreated or treated with (R)-9bMS and expression profiling was performed. We curated all the AR signatures from literature and generated a heat map. While the AR dependent cell lines C4-2B, VCaP and LAPC4 clustered together, the AR non-responsive cell lines PC3 and DU145 formed a separate cluster (Figure 5F), suggesting that (R)-9bMS may exert a distinct effect on AR-expressing PCs.
To determine whether the ectopic expression of AR can rescue the androgen-independent growth inhibition by (R)-9bMS, prostate cancer cells were co-transfected with GFP and vector or FLAG-tagged AR constructs (driven by CMV promoter and thus insensitive to ACK1 inhibition), followed by (R)-9bMS treatment. Exogenous AR-expressing cells exhibited a significant survival benefit despite (R)-9bMS treatment (Figure 5G and S4A), suggesting that exogenous AR can rescue the survival phenotype due to a functional loss of ACK1.
pY88-H4 dependent recruitment of MLL2/WDR5 chromatin remodeling complex upstream of AR
We assessed enrichment of ChIP-sequencing peaks at the specific sequence motifs which indicated that pY88-H4 deposition sites overlap with transcription factor binding motifs and other chromatin binding proteins (Figure S4B–D). To elucidate the molecular mechanism by which H4 Y88-phosphorylation regulates AR transcription, we performed an unbiased pull down to uncover potential ‘readers’ of the pY88-H4 marks. In brief, biotinylated histone H4-derived peptide with phosphorylated-Y88 residue at the center (80–96 aa) and an equivalent unphosphorylated peptide (as control) were immobilized onto streptavidin beads and bound proteins were analyzed by LC-MS/MS analysis. The top ‘hit’ in this proteomic analysis was MLL2 (KMT2D), a H3K4-methyltransferase (Table S4). In addition MLL2 interacting proteins WDR5 and ASH1L were also present in the pull down (Table S4 and Figure S5A and B). The WD40-repeat protein WDR5 is a key component of MLL2 H3K4-methyltransferase chromatin remodeling complex and is required for binding of the methyltransferase complex to the K4-dimethylated H3 tail facilitating global H3K4-trimethylation (Wysocka et al., 2005). The H3K4-trimethylation is associated with transcriptional activation in a number of contexts and the mono-, di- and trimethylated forms of H3K4 are known to be differentially enriched at promoter, enhancer and other regulatory sequences (Wysocka et al., 2005; Zhao et al., 2007). Thus, our proteomics analysis provided a key clue; the H4 Y88-phosphorylation marks may be recognized by WDR5 and could operate in trans by facilitating the recruitment of MLL2, followed by H3K4-tri-methylation. Towards verifying the relevance of these epigenetic interactions, we performed a pY88-H4 peptide pull down assay, followed by immunoblotting for WDR5. WDR5 exhibited preferential binding to the phosphorylated Y88-H4, as compared to the unphosphorylated H4 (Figure 6A).
Figure 6. Recruitment of MLL2/WDR5 and deposition of the H3K4me3 marks upstream of the AR gene.

(A) Peptide pull down assays was performed with either biotinylated pY88-H4 peptide or the biotinylated unphosphorylated H4 peptide and bound proteins were immunoblotted for WDR5. Total lysates were also immunoblotted with WDR5 antibody.
(B) Chromatin from LNCaP cells expressing FLAG-tagged H4 or Y88F-H4 were immunoprecipitated with FLAG beads followed by immunoblotting with H3K4me3 or H3K9me3 antibody.
(C–F) Chromatin prepared from C4-2B cells treated with vehicle or (R)-9bMS was subjected to ChIP using WDR5 (C), MLL2 (D), H3K4me3 (E), or RNA Pol II (F) antibodies followed by qPCR using primers corresponding to AREM1 or control (gene desert) region. Data are represented as mean ± SEM (n = 2, three replicates). *p<0.05, **p<0.01.
(G) Representative images of prostate TMA sections stained with H3K4me3 or AR antibody.
(H, I) Box plots summarize distributions of staining intensities for H3K4me3 and AR in prostate biopsies of BPH, PIN and different stages of prostate cancer. Each box has lines at the lower quartile (25%), median (50%), and upper quartile values (75%). Whiskers extend from each end of the box to the most extreme values within 1.5 times the inter quartile range from the ends of the box. The data with values beyond the ends of the whiskers, displayed as circles, are potential outliers.
(J) Venn diagrams summarizing overlap between sites marked by H3K4me3 and pY88-H4 in C4-2B cells.
See also Figures S4 and S5 and Tables S4–S7.
To further validate a direct role of pY88-H4 in the recruitment of the WDR5/MLL2 complex, chromatin prepared from either the FLAG-tagged H4 or the Y88F-H4 mutant transfected cells was immunoprecipitated with FLAG beads followed by analysis of the H3K4 trimethylation status. Chromatin extracted from the mutant Y88F-H4 expressing cells displayed a significant decrease in the H3K4me3 marks compared to the WT-H4 expressing cells (Figure 6B). Moreover, H3K9 trimethylation, known to be transcriptionally repressive, were significantly increased in the H4 Y88F mutant expressing cells (Figure 6B), suggesting that the interplay of activating/repressive methylation may fine-tune AR mRNA output in prostate cancer cells.
To determine the physiological relevance of pY88-H4 interaction with the WDR5/MLL2 complex, we analyzed the localization of WDR5, MLL2, H3K4me1, H3K4me2, H3K4me3, and RNA Pol II at the AREM1-3 sites. ChIP-qPCR analysis revealed a specific recruitment of WDR5, MLL2, RNA Pol II and enrichment of H3K4me3 at the AREM1-3, which was abolished following treatment with (R)-9bMS (Figure 6C–F and S5C–G). In contrast, the deposition of H3K4me1 and H3K4me2 was not impacted by the loss of pY88-H4 (Figure S5H). Collectively, these data indicate that activated ACK1 establishes a transcriptionally permissive chromatin landscape rich in H4-Y88 and H3K4me3 to promote AR mRNA transcription in the androgen-deficient environment of CRPCs.
