

Synopsis
The presence of FLT3 mutations in AML carries a particularly poor prognosis making the development of FLT3 inhibitors an imperative goal for these patients. The last decade has seen an abundance of clinical trials using these drugs alone or in combination with chemotherapy. This culminated with the imminent approval by the FDA of FLT3 inhibitors for the treatment of AML. Unfortunately, the initial success stories have been rapidly followed by the emergence of clinical resistance. While novel FLT3 inhibitors are actively being developed, studies into mechanisms of resistance to these drugs raise hope of new strategies to prevent emergence of resistance and eliminate minimal residual disease in this AML.
Keywords: FLT3 inhibitors, FLT3-ITD, AML, stem cell niche
Introduction
FLT3 mutated AML represents about a third of all cases of newly diagnosed AML1. Two classes of mutations are frequently found: activation loop or tyrosine kinase domain mutations (TKD, about 5-10% of patients) and in-frame, internal tandem duplication (ITD, about 23% of patients). While the prognostic impact of de novo FLT3-TKD mutations is usually minimal, the presence of FLT3-ITD mutations confers poor prognosis in AML2-4 and the frequency of the mutated allele (allelic ratio)4 as well as the length of the tandem repeat (ITD length) correlate with worse outcome5.
FLT3 is a class III receptor tyrosine kinase that dimerizes upon ligand binding and undergoes auto-phosphorylation to initiate multiple intracellular signaling programs6. These pathways, including PI3K/AKT, Jak/STAT and Ras/MAPK, transduce signals resulting in survival and proliferation of target cells (see Figure 1).
Figure 1. FLT3 signaling and mechanisms of resistance to FLT3 inhibitors.
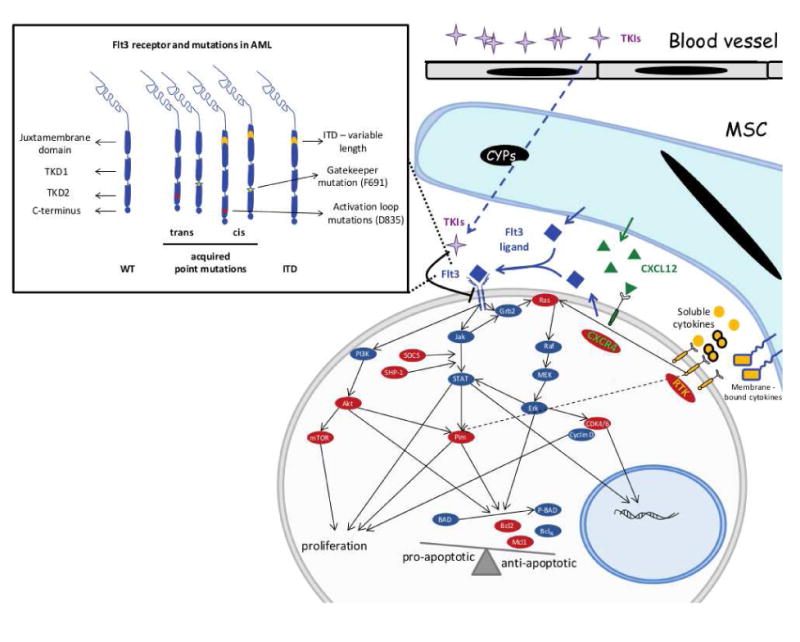
Wild type as well as FLT3 mutated receptor signal via Jak/STAT, PI3K/Akt and Ras/MAPK to provide anti-apoptotic as well as proliferative signaling to the leukemic blasts. Mechanisms that maintain these pathways active in the presence of FLT3 inhibitors create the conditions for the development of resistance. Combining FLT3 inhibitors with inhibitors of these pathways hold the promise of preventing development of resistance. Potential targets are highlighted in red. Point mutations in the FLT3 receptor are detailed in the rectangular insert.
During normal hematopoiesis, FLT3 is expressed in early progenitor cells and the receptor is down-regulated while cells differentiate down the myeloid lineage. Patients with FLT3 mutated AML not only have constitutively active signaling but given the lack of differentiation, these AML blasts continue to express high levels of this mutated protein in addition to the wild type FLT3 receptor7.
This sustained survival and proliferation signal is the hallmark of FLT3 mutated AML. The clinical presentation of these patients is dominated by hyper-leukocytosis, myelo/monoblastic differentiation and usually de novo (as opposed to secondary) AML. Compared to other poor prognostic factors in AML, the presence of a FLT3 ITD mutation does not have a major impact on achieving remission post-induction chemotherapy, but the remission is characteristically short-lived and relapse often occurs during cycles of consolidation (sometimes while an allogeneic donor search is underway). There is a consensus that prompt blood or marrow transplantation in first remission can improve the outcome in this disease even in the absence of FLT3 inhibitors. Nevertheless, the high rate of relapse even post-transplantation makes this approach alone suboptimal.
