

Abstract
The parathyroid glands are critical to maintaining calcium homeostasis through actions of parathyroid hormone (PTH). Recent clinical and molecular research has shown that direct and indirect actions of PTH also affect the heart and vasculature through downstream actions of G protein-coupled receptors in the myocardium and endothelial cells. Patients with disorders of the parathyroid gland have higher incidences of hypertension, arrhythmias, left ventricular hypertrophy, heart failure, and calcific disease which translate into increased cardiac morbidity and mortality. Importantly, clinical research also suggests that early treatment of parathyroid disorders through medical or surgical management may reverse cardiovascular remodeling and mitigate cardiac risk factors.
Keywords: parathyroid gland, parathyroid hormone, PTH, hypoparathyroidism, hyperparathyroidism
Introduction
The parathyroid glands are unique organs responsible for maintaining the critical function of calcium homeostasis. There are commonly four parathyroid glands that weigh approximately 40 grams each and are generally located posterior and inferior to the thyroid in the neck.1 These organs secrete parathyroid hormone (PTH), which controls calcium regulation. Secretion of PTH is modulated not only by serum calcium but also phosphorus and vitamin D through negative and positive feedback loops. In the bone, PTH binds to PTH type 1 receptors (PTH1R) to assist with calcium resorption. In the kidney, PTH acts to increase renal calcium, decrease phosphate reabsorption, and activate metabolism of vitamin D. In the intestine, PTH transcriptionally upregulates 1 alpha hydroxylase, leading to increased production of 1,25-dihydroxyvitamin D, which in turn enhances calcium and phosphorus reabsorption.2 These actions of PTH on the bones, kidneys, and intestines are a careful orchestration of interrelated processes driven by feedback loops. Subsequently, excessive or insufficient secretion of PTH can lead to disruption of these loops and, in turn, alterations in calcium homeostasis.
Both the direct action of PTH on the heart and alterations of calcium homeostasis (e.g., hypercalcemia or hypocalcemia) comprise the two primary mechanisms by which diseases of the parathyroid affect the cardiovascular system. In recent years, clinical and molecular research has bolstered awareness of several cardiovascular complications that are associated with parathyroid disorders—namely, hypertension, arrhythmias, heart failure, and calcific disease of vessels and valves.2 This review evaluates current studies and relationships between parathyroid disease and the cardiovascular system and highlights the important implications for mortality and morbidity stemming from these disorders.
Molecular Actions of PTH on the Cardiovascular System
Recent studies indicate that cardiomyocyte and smooth muscle physiology is impacted by PTH.3 Structurally, PTH is a peptide hormone composed of 84 amino acids, with the biologically active portion comprising the first 34.4 This moiety binds to target organ PTH1R, which is a G-protein-coupled receptor with seven transmembrane domains. Depending on the target organ, subsequent activation of this receptor usually involves either downstream activation of adenylyl cyclase and protein kinase A pathway or phospholipase C/protein kinase C (PKC) pathway (Figure 1).4
Figure 1.