H3K4 trimethylation and AR expression correlate with disease progression in metastatic prostate cancer
To determine the clinical relevance of H3K4me3 mark deposition on AR protein expression, we performed immunohistochemical staining (IHC) of clinically annotated prostate (n = 250) tissue microarrays (TMA) with respective antibodies. The H3K4me3 and AR levels increased as PCs progressed from the benign prostatic hyperplasia (BPH), Prostatic Intraepithelial Neoplasia (PINs), G6, G7, G8–10 PCs to the CRPC stage (Figure 6G–I). In addition, H3K4me3 and AR expression were found to positively correlate with each other during disease progression (Pearson correlation r = 0.66, p<0.0001). Moreover, the expression of H3K4me3 and AR differed significantly between the stages of the disease; a significant increasing trend of intensity across progression stages was detected (Mantel-Haenszel test, p = 0.01) (Table S5 and Table S6). Based on the pair-wise comparisons using Tukey’s multiple adjustment method G7, G8/9/10 and CRPC groups showed a higher mean value of H3K4me3 level compared to the benign prostatic hyperplasia (BPH) group. The expression of H3K4me3 in CRPC set was also significantly higher than PIN, G6 and G7 groups. Moreover, the mean AR expression in G7, G8/9/10 and CRPC groups were all significantly higher than BPH, PIN and G6 groups (Table S5 and Table S6). We also assessed whether the peaks corresponding to pY88-H4 epigenetic marks overlap with the previously reported H3K4me3 deposition in DHT or EtOH treated C4-2B cells and observed that 60 pY88-H4 peaks overlapped with H3K4me3 peaks (Figure 6J). In addition, pY88-H4 levels positively correlated with AR expression during disease progression (Pearson correlation r = 0.3, p<0.0001) (Table S7). Collectively, these data indicate that the expression of pY88-H4, H3K4me3 and AR progressively increase as disease progressed to later stages with the highest expression in CRPCs. Further, it indicates that H3K4me3 marks may have a direct impact on AR expression in metastatic PC.
Targeting the ACK1 epigenetic activity mitigates CRPC tumor growth
To investigate the anti-tumor activity of ACK1 inhibitor, we first analyzed the ability (R)-9bMS to suppress proliferation of PC cell lines. AR-positive cell lines C4-2B, VCaP, LAPC4 and LNCaP were found to be sensitive to the (R)-9bMS treatment with IC50 of 400 nM, 450 nM, 750 nM and 1.8 μM, respectively (Figure 7A). Interestingly, CRPC-forming C4-2B and VCaP were the most sensitive, while the ‘normal’ prostate cells, RWPE, were the most resistant to (R)-9bMS treatment (IC50 ~10 μM). To examine consequences of (R)-9bMS mediated AR mRNA transcription suppression, cell cycle analysis was performed. A significant increase in the number of cells in G2/M phase was seen following (R)-9bMS treatment (Figure 7B). To assess whether this G2/M arrest could lead to the activation of apoptotic pathways, tyrosine kinase inhibitor treated cells were stained with caspase-3/7 detection agent and SYTOX® dead cell stain. In contrast to the selective SRC inhibitor, saracatinib (22%), a significant increase in apoptotic cells (52%) was observed following treatment with (R)-9bMS (Figure S6A).
Figure 7. (R)-9bMS overcomes enzalutamide resistance and inhibits CRPC tumor growth.

(A) Cells were treated with (R)-9bMS or vehicle (10% DMSO) for 96 hr and number of viable cells counted by trypan blue exclusion assay (n = 2, three replicates).
(B) Flow cytometry based cell cycle analysis of LNCaP (labelled in red), LAPC4 (blue) and C4-2B (green) cells treated with vehicle or (R)-9bMS for 24 hr and stained with propidium iodide.
(C) Enzalutamide-resistant C4-2B cells were treated with enzalutamide or (R)-9bMS for 96 hr and number of viable cells were determined by trypan blue dye exclusion assay (n = 2, three replicates).
(D) Total RNA isolated from parental and Enzalutamide-resistant C4-2B cells was subjected to qRT-PCR with AR and actin primers (n = 2, three replicates).
(E) Enzalutamide-resistant C4-2B cells were treated with vehicle (−) or (R)-9bMS (+) and immunoblotting was performed with indicated antibodies.
(F) C4-2B cells were implanted subcutaneously in castrated male SCID mice. When tumors became palpable, mice were injected either with vehicle (10% DMSO in PBS) or (R)-9bMS (50 mg/kg of body weight) for 5 days a week for 4 weeks (n = 7 mice for each treatment). Tumor volumes were measured with calipers.
(G) The weights of the xenograft tumors in (F) are shown.
(H) Total RNA was prepared from xenograft tumors followed by qRT-PCR with AR (left), PSA (middle) and TMPRSS2 (right) primers (n = 2, three replicates).
(I) Heart, liver, spleen and kidneys were harvested from vehicle or (R)-9bMS treated mice, fixed and sectioned followed by hematoxylin and eosin staining.
Data are represented as mean ± SEM as in (A, C, D, F–H). **p<0.01, *p<0.05.
See also Figures S6 and S7
Among the various PC cell lines, C4-2B cells display high sensitivity to enzalutamide (Figure S6B). To examine the relative sensitivity of CRPCs to enzalutamide and (R)-9bMS, we generated an enzalutamide-resistant C4-2B cell line that grows in the presence of 25 μM of enzalutamide. Unexpectedly, enzalutamide at lower concentrations acted as an agonist and fueled the proliferation of enzalutamide-resistant C4-2B cells (Figure 7C). Interestingly, a significant increase in AR mRNA expression was observed in enzalutamide-resistant C4-2B cells, suggesting that CRPCs could overcome enzalutamide blockade by reinvigorating AR expression (Figure 7D). Although these cells exhibited considerable resistance to enzalutamide (IC50 >7.5 μM), cells retained sensitivity to (R)-9bMS (Figure 7C), by virtue of its ability to suppress AR expression (Figure 7E). Taken together, these data suggest that ACK1/AR signaling can overcome enzalutamide-resistance by downregulating AR transcription.
To investigate the effect of (R)-9bMS on CRPC xenograft tumor growth, C4-2B cells were implanted subcutaneously in castrated male SCID mice. When the tumors reached approximately 100 mm3 in size, the mice were randomized and injected with either the vehicle or (R)-9bMS. Although vehicle treated mice formed robust subcutaneous CRPC tumors, tumor growth was not observed in the (R)-9bMS injected mice (Figure 7F, G and S6C). Interestingly, metastatic tumors were observed at distant sites in some mice; two large tumors were seen attached to the prostate in the vehicle treated mice (Figure S6C). In contrast, a small tumor was found to be attached to the inner side of rib cage in (R)-9bMS injected mice (Figure S6C), suggesting that (R)-9bMS may also inhibit metastatic CRPC tumor growth. Further, total RNA was isolated from the xenograft tumors from the vehicle and (R)-9bMS treated group (3 small tumors were pooled from (R)-9bMS injected mice from one of the two repeats) to examine AR and its target genes expression. The CRPC tumors from (R)-9bMS treated mice exhibited a significant reduction in AR, PSA and TMPRSS2 mRNA levels compared to the vehicle treated group (Figure 7H).
We also assessed the effect of (R)-9bMS in another CRPC xenograft model, LNCaP-caAck (Mahajan et al., 2007). Similar to C4-2B, LNCaP-caAck CRPC tumors responded to (R)-9bMS treatment with a significant reduction in xenograft tumor growth (Figure S6D).
