

Abstract
Thyroid hormones have a significant impact on cardiac function and structure. Excess thyroid hormone affects cardiovascular hemodynamics, leading to high-output heart failure and, in late stages, dilated cardiomyopathy. In this review, we discuss how hyperthyroidism affects cardiovascular pathophysiology and molecular mechanisms and examine the complications caused by excess thyroid hormone, such as heart failure and atrial fibrillation.
Keywords: hyperthyroidism, heart failure, atrial fibrillation, subclinical hyperthyroidism
Role of Thyroid Hormone in Energy Homeostasis
Thyroid hormones greatly impact energy homeostasis in the heart, and excess thyroid hormone leads to a hypermetabolic state. The thyroid gland produces two hormones, thyroxine (T4) and triiodothyronine (T3). The major form of thyroid hormone is thyroxine, which acts mostly as a prohormone.1 The set point for thyroid hormone production and secretion by the thyroid gland is regulated by the hypothalamic thyrotropin-releasing hormone (TRH), which stimulates the production and secretion of thyroid stimulating hormone (TSH) that, in turn, controls thyroid hormone concentrations. Most of T4 is converted to biologically active T3 through the removal of an iodide by deiodinases. While there are three types of deiodinases, most of the circulating T3 is derived from Type 1; Type 1 activates thyroid hormone by converting T4 to active T3, and it deactivates thyroid hormone by converting T4 to inactive reverse T3 (rT3) or to T2.2 It is important to note that there is no significant intracellular deiodinase activity in cardiac cells; therefore, the heart relies mainly on the action of T3 since that is the hormone transported into the myocyte.3 Both T4 and T3 circulate in the blood almost entirely (> 95%) bound to thyroxine-binding globulin and a family of other hormone-binding proteins. The remaining unbound T3 is transported through a variety of membrane transport proteins and subsequently to the cell nucleus to regulate expression of selected genes.4
Molecular Mechanisms of Thyroid Hormone Action
The intracellular cardiac effects of thyroid hormone are exerted by two mechanisms: genomic and nongenomic. Several of the main effects are exerted through genomic actions, which consist of T3 linking to nuclear receptors that bind to thyroid-responsive elements (TREs) in the promoter of target genes.5 There are several key myocyte-specific genes regulated by this mechanism (Table 1).3 Binding of thyroid hormone to these TREs can either activate or repress gene expression, thereby regulating the expression of specific messenger RNA and translated proteins and producing different tissue-specific responses. Importantly, thyroid hormone-regulated genes are also involved in structural and regulatory proteins, and long-term exposure to high T3 levels can increase the synthesis of cardiac proteins, leading to cardiac hypertrophy and dysfunction.6 Extranuclear nongenomic activities provoke rapid changes in the cardiac myocyte plasma membrane and cytoplasmic organelles. These include changes in sodium, potassium, and calcium ion channels; changes in actin cytoskeleton polymerization; and changes to the intracellular signaling pathways in the heart and smooth muscle cells.
Table 1.
Hyperthyroidism and its effect on cardiac gene expression. Adapted from Klein et al.3
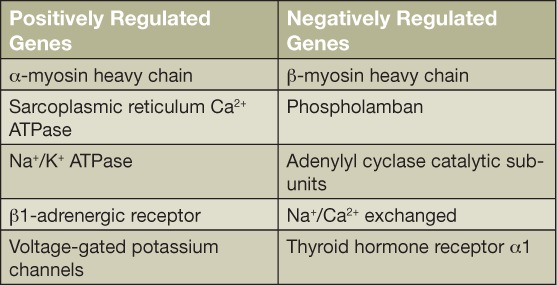
Both genomic and nongenomic mechanisms act together to regulate cardiac function and cardiovascular hemodynamics.2 For example, they upregulate expression of the sarcoplasmic reticulum calcium-activated ATPase and downregulate phospholamban expression, thereby enhancing myocardial relaxation. They also increase expression of the more rapid contractile isoforms of the myosin heavy chain (α isoforms), which contributes to enhanced systolic function. T3 also increases the rates of both depolarization and repolarization of the sinoatrial node, thus increasing heart rate. Consequently, thyroid hormones have positive inotropic and chronotropic effects on the heart that, along with heightened adrenergic sensitivity, account for the increased heart rate and contractility in hyperthyroidism.2
Hemodynamic Changes in Hyperthyroidism
Role of Catecholamines
