

Summary
Tumor evolution is an iterative process of selection for pro-oncogenic aberrations. This process can be accelerated by genomic instability, but how it interacts with different selection bottlenecks to shape the evolving genomic landscape remains understudied. Here, we assessed tumor initiation and therapy resistance bottlenecks in mouse models of melanoma, with or without genomic instability. At the initiation bottleneck, whole exome sequencing revealed that drug-naïve tumors were genomically silent, and this was surprisingly unaffected when genomic instability was introduced via telomerase inactivation. We hypothesize that the strong engineered alleles created low selection pressure. At the therapy resistance bottleneck, strong selective pressure was applied using a BRAF inhibitor. In the absence of genomic instability, tumors acquired a non-genetic drug resistance mechanism. By contrast, telomerase-deficient, drug resistant melanomas acquired highly recurrent copy number gains. These proof-of-principle experiments demonstrate how different selection pressures can interact with genomic instability to impact tumor evolution.
Keywords: Mouse models, genomic instability, selection pressure, tumor evolution, tumor genomics, melanoma, drug resistance, telomere dysfunction, evolution bottlenecks, copy number aberrations
Introduction
Tumor evolution is shaped by different forces, including genomic instability which increases the rate of genetic aberrations and selection pressures which create bottlenecks, together enhancing the enrichment for tumor cells harboring pro-oncogenic aberrations. During the course of melanoma development, several different bottlenecks can occur, including tumor initiation, progression to metastasis, and therapeutic resistance. Clarifying how genomic instability and selection pressures interact could help to understand risk factors and anticipate types of adaptive changes at each bottleneck.
As a prime example of iterative, pro-tumorigenic selection, high counts of somatic copy number aberrations (SCNAs) and mutations are a hallmark of late stage melanoma (Cancer Genome Atlas, 2015; Kabbarah et al., 2010). Indeed, analysis of biopsies from 37 patients revealed a monotonic increase in both mutation and SCNA burden over the course of intrapatient progression from nevus to malignant melanoma (Shain et al., 2015). Oncogenic SCNAs selected for over the course of tumor evolution include: well-established losses of the CDKN2A and PTEN tumor suppressors; broad gains of chromosomes 7, 8, 13, 17, 20, and 22 (Cancer Genome Atlas, 2015; Kabbarah et al., 2010); and gain of TERT, a gene that encodes a critical component of the telomerase holoenzyme (Shain et al., 2015). TERT gain, as well as frequently observed TERT promoter activating mutations (Horn et al., 2013; Huang et al., 2013) which are further boosted by BRAF signaling (Li et al., 2016), mark reactivation of telomerase as a common early event in human melanoma (Shain et al., 2015). The reason this can be critical for tumor progression is that in humans, while telomerase is widely active during fetal development and in some adult stem cells, it is non-expressed in most cells after birth (Blackburn et al., 2015). This loss of telomerase expression eventually results in the unrepaired degradation and shortening of telomere ends, which can lead to genomic instability and activation of senescence programs, which in turn can limit tumor progression (Jafri et al., 2016). Thus, mutational reactivation of TERT can relieve the negative selection pressure of senescence and thereby promote melanoma progression. However, this may also allow for accumulated telomerase deficiency-driven genomic aberrations to provide pro-oncogenic substrates for further evolutionary selection.
By contrast, mice express telomerase throughout life. Thus, in order to more accurately model human cancer, mouse cancer models deficient for telomerase can be used to mimic telomere dysfunction, genomic instability, and senescence induction. Similar to in human adults, these telomerase-deficient phenotypes can suppress tumor formation and progression in mice (Ding et al., 2012; Greenberg et al., 1999; Hu et al., 2012; Maser et al., 2007; Perera et al., 2008; Rudolph et al., 2001), and this suppression can be relieved through reactivation of telomerase (Ding et al., 2012; Hu et al., 2012) or, critically, through certain other oncogenic mutations such as loss of p53 (Maser et al., 2007; Perera et al., 2008). Once this suppression is relieved, selection for pro-tumorigenic DNA lesions can lead to aberrant genomic copy number profiles similar to that of human tumors (Hu et al., 2012; Maser et al., 2007) and can enhance tumor progression and metastasis (Ding et al., 2012; Perera et al., 2008), suggesting that persistent telomere dysfunction-induced genomic instability can continue to play a role in tumor evolution.
Here, using genetically engineered mouse models of melanoma with strong initiating oncogenes, we explore the interaction of genomic instability via telomerase loss and the selection pressure bottlenecks of tumor initiation and therapy resistance. We describe whole exome sequencing results that address how selection pressure strengths can interact with genomic instability to shape tumor genomic evolution.
Results
Genetically Engineered Mouse Melanomas are Genomically Silent
We have previously described two inducible mouse models of melanoma: (i) iBIP, driven by a human BRAFV600E transgene, inducibly null for PTEN, and germline homozygous null for CDKN2A (Figure 1A) (Kwong et al., 2015); and (ii) iNRAS, driven by a human NRASQ61K transgene and germline homozygous null for CDKN2A (Figure 1A) (Kwong et al., 2012). We confirmed the differential expression of PTEN, pAKT, and pERK in tumors from these mice by reverse phase protein array (Figure 1B) and western blot (Figure 1C). The mice exhibit different tumor latencies (Figure S1). To determine the mutational load of these mouse melanomas, we performed whole exome sequencing (WES) on 19 iBIP and 16 iNRAS snap-frozen tumors and paired normal tissue. Overall, the data produced an average 82X coverage on targeted regions and accurately identified deleted CDKN2A and PTEN alleles (Figure S2A).
Figure 1.
