

Abstract
Polychlorinated biphenyls (PCBs) are persistent environmental pollutants that disrupt hepatic xenobiotic and intermediary metabolism, leading to metabolic syndrome and nonalcoholic steatohepatitis (NASH).
Since phenobarbital indirectly activates Constitutive Androstane Receptor (CAR) by antagonizing growth factor binding to the epidermal growth factor receptor (EGFR), we hypothesized that PCBs may also diminish EGFR signaling.
The effects of the PCB mixture Aroclor 1260 on the protein phosphorylation cascade triggered by EGFR activation were determined in murine (in vitro and in vivo) and human models (in vitro). EGFR tyrosine residue phosphorylation was decreased by PCBs in all models tested.
The IC50 values for Aroclor 1260 concentrations that decreased Y1173 phosphorylation of EGFR were similar in murine AML-12 and human HepG2 cells (~2–4 μg/mL). Both dioxin and non-dioxin-like PCB congeners decreased EGFR phosphorylation in cell culture.
PCB treatment reduced phosphorylation of downstream EGFR effectors including Akt and mTOR, as well as other phosphoprotein targets including STAT3 and c-RAF in vivo.
PCBs diminish EGFR signaling in human and murine hepatocyte models and may dysregulate critical phosphoprotein regulators of energy metabolism and nutrition, providing a new mechanism of action in environmental diseases.
Introduction
Polychlorinated biphenyls (PCBs) are lipid-soluble, persistent organic pollutants that preferentially accumulate in visceral adipose tissue and liver (Kim and others 2014). Previously, PCBs were used in industrial applications such as heat exchangers and electrical capacitors. PCBs were manufactured as mixtures based on chlorine content, rather than as individual congeners. The PCB mixture, Aroclor 1260 (Monsanto Company, US), contains 60% chorine by weight, and its composition is similar to human adipose bioaccumulation patterns (McFarland and Clarke 1989; Wahlang and others 2014a). Daily human PCB consumption is estimated to be 30 ng/day (Schecter and others 2010), but is likely to be greater in people eating highly contaminated foods, such as fish from polluted waterways. Due to both the widespread contamination and the persistence of some congeners in vivo, 100% of US adult participants in the National Health and Nutrition Examination Survey (NHANES) had detectable levels of PCBs in serum (Cave and others 2010). Higher chlorinated PCB congeners such as PCB 180 have a biological half-life of 11.5 years and thus could chronically alter signaling pathways involved in metabolic disorders (Ritter and others 2011).
In humans, PCBs have been associated with metabolic diseases including nonalcoholic fatty liver disease (NAFLD) (Cave and others 2010; Kumar and others 2014; Serdar and others 2014; Yorita Christensen and others 2013), obesity (Ghosh and others 2014), hypertension (Goncharov and others 2010), diabetes (Patel and others 2010; Silverstone and others 2012; Wang and others 2008), and dyslipidemia (Aminov and others 2013). Animal models have documented the development of NAFLD and its more severe form, nonalcoholic steatohepatitis (NASH), following PCB treatment (Wahlang and others 2014b). In fact, PCBs were among the most potent environmental chemicals associated with NAFLD in rodents (Al-Eryani and others 2014). Determining mechanisms by which PCBs may contribute to NAFLD pathogenesis is the overall objective of this manuscript.
PCB congeners have been classified by structure-activity relationships based on their observed effects on hepatic xenobiotic metabolism (Safe and others 1985). “Dioxin-like” PCBs increase ethoxyresorufin O-deethylase activity (Sawyer and Safe 1982), while “phenobarbital-like” PCBs increase pentoxyresorufin O-dealkylase activity. PCBs were subsequently noted to activate xenobiotic receptors including the aryl hydrocarbon receptor (AhR), the pregnane X receptor (PXR), and the constitutive androstane receptor (CAR) leading to the induction of specific cytochrome P450 enzymes including CYP1A, CYP3A, and CYP2B, respectively (Al-Salman and Plant 2012; Wahlang and others 2014b). While PCB-nuclear receptor interactions influenced intermediary metabolism in an animal model of NASH (Wahlang and others 2016), exposure to PCBs alone was insufficient to cause NASH, but rather served as a ‘second hit’ in the transition of diet-induced steatosis to steatohepatitis (Wahlang and others 2014b). Aroclor 1260 treated Car−/− mice fed a high fat diet (HFD) had increased activity, decreased food consumption, and increased insulin sensitivity compared to wild type mice also fed HFD (Wahlang and others 2016). However, NASH pathology did not improve concordantly with these favorable metabolic changes in Car−/−. This suggests that while PCB-Car interactions are important in metabolic syndrome, other mechanisms must also be necessary for PCB-related NAFLD.
CAR may be activated either directly or indirectly by xenobiotics. The mechanism of CAR activation may be important, because indirect CAR activation could be associated with additional ‘off target’ effects influencing NAFLD. We observed that PCBs directly activated human CAR (hCAR) splice variants 2–3 but not hCAR1 (Wahlang and others 2014a). Although mouse CAR (mCAR) is homologous to hCAR1, mice treated with PCBs displayed increased Cyp2b10 expression (Wahlang and others 2014b). The reasons for the varied responses observed by these homologous proteins is most likely due to splice variants 2 and 3 having slightly larger ligand binding pockets allowing additional space for ligand binding. Phenobarbital is a prototypical Cyp2b10 inducer, and the indirect mechanism by which phenobarbital activates CAR has recently been discovered (Mutoh and others 2013). Phenobarbital prevented epidermal growth factor (EGF) binding to its receptor (EGFR), inhibiting the phosphorylation cascade leading to an increased abundance of dephosphorylated, active CAR.
EGFR is a transmembrane receptor tyrosine kinase that is ligand-activated by the epidermal growth factor (EGF) and other similar growth factors. Upon ligand binding, EGFR dimerises with itself or other family members. This induces activation of the receptor’s intrinsic kinase activity, resulting in transphosphorylation of tyrosine residues on its receptor pair. These newly formed phosphotyrosine residues serve as docking sites for downstream signaling proteins (effectors) that act to modulate metabolic gene expression. Normal EGFR signaling is essential in development and metabolism (Gschwind and others 2001). In addition to CAR, downstream targets include the phosphoinositol 3-kinase/serine/threonine kinase 1 or PKB/mechanistic target of rapamycin (PI3K/Akt/mTOR) pathway, signal transducer and activator of transcription 3 (STAT3), serine and threonine kinase (c-Raf) and extracellular signal-regulated kinase (ERK). Akt and mTOR are effector kinases downstream of EGFR that regulate lipid metabolism, and glycogen synthesis (Caron and others 2015; Taniguchi and others 2006). Decreased EGFR signaling has been implicated in metabolic diseases such as type II diabetes (Bernal-Mizrachi and others 2014; Miettinen and others 2008) and NASH (Collin de l’Hortet and others 2014; Deaciuc and others 2002; Komposch and Sibilia 2016; Scheving and others 2014; Scheving and others 2015), both of which have been associated with PCB exposures (Cave and others 2010; Li and others 2013; Taylor and others 2013; Wahlang and others 2016; Wahlang and others 2014b; Yu and others 1997). Thus, we hypothesised that if PCBs antagonised EGFR like phenobarbital, this mechanism could account for both indirect CAR activation and the associated ‘off target’ effects contributing to PCB-related NASH. Understanding how PCBs disrupt these pathways could provide mechanistic insight into how PCBs mediate metabolic syndrome and liver disease (Wahlang and others 2016; Wahlang and others 2014b). In this manuscript, we determine: (i) if PCB-mediated indirect CAR activation in mice is mediated by inhibition of EGFR phosphorylation; (ii) if PCB effects on EGFR phosphorylation in mice are modified by diet; (iii) if PCB-mediated hypophosphorylation of EGFR in mice impacts additional downstream phosphoprotein targets (including Akt and mTOR); and (iv) if PCBs also inhibit human EGFR phosphorylation. We conclude from our studies that PCBs decrease phosphorylation of EGFR and its downstream effector kinases.
Materials and Methods (Wahlang and others 2016; Wahlang and others 2014b)
Chemicals
1,4-Bis(3,5-dichloro-2-pyridyloxy)benzene (TCPOBOP) and androstanol were obtained from Sigma Chemical Co., St. Louis, MO. Aroclor 1260, Aroclor 1254, and PCB congeners, PCB 3, PCB 6, PCB 8, PCB 9, PCB 126, PCB 138, PCB 149, PCB 151, PCB 153, PCB 170, PCB 174, PCB 180, PCB 187, were obtained from AccuStandard, Inc., New Haven, CT. Epidermal growth factor (EGF) and EGFR inhibitor (EI, Cyclopropanecarboxylic acid-(3-(6-(3-trifluoromethyl-phenylamino)-pyrimidin-4-ylamino)-phenyl)-amide) were purchased from Millipore (Norwood, OH). The following antibodies were used for Western blot analysis of cell lysates from HepG2 and AML-12 cells: EGFR (Santa Cruz, Santa Cruz, CA), P-EGFR Y1173 (abcam, Cambridge, MA), P-EGFR Y845 (abcam, Cambridge, MA), and P-EGFR Y1068 (CST, Danvers, MA), mouse liver EGFR, P-EGFR Y845, P-EGFR Y1068, mTOR, P-mTOR, AKT, P-AKT, STAT3, cRaf, ERK, P-ERK, β-actin, GAPDH (CST, Danvers, MA).