In the setting of relapsed disease, the FLT3-ITD mutation is often present at a high allelic ratio8, and the mutated receptor in the malignant clone renders this disease resistant to chemotherapy and addicted to signaling downstream of this RTK and thus, sensitive to FLT3 inhibitors. Thus, the clinical development of small molecule inhibitors that target mutant FLT3 is an active area of research. More than 60 clinical trials are either open or completed testing different FLT3 inhibitors as single agents or in combination with other therapeutic approaches in AML (clinicaltrials.gov). Full updates on the clinical development of these strategies can be found in this issue by Garcia J and Stone R. Here, we will focus our attention on mechanisms of resistance to FLT3 inhibitors and strategies to overcome such resistance and achieve cure in FLT3 mutated AML.
Mechanisms that allow survival of FLT3 mutated AML cells during treatment with FLT3 inhibitors
Wisdom gathered from over seven decades of antimicrobial use to treat infections tells us that resistance is either acquired via genetic adaptation or due to already-present clones that are selected under survival pressure. In AML, resistance to chemotherapy can take either of the two forms. Elegant genetic studies have shown that AML at diagnosis is a polyclonal disease, while relapsed AML is usually more oligoclonal9. Most of the time the relapsed clone can be retrospectively found at presentation but at much lower frequency. This appears to be the case with FLT3 mutated AML relapsing after induction chemotherapy. Most of the initial malignant subclones are sensitive to treatment and the patient achieves a clean complete remission. Nevertheless some clones survive chemotherapy and are responsible for disease relapse. At relapse, the mutant allelic ratio is often higher than what was seen at diagnosis.
On the other hand, it is probable that the development of resistance to FLT3 inhibitors is at least in part dependent on some genetic or epigenetic events. In order for these genetic and epigenetic events to take place, some FLT3 mutated cells will need to have survived the initial treatment.
Initial studies using FLT3 inhibitors demonstrated clearance of circulating FLT3-ITD blasts, but there was little to no effect on bone marrow blasts10. More potent and selective FLT3 inhibitors are effective at differentiating most bone marrow blasts11 but since these drugs cannot eliminate minimal residual disease as single agents, some leukemia cells, perhaps residing in the stem cell niche, must survive the treatment with the inhibitor.
This early evidence pointed towards stromal mediated mechanisms of survival. To this end, co-culture of FLT3-ITD AML cells with bone marrow mesenchymal stroma also protects the blasts from Quizartinib12 as well as Fl-70013. In these settings, soluble as well as membrane bound cytokines that appear to play a major role include CXCRL12, angiopoietins, as well as VEGF/EGF/IGF and G-CSF/GM-CSF/TNF12,13. Similarly, patients with FLT3-ITD AML treated with Quizartinib develop high levels of stromal-derived FGF2 in bone marrow mesenchymal cells and signaling downstream of FGFR1 maintain active RAS/MAPK signaling in FLT3-ITD blasts treated with Quizartinib14. Most patients with FLT3-ITD continue to have a wildtype allele of FLT3. This receptor is rather resistant to FLT3 inhibitors but sensitive to FLT3 ligand. Since high levels of FLT3 ligand are found in the bone marrow microenvironment during induction therapy, ligand induced activation of WT FLT3 – MAPK pathway may provide survival signals to leukemic blats even in the presence of effective TKI treatment14,15.
Potent FLT3-ITD inhibition results in apoptosis of AML blasts in the absence of stroma. The presence of bone marrow mesenchymal stroma rescues the blasts from apoptosis through ERK mediated signaling but not STAT512. In this context, inhibition of FLT3-ITD induces G1 arrest, maybe via downregulation of cyclin D2/Cyclin D3 and subsequent dephosphorylation of Rb16. Individual FLT3 inhibitors may have differential effects, for instance, contact with niche cells expand FLT3-ITD blasts treated with SU5615 but not Sorafenib17.
Complete and sustained inhibition of FLT3-ITD is paramount for successful elimination of the malignant clone. Initial studies with Midostaurin clearly demonstrated the impact of hepatic drug metabolism and CYP3A4 activity in particular of this drug plasma PK's. More so, PD studies have shown wide variations between systemic concentrations and plasma inhibitory activity of various TKIs, likely due to unique protein binding affinities of specific drugs. PD-directed dose escalation clinical studies have mitigated these limitations for the most part and resulted in improved efficacy in clearing not only circulating blasts but also most bone marrow disease. Recent work proposes the existence of unique niches in the bone marrow, some of which are true biochemical sanctuaries where local drug levels may be significantly different from systemic plasma drug levels18. To this end, bone marrow mesenchymal stroma expresses similar levels of drug metabolizing enzymes compared to hepatocytes. They are able to metabolize CYP3A4 substrates creating potential biochemical spaces where FLT3 inhibitors achieve levels inadequate for potent inhibition of FLT3-ITD.
Emergence of resistance
Initially, survival in the setting of TKI therapy probably happens in remote and unique niches within the bone marrow, and thus relies on the presence of a minute population of leukemia stem cells. The emergence of clinical resistance is most likely preceded by cell intrinsic events that allow the new clone to leave the “nest” and dominate the organism. Accumulating knowledge from studying the emergence of resistance to imatinib in CML point towards two types of resistance: a) mutation in the target receptor or b) activation of alternative pathways that by-pass the mutant receptor.
a) Mutations in the target receptor
A variety of FLT3 mutations that could confer resistance to FLT3 inhibitors have been predicted based on in vitro models of resistance. Some of these predicted mutations have been confirmed in patients relapsing with FLT3 mutated disease during treatment with FLT3 inhibitors.