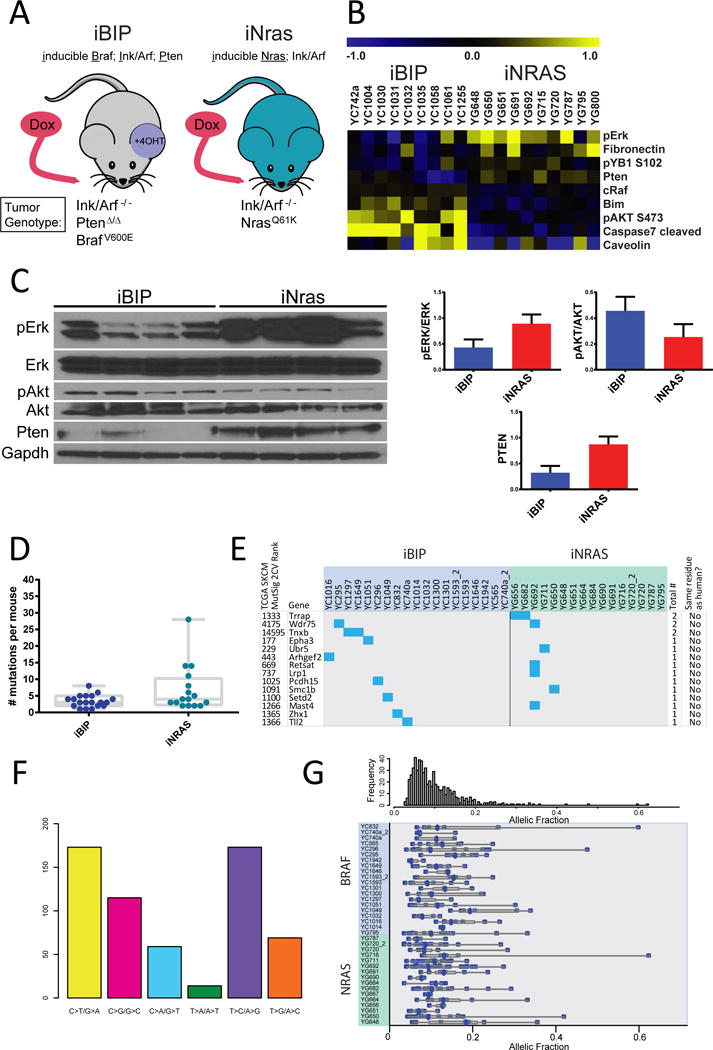
BRAF- and NRAS-driven mouse melanomas have low mutation rates and allelic fractions.
(A) Schematic of the alleles in the iBIP and iNRAS models.
(B) Significantly differentially expressed proteins from RPPA analysis of the two models.
(C) Western blot confirmation of selected RPPA results.
(D) Number of non-synonymous mutations per tumor.
(E) Mutation type spectrum for all mutations.
(F) Mutation matrix for selected genes, either recurrently mutated or significantly mutated in human melanoma (TCGA Mutsig p<0.05). Mutated mouse residues were compared to human mutated residues in the corresponding gene from three databases(Cancer Genome Atlas, 2015; Hodis et al., 2012; Krauthammer et al., 2012).
(G) Allelic fractions for all mutations, corrected for purity.
Next, using the MuTect program (Cibulskis et al., 2013) followed by manual review, we generated a high-confidence list of a total of 270 somatic mutations including 197 non-silent (Table S1) and 73 silent mutations (2.7:1 ratio) for an overall mutation rate of 0.2/Mbp. We then calculated the median number of non-synonymous coding events per tumor as 3 for iBIP and 4 for iNRAS (Figure 1D). Although iNras trended towards more mutations, the difference was only borderline significant (p=0.042). Overall, these mutation numbers are well below the median value for human melanomas (Cancer Genome Atlas, 2015). This was not surprising given that the mice were not exposed to years of UV radiation, consistent with the mutation profile being distinct from the predominant human melanoma C-to-T driven UV signature (Figure 1E).
We then asked whether iBIP and iNRAS mice require secondary mutation events for tumorigenesis, by assessing for additional known or likely oncogenic events. Analysis of the mutations and cross-comparison with human melanoma sequencing data (Cancer Genome Atlas, 2015; Hodis et al., 2012; Krauthammer et al., 2012) did not yield any obvious or known oncogenic events such as hotspot RAC1 or PPP6A mutations (Table S1). Furthermore, mutations in either recurrently mutated genes or genes ranked as significantly mutated in the human TCGA dataset (p<0.05) were not at homologous residues, e.g. not at S722 in TRRAP (Wei et al., 2011) (Figure 1F). Moreover, the allelic fractions of all the mutations are generally very low even after correction for tumor purity (Figure 1G and Table S1), suggesting a lack of strong selection pressure for additional driver mutations.
Next, we examined the genome copy numbers inferred from the whole exome sequencing data. The vast majority (32/35, 91%) of tumors had stable euploid genomes. Two iBIP and one iNRAS tumor showed a shallow gain of chromosome 15 (Figure 2). No recurrent focal copy number aberrations were observed. Interestingly, the two iBIP tumors showing chromosome 15 gain were the only tumors germline heterozygous rather than homozygous for knockout of CDKN2A and exhibited longer tumor latencies (Kwong et al., 2015). Notably, the CDKN2A locus experienced loss of heterozygosity in these two tumors (Figure S2B). Together, the mutation and copy number data suggest that the genetically engineered melanomas are largely genomically silent, which we hypothesize is due to the strength of the initiating oncogenic lesions and the consequent low selective pressure for additional cooperative driver events.
Figure 2.