Cell culture
Alpha mouse liver 12 (AML-12) cells of male origin obtained from ATCC (Mannassas, VA), were seeded at 1×104 per well in 12-well plates and grown to confluence for 24 hours in DMEM/F12 (GE Healthcare, Pittsburgh, PA) media supplemented with 10% fetal bovine serum, and 1% antimyotic/antibiotic solution. The cells were incubated in a 5% carbon dioxide atmosphere at 95% humidity and 37°C were sub-cultured every 2–3 days. Cells were treated for 30 minutes with either 20 ng/mL of epidermal growth factor (EGF) (Fisher Scientific, Pittsburgh, PA) as a positive control, 20 ng/mL of EGF and 20 μg/mL EGFR inhibitor (EI) (CAS 879127-07-8) (Millipore, Norwood, OH) as a negative control, or 20 ng/mL EGF and 20 μg/mL Aroclor 1260 (A1260). This Aroclor 1260 concentration was used based on previous work demonstrating that in vitro concentrations higher than 20 μg/ml were cytotoxic (Toxicity threshold = 26.7 ± 3.7 μg/mL in HepG2s) (Wahlang and others 2014a). The concentration of EGF was optimal to induce EGFR phosphorylation, and EGFR inhibitor was used at its IC50 concentration for EGFR dephosphorization. A 30-minute treatment period produced robust EGFR phosphorylation but had no effect on PCB-mediated inhibition of EGFR phosphorylation. The positive and negative controls were used to define phosphorylated EGFR’s dynamic range. Media was removed by aspiration and the cells were washed with PBS. Cells were lysed in modified radioimmunoprecipitation assay (RIPA) buffer (20 mM Tris, 150 mM NaCL, 1 mM EDTA/EGTA/β-glyceraphosphate/Na3VO4, and 1% Triton X-100) supplemented with protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO).
Human hepatoma-derived (HepG2) cells (male origin) obtained from American Type Culture Collection (ATCC, Manassas, MD) were plated 3×105 cells per well in 12-well plates and grown to confluence in DMEM High Glucose media (GE Healthcare, Pittsburgh, PA) supplemented with 10% fetal bovine serum (FBS) and 1% antimycotic/antibiotic solution (Mediatech, Manassas, VA). HepG2 cells were treated and collected in a similar manner as the AML-12 cells. The Aroclor 1260 concentration-dependence assay was conducted in AML-12 and HepG2 cells similarly to aforementioned experiments with varied concentrations of Aroclor 1260 ranging from (0, 0.38, 0.94, 1.88, 5, 10, 15, 20 μg/mL) at constant EGF concentration. Aroclor 1254 was used at a concentration of 20 μg/mL similarly to the Aroclor 1260 experiments in HepG2 cells. The experiments with individual PCB congeners (PCB 3, PCB 6, PCB 8, PCB 9, PCB 138, PCB 149, PCB 151, PCB 153, PCB 170, PCB 174, PCB 180, and PCB 187 were performed with HepG2 cells at congener concentrations of 10 μM.
Preparation of Expression vectors
A mCAR-actived luciferase reporter containing two AGGTCA repeats separated by 4 base pairs was generated as an expression construct for this receptor (Wahlang and others 2014a). Murine CAR cDNA was generously provided by T.H Rushmore (Merck, Westpoint, PA). The expression vector used was a modified version of pcDNA3.1 (Thermo Scientific, Waltham, MA) in which the CMV promoter was replaced with a weaker promoter, the minimal promoter from the rat glutathione S-transferase A2 gene (Falkner and others 1998) which produces lower expression levels of CAR.
Transient Transfection Assays
HepG2 cells were plated in Nunc 24-well plates (Thermo Scientific, Waltham, MA) and transfected at 40–60% confluence. Unless otherwise specified, the transfection mix per well contained 150 ng β-galactosidase expression plasmid pCMV-β (Stratagene, San Diego, CA) as a transfection control, 50 ng receptor expression plasmid, and 150 ng luciferase reporter plasmid. All cells were transfected using lipofectamine reagent (Thermo Scientific, Waltham, MA) according to the manufacturer’s instructions and Opti-MEM (Thermo Scientific, Waltham, MA) as the transfecting medium. After a 4 hour incubation, the medium was changed to the growth media DMEM (Thermo Scientific, Waltham, MA) supplemented with 10% FBS and 1% antimycotic/antibiotic solution. After 24 hurs, cells were treated with either Aroclor 1260 (0–20 μg/mL), androstanol (10 μM) or 1,4-Bis(3,5-dichloro-2-pyridyloxy)benzene (TCPOBOP, 10 μM) either alone or in combination. At 24 hours post-treatment, the cells were harvested using cell lysis buffer (Promega, Fitchburg, WI) and the β-galactosidase activity determined spectrophotometrically on a Biotek Synergy plate reader using chlorophenol red β-D-galactopyranoside as substrate. The luciferase activity was determined using a Berthold (Bad Wildbad, Germany) plate reading luminometer using the luciferase assay kit from Promega, (Fitchburg, WI) according to the manufacturer’s instructions. All data are presented as normalised luciferase activity/β-galactoside activity from 4 independent experiments and all comparisons were made to the untreated group.
Exposure of mice to Aroclor 1260
The mouse liver samples used in this study were obtained from archived (−80°C) tissues obtained during a previous study (Wahlang and others 2014b). The treatment protocol was approved by the University of Louisville Institutional Animal Care and Use Committee. Male C57Bl/6J mice (8 weeks old; The Jackson Laboratory, Bar Harbor, ME, USA) were divided into 4 study groups (n = 10) based on diet and Aroclor 1260 exposure in this 12-week study utilizing a 2 × 2 design. Mice were fed either a control diet (CD, 10.2% kCal from fat; TD.06416 Harlan Teklad, Indianapolis, IN) or a HFD (42% kCal from fat; TD.88137 Harlan Teklad, Indianapolis, IN). Aroclor 1260 (AccuStandard, CT, USA) was administered in corn oil by oral gavage (vs. corn oil alone) at a dose of 20 mg/kg early in week 1, at a dose similar to the maximum human PCB exposures seen in the Anniston cohort (Cave and others 2010). Mice were housed in a temperature- and light controlled-room (12 h light; 12 h dark) with food and water ad libitum. The animals were euthanised at the end of week 12 using ketamine/xylazine, 100/20 mg/kg body weight, i.p., respectively), followed by exsanguination to minimise blood in the liver sample. The mice fed a HFD and treated with Aroclor 1260 developed steatohepatitis as demonstrated by CAE and H&E staining. Aroclor 1260 treatment was associated with induction of Cyp2b10 consistent with Car activation (Wahlang and others 2014b).
Western Blot Analysis
Mouse liver samples and cell culture lysates were homogenised in RIPA Buffer (100 mg tissue/0.5 mL RIPA supplemented with protease and phosphatase inhibitors 1 μl/mL, (Sigma-Aldrich, St. Louis, MO). The protein concentration was determined by bicinchoninic acid protein assay (Sigma-Aldrich, St. Louis, MO). Protein (15 μg) was separated on 7.5% SDS Gel (BioRad, Hercules, CA), transferred to polyvinylidene difluoride membranes and blocked in Phosphate Buffered Saline (PBS), pH 7.5 containing 1% Tween 20 (PBS-T) and 5% fat free milk for 1 hour at room temperature. Membranes were incubated at 4° C overnight with primary antibody at a 1:1000 dilution in 5% bovine serum albumin in PBS-T, then washed 4 times with PBS-T for 5 minutes followed by incubation with secondary antibody 1:2000 in 5% milk in PBS-T (Cell Signaling Technology, Danvers, MA). After 4 PBS-T washes the membranes were incubated with ECL (Thermo Scientific, Waltham, MA) and luminescent signals were captured with BioRad Chemidoc Imaging System (Hercules, CA). Western blot protein bands were quantified using BioRad Image Software. For detection of phosphoproteins, the phospho-antibody blots were stripped for 15 minutes (Thermo Scientific, Waltham, MA) then re-probed using primary antibody against total protein overnight in 5% fat free milk in PBS-T. For phospho-Akt detection in mouse liver, duplicate protein samples were analyzed in parallel under identical conditions. One blot was probed for phosphoprotein and the other total protein and then both were re-probed for the loading control β-actin. The quantitation of band density was normalised to β-actin and then to total protein. This strategy was necessary due to the inability to strip the phospho antibodies for Akt.