Markedly, the (R)-9bMS treatment regimen was well tolerated by mice; a small but insignificant weight loss was noticed in (R)-9bMS treated mice as compared to the vehicle treated mice (Figure S6E). To further assess any pathological effect of (R)-9bMS treatment, heart, spleen, kidneys and livers from the mice were excised and stained with hematoxylin and eosin (H and E). The organs from vehicle and (R)-9bMS treated mice did not reveal histological abnormalities (Figure 7I).
As an initial response to ADT, the prostate tumors shrink with a significant decrease in PSA levels (Grossmann et al., 2001). Paradoxically, CRPC tumors ‘learn’ to thrive under castrate levels of androgen. A pervasive mechanism in CRPCs appears to be to overexpress AR, suggesting that CRPCs have eruditely dealt with dwindling androgen levels. But, how does AR sustain its own expression in the absence of ligand? Interestingly, androgen deprivation of androgen-sensitive LAPC4 cells for 10 days resulted in a significant upregulation of ACK1 levels (Figure S6F). Activation of ACK1 leading to H4 Y88-phosphorylation, resulting in AR transcriptional activation seem to be interlinked in androgen-deficient environment (Figure S6G and H). Importantly, expression of Y88F-H4 mutant in LNCaP cells significantly compromised growth in androgen-deficient media (Figure S7A). Moreover, castrated mice injected with the C4-2B cells expressing Y88F mutant of H4 exhibited a reduction in xenograft tumor growth and PSA levels as compared to H4 or vector expressing cells (Figure S7B–D). Although, cells seem to be equally adept in incorporating Y88F-H4 as well as H4 in chromatin, consistent with its reader function, MLL2/WDR5 recruitment was significantly compromised at AREM when mutant Y88F-H4 was overexpressed (Figure S7E–G). A frequent phenomenon in CRPC is that AR and ACK1 gene amplification is seen in ~70% and 22% of these tumors, respectively (Figure S7H–J); indeed, AR and ACK1 exhibit significant tendency towards co-occurrence in CRPCs (p value = 0.002, Log odds ratio 1.68) (Figure S7K). Taken together with data shown in Figure S6F, these results indicate that CRPCs adapt to androgen-deficiency by selectively favoring ACK1/AR expression.
DISCUSSION
Here we demonstrate chromatin directed transcriptome reprogramming of the cancer cells as a paradigm for the emergence of drug-resistant and lethal forms of prostate cancer. We uncovered that ACK1 directly reprograms the AR locus to promote castration and drug-resistance. Further, we have demonstrated that ACK1-AR signaling sustains expression of genes important for DNA repair, cell cycle by regulating E2F target genes and cell proliferation by MYC regulation to promote malignancy. This is consistent with earlier reports demonstrating a role for AR in the regulation of DNA repair genes in PCs (Goodwin et al., 2013; Mahajan et al., 2012a; Polkinghorn et al., 2013; Spratt et al., 2015). AR has been shown to regulate MYC transcription program in an androgen-independent manner (Gao et al., 2013), which explains the significant loss of MYC-target gene expression upon (R)-9bMS treatment. Intriguingly, these data also suggest a role for ACK1 in driving ER-mediated transcription which is consistent with our earlier observation wherein ACK1 interacts with ER and its co-activator histone demethylase KDM3A (Mahajan et al., 2014). Combined, these results illustrate targeting of major survival pathways by ACK1 inhibitor underlie its effectiveness in restraining the growth of PC cells.
Prostate cancer is considered as a cancer of the epigenome (Grasso et al., 2012; Robinson et al., 2015). Our results demonstrate that the interaction of AR with ACK1 drives the positive feedback epigenetic circuitry that is ultimately conducive to promote AR transcription. Further, this circuitry subjugates AR transcription in CRPCs when the H4-Y88 epigenetic regulation is terminated. An unexpected finding that has emerged from this study is the role of ACK1 as an ‘epigenetic writer’ that deposits the first layer of pY88-H4 marks, which are recognized by an ‘epigenetic reader’ WDR5. This in turn results in the recruitment of WDR5 partner MLL2, which acts as the ‘epigenetic scribe’ and introduces H3K4me3 activating marks, to drive AR expression. Interestingly, recent study has shown that AR interacts with menin, which is a component of MLL1/WDR5/ASH2L complex (Grasso et al., 2012; Malik et al., 2015). Whether H4 Y88-phosphorylation was important for the assembly of MLL1/WDR5/menin complex is not known, however, the presence of WDR5 in both MLL1 and MLL2 complexes does not rule out the possibility of global regulatory function of pY88-H4 marks in regulation of the AR-target genes. Significantly, our proteomics analysis revealed many other WD repeat containing proteins, including WDR55, WDR43, WDR19, WDR12, WDR46, and WDR75, also binding to pY88-H4, opening the possibility that WD40 repeat could be the pY88-H4 reader element and WD40-repeat containing proteins might be recruited to chromatin using these epigenetic marks.
In addition to WDR5, our assessment of pY88-H4 ChIP-sequencing peaks revealed the top match being the SOX8 (or sex determining region Y-box 8), a transcription factor involved in the regulation of embryonic development and determination of the cell fate, including male sex determination (Chaboissier et al., 2004). Whether pY88-H4 marks have an additional role in male gonad differentiation remains to be seen.
Prompt restoration of AR expression reinforces the notion that sustained androgen-deprivation may prompt CRPC tumors to use ACK1/pY88-H4 epigenetic signaling to overcome androgen-dependence. Although ACK1/pY88-H4/AR signaling could be the promising scheme for CRPCs to acquire above mentioned properties; unavailability of ACK1 small molecule inhibitor had precluded testing this hypothesis comprehensively. Our data not only provides evidence of CRPC-dependence on ACK1-AR signaling but also indicates that enzalutamide-non-responsive PCs may be sensitive to ACK1 inhibitor. Indeed, combining enzalutamide with (R)-9bMS might provide the best outcome-dampening the progression of early disease to castration-resistance. Overall, uncovering the epigenetic control of CRPCs has considerable potential to advance (R)-9bMS as a therapeutic option.
METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the corresponding author Nupam P. Mahajan (Nupam.Mahajan@moffitt.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
RWPE-1, LNCaP, VCaP, PC3 and DU145 cells were obtained from ATCC. LAPC4 cells were obtained from Dr. Robert Reiter’s Lab in UCLA and grown in IMDM media supplemented with 10% FBS. LNCaP-C4-2B cells were obtained from Dr. Evan Keller, University of Michigan. LNCaP, PC3 and DU145 cell lines were grown in RPMI 1640 supplemented with 10% FBS (Invitrogen). VCaP cells were grown in DMEM with 10% FBS (Invitrogen). RWPE-1 (ATCC) was grown in Keratinocyte serum-free media with L-glutamine (Invitrogen) supplemented with 2.5 mg EGF (Invitrogen) and 25 mg Bovine Pituitary Extract (Invitrogen). LNCaP-C4-2B cells were grown in Complete T-medium containing per Liter:150 ml of F12, 750 ml of DMEM low glucose, 2 ml of insulin (2.5 mg/ml stock soln.= 500X), 2 ml of T3 (triiodo-thyronine, 6.825 ng/ml stock solution in 0.1% BSA in PBS=500X), 2 ml of apo-transferin (2.5 mg/ml =500X), 2 ml of d-Biotin (1.22 mg/10 ml 0.1% BSA in PBS + NaOH = 500X), 4 ml of Adenine Hemi-Sulfate (12.5 mg/ml in 0.1% BSA in PBS), 10 ml of Pen-Strep, 100 ml FBS. All cultures were maintained with 50 units/ml of penicillin/streptomycin (Invitrogen) and cultured in 5% CO2 incubator.
