

Abstract
Medulloblastoma (MB) arising from the cerebellum is the most common pediatric brain malignancy, with leptomeningeal metastases often present at diagnosis and recurrence associated with poor clinical outcome. In this study, we employed mouse MB models to explore the relationship of tumor pathophysiology and dysregulated expression of the NOTCH pathway transcription factor ATOH1, which is present in aggressive MB subtypes driven by aberrant Sonic Hedgehog/Patched (SHH/PTCH) signaling. In experiments with conditional ATOH1 mouse mutants crossed to Ptch1+/− mice which develop SHH-driven MB, animals with Atoh1 transgene expression developed highly penetrant MB at a young age with extensive leptomeningeal disease and metastasis to the spinal cord and brain, resembling xenografts of human SHH MB. Metastatic tumors retained abnormal SHH signaling like tumor xenografts. Conversely, ATOH1 expression was detected consistently in recurrent and metastatic SHH MB. ChIP-seq and gene expression profiling identified candidate ATOH1 targets in tumor cells involved in development and tumorigenesis. Among these targets specific to metastatic tumors, there was an enrichment in those implicated in extracellular matrix remodeling activity, cytoskeletal network and interaction with microenvironment, indicating a shift in transcriptomic and epigenomic landscapes during metastasis. Treatment with bone morphogenetic protein (BMP) or SHH pathway inhibitors decreased tumor cell proliferation and suppressed metastatic tumor growth, respectively. Our work reveals a dynamic ATOH1-driven molecular cascade underlying MB metastasis that offers possible therapeutic opportunities.
Keywords: ATOH1, Sonic Hedgehog, medulloblastoma, leptomeningeal metastasis
Introduction
Medulloblastoma (MB) originates from the cerebellum through four molecular routes (WNT, Sonic Hedgehog [SHH], group 3, and group 4) and constitutes the most frequent pediatric brain cancer (1, 2). MB tends to metastasize via spread through the cerebrospinal fluid (CSF) to the meninge and subarachnoid space over the brain and spinal cord. Leptomeningeal metastasis, frequently found at diagnosis and associated with poor prognosis, requires radiation and cytotoxic chemotherapy associated with debilitating sequelae in survivors (3–5). Nonetheless, relapse after standard treatment occurs, often accompanied by metastasis, leading almost universally to fatal consequence (5, 6). Despite conserved subgroup identity, genetic divergence from the dominant clone in primary or naïve tumor is commonly observed at metastasis and relapse (local or metastatic recurrence) (7–9). Knowledge of molecular defects underlying metastatic disease is essential for the development of therapies specifically tailored to the biology of metastasis without detrimental effects on the developing brain.
One subgroup of MB arises from cerebellar granule neuron progenitor (GNP) following abnormal activation of the SHH pathway (10). SHH signaling are mediated by two membrane receptors: PATCHED (PTCH1) and SMOOTHENED (SMO) (11). Binding of SHH to PTCH1 relieves its suppression of SMO that subsequently orchestrates the activation of glioma-associated oncogene (GLI) transcription factors (GLI1, GLI2) and expression of GLI1, MYCN, and cyclin D1 (CCND1). PTCH1 loss or activating mutations of SMO causes SHH-driven MB, whereas several SHH pathway inhibitors are approved for the treatment of MB (12).
The NOTCH pathway transcription factor Atonal Homolog 1 (ATOH1, also known as Math1), a key regulator of cerebellar development, plays a crucial role in SHH MB (13–18). Conversely, treatment with Bone Morphogenetic Proteins (BMPs) causes post-translational degradation of ATOH1 and suppresses MB development (14–16). Here we show that metastatic and recurrent SHH MB displays abnormal ATOH1 expression, whereas ATOH1 overexpression accelerates MB development and promotes metastasis in tumor-prone Ptch1+/− mice. Using chromatin immuno-precipitation combined with high-throughput sequencing (ChIP-seq) and gene expression analyses, we identified candidate ATOH1 targets in MB. ATOH1 targets in metastatic tumor are greatly enriched for extracellular matrix remodeling activity, consistent with an enrichment for interaction with microenvironment in metastatic tumor. Treatment with BMP4 or SMO inhibitors, significantly suppresses tumor cell proliferation and metastatic tumor growth.