Statistical Analysis
Western blot densitometry values were statistically analyzed using Graphpad Prism Version 6 for Macintosh (San Diego, CA). The data are expressed as mean ± standard error of the mean. All in vitro datasets were compared using one-way ANOVA. p<0.05 was considered statistically significant. In vivo data sets were compared using two-way ANOVA and p<0.05 was considered statistically significant. The concentration dependence curves and IC50 values were generated by fitting data to a 4-parameter logistic equation using SigmaPlot (Systat Software Inc. San Jose, CA).
Results
Aroclor 1260 did not counter the inhibitory effect of the direct CAR antagonist, androstanol, in vitro
To establish whether PCBs can directly activate mCAR, HepG2 cells transfected with mCAR and a DR4 luciferase reporter gene were incubated with varied concentrations of Aroclor 1260 (0–20 μg/mL), and compared to control HepG2 cells treated with direct mCAR agonist, TCPOBOP with or without androstanol exposure, as shown in Figure 1. As anticipated, treatment with 10 μM androstanol reduced luciferase activity by 45%. The CAR ligand, TCPOBOP did not significantly increase luciferase expression above control, consistent with the concept that CAR is constitutively active when over-expressed in HepG2 cells. TCPOBOP treatment did, however, completely reverse the inhibitory effect of androstanol in TCPOBOP-treated cells; cells treated with both TCPOBOP and androstanol had the same activity as TCPOBOP-treated cells alone. In contrast, Aroclor 1260-treated cells failed to show reversal of the inhibitory effects of androstanol at any concentration of Aroclor 1260 tested (from 0–20 μg/mL). In fact, Aroclor 1260 exposure decreased luciferase activity either by itself or in combination with androstanol. Because the negative androstanol effect was observed at even the highest concentrations of Aroclor 1260 exposure, Aroclor 1260 cannot counter androstanol’s inhibition of CAR, therefore is unlikely to be a direct murine CAR ligand. Direct binding assays are required to conclude that PCBs cannot directly bind to murine CAR.
Figure 1. Effects of Aroclor 1260 exposure on a DR4-luciferase reporter gene activity in HepG2 cells transfected with an expression vector for murine CAR.

Cells were grown and after transfection with expression vector for mCAR and luciferase reporter DR4 luciferase treated with either androstanol, TCPOBOP, or increasing amounts of Aroclor 1260 (0–20 μg/mL). The results as presented represent n = 4 separate experiments. A one-way ANOVA statistical test was used to compare experimental data to non-treated cells. * = p < 0.05
Aroclor 1260 exposure decreased murine hepatic EGFR phosphorylation in vitro
To determine if Aroclor 1260 exposure could cause an indirect activation of CAR, the phosphorylation status of EGFR was examined in AML-12 cells, a murine cell line that expresses normal functioning EGFR. EGFR has multiple tyrosine residues within the receptor tyrosine kinase (RTK) domain that have specific downstream effects when autophosphorylated upon homo or heterodimerisation. EGFR phosphorylation status was measured at Y845, Y1068, and Y1173 due to their known role in regulating the specific downstream effects of CAR (Y845), STAT2, cRaf, and ERK (Y1068), and Akt (Y1273) by tyrosine phosphorylation (Mutoh and others 2013). In AML-12 cells, EGFR phosphorylation at Y1173 was diminished by co-exposure to EGF and Aroclor 1260 for 30 min (Figure 2A), relative to EGF alone. EGFR inhibitor also decreased phosphorylation at Y1173. EGFR phosphorylation at Y1173 was decreased 48% by Aroclor 1260 and EGF co-exposure as compared to EGF alone (p = 0.006) (EGF 0.87 ± 0.09 vs. A1260 0.45 ± 0.1), while EGFR inhibitor in the presence of EGF caused a 45% reduction in EGFR (0.39±0.03%) phosphorylation. Phosphorylation at Y1068 was also decreased with EGF and Aroclor 1260 co-exposure as shown in Figure 2B. In a similar manner, Y1068 EGFR phosphorylation was decreased 45% with Aroclor 1260 and EGF co-exposure for 30 min as compared to EGF alone (p = 0.003) (EGF 0.92 ± 0.04 vs. A1260 0.51 ± 0.08 vs. EGFR inhibitor 0.32 ± 0.02). Y845 phosphorylation also decreased in AML-12 cells with Aroclor 1260 and EGF co-exposure (Figure 2C). At Y845, EGFR phosphorylation was decreased 43% with Aroclor 1260 and EGF exposure as compared to EGF alone (p = 0.005) (EGF 0.61 ± 0.05 vs. A1260 0.35 ± 0.03 vs. EGFR inhibitor 0.12 ± 0.02). In the AML-12 cells, the concentration-dependence of the inhibition of EGFR Y1173 phosphorylation by Aroclor 1260 is shown in Figure 2D. The IC50 value obtained was 1.2 ± 0.3 μg/mL and the Hill slope coefficient is 1.4 ± 0.5 indicating one binding site at EGFR. These findings demonstrate that not only can PCBs diminish EGFR phosphorylation contributing to increased CAR activity (Y845) previously reported, but they may also affect many other downstream targets independent of CAR (Y1068, Y1173) based on the decreased phosphorylation of the EGFR tyrosine residues and the specific downstream targets.
Figure 2. Decreased EGFR phosphorylation at multiple sites in vitro with Aroclor 1260 exposure.

AML-12 cells treated with EGF and either EGFR inhibitor (EI), or Aroclor 1260 (A1260) for 30 minutes were lysed for western blot analysis as described in the Materials and Methods. 2A: EGFR phosphorylation at Y1173 in AML-12 cells was decreased by Aroclor 1260 and EGF co-exposure as compared to EGF alone (p = 0.006). 2B: EGFR phosphorylation at Y1068 was decreased with Aroclor 1260 and EGF co-exposure as compared to EGF alone (p = 0.003). 2C: EGFR phosphorylation at Y845 was decreased with Aroclor 1260 exposure as compared to EGF alone (p = 0.005). 2D: Aroclor 1260 displayed an IC50 of 1.18 ± 0.32 μg/mL for EGFR hypo-phosphorylation in AML-12 cells. The Hill slope coefficient was 1.39 ± 0.5. A representative blot of the effect was shown in each panel and the graph represents quantitation of normalised protein relative to EGF alone. The results are presented as mean ± SEM and represent n = 3 experiments. A one-way ANOVA statistical test was used to compare experimental data to the positive control data (EGF exposure alone), p < 0.05 was considered significant. * p < 0.05, ** p < 0.01
Aroclor 1260 exposure diminished murine hepatic EGFR phosphorylation in vivo
To validate the effect of Aroclor 1260 on hepatic EGFR phosphorylation, hepatic EGFR Y1173 phosphorylation was measured in archived liver tissue from a previous diet-induced obesity experiment, in which mice were exposed to 20 mg/kg Aroclor 1260 (Wahlang and others 2014b). Mice fed either a control diet (10.2% of calories as fat) or a high fat diet (43% of calories as fat) for 12 weeks were treated with Aroclor 1260 (20 mg/kg by gavage) or vehicle control early in week 1. Mice treated with Aroclor 1260 plus a high fat diet developed NASH (Wahlang and others 2014b). As shown in Figure 3A, Aroclor 1260 exposure resulted in a 43% and 48% decrease in hepatic EGFR phosphorylation at Y1173 in the control diet (CD 0.67 ± 0.12 vs. CD+ 0.39 ± 0.08) and high-fat diet (HFD 0.64 ± 0.12 vs. HFD+ 0.34 ± 0.1) groups, respectively (p = 0.02). In Figure 3B, Aroclor 1260 exposure resulted in a 60% and 41% decrease in hepatic EGFR phosphorylation at Y1068 in the control diet (CD 0.48 ± 0.14 vs. CD+ 0.19 ± 0.05) and high-fat diet (HFD 0.36 ± 0.07 vs. HFD+ 0.21 ± 0.06) groups, respectively (p = 0.03). In Figure 3C, Aroclor 1260 exposure resulted in a 46% and 49% decrease in hepatic EGFR phosphorylation at Y845 in the control diet (CD 0.64 ± 0.16 vs. CD+ 0.35 ± 0.08) and high-fat diet (HFD 0.53 ± 0.16 vs. HFD+ 0.27 ± 0.02) groups, respectively (p = 0.04). PCBs decreased phosphorylation of all three tyrosine residues suggesting that they alter the downstream effector protein kinases.