Mouse Xenograft studies
All animal experiments were performed using the standards for humane care in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Mice breeding and colony maintenance was performed according to IACUC protocols approved in writing by University of South Florida (USF) Division of Research Integrity and Compliance. 2×106 LNCaP-C4-2B cells were suspended in 200 μl of PBS with 50% matrigel (BD Biosciences) and were implanted subcutaneously into the dorsal flank of castrated six week old male SCID C. B17 mice. Once the tumors reach approximately 100 mm3 in size (about 4–5 weeks), mice were injected subcutaneously with (R)-9bMS (or 10% DMSO in PBS as vehicle) at the concentrations 50 mg/kg of body weight, six times a week, for 4–5 weeks. Tumor volumes were measured twice weekly using calipers. Formation of tumors was monitored over an entire 10 week period. At the end of the study, all mice were humanely euthanized, tumors extracted and weighed. Additionally, spleen, liver and spleen were harvested and stained with Hematoxylin and Eosin (H and E). For LNCaP-caACK xenograft study, similar strategy was used except that the cells were injected in male nude mice.
Prostate TMA
We used the prostate TMA built using prostate biopsies that were de-identified. This study is exempt from IRB approval as no personal information about patients is sought.
METHOD DETAILS
Proliferation assay
For experiments involving RNA extraction or ChIP, VCaP, LAPC4 and LNCaP cells were grown in media containing 5% charcoal-striped serum for 24–48 hr followed by treatment with 3.5, 5 or 7 μM (R)-9bMS, 53μM enzalutamide or stimulated with 103nM DHT for 163hr. LNCaP-C4-2B cells were grown in its own media. To assess the effect of various treatments on cell proliferation, the cells were treated with 0.125 to 7.5 μM of (R)-9bMS or enzalutamide for 96 hr and number of viable cells counted by trypan blue exclusion assay.
In vitro kinase assay
For the in vitro kinase assay, 250 ng of purified ACK1 and 1 μg of H2A/H2B/H3/H4 (New England Biolab) were incubated in the presence or absence of ACK1 inhibitor in kinase assay buffer containing 50 mM HEPES (pH 7.5), 15 mM MgCl2, 1 mM EGTA, 10% glycerol, 10 mM DTT and 0.1 mM ATP at 30°C. After 60 min, the reaction was separated on SDS-PAGE followed by immunoblotting with pY88-H4, total H2B, H2A, H3, H4 and ACK1 antibodies.
Peptide Pull down assay
Two human histone H4 peptides spanning amino acids 78–92 were synthesized with Tyr88 at middle of the peptide. The sequences are as follows:
| H4 (78–92): | RKTVTAMDVVYALKRQG |
| pY88-H4 (78–92): | RKTVTAMDVVpYALKRQG |
Both the peptides were biotinylated at C-terminus and immobilized on streptavidin-sepharose beads. The beads were incubated with cell lysates made from LAPC4 cells in low-salt receptor lysis buffer (LS-RLB) containing 20 mM HEPES (pH 7.5), 225 mM NaCl, 1% Triton X-100, 1 mM DTT, 10% glycerol, phosphatase inhibitors (50 mM NaF, 1 mM Na2VO4), and protease inhibitor mix (Roche). The beads were extensively washed with LS-RLB buffer and bound WDR5 was resolved by SDS-PAGE followed by immunoblotting with respective antibodies.
Western Blot Analysis
LNCaP, VCaP, LAPC4, LNCaP-C4-2B or PC3 cells were grown in T-75 flasks at 5×106 cells/ml. For AR, AR-V7, ACK1, pACK1, WDR5, H3K4me1, H3K4me2 and H3K4me3, MLL2, Actin, histones H2a, H2B, H3 and H4 and FLAG detection, treated cells were harvested and lysed by sonication in receptor lysis buffer (RLB) containing 20 mM HEPES (pH 7.5), 500 mM NaCl, 1% Triton X-100, 1 mM DTT, 10% glycerol, phosphatase inhibitors (50 mM NaF, 1 mM Na2VO4), and protease inhibitor mix (Roche). Lysates were quantitated and 20 to 50 mg of protein lysates were boiled in SDS sample buffer, size fractionated by SDS-PAGE, and transferred onto a PVDF membrane (Immobilon). After blocking in 5% nonfat dry milk (or 3% BSA), membranes were incubated with the following primary antibodies: AR mouse monoclonal antibody (1:3000), AR-V7 antibody (1:1000), ACK1 mouse monoclonal antibody (1:1000), pACK1 mouse monoclonal antibody (1:1000), WDR5 mouse monoclonal antibody (1:1000), H3K4me1, H3K4me2 and H3K4me3 mouse monoclonal antibody (1:1000), MLL2 mouse monoclonal antibody (1:1000), actin mouse monoclonal antibody (1:10,000), histone mouse monoclonal antibody (1:3000) or FLAG mouse monoclonal antibody (1:4000). Following three washes in PBS-T, the blots were incubated with horseradish peroxidase-conjugated secondary antibody. The blots were washed thrice and the signals visualized by enhanced chemiluminescence (ECL) system according to manufacturer’s instructions (GE Healthcare).
For ligand mediated histone H4 Y88-phosphorylation detection, cells were grown in absence of growth factors (-FBS) overnight. Cells were treated with IGF (LNCaP and LNCaP-C4-2B cells), EGF or PDGF (LAPC4) for indicated time points followed by harvesting. Cells were lysed by sonication in RLB, the lysates were quantitated and 0.5 to 1 mg of protein lysate was immunoprecipitated using 3–4 μg of pY88-H4 antibody coupled with protein A/G-sepharose (Santacruz) overnight, followed by washes with RLB and PBS buffers. The beads were boiled in sample buffer and immunoblotting was performed using anti-H4 antibody as described above. For detection of phospho-ACK1, immunoprecipitation was performed with 2 μg of ACK1 antibody followed by immunoblotting with pTyr antibody wherein after blocking in 3% BSA, membranes were incubated with anti-pTyr mouse monoclonal antibody (1:1000) with the rest of the procedure as described above.