Most FLT3 inhibitors are active against FLT3-ITD but have limited activity against TKD mutants. Even though a TKD mutation may have only minimal prognostic value when present at diagnosis, the appearance of point mutations in the FLT3 receptor is a major mechanism of resistance to FLT3 inhibitors. These point mutant can develop either in cis or in trans and newer FLT3 inhibitors have various degrees of activity against individual mutants19.
It is important to recognize that depending on the domain used to bind the receptor, FLT3 inhibitors can be segregated in two classes: type I inhibitors, like CEP-701, PKC-412 and crenolanib, bind to the “gatekeeper” domain adjacent to the activation loop or the ATP-binding domain; type II inhibitors, like Sorafenib, Quizartinib and MLN518, directly bind the ATP-binding domain. As expected, point mutants conferring resistance to one TKI show cross resistance within the class. To this end, patients with FLT3-ITD that relapse while treated with Quizartinib, if they are found to have point mutations in the activation loop (most frequent D835) or “gatekeeping” domain (i.e. F691) usually show resistance to Sorafenib, another type II TKI. Interestingly, these cells remain sensitive to type I TKIs such as PKC412 and crenolanib20. Similarly, some TKIs, like the type I inhibitor TTT-3002 demonstrate preclinical potential to target both type of mutations21.
b) Activation of alternative signaling pathways
While intensively studied, the accumulation of additional mutations in the FLT3 receptor represents a minority of cases developing resistance to FLT3 inhibitors. In a small study following 60 patients with FLT3-ITD alone treated with single agent TKI, two thirds of patients progressed on FLT3 inhibitor treatment even though they showed no additional mutations in FLT3 wt allele or FLT3-ITD. Only 22% of patients acquired additional mutations, all of them D835 or I83622. Thus, alternative mechanisms of resistance, independent of FLT3 receptor, must be playing a major role and recent studies have uncovered some of these pathways.
Generally, these pathways either provided survival signals independent of FLT3-ITD or they change the transcriptional factor network of the leukemic cell to a state where FLT3 signaling can be replaced by activation of other RTKs.
As mention FLT3-ITD can activate signaling cascades downstream of JAK/STAT, PI3K/AKT and MAPK pathways. Since blasts become addicted to this constitutively active signaling, FLT3 inhibitors induce rapid apoptosis. While microenvironmental factors may rescue these cells in the stem cell niche, development of cell intrinsic mechanisms that can protect these cells from apoptosis coincide with development of resistance to TKIs. FLT3-ITD changes the balance between anti-apoptotic proteins such as Bcl2/BclXL and pro-apoptotic BAD. Sustained activation of phospho-STAT5 by FLT3-ITD signaling, for instance, activates Pim kinases which in turn, by phosphorylating BAD, sequesters these proteins in the cytoplasm and allows anti-apoptotic activities of Bcl2 and BclXL23,24. Inhibition of FLT3-ITD results in rapid loss of phospho-STAT5 and downregulation of Pim-123. Cells resistant to FLT3-inhibitors show sustained activity of Pim-123 or Pim-225,26 and high levels of phospho-BAD and thus, protection from apoptosis. Thus, combined inhibitions of FLT3-ITD and Pim127 or Pim-226 are synergistic in inducing apoptosis in mutant blasts. Similarly, high levels of Bcl2 can also confer resistance to FLT3 inhibitors. In these settings the use of Bcl2 inhibitors such as ABT-737 (85) rescues FLT3 inhibitor – induced apoptosis of mutated cells. Interestingly, FLT3-ITD/TKD mutants that show sustained activation of phospho-STAT5 also exhibit elevated levels of anti-apoptotic signals mediated by BclXL28. In these models, inhibition of the mTOR pathway can rescue the sensitivity of these cells to both FLT3 inhibitors and anthracyclines28. Similarly, cells resistant to Sorafenib continue to have an active mTOR/PI3K/Akt pathway even in the presence of effective FLT3 inhibition29,30, and mTOR inhibitors can re-sensitize the blasts to TKI29. Some FLT3-ITD point mutations (D627E) can induce expression of Mcl-1 (a Bcl-2 family member) independent of kinase activity via a conformational change that favors Grb-2 docking31. Since Mcl-1, in addition to its anti-apoptotic roles, also impacts mitochondrial morphology and function32, it is not surprising that Sorafenib resistant cells adopt an abnormal mitochondrial respiratory chain and rely mostly on glycolysis for their energy demands33. Thus, glycolytic inhibitors like 2-deoxyglucose can re-sensitize cells to Sorafenib33. Of note, a major limitation to a predominantly glycolytic metabolism is a sustained drop in intracellular pH. Consistent with this concept, FLT3-ITD cells developing resistance to Sorafenib also upregulated tescalcin, a type I Na/H exchange channel. Downregulation of this protein or inhibition via amiloride reduced leukemia initiation in xenograft models of Sorafenib resistant FLT3-ITD AML34.
Maintaining an active MAPK/ERK pathway either by expression of constitutively Axl-135 or acquiring activating mutations in NRAS30 has also been shown to be potential mechanisms of resistance to FLT3 inhibitors.