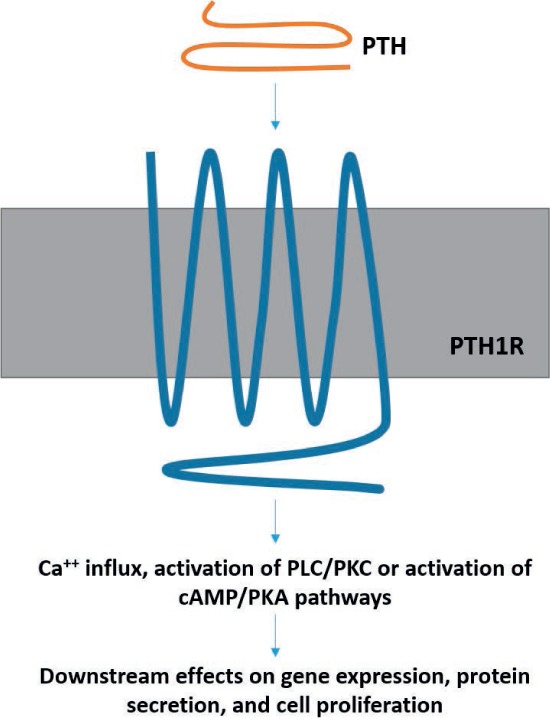
PTH binds to the 7-transmembrane PTH1R, which is a GPCR. Activation of this receptor can activate cAMP/PKA or PLC/PKC cascades depending on the end organ. In the heart, the PKC pathway is activated and leads to downstream effects such as cell proliferation/hypertrophy and increased chronotropy. In vascular smooth muscle cells, PTH activation of cAMP cascade decreases Ca++ influx and leads to vasodilation. PTH: pituitary thyroid hormone; GPCR: G-protein coupled receptors; cAMP: cyclic adenosine monophosphate; PKA: protein kinase A; PLC: phospholipase C; PKC: protein kinase C
In adult cardiomyocytes, PTH binding activates G-protein signaling and subsequent calcium influx into cardiac cells.3 This calcium influx, however, does not lead to direct contractile effects on cardiac myocytes but is thought to trigger several indirect effects on the myocardium. These include activation of PKC, which may attenuate contractility by interfering with B-adrenoceptor stimulation.3 Additionally, PKC's downstream actions on gene expression, protein secretion, and cell proliferation are thought to cause excessive growth and myocardial hypertrophy. Parathyroid hormone also exerts unique effects on the vascular system. Animal and molecular studies have revealed that PTH decreases calcium influx in vascular smooth muscle cells, leading to cAMP-dependent inhibition of L-type calcium channels and subsequent vasodilation.3 Thus, while the main function of PTH is to maintain calcium homeostasis, it has important effects on the cardiovascular system that likely contribute to various cardiac pathophysiologies when this hormonal pathway is disturbed.
Hypercalcemia and Heart Disease
Primary Hyperparathyroidism and Cardiac Mortality
One of the more common disorders of the parathyroid glands is primary hyperparathyroidism (PHPT), an overproduction of PTH that subsequently leads to hypercalcemia. This is most commonly due to a solitary parathyroid adenoma, but about 15% of cases can be caused by diffuse hyperplasia of the glands.5 While the typical complications and symptoms of PHPT are well known (e.g., nephrolithiasis, osteoporosis, constipation, and weakness), cardiovascular complications are increasingly gaining recognition. Indeed, patients with symptomatic PHPT have increased mortality due to myocardial infarction, stroke, and other cardiovascular causes, and they also have increased all-cause mortality.5 One mortality study looked at 4,461 patients who underwent parathyroidectomy between 1987 and 1994. The study found PHPT to have an increased risk ratio of 1.71 in men and 1.85 in women for both all-cause mortality and cardiovascular death. This suggests that long-standing PHPT may still carry significant mortality risk even after treatment with parathyroidectomy.5 However, with increasing recognition of PHPT at early and asymptomatic stages of disease, it is likely that intervention can reduce mortality in this population, although further studies are necessary. One prospective study of 123 people by Stefenelli et al. showed regression of left ventricular hypertrophy (LVH) after parathyroidectomy and 41 months of normal calcium and PTH levels. If correction of PHPT can reverse structural cardiac changes, this may indicate that the mortality and morbidity risk associated with the initial changes can be mitigated as well.
Exactly how does PHPT lead to cardiac disease and cardiovascular events? Several complications are thought to develop from sustained elevated PTH and calcium levels, including hypertension, cardiac arrhythmias, LVH, and valvular calcific disease (Table 1). These cardiac abnormalities and their relation to PHPT are reviewed in the following sections.
Table 1.