Hyperthyroidism is characterized by increases in resting heart rate, blood volume, stroke volume, myocardial contractility, and ejection fraction and an improvement in diastolic relaxation, which is similar to a state of increased adrenergic activity.2 In addition, the therapeutic benefits of β-blockers suggest that the cardiac manifestations of hyperthyroidism are caused by increased catecholamine action.6 In thyrotoxicosis, plasma catecholamines are unchanged or low, and the β-adrenergic receptor density is altered in a time- and tissue-dependent manner, resulting in increased tissue sensitivity to catecholamines.2 Accompanying the increased levels of β-1 adrenergic receptors and guanosine triphosphate binding proteins, thyroid hormone decreases the expression of cardiac-specific adenylyl cyclase catalytic subunit isoforms and maintains cellular response to β-1 adrenergic agonists within normal limits.4 Therefore, the overall sensitivity of the heart to adrenergic stimulation remains unchanged. This is supported by the fact that administering a β-adrenergic receptor antagonist to patients with hyperthyroidism slows the heart rate but does not alter systolic or diastolic contraction, suggesting that the positive inotropic effect of T3 is independent of adrenergic signaling pathways.6
Role of Renin-Angiotensin-Aldosterone System (RAAS)
Preload is increased in a state of hyperthyroidism, and the reduced peripheral vascular resistance and elevated heart rate lead to increased cardiac output. The reduction in systemic vascular resistance results in decreased renal perfusion pressure and activation of the renin-angiotensin-aldosterone system (RAAS), thereby increasing sodium reabsorption and blood volume. In turn, this leads to increased preload, decreased afterload, and ultimately a significant increase in stroke volume.2 In addition, there is evidence that T3 directly stimulates the synthesis of renin substrate in the liver and enhances the cardiac expression of renin mRNA, leading to increased cardiac levels of renin and angiotensin II that are independent of the circulating renin and angiotensin. The expression of angiotensin II receptors in the myocardium increases in the hyperthyroid state.6 These hemodynamic changes that cause atrial stretch trigger the secretion of atrial natriuretic peptide (ANP), causing more vasodilation.2 These changes suggest a central role of the myocardial RAAS in thyroxine-induced cardiac hypertrophy as well as potential therapeutic implications of agents that block this system.6
Pulmonary Hypertension
Approximately 20% of patients with pulmonary hypertension have thyroid disease as a comorbidity, which is more frequent than the general population.7 Pulmonary artery hypertension (PAH) is an increase in mean pulmonary arterial pressure ≥ 25 mm Hg at rest.4 Higher pressure in the left atrium increases pressure in the pulmonary veins, which stimulates baroreceptors and causes a reflex contraction in the arterioles. The resulting increase in pulmonary artery pressure increases the load on the right ventricle. The extra load causes the right ventricle to contract with greater force to eject blood into the pulmonary vasculature, ultimately leading to increased pulmonary resistance and PAH.2 Although the mechanism of PAH in hyperthyroidism is uncertain, its reversal once the euthyroid state is restored supports a causal relationship.8 A recent study suggests a correlation between TSH receptor antibodies and PAH, providing support for a possible autoimmune-mediated pulmonary vascular remodeling in this condition.9 All patients with PAH should be screened for hyperthyroidism, and all patients with hyperthyroidism and dyspnea should be screened for PAH.7
Heart Failure in Hyperthyroidism
Hemodynamic alterations due to hyperthyroidism decrease myocardial contractile reserve, precluding further increases in ejection fraction and cardiac output on exertion. As discussed in the previous section, hemodynamic changes caused by excess thyroid hormone predispose the patient to heart failure.
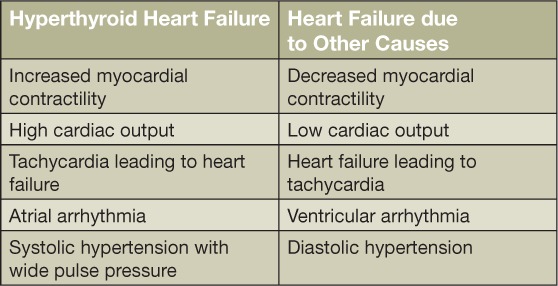
Hyperthyroid patients can manifest findings of congestive heart failure in the absence of prior cardiac injury. This state has inaccurately been called “high-output heart failure,” with paradoxical features such as enhanced cardiac output and contractility that are characterized by thyroid hormone excess.10 True heart failure manifests as decreased cardiac contractility, abnormal diastolic compliance, and pulmonary congestion, all of which can be consequences of severe and chronic hyperthyroidism, tachycardia, and atrial fibrillation.1,4,11 More specifically, thyrotoxic cardiomyopathy is defined as myocardial damage caused by toxic effects of excessive thyroid hormone, resulting in altered myocyte energy production, intracellular metabolism, and myofibril contractile function. Main manifestations are left ventricular hypertrophy, heart rhythm disturbances, primary atrial fibrillation, dilation of the heart chambers, heart failure, PAH, and diastolic dysfunction (Table 2).2 Patients with high-output heart failure can manifest with symptoms such as dyspnea on exertion, fatigue, and fluid retention with peripheral edema, pleural effusion, hepatic congestion, and PAH.11
Table 2.