Epigenetic events, particularly methylation of target genes have been proposed as potential mechanisms of resistance to FLT3 inhibitors. To this end, methylation of SHP-136 and silencing of SOCS proteins37 both negative regulators of JAK/STAT pathway have been implicated in resistance to FLT3-inhibitors. Treatment with DNMT inhibitors not only rescues expression of these proteins but also re-sensitizes the cells to the TKI36.
Strategies to prevent development of resistance or to sensitize cells to FLT3 inhibitors
Initial agents used to treat patients with FLT3-ITD had broader activity against multiple receptor tyrosine kinases but also less-than-ideal pharmacologic properties. Recently developed inhibitors are more specific and very potent. More targeted agents have fewer side effects and thus, higher doses can be used. On the other hand, agents that inhibit multiple RTKs may prevent emergence of resistance via these mechanisms. Plasma Inhibitory assay (PIA) directed studies have overcome some of the primary failure seen with single agent FLT3-inhibitors and helped optimize dosage for newer drugs.
Given the type of secondary mutations that can arise in the FLT3 receptor during treatment with FLT3 inhibitors, it was suggested that switching to a different class of inhibitor may prove beneficial in these patients. Ponatinib for instance is effective against gate keeper mutations (F691) that arise during treatment with Quizartinib but not against activation loop mutations (D835)38. In the same study, SAR302503, a dual Jak2/FLT3 inhibitor was highly effective in vitro against both types of mutants. On the same note, Crenolanib, a type I inhibitor is effective against FLT3-ITD expressing cells that became resistant to Sorafenib via accumulation of mutations in both activation loop39,40 and gate keeper mutations40. Similarly, G-749 a FLT3 inhibitor active against multiple activation loop as well as gate keeper mutations has shown great efficiency in xenograft models of FLT3-ITD AML that would be otherwise resistant to Quizartinib or PKC41241.
Though single agent FLT3 inhibitors can induce remission, the complete eradication of disease relies on combination therapy. There are a number of preclinical studies that investigate the efficiency of concomitant targeting of FLT3 signaling as well as pathways implicated in resistance. For instance, targeting the ERK/MAPK pathway either by inhibition of upstream RTK such as Axl135, inhibition of MEK42, MERTK43 or NRas30 with small molecules showed promising activity in preclinical models including xenografts of FLT3-ITD AML. This approach of multi kinase inhibition can likely by-pass the stromal protection against FLT3 inhibitors and decrease emergence of resistance12. To this end, a promising strategy is targeting Pim kinases (known to be upregulated in response to cytokines produced by the mesenchymal stroma). The combination of Pim1/Pim2 inhibitors with FLT3 inhibitors is active against FLT3-ITD AML in preclinical models26. Additionally, agents that target anti-apoptotic mechanisms important in FLT3-ITD signaling have shown activity in AML blasts in combination with FLT3 inhibitors. To this end, a dual inhibitor of Akt/FLT3-ITD, A674563, can overcome FLT3 ligand induced drug resistance in vitro and in xenograft models44. Similarly, mTOR inhibition can sensitize cells to FLT3 inhibition28 likely via targeting signaling downstream of PI3K29. An alternative approach that showed preclinical activity is to directly target Bcl2. In this regard, ABT-737 has shown synergistic effects with FLT3 inhibitors against FLT3-ITD AML45. Corroborated with preliminary data coming from clinical studies using Bcl2 inhibitors agents in other AML subtypes, it may be of interest to study the effects of FLT3 inhibitors in combination with Bcl2 inhibitors in clinical studies.
As mentioned above, a potential mechanism that is associated with resistance to FLT3 inhibitors relies on CDK4/6 activity and their impact on either cyclin D2/cyclin D3 or direct transcriptional activation of FLT3 and Pim kinases. Targeting this mechanism with either the dual CDK4/FLT3-ITD inhibitor, AMG92546,47 or by adding Palbociclib48, a CDK6 inhibitor showed promise to sensitize resistant cells to FLT3 inhibitors.
Since the survival of FLT3-ITD leukemia cells depends on absolute levels of FLT3-ITD, decreasing oncoprotein stability using Hsp90 inhibitors49 or activating autophagy via proteasome inhibitors like Bortezomib50 can also resensitize resistant FLT3-ITD AML cells to TKI. Lastly, in a small case series, concomitant treatment with CsA benefitted patients with FLT3-ITD AML who were being treated with FLT3 inhibitors, perhaps via inhibition of NFATc151. This mechanism of sensitization will need to be compared in larger patient studies before definitive conclusions can be drawn.