Peptide Synthesis
Synthetic peptides (including biotin conjugated peptides) were custom-synthesized and HPLC-purified by Moffitt Proteomics Facility. HPLC-MS was used to confirm 95% or higher purity for each peptide. Peptides were dissolved in sterile water and aliquots were stored in −20°C.
Mass Spectrometry
Histones were purified from frozen human CRPC samples using Histone Purification Kit (Active Motif). Samples were digested overnight with modified sequencing grade trypsin (Promega, Madison, WI), Glu-C (Worthington, Lakewood, NJ), or Arg-C (Roche, Switzerland). Phosphopeptides were enriched using Phospho Select IMAC resins (Sigma). A nanoflow ultra high performance liquid chromatograph (RSLC, Dionex, Sunnyvale, CA) coupled to an electrospray bench top orbitrap mass spectrometer (Q-Exactive plus, Thermo, San Jose, CA) was used for tandem mass spectrometry peptide sequencing experiments. The sample was first loaded onto a pre-column (2 cm × 100 μm ID packed with C18 reversed-phase resin, 5 μm, 100Å) and washed for 8 minutes with aqueous 2% acetonitrile and 0.04% trifluoroacetic acid. The trapped peptides were eluted onto the analytical column, (C18, 75 μm ID × 50 cm, 2 μm, 100Å, Dionex, Sunnyvale, CA). The 90-minute gradient was programmed as: 95% solvent A (2% acetonitrile + 0.1% formic acid) for 8 minutes, solvent B (90% acetonitrile + 0.1% formic acid) from 5% to 38.5% in 60 minutes, then solvent B from 50% to 90% B in 7 minutes and held at 90% for 5 minutes, followed by solvent B from 90% to 5% in 1 minute and re-equilibrate for 10 min. The flow rate on analytical column was 300 nl/min. Sixteen tandem mass spectra were collected in a data-dependent manner following each survey scan. Both MS and MS/MS scans were performed in Orbitrap to obtain accurate mass measurement using 60 second exclusion for previously sampled peptide peaks. Sequences were assigned using Sequest (Thermo) and Mascot (www.matrixscience.com) database searches against SwissProt protein entries of the appropriate species. Oxidized methionine, carbamidomethyl cysteine, and phosphorylated serine, threonine and tyrosine were selected as variable modifications, and as many as 3 missed cleavages were allowed. The precursor mass tolerance was 20 ppm and MS/MS mass tolerance was 0.05 Da. Assignments were manually verified by inspection of the tandem mass spectra and coalesced into Scaffold reports (www.proteomesoftware.com). ArgC digested peptide KTVTAMDVVYALKR was observed as triply charged, with m/z 558.9605, which represents a mass error of 1.2 ppm; MS/MS spectrum was identified using Mascot with a score of 32.1 in CRPC sample#1. Label-Free Quantification of phospho-Tyrosine 88 in human histone H4 in CRPC samples.
Chromatin immunoprecipitation (ChIP) and ChIP-sequencing
C4-2B cells (5×107 cells) were either untreated or treated with vehicle or (R)-9bMS. Cells were harvested, fixed in p-formaldehyde and nuclei were prepared. For native ChIP (Mahajan et al., 2012b), C4-2B cells (5×107 cells) were treated with (R)-9bMS, harvested and nuclei were prepared. Purified nuclei were resuspended in RLB buffer and sonicated for 25 seconds (Branson sonicator). The soluble chromatin was incubated overnight at 4°C with antibodies and protein-G and A magnetic beads. The soluble chromatin was processed in the same way without immunoprecipitation and termed input DNA. The amount of immunoprecipitated DNA was determined by real-time PCR. The complexes were washed with RLB buffer followed by ChIP buffer 1 and 2 (Active Motif), eluted with elution buffer and subjected to proteinase-K treatment. Fixed ChIP DNA was ‘reversed’ cross linked and subjected to proteinase-K treatment. ChIP DNA was purified using PCR DNA purification columns (Qiagen).
C4-2B cells were treated with IGF ligand and either untreated or treated with (R)-9bMS. ChIP was performed using pY88-H4 antibody or IgG and DNAs were subjected to sequencing. Chromatin immunoprecipitation sequencing (ChIP-seq) was performed by the Molecular Genomics Core Facility at the Moffitt Cancer Center. Fifty nanograms of immunoprecipitated DNA was fragmented to 300 base pairs using a Covaris M220 Focused-ultrasonicator (Covaris, Inc., Woburn, MA) and then used to generate sequencing libraries using the Kapa Hyper Prep Kit (Kapa Biosystems, Wilmington, MA). The size and quality of the library was evaluated using the Agilent BioAnalzyer, and the library was quantitated with the Kapa Library Quantification Kit. Each enriched DNA library was then sequenced on an Illumina NextSeq 500 sequencer. The sequencing yield was very good, with over 90 million reads in each sample, of which 82.8 (IGF) and 77.9 million (IgG), respectively, mapped uniquely to the human genome GRCh37.
Flow Cytometry Analysis
1–2 × 106 cells were seeded in 100 mM dishes. After treatment the media was aspirated, and the dishes were washed in 5 ml of 1X Phosphate Buffered Saline (PBS) via gentle rocking. After aspiration of the 1X PBS, 3 ml of Trypsin EDTA was added. Cells were harvested, washed in 1X PBS and pellet was stored in citrate storage buffer. Cells were trypsinized in 450 μl of trypsin solution at room temperature for 10 minutes, incubated in 375 μl trypsin inhibitor/RNase A (Sigma) solution for 10 min and stained with 250 μl of ice-cold propidium iodide solution for 10 min and kept in dark. Samples were analyzed using the FACS Calibur flow cytometer; 10,000 events were collected and cell cycle analysis was carried out using the ModFit program.
Immunofluorescence analysis
To visualize the intracellular distribution of pY88-H4, C42B prostate cancer cells were seeded on coverslips, transfected with HA-tagged ACK1 or vector. The cells were washed gently with 1X phosphate buffered saline (PBS) and treated with 0.5% TritonX-100 in 1X PBS for 8 min without rocking, followed by incubation in blocking buffer (3% BSA, 5% FBS in 1XPBS) for 1hr. Next, the cells were incubated with the primary antibodies, HA mouse monoclonal and H4 pY88 rabbit polyclonal at a dilution of 1:100 in blocking buffer for 1 hr. The excess antibody was washed off with 1X PBS. The cells were incubated with Alexaflour 488 conjugated anti-rabbit secondary antibody (1:500 dilution) and Alexaflour 594 conjugated anti-mouse secondary antibody (1:500 dilution) for 30 min. The cells were washed and coverslips were mounted on glass slides in DAPI (Vector Labs) containing mounting media. The cells were visualized using Axiovert Leica Microscope with a 40X oil immersion objective. Axiovision version 4.8 software suite was used to acquire and process images.