Proposed mechanisms of cardiac disease in parathyroid disorders. PTH: parathyroid hormone; RAAS: renin-angiotensin-aldosterone system; PTH1R: PTH type 1 receptors; PHPT: primary hyperparathyroidism

Primary Hyperparathyroidism and Hypertension
One of the ways in which PHPT may increase cardiovascular mortality is by contributing to the development of hypertension. Both in vitro and in vivo studies have demonstrated that PTH causes vasodilation by binding to vascular smooth muscle PTH1R.5 However, excess PTH secretion (as in PHPT) is associated with paradoxical blood pressure elevation. The exact mechanism of this phenomenon is not well understood. However, one hypothesis is that elevated PTH levels cause changes in the vascular endothelium that alter vasodilatory properties. This may be caused by PTH-stimulated increases in endothelin-1 and IL-6, which in turn stimulate smooth muscle secretion of collagen and β-1 integrin, leading to vascular remodeling.6 Another hypothesis is that hypercalcemia not only augments catecholamine release but also increases arterial response to catecholamines.7 For example, an older study by Vlachakis et al. found that patients with PHPT and normal renal function had higher levels of basal catecholamines as well as higher increases in mean arterial pressures after infusions of norepinephrine when compared to normotensive individuals. Other mechanisms point to increased atherosclerotic disease in PHPT driven directly by long-term severe hypercalcemia.5
Another important suggested mechanism underlying hypertensive disease in PHPT involves the complex interaction between PTH and the renin-angiotensin-aldosterone system (RAAS). There is increasing evidence of a bidirectional relationship between PTH and RAAS, whereby increased PTH leads to subsequent activation of RAAS (and therefore increased aldosterone levels) and vice versa.8 The proposed underlying pathophysiology involves direct PTH stimulation of zona glomerulosa cells when binding to PTH1R, which leads to a relative excess of aldosterone. This excess leads to renal and gastrointestinal calcium loss that in turn stimulates PTH secretion. Other observational studies have noted this relationship, whereby higher aldosterone concentrations are associated with higher serum PTH, and use of RAAS inhibitors such as angiotensin-converting enzyme inhibitor and angiotensin II receptor blockers are associated with lower PTH levels.8 Thus, while it may seem that treating PHPT patients with mineralocorticoid inhibitors such as eplerenone would mitigate this cycle and potentially lead to cardiovascular benefits, such studies have not yet been conducted. However, some studies are currently underway, including the Effects of Eplerenone in Patients with Primary Hyperparathyroidism (EPATH) trial, which will hopefully shed further light on the intervention implications of this bidirectional relationship.
A few epidemiologic and prospective studies have shown an increased incidence of hypertension in patients with PHPT.6 In a prospective observational study by Yao et al., increased PTH levels (defined as > 65 pg/mL) in blacks were associated with an increased risk of incident hypertension with a median follow-up of 6 years. However, other studies of PHPT have not found PTH to be an independent risk factor for hypertension. In a study by Veheyen et al. examining a cohort of patients with PHPT, PTH levels were not correlated with nocturnal hypertension. However, the angiotensin-renin ratio was independently predictive of higher nocturnal blood pressure. Again, such studies indicate that while PHPT has strong associations with hypertension, it is unclear if these are related directly to PTH or other physiological results of PHPT. Thus, further studies are required to elucidate the likely multifactorial etiologies and relationship between hypertension and PHPT.
Primary Hyperparathyroidism and Left Ventricular Hypertrophy
Left ventricular hypertrophy is an important predictor of cardiac morbidity and mortality, and its association with PHPT has grown stronger in recent years.5,9 Indeed, LVH is thought to be the most common pathology seen in patients with PHPT, independent of hypertension.9 In vitro studies suggest that this is possibly driven by the direct effects of PTH on cardiac PTH1R. As mentioned before, PTH activation of these receptors leads to increased intracellular calcium and subsequent upregulation of PKC, which activates hypertrophic development of cardiac myocytes.
Multiple studies have discovered an increased association between PHPT and LVH as confirmed with cardiac imaging and even regression of LVH after parathyroidectomy. In a prospective study by Stefenelli et al., 119 patients with PHPT underwent parathyroidectomy with baseline measurements of LV mass pre- and postoperatively. Of these patients, 81.6% had hypertrophy of the interventricular septum and 78.2% had hypertrophy of the posterior walls. Interestingly, at 41 months post-parathyroidectomy, the thickness of the interventricular and posterior walls was reduced on average by 0.62 mm and 2.21 mm, respectively. This was independent of changes in blood pressure. Furthermore, patients who were normotensive experienced significant reductions in LVH (1.29-mm and 2.32-mm reductions in the interventricular and posterior walls, respectively). Smaller studies have produced similar results, showing increased LVH at baseline in patients with PHPT controlled for age, blood pressure, and gender.5 Additionally, regression of LVH may be long-term and has been shown to persist between 4 and 5 years post-parathyroidectomy.9 While such studies support the notion that patients with PHPT, especially those who are normotensive at baseline, benefit from parathyroidectomy, other studies have been less conclusive. For example, in a study by Walker et al., patients with mild PHPT did not have evidence of increased LV mass. Thus, it remains unclear if earlier intervention in patients with PHPT results in significant cardiac mortality and morbidity benefits. Ultimately, larger studies that evaluate all-cause and cardiovascular mortality in patients with LVH and PHPT are required to better define the association between the two and the timing of intervention.