Our approach to resistant FLT3-ITD AML
In spite of all the potentially available strategies to overcome resistance to TKIs, the treatment of patients with FLT3-ITD AML relapsing while on FLT3 inhibitors remains a major clinical challenge. Our current strategy relies on enrollment in a clinical trial if available. If not available, one of the most promising approaches in our clinic is the use of the combination of FLT3 inhibitors (e.g., sorafenib) with hypomethylating agents. We prefer 5-azacitidine52,53 but other groups have shown similar results with decitabine. In patients receiving sorafenib and 5-azacitidine the leukemia undergoes differentiation. There is a gradual decrease of bone marrow blasts to the point where after three cycles 40-50% of these patients have achieved a morphological remission. To date, it remains unclear how 5-azacitidine or decitabine sensitizes FLT3-ITD cells to TKI but mechanisms may include re-expression of methylated genes such as SOCS1, SOCS2, SOCS337 or SHP-136 or even tumor de-bulking without the associated increased in FLT3 ligand seen with classical chemotherapy53. Lastly, it was suggested that DNMT inhibitors may sensitize cells to FLT3 inhibitors via their pro-differentiation effects52. Treatment with 5-azacitidine for instance, decreases total FLT3-ITD as cells differentiate and thus, make them more sensitive to a FLT3 inhibitor. To this end, differentiation agents such as homoharringtonine54 or all-trans retinoic acid55 have been shown to synergize with FLT3 inhibitors in inducing apoptosis in FLT3 mutated AML. To what extent these are viable approaches to not only control the bulk of the tumor but eliminate MRD and prevent resistance remains to be tested in clinical studies. Moreover, recent evidence suggests that some bone marrow niches may inactivate retinoids and thus, protect malignant cells from differentiation56-58.
For this reason, patients with FLT3-ITD that achieve remission after treatment with FLT3 inhibitor plus 5-azacitidine still go on to receive allogeneic transplantation in our center. In addition, it is our experience that treatment with a FLT3 inhibitor post-transplant helps maintain disease burden to undetectable levels. This may be mediated via both a direct effect on the leukemia clone as well as potential immune modulatory effect of FLT3 inhibitors. Nevertheless, many of these patients do experience various degrees of graft vs host symptoms. To what extent this approach will translate into a viable clinical option is currently being investigated in a BMT CTN clinical trial.
Key points.
Pharmacokinetics and pharmacodynamics don't always correlate and pharmacodynamics is a better predictor of efficacy when using FLT3 inhibitors.
FLT3 inhibitors are potent in clearing circulating blasts yet this does not always translate into similar effects on bone marrow blasts.
Single agent FLT3 inhibitors can induce remission without cure in FLT3-ITD AML but to eliminate the last bastion of minimal residual disease additional interventions are required.
Resistance to FLT3 inhibitors may be facilitated by signals from the microenvironment that allow survival of AML cells in the presence of FLT3 inhibitors.
Accumulation of additional mutations in the FLT3-ITD AML clone allows for emergence of clones with cell intrinsic resistance to FLT3 inhibitors.
Effective and complete elimination of the FLT3-ITD AML clones may be possible via a coordinated approach that targets not only signaling downstream of FLT3-ITD but also the microenvironment-dependent mechanisms of resistance and activate a potent and sustained immune response.
Acknowledgments
M.L. receives research funding from Novartis and Astellas. M.L. serves as a consultant for Novartis, Daiichi-Sankyo, Astellas, and Arog.
Footnotes
G.G. has nothing to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Gabriel Ghiaur, Assistant Professor of Oncology and Medicine, Johns Hopkins University, Sidney Kimmel Comprehensive Cancer Center, Division of Hematological Malignancies, Adult Leukemia Program, 650 Orleans St. CRB I, Room 243, Baltimore, MD, 21287, Ph: 1-410-502-3183, Fax: 1-410-614-7279
Mark Levis, Professor of Oncology and Medicine, Johns Hopkins University, Sidney Kimmel Comprehensive Cancer Center, Division of Hematological Malignancies, Adult Leukemia Program, 1650 Orleans St. CRB I, Room 2M44, Baltimore, MD, 21287, Ph: 1-410-502-3629, Fax: 1-410-614-1005
References
- 1.Levis M, Small D. FLT3: ITDoes matter in leukemia. Leukemia. 2003 Sep;17(9):1738–1752. doi: 10.1038/sj.leu.2403099. [DOI] [PubMed] [Google Scholar]
- 2.Kottaridis PD, Gale RE, Langabeer SE, Frew ME, Bowen DT, Linch DC. Studies of FLT3 mutations in paired presentation and relapse samples from patients with acute myeloid leukemia: implications for the role of FLT3 mutations in leukemogenesis, minimal residual disease detection, and possible therapy with FLT3 inhibitors. Blood. 2002 Oct 1;100(7):2393–2398. doi: 10.1182/blood-2002-02-0420. [DOI] [PubMed] [Google Scholar]