Materials and Methods
Animals
Animal study at Sanford Research was approved by Institutional Animal Care and Use Committee (IACUC). For animal studies performed at Institut Curie, all experimental procedures were approved by the reporting ethical committee IDFParis-Comité 1 (#2011–0012), and conducted in compliance with European and National regulatory standards (directive 86/609). Mouse strains used were described in Supplemental materials and methods. Animals were treated with 200 mg kg−1 of tamoxifen (Cayman Chemical) to induce Atoh1-creER transgene activity. Experimental animals were administered vismodegib (100 mg kg−1, LC laboratories) or vehicle daily for 14 days.
Human samples
Human samples for xenograft studies were obtained with informed consent of patients, and all experimental procedures were performed following guidelines from Institutional Review Board at Necker Hospital. Primary tumor samples were transplanted into immuno-compromised NSG mice as described (17). Cohorts of primary, recurrent, and metastatic MB samples were described previously (4, 5). All tissues were handled in compliance with International Ethical Guidelines for Biomedical Research Involving Human Subjects (CIOMS).
Animal imaging
Animals were given D-luciferin (Perkin Elmer) and imaged using In-Vivo Xtreme imaging system (Bruker) (19). For MRI, mice were scanned with 7 Tesla using vertical bore spectrometer with micro imaging accessories and 20 mm volume coil (Bruker) (17).
Cell culture
Tumor cells or GNPs were isolated as described (14). Cultured tumor cells were treated with BMP4 (100 ng/ml; R&D Systems), or cyclopamine (10 μM; LC laboratories).
X-Gal staining, immunohistochemistry, and immunofluorescence
Brains were processed by X-Gal staining as described (20). Immunostaining was carried out as described (21). Primary antibodies used include: anti-β-galactosidase (Promega), anti-GFP (Aves Lab), anti-Ki67 (BD Biosciences), anti-cyclin D1 and anti-CDKN1B (both from Santa Cruz), anti-HA, anti-Cleaved Caspase-3 and anti-Pyruvate kinase M2 (PKM2) (all from Cell Signaling Technology), anti-CD31 (abcam), anti-Atoh1 and anti-Pax6 (both from DSHB), anti-Tubulin β 3 (TUJ1, BioLegend), anti-cre, anti-NeuN and anti-GFAP (all from EMD Millipore).
Western blot
Western blot was performed as previously described (14). Antibodies used include: anti-β-actin (Sigma-Aldrich), anti-β-galactosidase (MP Cappel), anti-GFP (Aves Lab), anti-HA (abcam), and anti-ATOH1 (DSHB).
RT-qPCR, in situ hybridization, microarray, and sequencing
RT-qPCR was performed using gene-specific primers and probes (Supplemental Table 1). In situ hybridization, microarray and RNA-seq were performed as described (21). ChIP-seq was performed as described using tumor from mice (22). Experimental details and data analyses are described in Supplemental materials and methods. Array and sequencing data are available from NCBI (SuperSeries GSE98302 with the SubSeries GSE98298, GSE98299, GSE98300, and BioProject PRJNA384622).
Results
Generation of Atoh1 trangenic strains
To generate transgenic animals with inducible Atoh1 expression (referred to as CAG-A1Z and A1G), we first created several transgenic lines with the CAG-LSL-Atoh1-IRES-LacZ (CAG-A1Z) construct. In these mice, the CAG promoter drives Atoh1 and internal ribosome entry sequence/β-galactosidase (IRES/LacZ) expression after Cre-mediated removal of a loxP-flanked transcription termination (STOP) cassette between promoter and cDNA (Figure 1A; Supplemental Figure 1A). Secondly, we inserted the cDNA encoding ATOH1 with a c-terminal hemagglutinin tag (ATOH1-HA) into a Rosa26 targeting vector between the loxP-flanked Neomycin-STOP cassette and an frt-flanked cassette of IRES-driven enhanced green fluorescent protein (eGFP) (Figure 1B) (23). After electroporation of the targeting vector (A1G) into embryonic stem (ES) cells, clones with Atoh1-HA targeted into the Rosa26 locus were used to establish A1G transgenic line (Figure 1, B and C).