Primary Hyperparathyroidism and Heart Failure
While the direct relationship between heart failure (HF) and PHPT has not been studied extensively, several observational and population studies have discovered an association between elevated PTH and HF.10 Again, this relationship may be attributed to the direct effects of PTH on cardiac myocytes, endothelial cells, and vascular smooth muscles.10 However, a few prospective studies have demonstrated a temporal relationship between PTH and incident HF. In one of these studies, 3,731 men aged 60 through 79 years with no established cardiovascular disease were followed for 13 years. The study demonstrated a higher risk of incident HF with elevated PTH levels (defined as ≥ 55.6 pg/mL).11 This higher risk was seen even after the adjustment of other factors such as hypertension, renal dysfunction, and lung function. Interestingly, this correlation was independent of other markers of mineral metabolism such as calcium, phosphate, and 25(OH)D levels. Another prospective cohort study by Bansal et al. (MESA: Multi-Ethnic Study of Atherosclerosis) examined 6,459 participants aged 45 through 84 and with no prior CVD. The study found a 50% greater risk of incident HF in participants with PTH levels ≥ 65 pg/mL compared to those with PTH levels < 65 pg/mL. Interestingly, the association remained statistically significant after adjustment for patient characteristics, showing 44% greater incident risk of heart failure in participants with PTH levels ≥ 65 pg/mL. This association was especially strong in blacks.10 HF is an important predictor of cardiac morbidity and mortality, and there is increasing evidence that elevated PTH levels are an independent risk factor for incident HF. While many observational studies have documented this association, more prospective studies of diverse population groups are warranted to better characterize this connection.
It is worthwhile mentioning the independent relationship between vitamin D deficiency and HF. An estimated 90% of patients with HF have vitamin D deficiency, which is associated with increased incident chronic HF and more severe HF.12 The proposed mechanisms behind this effect include adverse cardiomyocyte remodeling due to interference with calcium transport and subsequent increase in hypertrophy, inflammation, and fibrosis. Another mechanism likely involves the increased RAAS activity seen with vitamin D deficiency.12 The recent VINDICATE study was a randomized, placebo-controlled, double-blind trial looking at vitamin D supplementation in patients with hypovitaminosis D and chronic HF on optimal medical therapy. While the primary end point of improved functionality based on a 6-minute walk test was not significant with vitamin D supplementation at 1 year, there was significant improvement in cardiac function.12 The improvement in cardiac function included increases in LVEF as well as decreases in LV end diastolic and systolic diameters. Thus, correction of vitamin D deficiency may reverse cardiac remodeling and would be an important therapeutic intervention to consider in this patient population.
Primary Hyperparathyroidism and Calcific Disease
Unsurprisingly, chronic hypercalcemia increases calcium deposition not only in cardiac myocardium but also in cardiac valves. Several studies have shown a higher incidence of calcific heart disease in patients with PHPT and secondary HPT, with reports showing up to 78% involvement of either myocardium or valves.5 Calcium deposition has been identified in valvular annuli and cusps, coronary artery media and intima, individual myocardial fibers, and the interventricular septum.9 More commonly, calcific valvular disease involves the mitral and aortic valves. Tricuspid valvular calcification is more uncommon and, if detected, could indicate the presence of parathyroid disease. Additionally, conditions such as bicuspid aortic valves, which are more prone to calcification in general, are even more susceptible to accelerated calcific disease in patients with PHPT. In younger patients especially, it has been suggested that the initial presentation of PHPT could be valvular stenosis from calcific disease; therefore, it should be an important consideration in the differential diagnosis.9
In a prospective study by Stefenelli et al., echocardiogram was used to determine calcific disease in 123 patients. The study found 46% of patients with aortic valvular calcifications, 39% with mitral calcifications, and 74% with myocardial deposits. Additionally, the patients were followed for 12 and 41 months after parathyroidectomy, with repeat echocardiograms showing no progression of valvular calcific disease.13 Importantly, while similar studies have reproduced the higher prevalence of calcific disease in patients with PHPT, none have been able to establish prognostic variables to correlate severity of PHPT (based on laboratory values) with the extent of calcific disease in an individual patient. However, results from the Stefenelli et al. study suggest that intervention in patients with PHPT can potentially halt progression of valvular disease, which carries its own risks of cardiac morbidity and mortality.