- 3.Schnittger S, Schoch C, Dugas M, et al. Analysis of FLT3 length mutations in 1003 patients with acute myeloid leukemia: correlation to cytogenetics, FAB subtype, and prognosis in the AMLCG study and usefulness as a marker for the detection of minimal residual disease. Blood. 2002 Jul 1;100(1):59–66. doi: 10.1182/blood.v100.1.59. [DOI] [PubMed] [Google Scholar]
- 4.Thiede C, Steudel C, Mohr B, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. 2002 Jun 15;99(12):4326–4335. doi: 10.1182/blood.v99.12.4326. [DOI] [PubMed] [Google Scholar]
- 5.Kayser S, Schlenk RF, Londono MC, et al. Insertion of FLT3 internal tandem duplication in the tyrosine kinase domain-1 is associated with resistance to chemotherapy and inferior outcome. Blood. 2009 Sep 17;114(12):2386–2392. doi: 10.1182/blood-2009-03-209999. [DOI] [PubMed] [Google Scholar]
- 6.Rosnet O, Buhring HJ, deLapeyriere O, et al. Expression and signal transduction of the FLT3 tyrosine kinase receptor. Acta haematologica. 1996;95(3-4):218–223. doi: 10.1159/000203881. [DOI] [PubMed] [Google Scholar]
- 7.Carow CE, Levenstein M, Kaufmann SH, et al. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood. 1996 Feb 1;87(3):1089–1096. [PubMed] [Google Scholar]
- 8.Pratz KW, Sato T, Murphy KM, Stine A, Rajkhowa T, Levis M. FLT3-mutant allelic burden and clinical status are predictive of response to FLT3 inhibitors in AML. Blood. 2010 Feb 18;115(7):1425–1432. doi: 10.1182/blood-2009-09-242859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012 Jan 11;481(7382):506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith BD, Levis M, Beran M, et al. Single-agent CEP-701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004 May 15;103(10):3669–3676. doi: 10.1182/blood-2003-11-3775. [DOI] [PubMed] [Google Scholar]
- 11.Sexauer A, Perl A, Yang X, et al. Terminal myeloid differentiation in vivo is induced by FLT3 inhibition in FLT3/ITD AML. Blood. 2012 Nov 15;120(20):4205–4214. doi: 10.1182/blood-2012-01-402545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang X, Sexauer A, Levis M. Bone marrow stroma-mediated resistance to FLT3 inhibitors in FLT3-ITD AML is mediated by persistent activation of extracellular regulated kinase. Br J Haematol. 2014 Jan;164(1):61–72. doi: 10.1111/bjh.12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kojima K, McQueen T, Chen Y, et al. p53 activation of mesenchymal stromal cells partially abrogates microenvironment-mediated resistance to FLT3 inhibition in AML through HIF-1alpha-mediated down-regulation of CXCL12. Blood. 2011 Oct 20;118(16):4431–4439. doi: 10.1182/blood-2011-02-334136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Traer E, Martinez J, Javidi-Sharifi N, et al. FGF2 from Marrow Microenvironment Promotes Resistance to FLT3 Inhibitors in Acute Myeloid Leukemia. Cancer Res. 2016 Sep 26; doi: 10.1158/0008-5472.CAN-15-3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen F, Ishikawa Y, Akashi A, Naoe T, Kiyoi H. Co-expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget. 2016 Jun 17; doi: 10.18632/oncotarget.10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang L, Wang J, Blaser BW, et al. Pharmacologic inhibition of CDK4/6: mechanistic evidence for selective activity or acquired resistance in acute myeloid leukemia. Blood. 2007 Sep 15;110(6):2075–2083. doi: 10.1182/blood-2007-02-071266. [DOI] [PubMed] [Google Scholar]
- 17.Parmar A, Marz S, Rushton S, et al. Stromal niche cells protect early leukemic FLT3-ITD+ progenitor cells against first-generation FLT3 tyrosine kinase inhibitors. Cancer Res. 2011 Jul 1;71(13):4696–4706. doi: 10.1158/0008-5472.CAN-10-4136. [DOI] [PubMed] [Google Scholar]
- 18.Alonso S, Su M, Jones JW, et al. Human bone marrow niche chemoprotection mediated by cytochrome P450 enzymes. Oncotarget. 2015 Jun 20;6(17):14905–14912. doi: 10.18632/oncotarget.3614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grundler R, Thiede C, Miething C, Steudel C, Peschel C, Duyster J. Sensitivity toward tyrosine kinase inhibitors varies between different activating mutations of the FLT3 receptor. Blood. 2003 Jul 15;102(2):646–651. doi: 10.1182/blood-2002-11-3441. [DOI] [PubMed] [Google Scholar]
- 20.Zhang W, Gao C, Konopleva M, et al. Reversal of acquired drug resistance in FLT3-mutated acute myeloid leukemia cells via distinct drug combination strategies. Clinical cancer research : an official journal of the American Association for Cancer Research. 2014 May 1;20(9):2363–2374. doi: 10.1158/1078-0432.CCR-13-2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma HS, Nguyen B, Duffield AS, et al. FLT3 kinase inhibitor TTT-3002 overcomes both activating and drug resistance mutations in FLT3 in acute myeloid leukemia. Cancer Res. 2014 Sep 15;74(18):5206–5217. doi: 10.1158/0008-5472.CAN-14-1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alvarado Y, Kantarjian HM, Luthra R, et al. Treatment with FLT3 inhibitor in patients with FLT3-mutated acute myeloid leukemia is associated with development of secondary FLT3-tyrosine kinase domain mutations. Cancer. 2014 Jul 15;120(14):2142–2149. doi: 10.1002/cncr.28705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kim KT, Baird K, Ahn JY, et al. Pim-1 is up-regulated by constitutively activated FLT3 and plays a role in FLT3-mediated cell survival. Blood. 2005 Feb 15;105(4):1759–1767. doi: 10.1182/blood-2004-05-2006. [DOI] [PubMed] [Google Scholar]