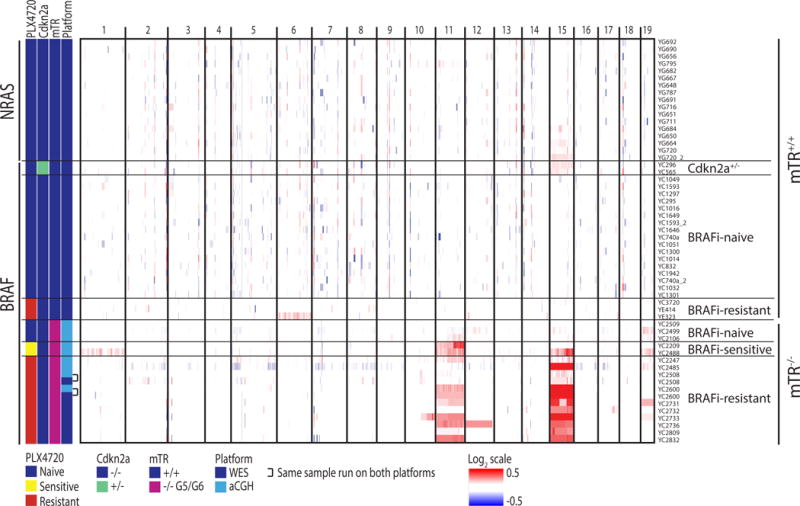
Copy number plots for all mice, determined from whole exome sequencing, array comparative genomic hybridization, or both. Samples that received both are annotated and show agreement between platforms. BRAFi-Sensitive and BRAFi-Resistant tumors are sampled while still on treatment.
We then examined the melanoma genomes at the bottleneck of drug resistance. We had previously treated iBIP mice with the BRAF inhibitor (hereafter “BRAFi”) PLX4720 continuously until they developed resistance at a median of 32 days (Kwong et al., 2015). These mice initially responded to treatment as evidenced by tumor regression (Kwong et al., 2015) and downregulation of pERK (Figure 3A and 3B). WES performed on 3 tumors that acquired resistance once again revealed low numbers of nonsynonymous mutations (5, 7, and 9) with low allelic frequencies (Table S2) and a stable copy number profile (Figure 2), with the exception of one sample harboring a shallow gain of chromosome 6. No previously described mutation that confers BRAFi resistance in humans was evident (Table S2). Protein analyses suggested that 2 of the 3 tumors became resistant through an elevated expression of BRAF resulting in partial reactivation of pERK and its downstream marker pS6 (Figure 3A–D), consistent with a previous study in a patient-derived xenograft model (Das Thakur et al., 2013). We confirmed a stepwise increase in BRAFV600E expression at the RNA level using an iBIP cell line xenograft that rapidly acquired PLX4720 resistance (Figure 3E and 3F). These results suggest that drug resistance does not select for DNA aberrations in the absence of genomic instability in the iBIP model.
Figure 3.
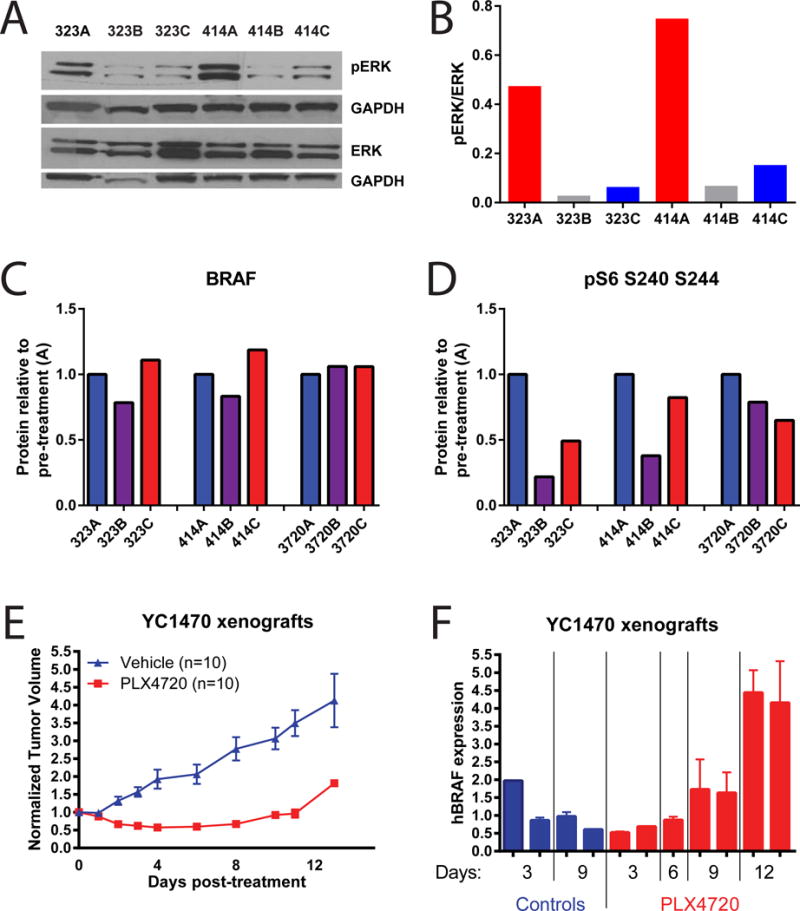
BRAF inhibitor resistance in the mTR+/+ iBIP mouse model is mediated by BRAF overexpression.
(A and B) Western blot for pERK and (B) its quantification on two sets of tumors that were sequenced and shown in Figure 2. The three samples per mouse are longitudinal biopsies taken pre-treatment “A”, on-treatment at 14d “B”, and post-resistance “C”. pERK/ERK was quantified by first normalizing each to GAPDH from its own blot (below each), then to each other.
(C and D) RPPA data for the same tumors from (A). (C) A Braf antibody that detects both endogenous mouse Braf and exogenous human BrafV600E. (D) A pS6 antibody that detects protein phosphorylated at serines 240 and 244.
(E) An iBIP xenograft model showing acquisition of BRAFi resistance by 12 days of treatment. All mice were injected identically using the same iBIP cell line.