Secondary Hyperparathyroidism and Cardiovascular Risk
Secondary hyperparathyroidism (SHPT) is seen early in chronic kidney disease (CKD) and is almost always present in ESRD. While the exact sequence of events leading to SHPT is not definitively established, it is generally thought to be driven early on by disturbances in renal phosphate handling and by the more recently discovered bone-derived fibroblast growth factor 23 (FGF23) (Figure 2). Studies have demonstrated that FGF23 levels rise early in CKD.14,15 Although the exact stimulus for this increase is unclear, it is evident that elevated FGF23 acts on proximal renal tubules to inhibit 1α-hydroxylase, which decreases 1,25-dihydroxyvitamin D.14 This decrease in 1,25-dihydroxyvitamin D disrupts feedback inhibition on the parathyroid gland and stimulates PTH secretion. Meanwhile, as CKD progresses, phosphate retention serves as another stimulus that leads to PTH secretion. In fact, even small decreases in calcium levels caused by these processes are enough to stimulate the parathyroid to secrete PTH.14 As CKD progresses, the chronic stimulation of the parathyroid by these various factors ultimately leads to parathyroid hyperplasia. This hyperplasia is associated with decreased expression of calcium-sensing receptors and resistance to feedback not only from FGF23 but also calcium and 1,25-dihydroxyvitamin D.16 Ultimately, the unregulated secretion of PTH results in hypercalcemia and hyperphosphatemia that are characteristic of CKD and ESRD and that contribute to the cardiovascular risk in these patients. Interestingly, newer studies also implicate FGF23 as an independent contributor to cardiac risk, but further discussion is outside of the scope of this review.15
Figure 2.
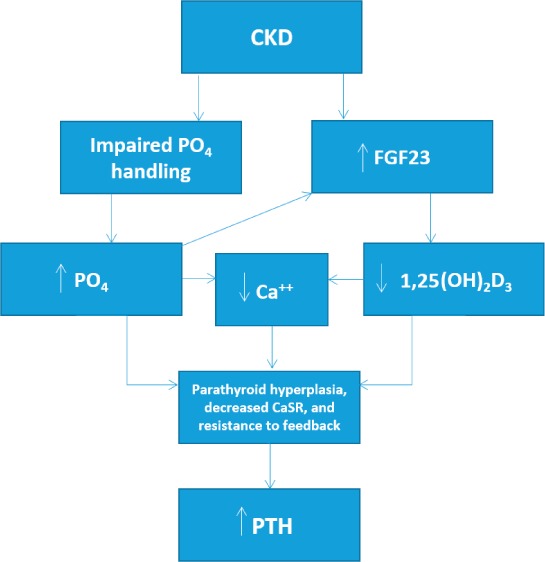
Simplified pathophysiology of secondary hyperparathyroidism in chronic kidney disease (CKD) leading to elevated parathyroid hormone (PTH) levels, which leads to downstream effects on the cardiovascular (CV) system and increased CV mortality in this patient population. PO4: plasma phosphate; FGF23: fibroblast growth factor 23; CaSR: calcium-sensing receptor; 1,25(OH)2D3: 1,25-dihydroxyvitamin D
Secondary hyperparathyroidism is an important contributor to cardiovascular mortality in ESRD and CKD, especially in more advanced stages. Indeed, in ESRD patients, the 5-year mortality is as high as 50%, with CVD as the leading cause of death, and is not explained solely by traditional risk factors such as age, diabetes, and smoking.17,18 Multiple cohort studies of ESRD patients have confirmed an association between high PTH levels (typically > 600 pg/mL) and increased mortality.17 Recent studies have also shown increased CV mortality in earlier stages of CKD. For example, in one retrospective cohort study looking at 196 veterans with CKD stages 3 and 4, there was an association between elevated intact PTH and CV events (e.g., myocardial infarction, cerebrovascular accident, revascularization procedures).18 While the exact mechanistic relationship between SHPT and increased CV mortality is unknown, it is likely multifactorial. The relationships seen with PHPT and hypertension, LVH, HF, calcific disease, and arrhythmias are likely contributors to the high mortality from CV events seen in this patient population.18