- 24.Kim KT, Levis M, Small D. Constitutively activated FLT3 phosphorylates BAD partially through pim-1. Br J Haematol. 2006 Sep;134(5):500–509. doi: 10.1111/j.1365-2141.2006.06225.x. [DOI] [PubMed] [Google Scholar]
- 25.Adam M, Pogacic V, Bendit M, et al. Targeting PIM kinases impairs survival of hematopoietic cells transformed by kinase inhibitor-sensitive and kinase inhibitor-resistant forms of Fms-like tyrosine kinase 3 and BCR/ABL. Cancer Res. 2006 Apr 1;66(7):3828–3835. doi: 10.1158/0008-5472.CAN-05-2309. [DOI] [PubMed] [Google Scholar]
- 26.Green AS, Maciel TT, Hospital MA, et al. Pim kinases modulate resistance to FLT3 tyrosine kinase inhibitors in FLT3-ITD acute myeloid leukemia. Science advances. 2015 Sep;1(8):e1500221. doi: 10.1126/sciadv.1500221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fathi AT, Arowojolu O, Swinnen I, et al. A potential therapeutic target for FLT3-ITD AML: PIM1 kinase. Leuk Res. 2012 Feb;36(2):224–231. doi: 10.1016/j.leukres.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bagrintseva K, Geisenhof S, Kern R, et al. FLT3-ITD-TKD dual mutants associated with AML confer resistance to FLT3 PTK inhibitors and cytotoxic agents by overexpression of Bcl-x(L) Blood. 2005 May 1;105(9):3679–3685. doi: 10.1182/blood-2004-06-2459. [DOI] [PubMed] [Google Scholar]
- 29.Lindblad O, Cordero E, Puissant A, et al. Aberrant activation of the PI3K/mTOR pathway promotes resistance to sorafenib in AML. Oncogene. 2016 Sep 29;35(39):5119–5131. doi: 10.1038/onc.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Piloto O, Wright M, Brown P, Kim KT, Levis M, Small D. Prolonged exposure to FLT3 inhibitors leads to resistance via activation of parallel signaling pathways. Blood. 2007 Feb 15;109(4):1643–1652. doi: 10.1182/blood-2006-05-023804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breitenbuecher F, Markova B, Kasper S, et al. A novel molecular mechanism of primary resistance to FLT3-kinase inhibitors in AML. Blood. 2009 Apr 23;113(17):4063–4073. doi: 10.1182/blood-2007-11-126664. [DOI] [PubMed] [Google Scholar]
- 32.Perciavalle RM, Stewart DP, Koss B, et al. Anti-apoptotic MCL-1 localizes to the mitochondrial matrix and couples mitochondrial fusion to respiration. Nature cell biology. 2012 Apr 29;14(6):575–583. doi: 10.1038/ncb2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang A, Ju HQ, Liu K, et al. Metabolic alterations and drug sensitivity of tyrosine kinase inhibitor resistant leukemia cells with a FLT3/ITD mutation. Cancer letters. 2016 Jul 28;377(2):149–157. doi: 10.1016/j.canlet.2016.04.040. [DOI] [PubMed] [Google Scholar]
- 34.Man CH, Lam SS, Sun MK, et al. A novel tescalcin-sodium/hydrogen exchange axis underlying sorafenib resistance in FLT3-ITD+ AML. Blood. 2014 Apr 17;123(16):2530–2539. doi: 10.1182/blood-2013-07-512194. [DOI] [PubMed] [Google Scholar]
- 35.Park IK, Mundy-Bosse B, Whitman SP, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia. 2015 Dec;29(12):2382–2389. doi: 10.1038/leu.2015.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Jamal HA, Mat Jusoh SA, Hassan R, Johan MF. Enhancing SHP-1 expression with 5-azacytidine may inhibit STAT3 activation and confer sensitivity in lestaurtinib (CEP-701)-resistant FLT3-ITD positive acute myeloid leukemia. BMC cancer. 2015 Nov 07;15:869. doi: 10.1186/s12885-015-1695-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou J, Bi C, Janakakumara JV, et al. Enhanced activation of STAT pathways and overexpression of survivin confer resistance to FLT3 inhibitors and could be therapeutic targets in AML. Blood. 2009 Apr 23;113(17):4052–4062. doi: 10.1182/blood-2008-05-156422. [DOI] [PubMed] [Google Scholar]