Figure 3. Effects of Aroclor 1260 exposure on mouse hepatic EGFR phosphorylation.

Mice placed on either a control (10.2% fat) or HFD (43% fat) for 12 weeks. Aroclor 1260 (20 μg/kg) was administered per os early in week 1. Phosphorylation was determined at Y1173, Y1068, and Y845 of EGFR by Western blot analysis as described in Materials and Methods. 3A: Hepatic Y1173 EGFR phosphorylation was diminished in mice independent of diet by Aroclor 1260 exposure (p = 0.015). 3B: EGFR phosphorylation at Y1068 was diminished in mice independent of diet with Aroclor 1260 exposure (p = 0.03). 3C: EGFR phosphorylation at Y845 was diminished in mice independent of diet with Aroclor 1260 exposure (p = 0.04). A two-way ANOVA statistical test was used to determine significant variation due to diet or Aroclor 1260 exposure. # denotes significance due to Aroclor 1260 exposure, p < 0.05. Displayed is a representative blot of the effect and the graph is a representation of the densitometry results from n = 5 mice in each group.
Aroclor 1260 exposure decreased phosphorylation of the downstream effectors of hepatic EGFR signaling, Akt and mTOR in vivo
The EGFR-AKT-mTOR pathway regulates hepatic regeneration and energy metabolism. Aroclor 1260 exposure was associated with decreased hepatic Akt and mTOR phosphorylation, independent of diet as shown in Figure 4A, B. In the control diet (CD 0.79 ± 0.11 vs. CD+ 0.32 ± 0.11) and high-fat diet (HFD 0.53 ± 0.1 vs. HFD+ 0.23 ± 0.07) groups, there was a 60% and 57% decrease in Akt phosphorylation due to Aroclor 1260 exposure, respectively (p = 0.002). For mTOR phosphorylation, there was a 70% and 25% decrease in the control diet (CD 0.65 ± 0.15 vs. CD+ 0.19 ± 0.05) and high-fat diet (HFD 0.6 ± 0.21 vs. HFD+ 0.45 ± 0.11) groups with Aroclor 1260 exposure, respectively (p = 0.04).
Figure 4. Effects of Aroclor 1260 exposure to mice for 12 weeks on hepatic Akt and mTOR phosphorylation.

Mice were exposed early in week 1 with Aroclor 1260 (20 μg/kg by gavage) and western blot analysis of mouse liver conducted as previously described in Figure 3 and Materials and Methods. 4A: Hepatic Akt phosphorylation was diminished in mice with Aroclor 1260 exposure independent of diet (p = 0.002). 4B: Hepatic mTOR phosphorylation was decreased in mice with Aroclor 1260 exposure independent of diet (p = 0.04). A two-way ANOVA statistical test was used to determine significant variation due to diet or Aroclor 1260 exposure. # denotes significance due to Aroclor 1260 exposure, p<0.05. A representative blot of the effect is displayed in each panel and the graph is a representation of the results from n = 5 mice in each group.
Aroclor 1260 exposure decreased hepatic protein expression of c-Raf and STAT3 while ERK activity was reduced by high-fat diet in vivo
Hepatic protein expression of STAT3 and cRaf were decreased in an Aroclor 1260-dependent manner as shown in Figure 5A, B, respectively. STAT3 expression was decreased 84% and 66% with Aroclor 1260 exposure in the control diet (CD 0.57 ± 0.18 vs. CD+ 0.09 ± 0.06) and high-fat diet (HFD 0.31 ± 0.06 vs. HFD+ 0.11 ± 0.04) groups, respectively (p = 0.007). c-Raf expression was decreased 56% and 77% with Aroclor 1260 exposure in the control diet (CD 0.57 ± 0.18 vs. CD+ 0.09 ± 0.06) and high-fat diet (HFD 0.31 ± 0.06 vs. HFD+ 0.11 ± 0.04) groups (p = 0.001). Due to unanticipated low expression of STAT3 and cRaf with Aroclor 1260 exposure, it was not possible to evaluate potential differences in the phosphorylation status of these proteins. ERK activity was decreased slightly in a high fat diet-dependent manner as shown in Figure 5c. Mice fed a high fat diet had a 29% decrease in ERK phosphorylation compared to the untreated group (CD 0.26 ± 0.08 vs. HFD 0.19 ± 0.05) and a 78% decrease in ERK phosphorylation in the Aroclor 1260 treated group (CD+ 0.61 ± 0.24 vs. HFD+ 0.13 ± 0.02) (p = 0.04). While ERK phosphorylation was increased with Aroclor 1260 exposure in the control diet group, the increase did not attain statistical significance (p = 0.22). Quantitation for ERK1 phosphorylation is shown, but similar findings were demonstrated in the quantitation of ERK2 phosphorylation as well. In summary, hepatic c-Raf and STAT3 protein expression were decreased in a PCB-dependent manner in vivo, while ERK activity was decreased in a high fat diet-dependent manner.
Figure 5. Effects of PCB exposure and high fat-diet for 12 weeks on mouse hepatic protein expression of downstream targets of EGFR and ERK activity.

Mice were exposed to Aroclor 1260 for 12 weeks as described above and western blot analysis conducted as previously described in Materials and Methods. 5A: STAT3 protein expression was decreased in an Aroclor 1260-dependent manner independent of diet (p = 0.007). 5B: cRaf protein expression was decreased with Aroclor 1260 exposure independent of diet (p = 0.001). 5C: Phosphorylated ERK was decreased in a high fat diet-dependent manner (p = 0.04). A two-way ANOVA statistical test was used to determine significant variation due to diet or Aroclor 1260 exposure. # = significant effect of Aroclor 1260 exposure p<0.05, ## = significant effect of high fat diet p < 0.05. A representative blot of the effect is displayed in each panel and the graph is a representation of the results from n=5 mice in each group.
Aroclor 1260 exposure decreased hepatic EGFR phosphorylation in a human in vitro model
We previously demonstrated that PCBs directly activated hCAR2 and hCAR3 (Wahlang and others 2014a), however, this does not exclude simultaneous indirect CAR activation through inhibition of EGFR signaling. In HepG2 cells, EGFR phosphorylation at Y1173, Y1068, and Y845 all decreased with EGF and Aroclor 1260 co-exposure for 30 min. as shown in Figure 6A–C. Y1173 phosphorylation was decreased 61% with Aroclor 1260 and EGF co-exposure as compared to EGF alone (p = 0.01) (EGF 0.67 ± 0.13 vs. A1260 0.26 ± 0.02); EGFR inhibitor also suppressed phosphorylation of EGFR (0.12 ± 0.02). EGFR phosphorylation at Y1068 was decreased 38% with Aroclor 1260 and EGF co-exposure as compared to EGF alone, (p = 0.04) (EGF 0.95 ± 0.05 vs. A1260 0.59 ± 0.18 vs. EGFR inhibitor 0.13 ± 0.03). EGFR phosphorylation at Y845 was decreased 40% with Aroclor 1260 and EGF co-exposure as compared to EGF alone (p =0.005), (EGF 0.81 ± 0.07 vs. A1260 0.49 ± 0.04 vs. EGFR inhibitor 0.36 ± 0.06). The concentration-dependence of the inhibition of EGFR phosphorylation at Y1173 is shown in Figure 6d. The IC50 value was 4.0 ± 1.5 μg/mL for EGFR hypo-phosphorylation by Aroclor 1260. The Hill slope coefficient is 1.37 ± 0.6.
Figure 6. Decreased EGFR phosphorylation at multiple tyrosines in HepG2 cells with Aroclor 1260 exposure.

HepG2 cells were grown, treated with EGF and either EGFR inhibitor (EI) or Aroclor 1260 (A1260) for 30 min, and lysed for western blot analysis as described in Materials and Methods. 6A: EGFR phosphorylation at Y1173 was decreased by Aroclor 1260 and EGF co-exposure as compared to EGF alone (p = 0.011). 6B: EGFR phosphorylation at Y1068 was decreased by Aroclor 1260 and EGF exposure as compared to EGF alone (p = 0.04). 6C: EGFR phosphorylation at Y845 was decreased by Aroclor 1260 and EGF co-exposure as compared to EGF alone (p = 0.005). 6D: the IC50 value for EGFR hypo-phosphorylation in HepG2 cells was 4.0 ± 1.5 μg/mL. The hill slope coefficient was 1.37 ± 0.6 indicative of one binding site. A representative blot of the effect is shown in each panel and the graph represents the mean ± SEM for n = 3 experiments for Y068, Y845, and 6 experiments for Y1173. The IC50 value for Aroclor inhibition of phosphorylation was determined from 3 experiments for Y1173. A one-way ANOVA statistical test was used to compare experimental data to the positive control data. * p < 0.05, ** p < 0.01.