(F) RTPCR analysis of the BRAFV600E transgene expression levels at the indicated times after BRAFi administration, from the same mice as in (D). Each column represents one tumor, taken at the indicated timepoint after administration of PLX4720. Error bars represent the samples run in triplicate.
Telomere Dysfunction Generates Recurrent SCNAs in the iBIP Model Only when Combined with Strong Selective Pressure
We then asked whether enforcing genomic instability would affect the iBIP phenotype. To determine this, we bred the iBIP mice onto a telomerase-deficient (mTR-knockout) background to the G5 or G6 generation which is known to engender chromosomal instability including nonreciprocal translocations and copy number aberrations (Artandi et al., 2000; Blasco et al., 1997; O’Hagan et al., 2002). These mice showed classical mTR-deficient phenotypes including reduced body weight, litter size (Lee et al., 1998), and significantly shortened telomeres (Figure S3), yet telomere dysfunction contributed only a modest improvement in long-term survival as expected on an intact DNA damage response background (Greenberg et al., 1999) (Figure 4A, p=0.048). These data contrast with the more robust anti-oncogenic effects reported for telomerase deficiency in mouse models of lymphoma (ATM−/−) (Hu et al., 2012), prostate (TP53−/−/PTEN−/−) (Ding et al., 2012), and colon cancer (APC+/−) (Rudolph et al., 2001), but is consistent with a lack of effect on virally-induced skin (HPV16) and pancreatic tumors (RIP1-Tag2) (Argilla et al., 2004), suggesting cell-type and/or driver mutation specificity. We also note that mTR loss differs phenotypically from TERT loss, such as their differential effects on Myc-induced lymphomagenesis (Koh et al., 2015).
Figure 4.
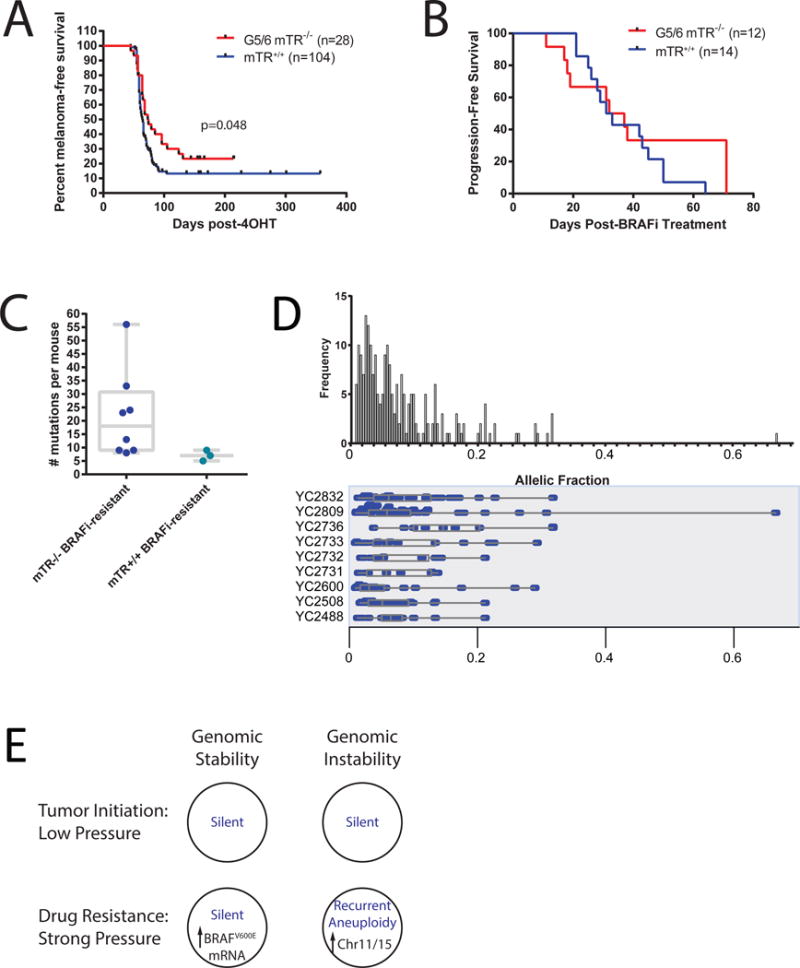
mTR deficiency does not majorly impact iBIP phenotypes.
(A) Melanoma latency in BRAF-mutant mice with or without mTR deficiency.
(B) Progression-free survival on the BRAF inhibitor PLX4720 in iBIP mice with or without mTR deficiency.
(C) Number of non-synonymous mutations in BRAFi-resistant iBIP mice with or without mTR deficiency.
(D) Allelic fractions in the 10 mTR-null, BRAFi-resistant tumors.
(E) Summarization of results. Blue text indicates genomic status (mutation + SCNAs).
Next, we generated BRAFi-resistant, mTR-deficient iBIP tumors. The median time to resistance in these samples was 32 days, identical to mTR-proficient iBIP mice (Figure 4B). We then sequenced 8 BRAFi-resistant, mTR-deficient melanomas with paired normal tissue. These did not show any known or obvious oncogenic or resistance-causing mutations (Table S3), and while the mutation load trended higher than mTR-proficient tumors (median = 18, Figure 4C), the difference was not statistically significant. These mutations once again had generally low allelic fractions (p=0.57 vs mTR-proficient iBIP tumors; Figure 4D and Table S3).