Unlike in PHPT, the management of SHPT is more nuanced. For example, just as very high iPTH levels are associated with increased mortality, observational studies suggest that very low iPTH levels (< 100 pg/mL) are also associated with higher mortality.17 This is likely related to concurrent factors such as uncontrolled diabetes, poor nutritional status, and steroid use. However, it does raise an important issue—namely, the level of PTH control in ESRD patients.17 For example, some more recent prospective cohort studies, including the UK Renal Registry of dialysis patients, did not show decreased mortality with maintenance of serum PTH levels between 150 and 300 pg/mL. Thus, the optimal PTH level in CKD and ESRD patients is controversial and not well-established. Indeed, the 2009 KDIGO (Kidney Disease: Improving Global Outcomes) guidelines state that an optimal PTH level is unknown for CKD stages 3A through 5. Meanwhile, for ESRD patients, a PTH range anywhere between 2 and 9 times the upper limit of normal is suggested, whereas recommendations for the management of SHPT are suggestive of a more case-by-case approach. For example, in patients with CKD stages 3A through 5 and in ESRD patients, rising iPTH should be evaluated and treated for modifiable factors such as hyperphosphatemia, hypocalcemia, and vitamin D deficiency.19 Such interventions not only include dietary restriction of phosphate and calcium/vitamin D supplementation but also include use of vitamin D analogs and calcimimetics such as cinacalcet to lower serum PTH levels. Additionally, parathyroidectomy is recommended only in severe HPT resistant to medical therapy or if PHPT is present. Furthermore, recent studies such as the EVOLVE trial, which did not find a significant reduction in death and nonfatal CV events in dialysis patients with moderate-to-severe SHPT, further confound the appropriate interventions in such patients.20 Thus, further studies are needed to examine the best interventions to mitigate CV morbidity and mortality and to better guide management of SHPT in patients with CKD and ESRD.
Hypoparathyroidism and Cardiovascular Disease
Hypoparathyroidism is an uncommon condition characterized by absent or low PTH levels, hypocalcemia, and hyperphosphatemia.21 The etiology of hypoparathyroidism is broad and can be congenital or acquired. For example, DiGeorge syndrome, which is due to a chromosomal deletion at 22q11.2, is characterized by parathyroid hypoplasia in addition to cardiac defects, thymic hypoplasia, neurocognitive problems, and renal and skeletal abnormalities, among others.22 Autoimmune polyendocrine syndrome type 1 (APS-1) is another genetic disorder that typically presents in childhood and adolescence with the classic triad of mucocutaneous candidiasis, adrenal insufficiency, and hypoparathyroidism. Isolated dysgenesis of the parathyroid glands can also be inherited, as is the case with familial hypoparathyroidism.22 Additionally, various genetic mutations of the preproPTH gene are some of the rarer causes of inherited hypoparathyroidism.22 In the adult population, the primary cause of hypoparathyroidism is acquired postoperatively (after thyroidectomy, neck dissection, parathyroidectomies).21 Autoimmune hypoparathyroidism is another less common cause in adults; it is characterized by antibodies to the parathyroid gland and occasional anti-CaSR antibodies, though the significance of the latter is not well-established.21 Autoimmune hypoparathyroidism can be idiopathic or part of an autoimmune polyglandular syndrome.21 Finally, isolated hypoparathyroidism, in which no genetic cause can be identified, is extremely rare and seen in adults.