Specific PCB congeners decreased hepatic EGFR phosphorylation in a human in vitro model
Different PCB congeners in Aroclor 1260 had varied effects on EGFR phosphorylation in HepG2 cells as shown in (Figure 7A, B). The “phenobarbital-like” PCBs 151, 153, 170, 180, and the “dioxin-like” PCBs 3, 6, 8, 9, 126 reduced EGFR phosphorylation. The PCBs 138, 149, 187, and 174 had no significant effect. With PCB 180 and EGF co-exposure for 30 min, EGFR phosphorylation decreased 54% (p = 0.01) in contrast to EGF alone (EGF 0.95 ± 0.05 vs. PCB 180 0.43 ± 0.14 vs. negative EGF control 0.17 ± 0.1). PCB 170 exposure decreased EGFR phosphorylation 51% (p = 0.03) (PCB 170 0.46 ± 0.12). PCB 153 decreased EGFR phosphorylation 49% (p = 0.04) (PCB 153 0.49 ± 0.14). PCB 151 exposure decreased EGFR phosphorylation 57%, (p = 0.02) (PCB 151 0.41 ± 0.12). PCB 126 decreased EGFR phosphorylation 68% (p = 0.004) (PCB 126 0.3 ± 0.01). PCB 3 decreased EGFR phosphorylation 50% (p= 0.002) in contrast to EGF alone (EGF 0.9 ± 0.06 vs. PCB 3 0.48 ± 0.07 vs. negative EGF control 0.16 ± 0.07). PCB 6 decreased EGFR phosphorylation 46% (p= 0.003) (PCB 6 0.49 ± 0.1). PCB 8 decreased EGFR phosphorylation 46% (p= 0.003) (PCB 8 0.48 ± 0.07). PCB 9 decreased EGFR phosphorylation 52% (p= 0.001) (PCB 9 0.43 ± 0.05). These results demonstrate that treatment with individual PCBs found in Aroclor 1260 and some not found in the mixture (PCB 3, 9) all blunt EGFR Y1173 phosphorylation, suggesting that this may be a phenomenon caused by many PCB congeners.
Figure 7. PCB congeners 3, 6, 8, 9, 126, 151, 153, 170, and 180 elicit the greatest decrease in EGFR phosphorylation in HepG2 cells.

HepG2 cells were treated with EGF and various PCB congeners for 30 min, and subsequently lysed for western blot analysis as described in Materials and Methods. 7A: Co-exposure with PCB 180 and EGF decreased EGFR phosphorylation at Y1173 as compared to EGF alone (p = 0.02), PCB 170 (p = 0.03), PCB 153 (p = 0.04), PCB 151 (p = 0.016), PCB 126 (p = 0.004). A representative blot representation of the results of the effect is shown with the graph. The results are presented as mean ± SEM and represent the average intensities from 3 experiments. 7B: Co-exposure with PCB 3 and EGF decreased EGFR phosphorylation at Y1173 as compared to EGF alone (p = 0.002), PCB 6 (p = 0.003), PCB 8 (p = 0.003), and PCB 9 (p = 0.001). 7C: Table comparing PCB congeners characteristics and EGFR hypo-phosphorylation percentage as compared to EGF alone control. CP denotes co-planar; NCP; non co-planar. A one-way ANOVA statistical test was used to compare experimental data to the positive control data * p <0.05, ** p < 0.01.
Discussion
In this study, Aroclor 1260 exposure did not counter androstanol’s inhibitory effect on activation of mCAR in transient transfection assays. Unlike humans, mice do not express multiple splice variants of CAR but rather produce a single transcript (Lamba and others 2004). Humans express at least three splice variants, and we have previously shown that only some of these splice variants (CAR2 and CAR3) were directly activated by PCB congeners in cell-based assays (Wahlang and others 2014a). Murine CAR is homologous to human CAR1, which apparently is not directly activated by PCBs in cell-based transactivation assays (Wahlang and others 2014a). Thus, PCB-induced mCAR activation must occur solely by indirect mechanisms. Furthermore, the concept that PCBs act as indirect CAR activators would explain how PCBs elicit a CAR activation response in vivo, but cannot lessen androstanol’s inhibitory effect in vitro. This may explain why PCBs worsened NAFLD in WT or Car/Pxr−/− mice (Wahlang and others 2016), while the direct CAR activator, TCPOBOP, improved NAFLD in a diet-induced liver disease animal model (Gao and others 2009).
To study the effects of PCBs on EGFR phosphorylation, we modeled our experimental design on Negishi’s approach to investigate mechanisms of phenobarbital-induced indirect CAR activation (Mutoh and others 2013). In that study, EGFR phosphorylation was maximal 30 minutes after EGF treatment in vitro, and phenobarbital inhibited EGF binding, leading to downstream CAR activation. Likewise, we determined the effects of PCBs by adding EGF and either EGFR inhibitor or PCB co-treatments. Similar to Negishi’s group, we found that EGFR phosphorylation was determined at 30 minutes post-exposure in mouse and human hepatocyte in vitro models. Aroclor 1260 and specific PCBs, including the classically described ‘phenobarbital-like’ congener, PCB 153, reduced EGFR phosphorylation just like phenobarbital (Mutoh and others 2013). The 30 minute time course and the Hill slopes for the concentration dependence curves were consistent with PCBs being simple EGFR antagonists, again similar to phenobarbital (Mutoh and others 2013). It is important to note that human PCB poisonings from the Yucheng incident were previously linked to decreased placental EGF-stimulated EGFR phosphorylation and low birth weights associated with exposures to PCBs 153 and 170 (Sunahara and others 1987). Preliminary radiolabeled EGF binding kinetic studies indicated possible competition for a low affinity EGF binding site in PCB-exposed Yucheng vs. unexposed placentas (Sunahara and others 1987). Also within the Yucheng cohort there is a substantial increase in mortality due to chronic liver disease and diabetes in both the 13 and 30 year follow up (Li and others 2013; Yu and others 1997). These proof of concept data document the potential human relevance of our findings.
The IC50 for PCB-mediated EGFR Y1173 hypophosphorylation occurred at 1.2±0.3 μg/ml in the murine AML-12 cell line and 3.9±1.9 μg/mL in the human derived HepG2 cell line. These PCB concentrations were similar to those that directly activated hCAR2 and hCAR3 as determined by a luciferase reporter construct in transient transfection assays (Wahlang and others 2014a). These exposures were also similar to the hepatic PCB levels achieved when rodents were exposed to PCB 153 at an environmentally relevant cumulative dose of 20 mg/kg (3.7 μg/g) (NTP 2006). However, because PCBs are lipophilic, they may concentrate in a cultured cell monolayer leading to significantly higher cellular PCB concentrations than in the surrounding media. Because AML-12 cells were 3.25-fold more sensitive to EGFR inhibition by PCBs than HepG2 cells, it is also possible that PCBs are stronger indirect CAR activators in mice than in humans. In the 12-week mouse study, Aroclor 1260 (20 mg/kg) robustly induced Cyp2b10 in mice fed control diet (1000-fold), but was a much weaker inducer of Cyp2b10 in mice fed a high fat diet (4-fold) (Wahlang and others 2014b). However, basal expression of Cyp2b10 was increased in livers of mice fed high fat diet. Our results show that the decrease in EGFR phosphorylation associated with Aroclor 1260 treatment was not diet-dependent, and other factors must regulate the basal and inducible CAR activity in the mouse liver. However, the in vivo EGFR phosphorylation results are consistent with the in vitro findings. While PCBs appear to simultaneously activate hCAR by both direct and indirect mechanisms, PCBs activate mCAR only indirectly. To fully characterise the observed EGFR inhibition, ligand-binding studies with PCB congeners and the extracellular domain of EGFR should be performed in the future.