Figure 1. Generation of Atoh1 transgenic mice.
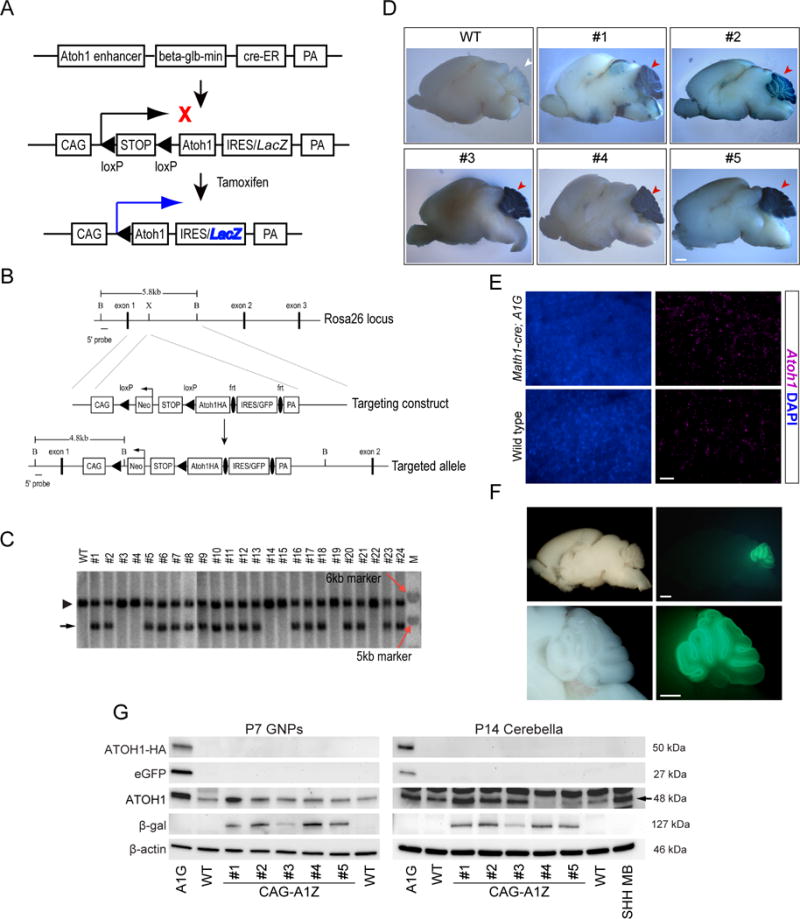
Schematic diagram of the CAG-LSL-Atoh1-IRES-LacZ vector (CAG-A1Z) (A), and targeted insertion of Atoh1-HA transgene into the Rosa26 locus (A1G) (B). PA: polyadenylation signal. Black bar: 5′ end probe used for Southern hybridization. B: BamHI; X: XbaI. (C) Southern hybridization of ES cell clones. Positive bands for wild type (WT, 5.8 kb, arrowhead) and targeted allele (4.8 kb, black arrow) are shown. M: DNA size marker (red arrows mark 5 and 6 kb bands, respectively). (D) Math1-cre;CAG-A1Z (lines #1, #2), Atoh1-creER;CAG-A1Z mice (lines #3, #4, #5) and wild type (WT) animals were treated with tamoxifen from postnatal day 1 (P1) to P3. Representative images of LacZ expression in the cerebellum at P23 were shown (red arrowheads). Scale bar, 1 mm. (E) Representative images of Atoh1 mRNA expression in the cerebella (P7) of Math1-cre;A1G and wild type (WT) animals. DAPI staining (blue) labels nuclei. Scale bar, 5 μm. (F) Whole-mount bright field (left) and fluorescent (right) images of brain from a representative Math1-cre;A1G mouse at P7. Scale bars, 1 mm. (G) Western blot analysis of transgene expression is shown in GNP (P7) or cerebella (P14) of Math1-cre;A1G (A1G), Math1-cre;CAG-A1Z (labeled CAG-A1Z, lines #1 - #5), and wild type (WT) mice. A Ptch1+/− tumor is used a positive control, whereas β-actin serves as loading control.