Differences between hyperthyroid and nonhyperthyroid heart failure.
Effect of Hyperthyroidism Treatment on the Heart
An untreated high-output state and hyperthyroidism can lead to ventricular dilation, persistent tachycardia, and eventual chronic heart failure that can result in a fatal event.11 In a study by Mitchell et al., heart failure patients with abnormal thyroid function had a 60% higher risk of mortality compared to euthyroid patients with heart failure.12 This highlights the importance of prompt diagnosis and treatment of cardiac dysfunction secondary to hyperthyroidism. Treatment with β-adrenergic blockade to reduce heart rate and diuretics to improve congestive symptoms are important aspects of medical management.3 Correction of the thyroid dysfunction is also crucial, although there is some controversy over the most appropriate treatment method. Antithyroid medications can improve thyroid function but normally require weeks to control thyroid hormone excess. Frequently, definitive treatment such as radioactive iodine ablation or thyroidectomy is performed to recover cardiac function.11,13 Heart failure secondary to hyperthyroidism has been traditionally considered a reversible cause of cardiomyopathy. There are several reports of patients with symptomatic heart failure who demonstrated reversal of echocardiographic measurements and clinical symptoms after achieving a euthyroid state.14,15 However, some studies have suggested that cardiovascular symptoms and signs, abnormal hemodynamics, and cardiac dysrhythmias can be persistent.16 It is probable that individual patient characteristics such as age, comorbidities, and underlying risk factors for cardiac dysfunction significantly influence how one responds to treatment of thyrotoxic cardiomyopathy.
Atrial Fibrillation in Hyperthyroidism
Palpitations are one of the most common symptoms of hyperthyroidism. Between 10% and 25% of hyperthyroid patients have atrial fibrillation (AF), with the higher end of that range accounting for hyperthyroid patients (primary males) aged 60 and older; conversely, only 5% of hyperthyroid patients under age 60 have AF.1 The type of AF is usually persistent rather than paroxysmal.2 Given the high incidence of AF in patients over 60 years of age with thyrotoxicosis, early screening for TSH, free T4, and total T3 is especially important for detecting thyroid dysfunction in this patient demographic.17 The risk factors for AF in patients with hyperthyroidism are similar to those in the general population: age, ischemic heart disease, congestive heart failure, male gender, and valvular heart disease.2 Other factors also have been associated with the presence of AF in hyperthyroidism, including obesity, chronic kidney disease, proteinuria, female gender, serum-free T4 concentration, and transaminase concentrations.18 While there is an increased risk of AF when free T4 levels reach the high end of the normal range, especially in younger patients, TSH levels have not been associated with AF.19 The development of AF may be due to multiple mechanisms, including elevated left atrial pressure that leads to increased left ventricular mass and impaired ventricular relaxation,20 ischemia resulting from raised resting heart rate,21 and increased atrial ectopic activity.22 Atrial fibrillation starts with premature complexes that originate in the pulmonary veins, and its persistence requires a pathway of reentry.17 Hyperthyroidism is associated with coagulation abnormalities such as shortened activated partial thromboplastin time, increased fibrinogen levels, and increased factor VII and factor X activity in patients in sinus rhythm with thyrotoxicosis,2 all of which contribute to the risk of cardiac blood clot formation in these patients. Approximately 55% to 75% of patients with AF due to hyperthyroidism and no other underlying cardiac disease will return to normal sinus rhythm within 3 to 6 months after treatment of their thyrotoxic state.4
Amiodarone-Induced Hyperthyroidism