We next assessed copy number changes in 3 BRAFi-naïve, 2 BRAFi-sensitive, and 10 BRAFi-resistant mTR-deficient iBIP melanomas by the aforementioned WES data, by array comparative genomic hybridization (aCGH), or both. For samples analyzed by both methodologies (n=2), we noted agreement between the two platforms (Figure 2). First, we observed that all 3 BRAFi-naïve samples were largely genomically silent, suggesting a lack of selection for mTR-deficiency-induced structural aberrations in the absence of strong selective pressures (Figure 2). In striking contrast, the BRAFi-resistant melanomas exhibited whole-chromosome 15 gain in all 10 samples and whole-chromosome 11 gain in 6 samples, to varying degrees (Figure 2). Interestingly, in the 2 BRAFi-sensitive samples treated for 30 days, gain of chromosomes 15 and/or 11 were already evident, suggesting that potentially resistant subpopulations may have already been selected for prior to phenotypic resistance. Together, these results imply that although telomerase deficiency did not dramatically alter tumor latency, growth, progression-free survival, or genomic silence at the tumor initiation bottleneck, it drastically changed the genomic profile at the targeted therapy bottleneck towards recurrent aneuploidy.
Finally, although the purpose of the study is not to identify novel BRAF inhibitor resistance mechanisms, which are already well-described (Johnson et al., 2015), we nonetheless tested specific hypotheses about the potential underlying causes of the selection for gain of chromosomes 11 and 15. We first hypothesized that the human BRAFV600E transgene may have integrated into either chromosome and thus served as the target for gain. However, analysis of human BRAF transgene reads in the exome data did not indicate an increase in transgene copy number when chromosome 11 or 15 was gained (Figure S4A), ruling out this explanation. Next, we found by immunohistochemistry that resistant tumors with Chr 11 or 15 gain had reactivated pERK (Figure S4B). This is consistent with MAPK reactivation being the major mechanism of BRAF inhibitor resistance in human patients (Van Allen et al., 2014) and suggests potential MAPK regulator(s) on mouse Chr 11 and 15. We note that these regions are largely syntenic to human chromosomes 17 and 8, respectively, regions commonly gained in human melanoma (Cancer Genome Atlas, 2015; Kabbarah et al., 2010).
Discussion
In this study, we assessed somatic mutations and copy number aberrations in engineered mouse models of melanoma, under conditions of low selective pressure (tumor initiation given strong oncogenic lesions) or high selective pressure (targeted pharmacological inhibition), with or without genomic instability (Figure 4E). We first assessed the selection bottleneck of tumor initiation. In drug-naïve tumors, our whole exome sequencing results revealed low mutation rates without clearly selected-for secondary oncogenic mutations, and a stable genomic copy number profile even with telomerase inactivation. These results stand in contrast to the high mutation and SCNA burden characteristic of human melanoma, as well as to recurrent secondary oncogenic aberrations seen in other mouse cancer sequencing studies. For example, recent studies have discovered TP53 inactivation in a PIK3CAH1047R knock-in breast cancer model (Yuan et al., 2013), PTEN inactivation in a TP53/RB1-null small cell lung cancer model (McFadden et al., 2014), and gain of chromosome 6 in RAS- or EGFR-activated lung cancer (McFadden et al., 2016; Westcott et al., 2015) and squamous cell carcinoma models (McCreery et al., 2015; Nassar et al., 2015), among other recurrent aberrations. We hypothesize that because iBIP tumors are engineered with a set of potent oncogenic lesions efficient at inducing tumorigenesis, there is little or no need to select for additional oncogenic mutations or SCNAs, even when genomic instability is introduced (Figure 4E). Moreover, given the minimal impact of telomerase loss on iBIP tumor phenotypes including latency, the strong initiating lesions also likely bypass the need for telomerase reactivation. By contrast, in human melanoma or in mouse models with less efficient initiating oncogenic lesions, the necessary acquisition of additional oncogenic aberrations requires random mutational processes and tumor evolutionary selection. By the time the full complement of lesions for tumorigenesis is achieved, the tumor sequencing snapshot records the accumulated history of genomic aberrations necessary to reach that point.
In this context, it was surprising that the iNRAS melanomas did not show additional oncogenic lesions, given that the tumor latency and penetrance are significantly longer and lower than in the iBIP model, respectively. This suggests that the iNRAS melanomas may have selected for non-genetic mechanisms for tumor initiation. Intriguingly, a study in zebrafish (Kaufman et al., 2016) showed that a BRAF;p53 melanoma model initiated tumors through a stochastic, epigenetic dedifferentiation program marked by the expression of the crestin gene. We hypothesize that a similar mechanism may underlie iNRAS tumor initiation, and note that NRASQ61K expression was driven in terminally differentiated melanocytes by the Tyrosinase promoter. We speculate that PI3K activation in iBIP mice efficiently drove dedifferentiation (Koren et al., 2015; Van Keymeulen et al., 2015), while iNRAS mice required a secondary stochastic event that was non-genetic. This is difficult to test directly given that the crestin gene has no mouse orthologue, but the hypothesis is an appealing one that could be explored further with more advanced engineering including determining the effect of telomerase deficiency on the iNRAS model phenotype.
We also examined the selection bottleneck imposed by targeted therapy, in this case pharmacological BRAF inhibitor. Compared to tumor initiation, BRAF inhibition suppresses one of the key oncogenic lesions needed to maintain tumor survival, fundamentally changing the selection pressure strength. In telomerase-proficient melanomas, we observed selection for non-genetic means of drug resistance, which includes high expression of the BRAF transgene allele in two of three tumors. By contrast, telomerase-deficient tumors exhibited an apparently genetic means of drug resistance: recurrent gain of the entire chromosomes 15 and/or 11 in all 10 assayed tumors (Figure 4E). We note that a mouse model of telomerase-deficient prostate cancer also showed gain of the whole chromosome 15 in 12/18 mice, suggesting a common recurrent target of gain (Ding et al., 2012). Thus, although telomere dysfunction did not increase aberrations at tumor initiation and did not accelerate drug resistance in the iBIP model, it drastically altered the drug-resistant genomic profile, indicating that evolutionary bottlenecks make use of pro-oncogenic aberration substrates that are most readily available for selection. Overall, these results demonstrate that an underlying genomic instability will not necessarily manifest as recurrent genomic derangement, depending on the strength of the selection pressure. The corollary is that a tumor that appears genomically silent may simply have a low selective pressure, and does not necessarily imply the absence of an underlying genomic instability.