In cell culture experiments, reduction of EGFR phosphorylation occurred with Aroclor 1260, the dioxin-like congener, PCB 126, and the non-dioxin-like PCB congeners 151, 153, 170 and 180. While all the PCB congeners tested diminished EGFR phosphorylation, the most significant were among the higher chlorinated, non-coplanar congeners tested. The congener specific effect on EGFR phosphorylation is most likely due to the differences in binding affinity between these congeners to EGFR which will be evaluated in future studies. PCBs have similar structures to phenobarbital which may explain their similar modes of action through EGFR. While Aroclor 1260 also reduced EGFR phosphorylation in vivo, it did not increase expression of Cyp1a2 (Wahlang and others 2014b), suggesting that AhR was not activated at the level of Aroclor exposure in this model. While the related persistent organic pollutant, 2,3,7,8-tetrachlorodibenzodioxin (TCDD), reduced EGFR phosphorylation in an AhR-dependent manner (Lin and others 1991), this process required 12–14 hours to reach maximal effect in vitro. In contrast, our cell culture experiments employed a shorter 30-minute study, making a receptor-dependent transcriptional response unlikely. These, and other data, (Mutoh and others 2013; Sunahara and others 1987) suggest that AhR activation is not required to reduce EGFR phosphorylation. However, this does not exclude the possibility that AhR-dependent pathways could also exist for PCBs, as they do for TCDD. In fact, among tested PCB congeners, the strongest observed effect occurred for PCB 126, a potent AhR activator. Previously PCB 104 was shown to activate EGFR demonstrating that there are varied affects on EGFR based on the congener used (Eum and others 2006). However, PCB 104 was not used because it is not present in Aroclor 1260 and was detected in the serum of only 1 of 26 industrial workers (CDC 2005).
Loss of EGFR activity is implicated in both the progression of liver disease and diabetes (Bernal-Mizrachi and others 2014; Komposch and Sibilia 2016). For example, 39.6% of chemotherapy patients on gefitinib, a small molecule EGFR inhibitor, developed abnormal liver function (Wang and others 2016). Further, loss of EGFR function in humans and mice is implicated in the development of steatosis (Collin de l’Hortet and others 2014; Scheving and others 2014). EGFR activation was shown to be hepatoprotective against chemical-induced liver injury and EGF supplementation attenuated alcohol-induced liver disease (Deaciuc and others 2002; Scheving and others 2015). Loss of EGFR function has also been implicated in diabetes, as it diminishes insulin production, islet cell mass, and proliferation (Bernal-Mizrachi and others 2014; Miettinen and others 2008). The diabetes and steatohepatitis associated with Aroclor 1260-HFD co-exposures were recently characterised in mice (Wahlang and others 2016).
Decreased PI3K/AKT/mTOR signaling was also associated with type-II diabetes (Karlsson and others 2005) and increased gluconeogenesis, a common feature in NASH. mTOR is an energy sensor kinase critical in regulating hepatic energy homeostasis. PI3K/Akt are involved in the development of steatosis via their control of lipid efflux (Angrish and others 2016). It appears that alterations in the Akt/mTOR pathway, independent of CAR and PXR, may contribute to the abnormal hepatic glucose and lipid metabolism associated with steatohepatitis, and this mechanism requires further investigation. Our current results demonstrated that PCB exposure decreased Akt and mTOR phosphorylation, a finding consistent with the observation that EGFR is directly involved in PI3K phosphorylation, independent of insulin receptor signaling.
STAT3 and cRaf are other downstream targets of EGFR that were impacted by PCB treatments in the animal model. The phosphorylation status of these targets could not be determined as their expression was decreased significantly by PCBs through yet unknown mechanisms. STAT3, a critical second messenger in interleukin 6 and leptin signaling, also regulates hepatic intermediary metabolism and the mitogenic response during liver regeneration. In our HFD-fed mice, Aroclor 1260 increased interleukin 6, while leptin was increased regardless of PCB exposure (Wahlang and others 2014b). The STAT3 pathway is activated in NAFLD, worsening insulin resistance while protecting against lipotoxicity by increasing autophagy and decreasing endoplasmic stress (Min and others 2015). STAT3 polymorphisms have been associated with human NAFLD (Sookoian and others 2008). However, decreased STAT3 levels may have induced adipocytokine resistance due to disrupted intracellular signaling. This warrants further study because IL-6 deficient mice fed HFD displayed worsened NASH (Miller and others 2011), while leptin resistance is implicated in the pathogenesis of obesity and metabolic syndrome.
While cRaf protein levels were reduced by PCB exposure, phosphorylation of its downstream target, ERK, was reduced by HFD. This is a novel nutrient-toxicant interaction. In the literature, this pathway has been linked to liver regeneration and carcinogenesis, but more recently it has been found to regulate lipid metabolism through sterol regulatory element binding protein-1 (SREBP-1) phosphorylation (Knebel and others 2014). PCBs have been demonstrated to induce oxidative stress in rats and ERK activity is increased in response to oxidative stress (Czaja and others 2003; Hong and others 2015). We demonstrate that hepatic ERK activity is diminished on a HFD demonstrating that with PCB exposure and a HFD, the liver cannot properly respond to oxidative stress potentially promoting steatohepatitis. In rats exposed to 32 mg/kg PCB 153 for 5 days, Akt and ERK activity increased which is characteristic of hepatocellular carcinoma (Liu and others 2014), while we observed decreased Akt activity in mice 12 weeks after a single exposure of 20 mg/kg Aroclor 1260 per os in mice in week 1. More research is required to understand the possible congener dose, time and species-specific responses of Akt, mTOR, and ERK to PCB treatments. Furthermore, while this manuscript identifies new pathways beyond hepatic nuclear receptors by which PCBs can impact hepatic cell signaling, future studies are required to demonstrate if PCB-mediated EGFR inhibition promotes NAFLD and diabetes. Likewise, the precise mechanism by which PCBs inhibit EGFR activation is the focus of ongoing in our laboratory.
In conclusion, polychlorinated biphenyls diminish epidermal growth factor receptor signaling in both humans and mice, leading to dysregulation of critical effector kinases and transcription factors implicated in hepatic xenobiotic, glucose, and lipid metabolism (Figure 8).
Figure 8. Proposed PCB-mediated EGFR Hypo-phosphorylation Model.
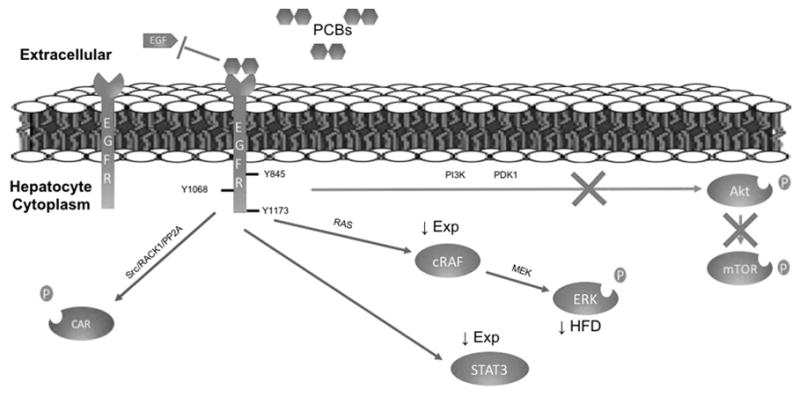
The figure represents the hypothesis that PCBs bind to EGFR preventing EGF binding, thereby preventing phosphorylation. Decreased EGF binding to EGFR leads to decreased Akt and mTOR phosphorylation downstream of EGFR. STAT3, cRaf, and ERK are downstream targets of EGFR. Chronic hypo-phosphorylation of STAT3 and cRaf leads to diminished expression either through transcription regulation or protein ubiquitination. ERK activity decreased on HFD. Phenobarbital was recently found to activate CAR through EGFR-RACK1 and PP2A (Mutoh and others 2013) and Aroclor 1260 acts in a similar manner, leading to increased xenobiotic metabolism. HFD denotes high fat diet.
Supplementary Material
Acknowledgments
The Authors thank Tom Rushmore for providing the vector containing cDNA for murine CAR.
Grant Support: Supported in part by the National Institute of Environmental Health Sciences [1R01ES021375, 1R13ES024661, F30ES025099], The National Institute of Alcohol Abuse and Alcoholism [K23AA18399] and the Centers for Disease Control and Prevention/Agency for Toxic Substances and Disease Registry [200-2013-M-57311].
Abbreviations
- Akt
protein kinase B
- AhR
aryl hydrocarbon receptor
- CAR
constitutive androstane receptor
- cRaf
serine/threonine-specific protein kinase
- DR4
direct repeat spaced by 4 nucleotides
- EGF
epidermal growth factor
- EGFR
Epidermal Growth Factor Receptor
- EI
EGFR Inhibitor
- ERK
extracellular signal-regulated kinase
- hCAR
human constitutive androstane receptor
- HFD
high fat diet
- mCar
mouse constitutive androstane receptor
- MEK
mitogen activated protein kinase
- mTOR
mechanistic target of rapamycin
- NAFLD
non-alcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NHANES
National Health and Nutrition Examination Survey
- PCB
polychlorinated biphenyl
- PI3K
phosphatidylinositol-4,5-bisphosphate 3-kinase
- PXR
pregnane X receptor
- RTK
receptor tyrosine kinase
- STAT3
signal transducer and activator of transcription 3
- TCPOBOP
1,4-Bis-[2-(3,5-dichloropyridyloxy)]benzene
Footnotes
Authorship Contributions:
Research Design: Cave, Prough, Ceresa, Falkner, Hardesty.