- 38.Kesarwani M, Huber E, Azam M. Overcoming AC220 resistance of FLT3-ITD by SAR302503. Blood cancer journal. 2013 Aug 30;3:e138. doi: 10.1038/bcj.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Galanis A, Ma H, Rajkhowa T, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance-conferring point mutants. Blood. 2014 Jan 2;123(1):94–100. doi: 10.1182/blood-2013-10-529313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zimmerman EI, Turner DC, Buaboonnam J, et al. Crenolanib is active against models of drug-resistant FLT3-ITD-positive acute myeloid leukemia. Blood. 2013 Nov 21;122(22):3607–3615. doi: 10.1182/blood-2013-07-513044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee HK, Kim HW, Lee IY, et al. G-749, a novel FLT3 kinase inhibitor, can overcome drug resistance for the treatment of acute myeloid leukemia. Blood. 2014 Apr 3;123(14):2209–2219. doi: 10.1182/blood-2013-04-493916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang W, Borthakur G, Gao C, et al. The Dual MEK/FLT3 Inhibitor E6201 Exerts Cytotoxic Activity against Acute Myeloid Leukemia Cells Harboring Resistance-Conferring FLT3 Mutations. Cancer Res. 2016 Mar 15;76(6):1528–1537. doi: 10.1158/0008-5472.CAN-15-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minson KA, Smith CC, DeRyckere D, et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI insight. 2016 Mar;1(3):e85630. doi: 10.1172/jci.insight.85630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang A, Wu H, Chen C, et al. Dual inhibition of AKT/FLT3-ITD by A674563 overcomes FLT3 ligand-induced drug resistance in FLT3-ITD positive AML. Oncotarget. 2016 May 17;7(20):29131–29142. doi: 10.18632/oncotarget.8675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kohl TM, Hellinger C, Ahmed F, et al. BH3 mimetic ABT-737 neutralizes resistance to FLT3 inhibitor treatment mediated by FLT3-independent expression of BCL2 in primary AML blasts. Leukemia. 2007 Aug;21(8):1763–1772. doi: 10.1038/sj.leu.2404776. [DOI] [PubMed] [Google Scholar]
- 46.Keegan K, Li C, Li Z, et al. Preclinical evaluation of AMG 925, a FLT3/CDK4 dual kinase inhibitor for treating acute myeloid leukemia. Molecular cancer therapeutics. 2014 Apr;13(4):880–889. doi: 10.1158/1535-7163.MCT-13-0858. [DOI] [PubMed] [Google Scholar]
- 47.Li C, Liu L, Liang L, et al. AMG 925 is a dual FLT3/CDK4 inhibitor with the potential to overcome FLT3 inhibitor resistance in acute myeloid leukemia. Molecular cancer therapeutics. 2015 Feb;14(2):375–383. doi: 10.1158/1535-7163.MCT-14-0388. [DOI] [PubMed] [Google Scholar]
- 48.Uras IZ, Walter GJ, Scheicher R, et al. Palbociclib treatment of FLT3-ITD+ AML cells uncovers a kinase-dependent transcriptional regulation of FLT3 and PIM1 by CDK6. Blood. 2016 Jun 9;127(23):2890–2902. doi: 10.1182/blood-2015-11-683581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu C, Kancha RK, Duyster J. Targeting oncoprotein stability overcomes drug resistance caused by FLT3 kinase domain mutations. PloS one. 2014;9(5):e97116. doi: 10.1371/journal.pone.0097116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Larrue C, Saland E, Boutzen H, et al. Proteasome inhibitors induce FLT3-ITD degradation through autophagy in AML cells. Blood. 2016 Feb 18;127(7):882–892. doi: 10.1182/blood-2015-05-646497. [DOI] [PubMed] [Google Scholar]
- 51.Metzelder SK, Michel C, von Bonin M, et al. NFATc1 as a therapeutic target in FLT3-ITD-positive AML. Leukemia. 2015 Jul;29(7):1470–1477. doi: 10.1038/leu.2015.95. [DOI] [PubMed] [Google Scholar]
- 52.Chang E, Ganguly S, Rajkhowa T, Gocke CD, Levis M, Konig H. The combination of FLT3 and DNA methyltransferase inhibition is synergistically cytotoxic to FLT3/ITD acute myeloid leukemia cells. Leukemia. 2016 May;30(5):1025–1032. doi: 10.1038/leu.2015.346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ravandi F, Alattar ML, Grunwald MR, et al. Phase 2 study of azacytidine plus sorafenib in patients with acute myeloid leukemia and FLT-3 internal tandem duplication mutation. Blood. 2013 Jun 6;121(23):4655–4662. doi: 10.1182/blood-2013-01-480228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu G, Mao L, Liu H, Yang M, Jin J, Qian W. Sorafenib in combination with low-dose-homoharringtonine as a salvage therapy in primary refractory FLT3-ITD-positive AML: a case report and review of literature. International journal of clinical and experimental medicine. 2015;8(11):19891–19894. [PMC free article] [PubMed] [Google Scholar]
- 55.Ma HS, Greenblatt SM, Shirley CM, et al. All-trans retinoic acid synergizes with FLT3 inhibition to eliminate FLT3/ITD+ leukemia stem cells in vitro and in vivo. Blood. 2016 Jun 9;127(23):2867–2878. doi: 10.1182/blood-2015-05-646786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alonso S, Hernandez D, Chang YT, et al. Hedgehog and retinoid signaling alters multiple myeloma microenvironment and generates bortezomib resistance. The Journal of clinical investigation. 2016 Oct 24; doi: 10.1172/JCI88152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghiaur G, Yegnasubramanian S, Perkins B, Gucwa JL, Gerber JM, Jones RJ. Regulation of human hematopoietic stem cell self-renewal by the microenvironment's control of retinoic acid signaling. Proceedings of the National Academy of Sciences of the United States of America. 2013 Oct 1;110(40):16121–16126. doi: 10.1073/pnas.1305937110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Su M, Alonso S, Jones JW, et al. All-Trans Retinoic Acid Activity in Acute Myeloid Leukemia: Role of Cytochrome P450 Enzyme Expression by the Microenvironment. PloS one. 2015;10(6):e0127790. doi: 10.1371/journal.pone.0127790. [DOI] [PMC free article] [PubMed] [Google Scholar]