In summary, we have leveraged genetically engineered mouse models to examine in vivo how genomic instability and selection pressures interact to alter the genome at different tumor evolution bottlenecks. The melanoma mouse models described here are genomically silent, providing clean genomic slates at baseline to test such hypotheses. In other words, the low background mutation and SCNA rates in these melanomas mean that the effects of new evolutionary forces – such as telomerase loss or targeted therapy – on genomic stability can be cleanly discerned. We suggest that these models could therefore be useful for assaying the genomic effects of other types of bottlenecks such as metastasis and immunotherapy resistance (Gao et al., 2016) and their combination with other sources of melanoma-relevant genomic instability such as chronic UV radiation exposure (Cancer Genome Atlas, 2015). At the same time, we caution that the low mutation burden means that mechanisms underlying drug resistance and genomic instability in these models per se are not the focus, but rather they are used as tools to understand how different evolutionary forces act on a tumor genome. Overall, the results of this study provide insight into how selection pressures at different bottlenecks can act together with genomic instability to differentially alter the tumor genome over the course of its evolution.
Experimental Procedures
Animal husbandry and drug studies
iBIP and iNRAS mice were maintained and treatment of iBIP mice with PLX4720 chow were as previously described1,2. iBIP mice were bred onto a mTR-deficient background (Blasco et al., 1997), then homozygous mice were intercrossed to produce G5 and G6 mTR-deficient iBIP mice. Mice of both genders were used as no phenotypic difference has been noted. 90% of mice were within 3–7 months of age; 4 mice were 12–14 months of age due to a long tumor latency. No correlation was seen between age and number of mutations. The University of Texas MD Anderson Cancer Center Institutional Animal Care and Use Committee approved all animal studies and procedures.
Reverse phase protein array
iBIP and iNRAS tumors were homogenized to extract protein in radioimmunoprecipitation assay (RIPA) buffer plus phosphatase and protease inhibitors (Sigma) and sonicated to shear genomic DNA. Samples were diluted to 1 μg/μl in RIPA buffer. RPPA was performed by the MD Anderson RPPA Core using 30ug of protein per sample. All antibodies are validated by western blot (Tibes et al., 2006) by the RPPA core. Mouse-incompatible antibodies are filtered out.
Sequencing of whole exome
Whole exome sequencing was performed as previously described (Gnirke et al., 2009). Mutations were identified using MuTect (Cibulskis et al., 2013). All somatic mutations were reviewed manually. DNA copy number alterations were identified from exome data using CapSeg (https://github.com/aaronmck/CapSeg), and GISTIC 2.0 was used to identify recurrent alterations (Mermel et al., 2011). IGV (Integrative Genomics Viewer, https://www.broadinstitute.org/igv) was used to visualize genomic data.
Array comparative genomic hybridization
aCGH was performed as previously described (Ding et al., 2012). iBIP tumors were profiled against matched normal kidney DNA. Labeled DNAs were hybridized onto Agilent mouse 244K CGH arrays, and scanning and data processing were performed per manufacturer’s protocol.
Pathway analysis
DAVID (Huang da et al., 2009) was used to calculate and display the enriched pathway(s) of genes located on chromsome 15 of mouse genome. The gene annotations for mouse reference (mm9) were retrieved from UCSC genome browser.
Counting BRAFV600E transgene reads
We extracted all unmapped reads from mouse aligned BAM files. In order to detect the human BRAF transgene reads, we aligned the unmapped mouse reads to Human Chromosome 7 (containing BRAF) using BWA aligner with default settings. From the Human chromosome 7 aligned BAM file, we used Samtools to count all the reads that have mapping quality score of 40. These BRAF reads were then normalized against the total number of sequencing reads per sample.
Estimation of telomere length
Telomere length was estimated from whole exome sequencing data using the program TelSeq, as previously described (Barthel et al., 2017).
Immunohistochemistry
Immunohistochemistry was performed as previously described (Kwong et al., 2012), using pERK antibody CST #9101.
Statistical methods
All p values were generated through either a student’s T test for number of mutations or a log rank Mantel-Cox test for survival curves using the GraphPad Prism software.
Supplementary Material
Nonsysnonymous mutations identified by whole exome sequencing in 19 iBIP and 18 iNras tumors.
Nonsysnonymous mutations identified by whole exome sequencing in 3 BRAF inhibitor resistant, Tert-wild type iBIP tumors.
Nonsysnonymous mutations identified by whole exome sequencing in 8 Tert-null iBIP tumors.
Acknowledgments
The authors would like to thank Ron Depinho for critical reading of the manuscript. The study was supported by the following: NIH 1P01 CA163222, NIH 5U54 CA163125, and the University of Texas Rising STARS Award (L.N.K).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions. L.N.K. performed mouse experiments, designed the study, and wrote the manuscript. L.Z. processed the genomic data including manual review of mutations and DAVID analysis. L.N.K. and L.Z. together analyzed the genomic data. S.C. performed western blotting. C.S.P. calculated the read counts of the human BRAF transgene. M.L. and S.J. provided technical assistance. A.P. performed array comparative genomic hybridization. J.H.Z. assisted with whole exome data including performing TelSeq analysis. G.G. and L.C. supervised the project.
Accession Numbers. The NCBI BioProject accession number for the sequences reported in this paper is PRJNA381592.