Conducted Experiments: Hardesty, Wahlang.
Performed data analysis: Hardesty, Wahlang.
Data Interpretation: Hardesty, Cave, Prough, Clark, Ceresa, Falkner, Clair.
Wrote or contributed to the writing of the manuscript: Hardesty, Cave, Prough, Falkner, Clair, Clark, Ceresa, Wahlang.
Conflicts of Interest: No conflicts of interest
References
- Al-Eryani L, Wahlang B, Falkner KC, Guardiola JJ, Clair HB, Prough RA, Cave M. Identification of Environmental Chemicals Associated with the Development of Toxicant-associated Fatty Liver Disease in Rodents. Toxicologic pathology. 2014 doi: 10.1177/0192623314549960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Salman F, Plant N. Non-coplanar polychlorinated biphenyls (PCBs) are direct agonists for the human pregnane-X receptor and constitutive androstane receptor, and activate target gene expression in a tissue-specific manner. Toxicol Appl Pharmacol. 2012;263:7–13. doi: 10.1016/j.taap.2012.05.016. [DOI] [PubMed] [Google Scholar]
- Aminov Z, Haase RF, Pavuk M, Carpenter DO Anniston Environmental Health Research C. Analysis of the effects of exposure to polychlorinated biphenyls and chlorinated pesticides on serum lipid levels in residents of Anniston, Alabama. Environmental health: a global access science source. 2013;12:108. doi: 10.1186/1476-069X-12-108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angrish MM, Kaiser JP, McQueen CA, Chorley BN. Tipping the Balance: Hepatotoxicity and the 4 Apical Key Events of Hepatic Steatosis. Toxicological sciences: an official journal of the Society of Toxicology. 2016;150:261–8. doi: 10.1093/toxsci/kfw018. [DOI] [PubMed] [Google Scholar]
- Bernal-Mizrachi E, Kulkarni RN, Scott DK, Mauvais-Jarvis F, Stewart AF, Garcia-Ocana A. Human beta-cell proliferation and intracellular signaling part 2: still driving in the dark without a road map. Diabetes. 2014;63:819–31. doi: 10.2337/db13-1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caron A, Richard D, Laplante M. The Roles of mTOR Complexes in Lipid Metabolism. Annual review of nutrition. 2015;35:321–48. doi: 10.1146/annurev-nutr-071714-034355. [DOI] [PubMed] [Google Scholar]
- Cave M, Appana S, Patel M, Falkner KC, McClain CJ, Brock G. Polychlorinated biphenyls, lead, and mercury are associated with liver disease in American adults: NHANES 2003–2004. Environ Health Perspect. 2010;118:1735–42. doi: 10.1289/ehp.1002720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CDC. National Report on Human Exposures to Environmental Chemicals. Centers for Disease Control; 2005. (Methylmercury results have been compared to total mercury in CDC biomonitoring.) [Google Scholar]
- Collin de l’Hortet A, Zerrad-Saadi A, Prip-Buus C, Fauveau V, Helmy N, Ziol M, Vons C, Billot K, Baud V, Gilgenkrantz H, et al. GH administration rescues fatty liver regeneration impairment by restoring GH/EGFR pathway deficiency. Endocrinology. 2014;155:2545–54. doi: 10.1210/en.2014-1010. [DOI] [PubMed] [Google Scholar]
- Czaja MJ, Liu H, Wang Y. Oxidant-induced hepatocyte injury from menadione is regulated by ERK and AP-1 signaling. Hepatology (Baltimore, Md) 2003;37:1405–13. doi: 10.1053/jhep.2003.50233. [DOI] [PubMed] [Google Scholar]
- Deaciuc IV, D’Souza NB, Burikhanov R, Lee EY, Tarba CN, McClain CJ, de Villiers WJ. Epidermal growth factor protects the liver against alcohol-induced injury and sensitization to bacterial lipopolysaccharide. Alcoholism, clinical and experimental research. 2002;26:864–74. [PubMed] [Google Scholar]
- Eum SY, Lee YW, Hennig B, Toborek M. Interplay between epidermal growth factor receptor and Janus kinase 3 regulates polychlorinated biphenyl-induced matrix metalloproteinase-3 expression and transendothelial migration of tumor cells. Molecular cancer research: MCR. 2006;4:361–70. doi: 10.1158/1541-7786.MCR-05-0119. [DOI] [PubMed] [Google Scholar]
- Falkner KC, Rushmore TH, Linder MW, Prough RA. Negative regulation of the rat glutathione S-transferase A2 gene by glucocorticoids involves a canonical glucocorticoid consensus sequence. Mol Pharmacol. 1998;53:1016–26. [PubMed] [Google Scholar]
- Gao J, He J, Zhai Y, Wada T, Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. The Journal of biological chemistry. 2009;284:25984–92. doi: 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Murinova L, Trnovec T, Loffredo CA, Washington K, Mitra PS, Dutta SK. Biomarkers linking PCB exposure and obesity. Current pharmaceutical biotechnology. 2014;15:1058–68. doi: 10.2174/1389201015666141122203509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncharov A, Bloom M, Pavuk M, Birman I, Carpenter DO. Blood pressure and hypertension in relation to levels of serum polychlorinated biphenyls in residents of Anniston, Alabama. J Hypertens. 2010;28:2053–60. doi: 10.1097/HJH.0b013e32833c5f3e. [DOI] [PubMed] [Google Scholar]
- Gschwind A, Zwick E, Prenzel N, Leserer M, Ullrich A. Cell communication networks: epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene. 2001;20:1594–600. doi: 10.1038/sj.onc.1204192. [DOI] [PubMed] [Google Scholar]
- Hong MY, Lumibao J, Mistry P, Saleh R, Hoh E. Fish Oil Contaminated with Persistent Organic Pollutants Reduces Antioxidant Capacity and Induces Oxidative Stress without Affecting Its Capacity to Lower Lipid Concentrations and Systemic Inflammation in Rats. The Journal of nutrition. 2015;145:939–44. doi: 10.3945/jn.114.206607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson HK, Zierath JR, Kane S, Krook A, Lienhard GE, Wallberg-Henriksson H. Insulin-stimulated phosphorylation of the Akt substrate AS160 is impaired in skeletal muscle of type 2 diabetic subjects. Diabetes. 2005;54:1692–7. doi: 10.2337/diabetes.54.6.1692. [DOI] [PubMed] [Google Scholar]
- Kim KS, Lee YM, Kim SG, Lee IK, Lee HJ, Kim JH, Kim J, Moon HB, Jacobs DR, Jr, Lee DH. Associations of organochlorine pesticides and polychlorinated biphenyls in visceral vs. subcutaneous adipose tissue with type 2 diabetes and insulin resistance. Chemosphere. 2014;94:151–7. doi: 10.1016/j.chemosphere.2013.09.066. [DOI] [PubMed] [Google Scholar]
- Knebel B, Lehr S, Hartwig S, Haas J, Kaber G, Dicken HD, Susanto F, Bohne L, Jacob S, Nitzgen U, et al. Phosphorylation of sterol regulatory element-binding protein (SREBP)-1c by p38 kinases, ERK and JNK influences lipid metabolism and the secretome of human liver cell line HepG2. Archives of physiology and biochemistry. 2014;120:216–27. doi: 10.3109/13813455.2014.973418. [DOI] [PubMed] [Google Scholar]
- Komposch K, Sibilia M. EGFR Signaling in Liver Diseases. International journal of molecular sciences. 2016:17. doi: 10.3390/ijms17010030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar J, Lind L, Salihovic S, van Bavel B, Ingelsson E, Lind PM. Persistent organic pollutants and liver dysfunction biomarkers in a population-based human sample of men and women. Environmental research. 2014;134:251–6. doi: 10.1016/j.envres.2014.07.023. [DOI] [PubMed] [Google Scholar]
- Lamba JK, Lamba V, Yasuda K, Lin YS, Assem M, Thompson E, Strom S, Schuetz E. Expression of constitutive androstane receptor splice variants in human tissues and their functional consequences. The Journal of pharmacology and experimental therapeutics. 2004;311:811–21. doi: 10.1124/jpet.104.069310. [DOI] [PubMed] [Google Scholar]
- Li MC, Tsai PC, Chen PC, Hsieh CJ, Leon Guo YL, Rogan WJ. Mortality after exposure to polychlorinated biphenyls and dibenzofurans: 30 years after the “Yucheng accident”. Environmental research. 2013;120:71–5. doi: 10.1016/j.envres.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin FH, Clark G, Birnbaum LS, Lucier GW, Goldstein JA. Influence of the Ah locus on the effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on the hepatic epidermal growth factor receptor. Mol Pharmacol. 1991;39:307–13. [PubMed] [Google Scholar]
- Liu C, Yang J, Fu W, Qi S, Wang C, Quan C, Yang K. Coactivation of the PI3K/Akt and ERK signaling pathways in PCB153-induced NF-kappaB activation and caspase inhibition. Toxicol Appl Pharmacol. 2014;277:270–8. doi: 10.1016/j.taap.2014.03.027. [DOI] [PubMed] [Google Scholar]