Amiodarone is a benzofuranic iodine-rich antiarrhythmic drug used to treat ventricular and atrial tachyarrhythmias.23 Approximately 37% of amiodarone by weight is organic iodine24; this causes abnormalities in thyroid function tests in 15% to 20% of patients treated with this drug.23 Amiodarone inhibits the 5′mono-deiodination of T4 in the liver and pituitary, thereby decreasing serum T3 and mildly increasing serum T4 levels without altering TSH concentrations. This effect is short-lived, perhaps in terms of a few weeks. Amiodarone-induced hyperthyroidism (AIT), on the other hand, is associated with a 3-fold increase for major adverse cardiovascular events,4 and the onset of AIT is often sudden and explosive.23 Type 1 AIT is due to the high iodine content of amiodarone. It occurs in areas of iodine deficiency and in patients with underlying thyroid disorders, such as multinodular goiter. In Type 1 AIT, the thyroid gland produces and releases excessive amounts of thyroid hormone. In contrast, Type 2 AIT results from a destructive process in the thyroid gland in which preformed thyroid hormones leak from the damaged follicular cells in patients without underlying thyroid disease.4 Color-flow Doppler sonography sometimes shows decreased vascularity in Type 2 AIT and increased vascularity in Type 1 AIT, helping to distinguish between the two. Thyroidal 131I uptake, determined by the radioactive iodine uptake test (RAIU), is usually low in type 2 (< 3%) due to the inflammatory process and destruction of thyroid tissue.23 In Type 1 AIT, iodine uptake may be high (> 10%).4 Mixed forms of AIT also occur, posing a diagnostic and therapeutic challenge. Type 1 AIT is treated with thionamides, and type 2 AIT is treated with oral glucocorticoids.3 Mixed forms may require a combination of thionamides and steroids. RAIU testing is not advised in AIT and is ineffective. Thyroidectomy is a valid option in cases resistant to medical therapy.13,23 Patients with AIT have a high event rate during follow-up, suggesting the need for close monitoring.25 The decision of whether or not amiodarone therapy can be discontinued requires interaction between the cardiologist and endocrinologist.23
Subclinical Hyperthyroidism and Heart
An increasing interest has emerged in recent years regarding the role of subclinical thyroid dysfunction, especially subclinical hyperthyroidism, and its impact on cardiovascular health. The definition of subclinical hyperthyroidism, based on biochemical findings, is a subnormal serum TSH level along with serum free T4 and T3 concentrations within the normal reference ranges.26 Its prevalence ranges from 0.6% to 16%.27 Persistent subnormal TSH values need to be confirmed within 2 to 3 months from initial values.28 Subclinical hyperthyroidism is further classified into two categories: Grade 1, with mildly low but detectable serum TSH (0.1–0.45 mIU/L), and Grade 2, with lower levels of serum TSH (< 0.1 mIU/L). The etiology of subclinical hyperthyroidism is broadly classified as exogenous or endogenous. Major exogenous causes are TSH suppressive therapy (or excessive administration of levothyroxine) in patients with thyroid carcinoma. Major endogenous causes are similar to overt hyperthyroidism and include mild Graves' disease, multinodular goiter, and autonomous functioning thyroid nodule. Several recent studies have demonstrated an association between subclinical hyperthyroidism and adverse effects on cardiovascular and bone health, especially in older populations. Nanchen et al. analyzed a large group of patients with subclinical hyperthyroidism and found a higher incidence of heart failure hospitalizations in older patients, particularly in those with grade 2 subclinical hyperthyroidism.29 Other studies discovered a significant association between AF and subclinical hyperthyroidism and observed an inverse correlation between TSH level and the risk of AF (Figure 1).30 More importantly, results from recent large retrospective studies have shown a significant association between subclinical hyperthyroidism and all-cause mortality and cardiovascular events, with heart failure as the leading cause of increased major adverse cardiac events.31 In their recently released clinical guidelines, the European Thyroid Association recommends treating grade 2 subclinical hyperthyroidism in patients older than 65 years and to consider treating milder grades in the presence of heart disease or other significant comorbidities or risk factors.28
Figure 1.