CRISPR-CAS9 mediated knockdown of ACK1 or AREMs
For ACK1 knockdown, CRISPR-CAS9 gene editing constructs were obtained from SantaCruz, while for AREM1 and AREM2 deletion, the constructs were custom synthesized by Sigma-Aldrich. PC cells were electroporated with constructs and positive cells were selected based on RFP expression using flow-sorting.
QUANTIFICATION AND STATISTICAL ANALYSIS
Quantitative RT-PCR and ChIP-qPCR
All RT reactions were done at the same time so that the same reactions could be used for all gene studies. For the construction of standard curves, serial dilutions of pooled sample RNA were used (50, 10, 2, 0.4, 0.08, and 0.016 ng) per reverse transcriptase reaction. One “no RNA” control and one “no Reverse Transcriptase” control were included for the standard curve. Three reactions were performed for each sample: 10 ng, 0.8 ng, and a NoRT (10 ng) control. Real-time quantitative PCR analyses were performed using the ABI PRISM 7900HT Sequence Detection System (Applied Biosystems). All standards, the no template control (H2O), the No RNA control, the no Reverse Transcriptase control, and the no amplification control (Bluescript plasmid) were tested in six wells per gene (2 wells/plate x 3 plates/gene). All samples were tested in triplicate wells each for the 10 ng and 0.8 ng concentrations. The no RT controls were tested in duplicate wells. PCR was carried out with SYBR Green PCR Master Mix (Applied Biosystems) using 2 μl of cDNA (or ChIP DNA) and the primers in a 20 μl final reaction mixture. After 2 min incubation at 50°C, AmpliTaq Gold was activated by 10 min incubation at 95°C, followed by 40 PCR cycles consisting of 15 s of denaturation at 95°C and hybridization of primers for 1 min at 55°C. Dissociation curves were generated for each plate to verify the integrity of the primers. Data were analyzed using SDS software version 2.2.2 and exported into an Excel spreadsheet. The actin data were used for normalizing the gene values.
Gene expression array analysis
C4-2B, VCaP, LAPC4, LNCaP were grown in androgen-deprived media and were treated with 3.5 uM (R)-9bMS for 243hr and total RNA was extracted using RNeasy Mini Kit (Qiagen) for gene expression array analysis. Expression profiling was performed using the Agilent Whole Human Genome Four-Plex 44K Microarray according to the manufacturer’s protocol. All samples were run in technical duplicates or quadruplicates against control. Differential gene expression was analyzed using Nexus expression 3.0 software. Over- and under-expressed gene sets were generated by filtering to include only data points that showed either over- or underexpression by at least two-folds (log ratio with p3<30.001) in all hybridizations. GSEA was performed using the JAVA program (http://www.broadinstitute.org/gsea) as described previously (Subramanian et al., 2005). Specifically, genes were ranked based on log fold change and pre-ranked setting was used to perform the analyses.
Computational analysis of ChIP-Seq data: Sequence analysis
The 75-nt paired end sequence reads were mapped to the genome using the BWA-MEM algorithm. Alignment information for each read was stored in the output file *.bam. Only reads that mapped uniquely with proper pairing were used in the subsequent analysis.
Determination of fragment density
Because the 5′ ends of the sequence reads represent the end of the ChIP or immunoprecipitation fragments, the reads were extended in silico (using MAC2) at their 3′ ends to a length of 173–244 bp, based on the fragment length calculated from the read pairs.
Peak finding
Peak regions were called using the MACS2 software with the following options-f BAMPE-SPMR-q 0.01-broad. The “BAMPE” option was used for calculating fragment lengths from the paired end reads, “SPMR” for normalizing read depths to number of fragments per million reads, “broad” for compositing broad regions from nearby peak regions, and the q value (FDR) cutoff was set to 0.01. A total of 370 peak regions were called between the IGF sample and the IgG control.
Motif analysis
HOMER (v4.7, 8-25-2014) program (http://homer.salk.edu/homer/) was used to identify de novo motifs in pY88-H4 ChIP-seq data.
Tissue Microarray (TMA) Analysis
For assessment of pY88-H4, H3K4me3 and AR expression in prostate cancer, immunohistochemistry was carried out on a high-density prostate TMAs (n = 250 cores) containing samples of different stages of disease as described earlier (Mahajan et al., 2010b).
Statistical Analysis
The Mantel-Haenszel χ2 test was performed to examine if there is an increasing trend for H3K4me3 and AR with respect to different progression stages of prostate cancer. The ordinal intensity levels of H3K4me3 and AR 0, 1, 2, 3, 4, 6, 9 were pooled into 6 levels (as 0, 1, 2, 3, 4, and 6 and above) to accommodate the rare observations in the highest intensity level in most stages. Analysis of variance was performed to examine whether the expression levels of H3K4me3 and AR differ among different tumor stages. Boxplots were used to summarize the intensity distribution at each progression stage. Furthermore, Tukey-Kramer method was performed to examine between which pairs of stages the expression levels are different. This post-hoc procedure adjusts for all pairwise comparisons and simultaneous inference. When more than one sample was obtained from a patient, the intensity of the most progressed stage was used for the analysis. Correlation between H3K4me3 and AR was explored using Spearman ranked correlation analysis. Statistical differences between the groups were determined using log-rank test.
Mass Spectrometry Data Analysis
Xcalibur software (Thermo) was used to extract ion chromatogram of H4 pY88-containing Arg-C digested peptide KTVTAMDVVYALKR. m/z tolerance was set to +/− 0.02 Th and retention time (RT) tolerance was set to +/− 60 seconds. Area under the curve (AUC) values were used to quantify relative intensities between samples.
DATA AND SOFTWARE AVAILABILITY
Links to GEP Database
The ChiP-Seq as well as microarray data has been submitted to GEO database, the accession numbers are GSE79689 and GSE79658, respectively.
Supplementary Material
Table S2 (related to Figure 1). Percent enzyme activity upon (R)-9b treatment relative to DMSO controls.
Provided as an Excel file.
Table S3 (related to Figure 2). The pY88-H4 binding sites in LNCaP-C4-2B cells identified by ChIP-sequencing.
Provided as an Excel file.
Significance.
Castration resistant prostate cancer (CRPCs) defines the incurable stage of disease. While the second generation anti-androgen enzalutamide and the androgen-synthesis inhibitor abiraterone stall CRPC progression for a limited time, tumors inevitably develop resistance by replenishing either the androgen receptor (AR) or its spliced variant AR-V7 that lack the ligand binding region. We uncovered ACK1 kinase as an epigenetic modifier hijacked by AR to modify its own gene locus, reactivating AR mRNA synthesis. Significantly, targeting this epigenetic deregulation with the ACK1 inhibitor (R)-9bMS not only suppressed AR and AR-V7 transcription but also overcame enzalutamide-resistance and mitigated CRPC tumor growth. This study demonstrates an epigenetic mechanism of tyrosine kinase driven CRPC growth and suggests a therapeutic strategy.
Highlights.
Characterization of a histone H4-Y88 phosphorylation in CRPCs.