To direct transgene expression, we utilized Math1-cre or Atoh1-creER lines that express Cre or tamoxifen-inducible CreER, respectively, in Atoh1+ GNPs (Figure 1A) (24, 25). When crossed with CAG-A1Z or A1G mice, the resulting Math1-cre;CAG-A1Z or tamoxifen-treated Atoh1-creER;CAG-A1Z mice exhibited ATOH1/LacZ expression, whereas ATOH1-HA/eGFP expression was detected in Math1-cre;A1G mice (Figure 1, D–G; Supplemental Figure 1B). Though Math1-cre;A1G mice exhibited 3–4 fold increase in ATOH1 expression, transgene expression was more variable among CAG-A1Z strains, likely due to strain-dependent variation in transgene insertion. Nonetheless, gene expression and morphology of cerebellum in these mice are comparable to those of control animal (Supplemental Figure 2, A–C).
Atoh1 overexpression promotes MB development and metastasis
We crossed Math1-cre;A1G or Math1-cre;CAG-A1Z (CAG-A1Z line #2) mice with Ptch1+/− mice which develop partially penetrant SHH MB in 24 months (26). Interestingly, 5 out of 8 Math1-cre;CAG-A1Z;Ptch1+/− animals developed MB within 8 months that infiltrates the leptomeningeal space of the spinal cord (Figure 2A). Consistently, all Math1-cre;A1G;Ptch1+/− (MAP) mice succumbed to MB and tumors along the entire spinal cord and in other brain regions, whereas only small pockets of metastatic tumor cells were present at lumbar and sacral levels of the spinal cord in ~50% of Ptch1+/− mice with MB (Figure 2, A–C; Supplemental Figure 3, A and B; Table 1) (27). MB and tumor at other sites in Math1-cre;CAG-A1Z;Ptch1+/− and MAP mice express PAX6, Ki-67, and NeuN at levels comparable to Ptch1+/− MB (Supplemental Figure 3, A and C), indicating that Atoh1 overexpression promotes MB development and metastasis in Ptch1+/− mice.
Figure 2. Atoh1 over-expression enhances MB development and metastasis in Ptch1+/− mice.
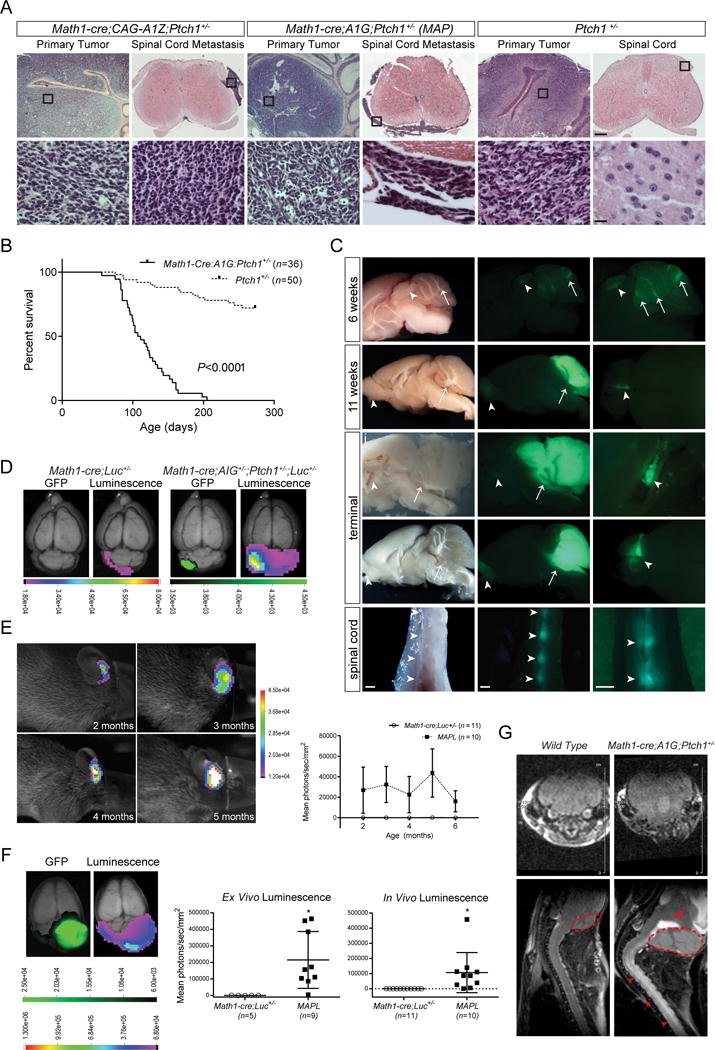
(A) Hematoxylin and eosin (H&E) staining of the cerebellum and spinal cord in Math1-cre;CAG-A1Z;Ptch1+/− (CAG-A1Z line #2), Math1-cre;A1G;Ptch1+/− (MAP), and Ptch1+/− mice. Boxed regions in representative images are magnified in bottom row. Scale bars, 250 μm (top row) and 12.5 μm (bottom row). (B) Kaplan-Meier curve depicting the survival