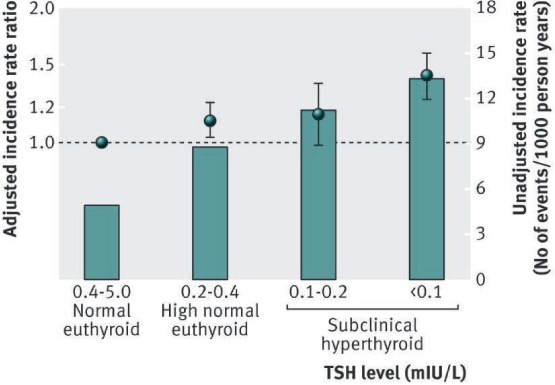
The spectrum of thyroid disease and risk of new onset atrial fibrillation: a large population cohort study. Reprinted from Selmer C et al.30
Conclusion
Hyperthyroidism causes high cardiac output and left ventricular hypertrophy in the early stage and biventricular dilatation and congestive heart failure in the late stage. Atrial fibrillation and PAH also add to the increased morbidity of untreated hyperthyroidism. Early and effective treatment of hyperthyroidism is key in preventing thyrotoxic cardiomyopathy.
Key Points:
Hyperthyroidism affects cardiovascular hemodynamics and leads to high-output heart failure and, in late stages, dilated cardiomyopathy
Early and effective treatment of hyperthyroidism can prevent congestive heart failure.
Controlling atrial fibrillation and preventing thromboembolic events are very important aspects of hyperthyroidism treatment.
Conflict of Interest Disclosure
The authors have completed and submitted the Methodist DeBakey Cardiovascular Journal Conflict of Interest Statement and none were reported.
References
- 1. Danzi S, Klein I. Thyroid disease and the cardiovascular system. Endocrinol Metab Clin North Am. 2014. June; 43 2: 517– 28. [DOI] [PubMed] [Google Scholar]
- 2. Vargas-Uricoechea H, Bonelo-Perdomo A, Sierra-Torres CH.. Effects of thyroid hormones on the heart. Clin Investig Arterioscler. 2014. Nov-Dec; 26 6: 296– 309. [DOI] [PubMed] [Google Scholar]
- 3. Klein I, Danzi S. Thyroid disease and the heart. Circulation. 2007. October 9; 116 15: 1725– 35. [DOI] [PubMed] [Google Scholar]
- 4. Klein I, Danzi S. Thyroid disease and the heart. Curr Probl Cardiol. 2016. February; 41 2: 65– 92. [DOI] [PubMed] [Google Scholar]
- 5. Gardner D, Shoback D. Greenspan's basic and clinical endocrinology. 9th ed New York: McGraw-Hill Education; 2011. 896 p. [Google Scholar]
- 6. Nabbout LA, Robbins RJ. The cardiovascular effects of hyperthyroidism. Methodist DeBakey Cardiovasc J. 2010. Apr-Jun; 6 2: 3– 8. [DOI] [PubMed] [Google Scholar]
- 7. Vallabhajosula S, Radhi S, Cevik C, Alalawi R, Raj R, Nugent K.. Hyperthyroidism and pulmonary hypertension: an important association. Am J Med Sci. 2011. December; 342 6: 507– 12. [DOI] [PubMed] [Google Scholar]
- 8. Siu CW, Zhang XH, Yung C, Kung AW, Lau CP, Tse HF.. Hemodynamic changes in hyperthyroidism-related pulmonary hypertension: a prospective echocardiographic study. J Clin Endocrinol Metab. 2007. May; 92 5: 1736– 42. [DOI] [PubMed] [Google Scholar]
- 9. Sugiura T, Yamanaka S, Takeuchi H, Morimoto N, Kamioka M, Matsumura Y.. Autoimmunity and pulmonary hypertension in patients with Graves' disease. Heart Vessels. 2015. September; 30 5: 642– 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001. February 15; 344 7: 501– 9. [DOI] [PubMed] [Google Scholar]
- 11. Biondi B. Mechanisms in endocrinology: Heart failure and thyroid dysfunction. Eur J Endocrinol. 2012. November; 167 5: 609– 18. [DOI] [PubMed] [Google Scholar]
- 12. Mitchell JE, Hellkamp AS, Mark DB, . et al. Thyroid function in heart failure and impact on mortality. JACC Heart Fail. 2013. February; 1 1: 48– 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tomisti L, Materazzi G, Bartalena L, . et al. Total thyroidectomy in patients with amiodarone-induced thyrotoxicosis and severe left ventricular systolic dysfunction. J Clin Endocrinol Metab. 2012. October; 97 10: 3515– 21. [DOI] [PubMed] [Google Scholar]