Androgen Receptor is a key epigenetic target of the H4-Y88 phosphorylation in CRPCs.
Targeting H4-Y88 phosphorylation subdues the expression of AR and AR splice variant.
The epigenetic inhibitor (R)-9bMS mitigates enzalutamide resistance.
Acknowledgments
We thank John Cleveland for critical reading of the manuscript and various suggestions. N.P.M. is a recipient of NIH/NCI grant (1R01CA135328), Department of Defense (W81XWH-14-1-0002, W81XWH-14-1-0003 and W81WXH-15-1-0312), Bankhead-Coley (6BC08) and Miles-for-Moffitt Award (09-33661-15-13). KM is supported by Department of Defense awards (W81XWH-12-1-0248, W81XWH-14-1-0251 and W81XWH-15-1-0059). This work was supported in part by the Core Facilities at Moffitt Cancer Center and by Cancer Center Support Grant P30 CA076292. The Moffitt Cancer Center has filed patent applications as follows: ‘Antibodies specific for phosphorylated histones and uses thereof’ (U.S. Patent No. 9,594,084; Serial#13700955.1; 14/124,662; 15/457,269), and ‘Inhibitors of ACK1/TNK2 Tyrosine Kinase’ (patent application no. 14/910,486 and PCT/US2016/045096). K.M. and N.P.M. are named as inventors on a patent for antibodies specific for phosphorylated histones and K.M., H.R.L, N.J.L and N.P.M. are named as inventors on ACK1/TNK2 kinase inhibitors. Both the patents have been licensed by TechnoGenesys, Inc. K.M. and N.P.M. are co-founders of TechnoGenesys, Inc, own stock, and serve as consultants for TechnoGenesys, Inc.
Footnotes
AUTHOR CONTRIBUTIONS
Conceptualization, K.M. and N.P.M.; Methodology, K.M., H.R.L., N.J.L. and N.P.M.; Investigation, K.M., P.M., N.P.M.; Formal Analysis, Z.C., J.K. and D.C.; Resources, K.M., H.R.L., N.J.L. and N.P.M.; Data Curation, C.K.S., R.M., S.S.; Original Draft, K.M. and N.P.M.; Review & Editing, K.M. and N.P.M.; Supervision, K.M. and N.P.M.; Project Administration, N.P.M.; Funding Acquisition, K.M. and N.P.M.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antonarakis ES, Lu C, Wang H, Luber B, Nakazawa M, Roeser JC, Chen Y, Mohammad TA, Chen Y, Fedor HL, et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. The New England journal of medicine. 2014;371:1028–1038. doi: 10.1056/NEJMoa1315815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arora VK, Schenkein E, Murali R, Subudhi SK, Wongvipat J, Balbas MD, Shah N, Cai L, Efstathiou E, Logothetis C, et al. Glucocorticoid receptor confers resistance to antiandrogens by bypassing androgen receptor blockade. Cell. 2013;155:1309–1322. doi: 10.1016/j.cell.2013.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balbas MD, Evans MJ, Hosfield DJ, Wongvipat J, Arora VK, Watson PA, Chen Y, Greene GL, Shen Y, Sawyers CL. Overcoming mutation-based resistance to antiandrogens with rational drug design. eLife. 2013;2:e00499. doi: 10.7554/eLife.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett LL, Ingason A. Enzalutamide (Xtandi) for patients with metastatic, resistant prostate cancer. The Annals of pharmacotherapy. 2014;48:530–537. doi: 10.1177/1060028013518899. [DOI] [PubMed] [Google Scholar]
- Burnstein KL. Regulation of androgen receptor levels: implications for prostate cancer progression and therapy. Journal of cellular biochemistry. 2005;95:657–669. doi: 10.1002/jcb.20460. [DOI] [PubMed] [Google Scholar]
- Chaboissier MC, Kobayashi A, Vidal VI, Lutzkendorf S, van de Kant HJ, Wegner M, de Rooij DG, Behringer RR, Schedl A. Functional analysis of Sox8 and Sox9 during sex determination in the mouse. Development. 2004;131:1891–1901. doi: 10.1242/dev.01087. [DOI] [PubMed] [Google Scholar]
- Dehm SM, Schmidt LJ, Heemers HV, Vessella RL, Tindall DJ. Splicing of a novel androgen receptor exon generates a constitutively active androgen receptor that mediates prostate cancer therapy resistance. Cancer research. 2008;68:5469–5477. doi: 10.1158/0008-5472.CAN-08-0594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Schwartzman J, Gibbs A, Lisac R, Kleinschmidt R, Wilmot B, Bottomly D, Coleman I, Nelson P, McWeeney S, et al. Androgen receptor promotes ligand-independent prostate cancer progression through c-Myc upregulation. PloS one. 2013;8:e63563. doi: 10.1371/journal.pone.0063563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin JF, Schiewer MJ, Dean JL, Schrecengost RS, de Leeuw R, Han S, Ma T, Den RB, Dicker AP, Feng FY, et al. A hormone-DNA repair circuit governs the response to genotoxic insult. Cancer discovery. 2013;3:1254–1271. doi: 10.1158/2159-8290.CD-13-0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasso CS, Wu YM, Robinson DR, Cao X, Dhanasekaran SM, Khan AP, Quist MJ, Jing X, Lonigro RJ, Brenner JC, et al. The mutational landscape of lethal castration-resistant prostate cancer. Nature. 2012;487:239–243. doi: 10.1038/nature11125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossmann ME, Huang H, Tindall DJ. Androgen receptor signaling in androgen-refractory prostate cancer. Journal of the National Cancer Institute. 2001;93:1687–1697. doi: 10.1093/jnci/93.22.1687. [DOI] [PubMed] [Google Scholar]
- Guo Z, Dai B, Jiang T, Xu K, Xie Y, Kim O, Nesheiwat I, Kong X, Melamed J, Handratta VD, et al. Regulation of androgen receptor activity by tyrosine phosphorylation. Cancer cell. 2006;10:309–319. doi: 10.1016/j.ccr.2006.08.021. [DOI] [PubMed] [Google Scholar]
- Guo Z, Yang X, Sun F, Jiang R, Linn DE, Chen H, Chen H, Kong X, Melamed J, Tepper CG, et al. A novel androgen receptor splice variant is up-regulated during prostate cancer progression and promotes androgen depletion-resistant growth. Cancer research. 2009;69:2305–2313. doi: 10.1158/0008-5472.CAN-08-3795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, Han M, Partin AW, Vessella RL, Isaacs WB, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer research. 2009;69:16–22. doi: 10.1158/0008-5472.CAN-08-2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, et al. A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509. Cancer discovery. 2013;3:1020–1029. doi: 10.1158/2159-8290.CD-13-0226. [DOI] [PubMed] [Google Scholar]
- Korpal M, Korn JM, Gao X, Rakiec DP, Ruddy DA, Doshi S, Yuan J, Kovats SG, Kim S, Cooke VG, et al. An F876L mutation in androgen receptor confers genetic and phenotypic resistance to MDV3100 (enzalutamide) Cancer discovery. 2013;3:1030–1043. doi: 10.1158/2159-8290.CD-13-0142. [DOI] [PubMed] [Google Scholar]