- McFarland VA, Clarke JU. Environmental occurrence, abundance, and potential toxicity of polychlorinated biphenyl congeners: considerations for a congener-specific analysis. Environ Health Perspect. 1989;81:225–39. doi: 10.1289/ehp.8981225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miettinen P, Ormio P, Hakonen E, Banerjee M, Otonkoski T. EGF receptor in pancreatic beta-cell mass regulation. Biochemical Society transactions. 2008;36:280–5. doi: 10.1042/BST0360280. [DOI] [PubMed] [Google Scholar]
- Miller AM, Wang H, Bertola A, Park O, Horiguchi N, Ki SH, Yin S, Lafdil F, Gao B. Inflammation-associated interleukin-6/signal transducer and activator of transcription 3 activation ameliorates alcoholic and nonalcoholic fatty liver diseases in interleukin-10-deficient mice. Hepatology (Baltimore, Md) 2011;54:846–56. doi: 10.1002/hep.24517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min HK, Mirshahi F, Verdianelli A, Pacana T, Patel V, Park CG, Choi A, Lee JH, Park CB, Ren S, et al. Activation of the GP130-STAT3 axis and its potential implications in nonalcoholic fatty liver disease. American journal of physiology Gastrointestinal and liver physiology. 2015;308:G794–803. doi: 10.1152/ajpgi.00390.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutoh S, Sobhany M, Moore R, Perera L, Pedersen L, Sueyoshi T, Negishi M. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci Signal. 2013;6:ra31. doi: 10.1126/scisignal.2003705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NTP. Toxicology and carcinogenesis studies of a binary mixture of 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126) (Cas No. 57465-28-8) and 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB 153) (CAS No. 35065-27-1) in female Harlan Sprague-Dawley rats (gavage studies) National Toxicology Program technical report series. 2006:1–258. [PubMed] [Google Scholar]
- Patel CJ, Bhattacharya J, Butte AJ. An Environment-Wide Association Study (EWAS) on type 2 diabetes mellitus. PLoS One. 2010;5:e10746. doi: 10.1371/journal.pone.0010746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritter R, Scheringer M, MacLeod M, Moeckel C, Jones KC, Hungerbuhler K. Intrinsic human elimination half-lives of polychlorinated biphenyls derived from the temporal evolution of cross-sectional biomonitoring data from the United Kingdom. Environ Health Perspect. 2011;119:225–31. doi: 10.1289/ehp.1002211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safe S, Bandiera S, Sawyer T, Robertson L, Safe L, Parkinson A, Thomas PE, Ryan DE, Reik LM, Levin W, et al. PCBs: structure-function relationships and mechanism of action. Environ Health Perspect. 1985;60:47–56. doi: 10.1289/ehp.856047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawyer T, Safe S. PCB isomers and congeners: induction of aryl hydrocarbon hydroxylase and ethoxyresorufin O-deethylase enzyme activities in rat hepatoma cells. Toxicology letters. 1982;13:87–93. doi: 10.1016/0378-4274(82)90142-4. [DOI] [PubMed] [Google Scholar]
- Schecter A, Colacino J, Haffner D, Patel K, Opel M, Papke O, Birnbaum L. Perfluorinated Compounds, Polychlorinated Biphenyl, and Organochlorine Pesticide Contamination in Composite Food Samples from Dallas, Texas. Environ Health Perspect. 2010 doi: 10.1289/ehp.0901347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheving LA, Zhang X, Garcia OA, Wang RF, Stevenson MC, Threadgill DW, Russell WE. Epidermal growth factor receptor plays a role in the regulation of liver and plasma lipid levels in adult male mice. American journal of physiology Gastrointestinal and liver physiology. 2014;306:G370–81. doi: 10.1152/ajpgi.00116.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheving LA, Zhang X, Stevenson MC, Threadgill DW, Russell WE. Loss of hepatocyte EGFR has no effect alone but exacerbates carbon tetrachloride-induced liver injury and impairs regeneration in hepatocyte Met-deficient mice. American journal of physiology Gastrointestinal and liver physiology. 2015;308:G364–77. doi: 10.1152/ajpgi.00364.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serdar B, LeBlanc WG, Norris JM, Dickinson LM. Potential effects of polychlorinated biphenyls (PCBs) and selected organochlorine pesticides (OCPs) on immune cells and blood biochemistry measures: a cross-sectional assessment of the NHANES 2003–2004 data. Environmental health: a global access science source. 2014;13:114. doi: 10.1186/1476-069X-13-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverstone AE, Rosenbaum PF, Weinstock RS, Bartell SM, Foushee HR, Shelton C, Pavuk M. Polychlorinated biphenyl (PCB) exposure and diabetes: results from the Anniston Community Health Survey. Environmental health perspectives. 2012;120:727–32. doi: 10.1289/ehp.1104247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookoian S, Castano G, Gianotti TF, Gemma C, Rosselli MS, Pirola CJ. Genetic variants in STAT3 are associated with nonalcoholic fatty liver disease. Cytokine. 2008;44:201–6. doi: 10.1016/j.cyto.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Sunahara GI, Nelson KG, Wong TK, Lucier GW. Decreased human birth weights after in utero exposure to PCBs and PCDFs are associated with decreased placental EGF-stimulated receptor autophosphorylation capacity. Mol Pharmacol. 1987;32:572–8. [PubMed] [Google Scholar]
- Taniguchi CM, Emanuelli B, Kahn CR. Critical nodes in signalling pathways: insights into insulin action. Nature reviews Molecular cell biology. 2006;7:85–96. doi: 10.1038/nrm1837. [DOI] [PubMed] [Google Scholar]
- Taylor KW, Novak RF, Anderson HA, Birnbaum LS, Blystone C, Devito M, Jacobs D, Kohrle J, Lee DH, Rylander L, et al. Evaluation of the association between persistent organic pollutants (POPs) and diabetes in epidemiological studies: a national toxicology program workshop review. Environ Health Perspect. 2013;121:774–83. doi: 10.1289/ehp.1205502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Falkner KC, Clair HB, Al-Eryani L, Prough RA, States JC, Coslo DM, Omiecinski CJ, Cave MC. Human receptor activation by aroclor 1260, a polychlorinated biphenyl mixture. Toxicological sciences: an official journal of the Society of Toxicology. 2014a;140:283–97. doi: 10.1093/toxsci/kfu083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Prough RA, Falkner KC, Hardesty JE, Song M, Clair HB, Clark BJ, States JC, Arteel GE, Cave MC. Polychlorinated Biphenyl-Xenobiotic Nuclear Receptor Interactions Regulate Energy Metabolism, Behavior, and Inflammation in Non-alcoholic-Steatohepatitis. Toxicological sciences: an official journal of the Society of Toxicology. 2016;149:396–410. doi: 10.1093/toxsci/kfv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlang B, Song M, Beier JI, Cameron Falkner K, Al-Eryani L, Clair HB, Prough RA, Osborne TS, Malarkey DE, Christopher States J, et al. Evaluation of Aroclor 1260 exposure in a mouse model of diet-induced obesity and non-alcoholic fatty liver disease. Toxicol Appl Pharmacol. 2014b;279:380–90. doi: 10.1016/j.taap.2014.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Wu Y, Dong M, He X, Wang Z, Li J, Wang Y. Observation of hepatotoxicity during long-term gefitinib administration in patients with non-small-cell lung cancer. Anti-cancer drugs. 2016;27:245–50. doi: 10.1097/CAD.0000000000000323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SL, Tsai PC, Yang CY, Leon Guo Y. Increased risk of diabetes and polychlorinated biphenyls and dioxins: a 24-year follow-up study of the Yucheng cohort. Diabetes Care. 2008;31:1574–9. doi: 10.2337/dc07-2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yorita Christensen KL, Carrico CK, Sanyal AJ, Gennings C. Multiple classes of environmental chemicals are associated with liver disease: NHANES 2003–2004. International journal of hygiene and environmental health. 2013 doi: 10.1016/j.ijheh.2013.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu ML, Guo YL, Hsu CC, Rogan WJ. Increased mortality from chronic liver disease and cirrhosis 13 years after the Taiwan “yucheng” (“oil disease”) incident. American journal of industrial medicine. 1997;31:172–5. doi: 10.1002/(sici)1097-0274(199702)31:2<172::aid-ajim6>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.