The cardiac effects from hypoparathyroidism stem from the resulting hypocalcemia. For example, hypocalcemia causes QT prolongation, which can predispose patients to potentially life-threatening arrhythmias.23 Additionally, dilated cardiomyopathy from chronic hypocalcemia is a well-known but uncommon complication.24 Although the exact mechanism is not well-established, it is thought that hypocalcemia reduces cardiac contractility. PTH also appears to have an independent effect on cardiac cells. Recall that molecular and animal research shows the role of PTH in maintaining normal cardiac contractility by having a positive chronotropic effect in cardiomyocytes.24 Indeed, case reports have found that systolic dysfunction can be reversed in adults with isolated hypoparathyroidism upon treatment of the hypocalcemia.
Conclusion
The parathyroid glands are not only vital in maintaining bone metabolism but are also central to calcium homeostasis as it relates to the cardiovascular system. While the exact mechanisms are not fully established, it is clear that calcium, phosphorus, vitamin D, and PTH have direct and indirect effects on the heart. Disruptions in parathyroid function are increasingly recognized as contributing to hypertension, LVH, heart failure, and calcific disease, all of which increase cardiac morbidity and mortality. Data increasingly show that identifying and treating parathyroid disease in patients early in the disease course can potentially slow, or in some cases even reverse, adverse effects on the cardiovascular system. In the meantime, research involving larger cohorts and longer follow-up of people with parathyroid disorders is needed to better understand long-term cardiac effects of untreated disease.
Key Points:
Parathyroid hormone (PTH) acts on G-protein coupled receptors in the heart to exert changes in cardiac myocyte contractility, proliferation, and hypertrophy. In the vasculature, PTH alters the endothelium.
Excess PTH (as seen in primary and secondary hyperparathyroidism) is associated with a higher incidence of hypertension, left ventricular hypertrophy, heart failure, cardiac arrhythmias, and valvular calcific disease, which may contribute to higher cardiac morbidity and mortality.
Low PTH states (as seen in congenital and acquired disorders of the parathyroid glands) are associated with cardiac arrhythmias and dilated cardiomyopathy.
Early medical and/or surgical treatment of parathyroid disorders can reverse detrimental structural and functional changes in the cardiovascular system such as left ventricular mass, conduction abnormalities, atherosclerotic disease, and valvular calcifications. This may result in reduced cardiac morbidity and mortality.
Conflict of Interest Disclosure
The authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
References
- 1. Shoback D, Sellmeyer D, Bikle DD.. Metabolic bone disease. : Gardner DG, Shoback D, . Greenspan's basic & clinical endocrinology. 9th ed. New York, NY: The McGraw-Hill Companies; 2011. p 227– 284. [Google Scholar]
- 2. Pyram R, Mahajan G, Gliwa A.. Primary hyperparathyroidism: Skeletal and non-skeletal effects, diagnosis and management. Maturitas. 2011. November; 70 3: 246– 55. [DOI] [PubMed] [Google Scholar]
- 3. Schlüter KD, Piper HM. Cardiovascular actions of parathyroid hormone and parathyroid hormone-related peptide. Cardiovasc Res. 1998. January; 37 1: 34– 41. [DOI] [PubMed] [Google Scholar]
- 4. Schlüter KD. PTH and PTHrP: Similar Structures but Different Functions. News Physiol Sci. 1999. December; 14: 243– 9. [DOI] [PubMed] [Google Scholar]
- 5. Andersson P, Rydberg E, Willenheimer R.. Primary hyperparathyroidism and heart disease--a review. Eur Heart J. 2004. October; 25 20: 1776– 87. [DOI] [PubMed] [Google Scholar]
- 6. Yao L, Folsom AR, Pankow JS, . et al. Parathyroid hormone and the risk of incident hypertension: the Atherosclerosis Risk in Communities study. J Hypertens. 2016. February; 34 2: 196– 203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vlachakis ND, Frederics R, Valasquez M, Alexander N, Singer F, Maronde RF.. Sympathetic system function and vascular reactivity in hypercalcemic patients. Hypertension. 1982. May-Jun; 4 3: 452– 8. [DOI] [PubMed] [Google Scholar]