References
- Argilla D, Chin K, Singh M, Hodgson JG, Bosenberg M, de Solorzano CO, Lockett S, DePinho RA, Gray J, Hanahan D. Absence of telomerase and shortened telomeres have minimal effects on skin and pancreatic carcinogenesis elicited by viral oncogenes. Cancer cell. 2004;6:373–385. doi: 10.1016/j.ccr.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Artandi SE, Chang S, Lee SL, Alson S, Gottlieb GJ, Chin L, DePinho RA. Telomere dysfunction promotes non-reciprocal translocations and epithelial cancers in mice. Nature. 2000;406:641–645. doi: 10.1038/35020592. [DOI] [PubMed] [Google Scholar]
- Barthel FP, Wei W, Tang M, Martinez-Ledesma E, Hu X, Amin SB, Akdemir KC, Seth S, Song X, Wang Q, et al. Systematic analysis of telomere length and somatic alterations in 31 cancer types. Nat Genet. 2017;49:349–357. doi: 10.1038/ng.3781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn EH, Epel ES, Lin J. Human telomere biology: A contributory and interactive factor in aging, disease risks, and protection. Science (New York, NY) 2015;350:1193–1198. doi: 10.1126/science.aab3389. [DOI] [PubMed] [Google Scholar]
- Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere Shortening and Tumor Formation by Mouse Cells Lacking Telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas, N. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013;31:213–219. doi: 10.1038/nbt.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das Thakur M, Salangsang F, Landman AS, Sellers WR, Pryer NK, Levesque MP, Dummer R, McMahon M, Stuart DD. Modelling vemurafenib resistance in melanoma reveals a strategy to forestall drug resistance. Nature. 2013;494:251–255. doi: 10.1038/nature11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Z, Wu CJ, Jaskelioff M, Ivanova E, Kost-Alimova M, Protopopov A, Chu GC, Wang G, Lu X, Labrot ES, et al. Telomerase reactivation following telomere dysfunction yields murine prostate tumors with bone metastases. Cell. 2012;148:896–907. doi: 10.1016/j.cell.2012.01.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell. 2016;167:397–404 e399. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, et al. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature biotechnology. 2009;27:182–189. doi: 10.1038/nbt.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg RA, Chin L, Femino A, Lee KH, Gottlieb GJ, Singer RH, Greider CW, DePinho RA. Short Dysfunctional Telomeres Impair Tumorigenesis in the INK4aΔ2/3 Cancer-Prone Mouse. Cell. 1999;97:515–525. doi: 10.1016/s0092-8674(00)80761-8. [DOI] [PubMed] [Google Scholar]
- Hodis E, Watson IR, Kryukov GV, Arold ST, Imielinski M, Theurillat JP, Nickerson E, Auclair D, Li L, Place C, et al. A landscape of driver mutations in melanoma. Cell. 2012;150:251–263. doi: 10.1016/j.cell.2012.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Figl A, Rachakonda PS, Fischer C, Sucker A, Gast A, Kadel S, Moll I, Nagore E, Hemminki K, et al. TERT promoter mutations in familial and sporadic melanoma. Science (New York, NY) 2013;339:959–961. doi: 10.1126/science.1230062. [DOI] [PubMed] [Google Scholar]
- Hu J, Hwang SS, Liesa M, Gan B, Sahin E, Jaskelioff M, Ding Z, Ying H, Boutin AT, Zhang H, et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. 2012;148:651–663. doi: 10.1016/j.cell.2011.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Huang FW, Hodis E, Xu MJ, Kryukov GV, Chin L, Garraway LA. Highly recurrent TERT promoter mutations in human melanoma. Science (New York, NY) 2013;339:957–959. doi: 10.1126/science.1229259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jafri MA, Ansari SA, Alqahtani MH, Shay JW. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Medicine. 2016;8:69. doi: 10.1186/s13073-016-0324-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DB, Menzies AM, Zimmer L, Eroglu Z, Ye F, Zhao S, Rizos H, Sucker A, Scolyer RA, Gutzmer R, et al. Acquired BRAF inhibitor resistance: A multicenter meta-analysis of the spectrum and frequencies, clinical behaviour, and phenotypic associations of resistance mechanisms. Eur J Cancer. 2015;51:2792–2799. doi: 10.1016/j.ejca.2015.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabbarah O, Nogueira C, Feng B, Nazarian RM, Bosenberg M, Wu M, Scott KL, Kwong LN, Xiao Y, Cordon-Cardo C, et al. Integrative Genome Comparison of Primary and Metastatic Melanomas. PLoS ONE. 2010;5:e10770. doi: 10.1371/journal.pone.0010770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman CK, Mosimann C, Fan ZP, Yang S, Thomas AJ, Ablain J, Tan JL, Fogley RD, van Rooijen E, Hagedorn EJ, et al. A zebrafish melanoma model reveals emergence of neural crest identity during melanoma initiation. Science (New York, NY) 2016;351:aad2197. doi: 10.1126/science.aad2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh CM, Khattar E, Leow SC, Liu CY, Muller J, Ang WX, Li Y, Franzoso G, Li S, Guccione E, Tergaonkar V. Telomerase regulates MYC-driven oncogenesis independent of its reverse transcriptase activity. The Journal of Clinical Investigation. 2015;125:2109–2122. doi: 10.1172/JCI79134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S, Reavie L, Couto JP, De Silva D, Stadler MB, Roloff T, Britschgi A, Eichlisberger T, Kohler H, Aina O, et al. PIK3CAH1047R induces multipotency and multi-lineage mammary tumours. Nature. 2015;525:114–118. doi: 10.1038/nature14669. [DOI] [PubMed] [Google Scholar]