of MAP (n=36) and Ptch1+/− (n=50) mice. (C) Representative images of whole-mount bright field (left column) and fluorescent images (middle, right columns) of brain and spinal cord from MAP mice at different time points reveal eGFP+ tumor cells in the cerebellum (arrows), and outside the cerebellum (arrowheads, magnified in the right column). Scale bars, 1 mm. (D) Ex vivo luminescence and fluorescence of representative Math1-cre;A1G+/−;Ptch1+/−;Rosa26-Luc+/− (MAPL) and control Math1-cre;Rosa26-Luc+/− (Math1-cre;Luc+/−) animals. Scale bar, photon/sec/mm2. (E) In vivo bioluminescence assay of a representative MAPL mouse at different time points. The intensity of luminescence signal is shown (MAPL mice: n=10; Math1-cre;Luc+/− mice: n=11; mean ± s.e.m.). Scale bar, photons/sec/mm2. (F) Ex vivo luminescence and fluorescence of brain from a representative terminally ill MAPL animal are shown. Scale bar,photons/sec/mm2. The intensity of luminescence is shown (MAPL mice: n=10 [ex vivo] and 9 [in vivo], Math1-cre;Luc+/− mice: n=5; mean ± s.e.m., two-tailed unpaired t-test, *P <0.05). (G) MRI of a representative MAP and an age-matched wild type animal. Transverse images of the cerebellum (top row) and sagittal images of the brain and spinal cord (bottom row) are shown. Primary tumor in the cerebellum (dotted lines) displays hemorrhage (arrow), while spinal tumor (arrowheads) is shown as enhanced signals (white) over the spinal cord.
Table 1.
Incidence of MB, leptomeningeal disease, and metastasis in mouse models.
| Strain Name | Primary Tumor (#) incidence | Leptomeningeal Disease (brain) (#) incidenceb | Brain Metastasis (#) incidenceb | Spinal Cord Metastasis (#) incidenceb | Average Age (days) |
|---|---|---|---|---|---|
| Math1-cre;A1G;Ptch1+/− (MAP) | 100% (36/36) **** | 89% (32/36) **** | 67% (24/36) **** | 100% (36/36) ****; ††† | 118 |
| Lmx1a-cre;A1G;Ptch1+/− (LAP) | 90% (17/19) **** | 88% (15/17) **** | 47% (8/17) ** | 94% (16/17) *; †† | 126 |
| Atoh1-creER;A1G;Ptch1+/− (AAP)a | 100% (7/7) *** | 57% (4/7) ns | 43% (3/7) * | 100% (7/7) *, ns | 198 |
|
| |||||
| Ptch1+/− | 28% (14/50) | 14% (2/14) | 0% (0/14) | 50% (7/14)†† | 165 |
AAP mice were treated with tamoxifen between 4–6 weeks of age.