- Lawrence HR, Mahajan K, Luo Y, Zhang D, Tindall N, Huseyin M, Gevariya H, Kazi S, Ozcan S, Mahajan NP, et al. Development of novel ACK1/TNK2 inhibitors using a fragment-based approach. J Med Chem. 2015;58:2746–2763. doi: 10.1021/jm501929n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonergan PE, Tindall DJ. Androgen receptor signaling in prostate cancer development and progression. Journal of carcinogenesis. 2011;10:20. doi: 10.4103/1477-3163.83937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, der Steen TV, Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor? Nature reviews Urology. 2015;12:137–144. doi: 10.1038/nrurol.2015.13. [DOI] [PubMed] [Google Scholar]
- Mahajan K, Challa S, Coppola D, Lawrence H, Luo Y, Gevariya H, Zhu W, Chen YA, Lawrence NJ, Mahajan NP. Effect of Ack1 tyrosine kinase inhibitor on ligand-independent androgen receptor activity. The Prostate. 2010a;70:1274–1285. doi: 10.1002/pros.21163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Coppola D, Challa S, Fang B, Chen YA, Zhu W, Lopez AS, Koomen J, Engelman RW, Rivera C, et al. Ack1 mediated AKT/PKB tyrosine 176 phosphorylation regulates its activation. PloS one. 2010b;5:e9646. doi: 10.1371/journal.pone.0009646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Coppola D, Rawal B, Chen YA, Lawrence HR, Engelman RW, Lawrence NJ, Mahajan NP. Ack1-mediated androgen receptor phosphorylation modulates radiation resistance in castration-resistant prostate cancer. The Journal of biological chemistry. 2012a;287:22112–22122. doi: 10.1074/jbc.M112.357384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Fang B, Koomen JM, Mahajan NP. H2B Tyr37 phosphorylation suppresses expression of replication-dependent core histone genes. Nature structural & molecular biology. 2012b;19:930–937. doi: 10.1038/nsmb.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Lawrence HR, Lawrence NJ, Mahajan NP. ACK1 tyrosine kinase interacts with histone demethylase KDM3A to regulate the mammary tumor oncogene HOXA1. The Journal of biological chemistry. 2014;289:28179–28191. doi: 10.1074/jbc.M114.584425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Mahajan NP. Shepherding AKT and androgen receptor by Ack1 tyrosine kinase. Journal of cellular physiology. 2010;224:327–333. doi: 10.1002/jcp.22162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan K, Mahajan NP. ACK1/TNK2 tyrosine kinase: molecular signaling and evolving role in cancers. Oncogene. 2015;34:4162–4167. doi: 10.1038/onc.2014.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan NP, Liu Y, Majumder S, Warren MR, Parker CE, Mohler JL, Earp HS, Whang YE. Activated Cdc42-associated kinase Ack1 promotes prostate cancer progression via androgen receptor tyrosine phosphorylation. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:8438–8443. doi: 10.1073/pnas.0700420104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan NP, Whang YE, Mohler JL, Earp HS. Activated tyrosine kinase Ack1 promotes prostate tumorigenesis: role of Ack1 in polyubiquitination of tumor suppressor Wwox. Cancer research. 2005;65:10514–10523. doi: 10.1158/0008-5472.CAN-05-1127. [DOI] [PubMed] [Google Scholar]
- Malik R, Khan AP, Asangani IA, Cieslik M, Prensner JR, Wang X, Iyer MK, Jiang X, Borkin D, Escara-Wilke J, et al. Targeting the MLL complex in castration-resistant prostate cancer. Nature medicine. 2015;21:344–352. doi: 10.1038/nm.3830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polkinghorn WR, Parker JS, Lee MX, Kass EM, Spratt DE, Iaquinta PJ, Arora VK, Yen WF, Cai L, Zheng D, et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer discovery. 2013;3:1245–1253. doi: 10.1158/2159-8290.CD-13-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM, Montgomery B, Taplin ME, Pritchard CC, Attard G, et al. Integrative clinical genomics of advanced prostate cancer. Cell. 2015;161:1215–1228. doi: 10.1016/j.cell.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spratt DE, Evans MJ, Davis BJ, Doran MG, Lee MX, Shah N, Wongvipat J, Carnazza KE, Klee GG, Polkinghorn W, et al. Androgen Receptor Upregulation Mediates Radioresistance after Ionizing Radiation. Cancer research. 2015;75:4688–4696. doi: 10.1158/0008-5472.CAN-15-0892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010;18:11–22. doi: 10.1016/j.ccr.2010.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, et al. Development of a second-generation antiandrogen for treatment of advanced prostate cancer. Science (New York, NY. 2009;324:787–790. doi: 10.1126/science.1168175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Horst EH, Degenhardt YY, Strelow A, Slavin A, Chinn L, Orf J, Rong M, Li S, See LH, Nguyen KQ, et al. Metastatic properties and genomic amplification of the tyrosine kinase gene ACK1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15901–15906. doi: 10.1073/pnas.0508014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Arora VK, Sawyers CL. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nature reviews. 2015;15:701–711. doi: 10.1038/nrc4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson PA, Chen YF, Balbas MD, Wongvipat J, Socci ND, Viale A, Kim K, Sawyers CL. Constitutively active androgen receptor splice variants expressed in castration-resistant prostate cancer require full-length androgen receptor. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:16759–16765. doi: 10.1073/pnas.1012443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD. WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell. 2005;121:859–872. doi: 10.1016/j.cell.2005.03.036. [DOI] [PubMed] [Google Scholar]
- Xu K, Shimelis H, Linn DE, Jiang R, Yang X, Sun F, Guo Z, Chen H, Li W, Chen H, et al. Regulation of androgen receptor transcriptional activity and specificity by RNF6-induced ubiquitination. Cancer cell. 2009;15:270–282. doi: 10.1016/j.ccr.2009.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao XD, Han X, Chew JL, Liu J, Chiu KP, Choo A, Orlov YL, Sung WK, Shahab A, Kuznetsov VA, et al. Whole-genome mapping of histone H3 Lys4 and 27 trimethylations reveals distinct genomic compartments in human embryonic stem cells. Cell stem cell. 2007;1:286–298. doi: 10.1016/j.stem.2007.08.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2 (related to Figure 1). Percent enzyme activity upon (R)-9b treatment relative to DMSO controls.
Provided as an Excel file.
Table S3 (related to Figure 2). The pY88-H4 binding sites in LNCaP-C4-2B cells identified by ChIP-sequencing.
Provided as an Excel file.