- 14. Al-Ghamdi AS, Aljohani N. Graves' thyrotoxicosis-induced reversible cardiomyopathy: a case report. Clin Med Insights Case Rep. 2013. March 27; 6: 47– 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Choudhury RP, MacDermot J. Heart failure in thyrotoxicosis, an approach to management. Br J Clin Pharmacol. 1998. November; 46 5: 421– 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Osman F, Franklyn JA, Holder RL, Sheppard MC, Gammage MD.. Cardiovascular manifestations of hyperthyroidism before and after antithyroid therapy: a matched case-control study. J Am Coll Cardiol. 2007. January 2; 49 1: 71– 81. [DOI] [PubMed] [Google Scholar]
- 17. Bielecka-Dabrowa A, Mikhailidis DP, Rysz J, Banach M.. The mechanisms of atrial fibrillation in hyperthyroidism. Thyroid Res. 2009. April 2; 2 1: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tănase DM, Ionescu SD, Ouatu A, Ambăruş V, Arsenescu-Georgescu C.. Risk assessment in the development of atrial fibrillation at patients with associate thyroid dysfunctions. Rev Med Chir Soc Med Nat Iasi. 2013. Jul-Sep; 117 3: 623– 9. [PubMed] [Google Scholar]
- 19. Chaker L, Heeringa J, Dehghan A, . et al. Normal Thyroid Function and the Risk of Atrial Fibrillation: the Rotterdam Study. J Clin Endocrinol Metab. 2015. October; 100 10: 3718– 24. [DOI] [PubMed] [Google Scholar]
- 20. Staffurth JS, Gibberd MC, Fui SN.. Arterial embolism in thyrotoxicosis with atrial fibrillation. Br Med J. 1977. September 10; 2 6088: 688– 90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Frost L, Vestergaard P, Mosekilde L.. Hyperthyroidism and risk of atrial fibrillation or flutter: a population-based study. Arch Intern Med. 2004. August 9–23; 164 15: 1675– 8. [DOI] [PubMed] [Google Scholar]
- 22. Sawin CT, Geller A, Wolf PA, . et al. Low serum thyrotropin concentrations as a risk factor for atrial fibrillation in older persons. N Engl J Med. 1994. November 10; 331 19: 1249– 52. [DOI] [PubMed] [Google Scholar]
- 23. Bogazzi F, Bartalena L, Martino E.. Approach to the patient with amiodarone-induced thyrotoxicosis. J Clin Endocrinol Metab. 2010. June; 95 6: 2529– 35. [DOI] [PubMed] [Google Scholar]
- 24. Kahaly GJ, Dillmann WH. Thyroid hormone action in the heart. Endocr Rev. 2005. August; 26 5: 704– 28. [DOI] [PubMed] [Google Scholar]
- 25. Conen D, Melly L, Kaufmann C, . et al. Amiodarone-induced thyrotoxicosis: clinical course and predictors of outcome. J Am Coll Cardiol. 2007. June 19; 49 24: 2350– 5. [DOI] [PubMed] [Google Scholar]
- 26. Surks MI, Ortiz E, Daniels GH, . et al. Subclinical thyroid disease: scientific review and guidelines for diagnosis and management. JAMA. 2004. January 14; 291 2: 228– 38. [DOI] [PubMed] [Google Scholar]
- 27. Biondi B, Palmieri EA, Lombardi G, Fazio S.. Effects of subclinical thyroid dysfunction on the heart. Ann Intern Med. 2002. December 3; 137 11: 904– 14. [DOI] [PubMed] [Google Scholar]
- 28. Biondi B, Bartalena L, Cooper DS, Hegedus L, Laurberg P, Kahaly GJ.. The 2015 European Thyroid Association Guidelines on Diagnosis and Treatment of Endogenous Subclinical Hyperthyroidism. Eur Thyroid J. 2015. September; 4 3: 149– 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nanchen D, Gussekloo J, Westendorp RG, . et al. Subclinical thyroid dysfunction and the risk of heart failure in older persons at high cardiovascular risk. J Clin Endocrinol Metab. 2012. March; 97 3: 852– 61. [DOI] [PubMed] [Google Scholar]
- 30. Selmer C, Olesen JB, Hansen ML, . et al. The spectrum of thyroid disease and risk of new onset atrial fibrillation: a large population cohort study. BMJ. 2012. November 27; 345: e7895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Selmer C, Olesen JB, Hansen ML, . et al. Subclinical and overt thyroid dysfunction and risk of all-cause mortality and cardiovascular events: a large population study. J Clin Endocrinol Metab. 2014. July; 99 7: 2372– 82. [DOI] [PubMed] [Google Scholar]