- Krauthammer M, Kong Y, Ha BH, Evans P, Bacchiocchi A, McCusker JP, Cheng E, Davis MJ, Goh G, Choi M, et al. Exome sequencing identifies recurrent somatic RAC1 mutations in melanoma. Nat Genet. 2012;44:1006–1014. doi: 10.1038/ng.2359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, et al. Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest. 2015;125:1459–1470. doi: 10.1172/JCI78954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LN, Costello JC, Liu H, Jiang S, Helms TL, Langsdorf AE, Jakubosky D, Genovese G, Muller FL, Jeong JH, et al. Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med. 2012;18:1503–1510. doi: 10.1038/nm.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HW, Blasco MA, Gottlieb GJ, Horner JW, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- Li Y, Cheng HS, Chng WJ, Tergaonkar V. Activation of mutant TERT promoter by RAS-ERK signaling is a key step in malignant progression of BRAF-mutant human melanomas. Proceedings of the National Academy of Sciences. 2016;113:14402–14407. doi: 10.1073/pnas.1611106113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007;447:966–971. doi: 10.1038/nature05886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCreery MQ, Halliwill KD, Chin D, Delrosario R, Hirst G, Vuong P, Jen KY, Hewinson J, Adams DJ, Balmain A. Evolution of metastasis revealed by mutational landscapes of chemically induced skin cancers. Nat Med. 2015;21:1514–1520. doi: 10.1038/nm.3979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Papagiannakopoulos T, Taylor-Weiner A, Stewart C, Carter SL, Cibulskis K, Bhutkar A, McKenna A, Dooley A, Vernon A, et al. Genetic and clonal dissection of murine small cell lung carcinoma progression by genome sequencing. Cell. 2014;156:1298–1311. doi: 10.1016/j.cell.2014.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden DG, Politi K, Bhutkar A, Chen FK, Song X, Pirun M, Santiago PM, Kim-Kiselak C, Platt JT, Lee E, et al. Mutational landscape of EGFR-, MYC-, and Kras-driven genetically engineered mouse models of lung adenocarcinoma. Proceedings of the National Academy of Sciences of the United States of America. 2016 doi: 10.1073/pnas.1613601113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mermel CH, Schumacher SE, Hill B, Meyerson ML, Beroukhim R, Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome biology. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassar D, Latil M, Boeckx B, Lambrechts D, Blanpain C. Genomic landscape of carcinogen-induced and genetically induced mouse skin squamous cell carcinoma. Nat Med. 2015;21:946–954. doi: 10.1038/nm.3878. [DOI] [PubMed] [Google Scholar]
- O’Hagan RC, Chang S, Maser RS, Mohan R, Artandi SE, Chin L, DePinho RA. Telomere dysfunction provokes regional amplification and deletion in cancer genomes. Cancer cell. 2002;2:149–155. doi: 10.1016/s1535-6108(02)00094-6. [DOI] [PubMed] [Google Scholar]
- Perera SA, Maser RS, Xia H, McNamara K, Protopopov A, Chen LF, Hezel A, Kim CF, Bronson RT, Castrillon DH, et al. Telomere dysfunction promotes genome instability and metastatic potential in a K-ras p53 mouse model of lung cancer. Carcinogenesis. 2008;29:747–753. doi: 10.1093/carcin/bgn050. [DOI] [PubMed] [Google Scholar]
- Rudolph KL, Millard M, Bosenberg MW, DePinho RA. Telomere dysfunction and evolution of intestinal carcinoma in mice and humans. Nat Genet. 2001;28:155–159. doi: 10.1038/88871. [DOI] [PubMed] [Google Scholar]
- Shain AH, Yeh I, Kovalyshyn I, Sriharan A, Talevich E, Gagnon A, Dummer R, North J, Pincus L, Ruben B, et al. The Genetic Evolution of Melanoma from Precursor Lesions. N Engl J Med. 2015;373:1926–1936. doi: 10.1056/NEJMoa1502583. [DOI] [PubMed] [Google Scholar]
- Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, Kornblau SM. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther. 2006;5:2512–2521. doi: 10.1158/1535-7163.MCT-06-0334. [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Wagle N, Sucker A, Treacy DJ, Johannessen CM, Goetz EM, Place CS, Taylor-Weiner A, Whittaker S, Kryukov GV, et al. The genetic landscape of clinical resistance to RAF inhibition in metastatic melanoma. Cancer Discovery. 2014;4:94–109. doi: 10.1158/2159-8290.CD-13-0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Keymeulen A, Lee MY, Ousset M, Brohee S, Rorive S, Giraddi RR, Wuidart A, Bouvencourt G, Dubois C, Salmon I, et al. Reactivation of multipotency by oncogenic PIK3CA induces breast tumour heterogeneity. Nature. 2015;525:119–123. doi: 10.1038/nature14665. [DOI] [PubMed] [Google Scholar]
- Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, Davis S, Program NCS, Stemke-Hale K, Davies MA, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–446. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westcott PM, Halliwill KD, To MD, Rashid M, Rust AG, Keane TM, Delrosario R, Jen KY, Gurley KE, Kemp CJ, et al. The mutational landscapes of genetic and chemical models of Kras-driven lung cancer. Nature. 2015;517:489–492. doi: 10.1038/nature13898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan W, Stawiski E, Janakiraman V, Chan E, Durinck S, Edgar KA, Kljavin NM, Rivers CS, Gnad F, Roose-Girma M, et al. Conditional activation of Pik3ca(H1047R) in a knock-in mouse model promotes mammary tumorigenesis and emergence of mutations. Oncogene. 2013;32:318–326. doi: 10.1038/onc.2012.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Nonsysnonymous mutations identified by whole exome sequencing in 19 iBIP and 18 iNras tumors.
Nonsysnonymous mutations identified by whole exome sequencing in 3 BRAF inhibitor resistant, Tert-wild type iBIP tumors.
Nonsysnonymous mutations identified by whole exome sequencing in 8 Tert-null iBIP tumors.