Incidence of leptomeningeal disease and metastasis calculated as a percentage of mice with histologically verified MB in the cerebellum.
P values were calculated using Fisher’s Exact Test to compare incidence of primary tumor, leptomeningeal disease, as well as metastasis among different mouse strains
P ≤0.05;
P ≤0.01,
P ≤0.001;
P ≤0.0001; ns, non-significant.
P values were calculated using Fisher’s Exact Test to compare the incidence of brain and spinal cord metastasis in each strain
P ≤0. 01;
P ≤0.001; ns, non-significant.
At 6 weeks of age, when nascent tumor was forming in MAP mice, small clumps of Atoh1-HA+/eGFP+ tumor cells were present in the CSF (Figure 2C; Supplemental Figure 4A), indicative of leptomeningeal disease. At terminal stage, tumor in the spinal cord and brain became more established (Figure 2C; Supplemental Figure 4A). The expression of PAX6, Ki-67, NeuN, Cyclin-dependent kinase inhibitor 1B (CDKN1B), CCND1, Pyruvate Kinase M2 (PKM2), Neuron-specific class III beta-tubulin (TUJ1), Glial fibrillary acidic protein (GFAP), and cleaved caspase 3 was comparable among tumors from MAP, Ptch1+/− and Smo/Smo mice, with the latter developing MB driven by a SMO mutant (Supplemental Figure 4, B–E; Supplemental Figure 5) (28).
To monitor tumor in vivo, MAP mice were crossed with Rosa26-Luc (luciferase) reporter mice, with tumor in the resulting Math1-cre;A1G+/−;Ptch1+/−;Rosa26-Luc+/− (MAPL) mice dually labeled with eGFP and luciferase (19). Though undetectable in early tumor in vivo, luciferase signals were observed ex vivo that co-localized with eGFP (Figure 2D). Gradually, tumor-bearing mice started to exhibit luminescence in the hindbrain, whereas no signals were detected from spinal tumor identified by magnetic resonance imaging (MRI) (Figure 2, E–G).
Metastatic tumors arise from primary tumor in the cerebellum
Previous studies showed that enforced expression of ATOH1 and GLI1 promotes MB development from GNPs (17). However, normal cerebellar development in MAP mice raises question about the origin of MB and tumor in the spinal cord and brain. To address this, we treated Atoh1-creER;A1G;Ptch1+/− (AAP) mice with tamoxifen between 4–6 weeks of age to direct transgene to pre-neoplastic cells in the cerebellum of Ptch1+/− mice, the only Atoh1+ tumorigenic cells in the cerebellum, brain and spinal cord in these mice between 3 to 8 weeks of age (Supplemental Figure 6A) (26). At 10 days after tamoxifen administration, nascent ATOH1-HA+/Ki-67+ tumor cells were present in the cerebellum of AAP mice but absent from Atoh1-creER;A1G mice, whereas no ATOH1-HA+ proliferative cells were detected in the spinal cord and brain in these mice (Figure 3, A and B, and data not shown). Consistently, all tamoxifen-treated AAP mice developed MB and spinal tumor, with frequent leptomeningeal involvement and lesions in the brain (Figure 3, C and D; Supplemental Figure 6, B and C; Table 1), indicating that ATOH1 drives MB that metastasizes to secondary sites in Ptch1+/− mice.
Figure 3. Metastatic tumor originates from primary tumor in the cerebellum.