- 8. Brown J, de Boer IH, Robinson-Cohen C, . et al. Aldosterone, parathyroid hormone, and the use of renin-angiotensin-aldosterone system inhibitors: the multi-ethnic study of atherosclerosis. J Clin Endocrinol Metab. 2015. February; 100 2: 490– 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kiernan TJ, O'Flynn AM, McDermott JH, Kearney P.. Primary hyperparathyroidism and the cardiovascular system. Int J Cardiol. 2006. November 18; 113 3: E89– 92. [DOI] [PubMed] [Google Scholar]
- 10. Bansal N, Zelnick L, Robinson-Cohen C, . et al. Serum parathyroid hormone and 25-hydroxyvitamin D concentrations and risk of incident heart failure: the Multi-Ethnic Study of Atherosclerosis. J Am Heart Assoc. 2014. December 2; 3 6: e001278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wannamethee SG, Welsh P, Papacosta O, Lennon L, Whincup PH, Sattar N.. Elevated parathyroid hormone, but not vitamin D deficiency, is associated with increased risk of heart failure in older men with and without cardiovascular disease. Circ Heart Fail. 2014. September; 7 5: 732– 9. [DOI] [PubMed] [Google Scholar]
- 12. Witte KK, Byrom R, Gierula J, . et al. Effects of Vitamin D on Cardiac Function in Patients With Chronic HF: The VINDICATE Study. J Am Coll Cardiol. 2016. June 7; 67 22: 2593– 603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stefenelli T, Abela C, Frank H, . et al. Cardiac abnormalities in patients with primary hyperparathyroidism: Implications for follow-up. J Clin Endocrinol Metab. 1997. January; 82 1: 106– 12. [DOI] [PubMed] [Google Scholar]
- 14. Silver J, Naveh-Many T. FGF-23 and secondary hyperparathyroidism in chronic kidney disease. Nat Rev Nephrol. 2013. November; 9 11: 641– 9. [DOI] [PubMed] [Google Scholar]
- 15. Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. 2012. October; 82 7: 737– 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cunningham J, Locatelli F, Rodriguez M.. Secondary hyperparathyroidism: pathogenesis, disease progression, and therapeutic options. Clin J Am Soc Nephrol. 2011. April; 6 4: 913– 21. [DOI] [PubMed] [Google Scholar]
- 17. Zand L, Kumar R. Serum parathyroid hormone concentrations and clinical outcomes in ESRD: A call for targeted clinical trials. Semin Dial. 2016. May; 29 3: 184– 8. [DOI] [PubMed] [Google Scholar]
- 18. Lishmanov A, Dorairajan S, Pak Y, Chaudhary K, Chockalingam A.. Elevated serum parathyroid hormone is a cardiovascular risk factor in moderate chronic kidney disease. Int Urol Nephrol. 2012. April; 44 2: 541– 7. [DOI] [PubMed] [Google Scholar]
- 19. Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. . KDIGO clinical practice guideline for the diagnosis, evaluation, prevention, and treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Kidney Int Suppl. 2009. August; 113: S1– 130. [DOI] [PubMed] [Google Scholar]
- 20. EVOLVE Trial Investigators, Chertow GM, Block GA, . et al. Effect of cinacalcet on cardiovascular disease in patients undergoing dialysis. N Engl J Med. 2012. December 27; 367 26: 2482– 94. [DOI] [PubMed] [Google Scholar]
- 21. Bilezikian JP, Khan A, Potts JT Jr, . et al. Hypoparathyroidism in the adult: epidemiology, diagnosis, pathophysiology, target-organ involvement, treatment, and challenges for future research. J Bone Miner Res. 2011. October; 26 10: 2317– 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Shoback D. Clinical practice. Hypoparathyroidism. N Engl J Med. 2008. July 24; 359 4: 391– 403. [DOI] [PubMed] [Google Scholar]
- 23. Part 10. 1: Life-threatening electrolyte abnormalities. Circulation. 2005. December; 112 24 suppl: IV-121– 5. [Google Scholar]
- 24. Bansal B, Bansal M, Bajpai P, Garewal HK.. Hypocalcemic cardiomyopathy-different mechanisms in adult and pediatric cases. J Clin Endocrinol Metab. 2014. August; 99 8: 2627– 32. [DOI] [PubMed] [Google Scholar]