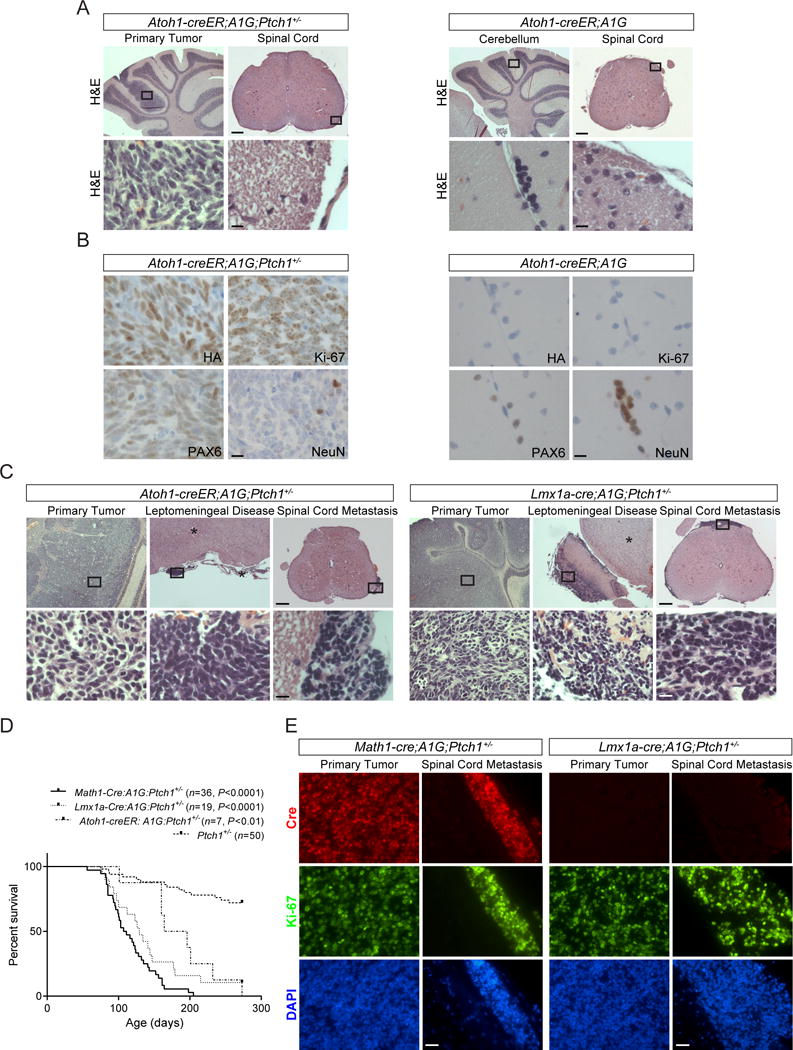
(A) H&E staining is shown for the cerebellum and spinal cord of representative Atoh1-creER;A1G;Ptch1+/− (AAP, left panel) and Atoh1-creER;A1G mice (right panel) treated with tamoxifen between 4–6 weeks of age and sacrificed 10 days later. Boxed are regions magnified in bottom rows. Scale bars, 250 μm (top rows) and 12.5 μm (bottom rows). (B) The expression of ATOH1-HA transgene (HA), PAX6, Ki-67, and NeuN in the cerebellum shown in (A). Scale bars, 12.5 μm. (C) H&E staining of the cerebellum, brain (*), and spinal cord from tamoxifen-treated AAP mice as shown in (A) but sacrificed at terminal stage (left panel), and terminally ill Lmx1a-cre;A1G;Ptch1+/− (LAP) mice (right panel). Boxed regions in representative images are shown in higher magnification (bottom rows). Scale bars, 250 μm (top rows) and 12.5 μm (bottom rows). (D) Kaplan-Meier curve depicting the survival of tamoxifen-treated AAP (n=7), LAP (n=19), MAP (n=36), and Ptch1+/− (n=50) mice. (E) Representative images of Cre (red) and Ki-67 (green) expression is shown in tumors from MAP (left panel) and LAP mice (right panel), respectively. DAPI staining (blue) labels nuclei. Scale bars, 12.5 μm.
We also utilized Lmx1a-cre transgenic mice that express Cre in roof plate adjoining Atoh1+ rhombic lip (29). Though progenitors in hindbrain roof plate expressing the transcription factor Lmx1a contribute mainly to the choroid plexus, Lmx1a+ cells comprise a source for rhombic lip progenitors (30). Consistently, Lmx1a-cre;A1G;Ptch1+/− (LAP) mice succumbed to MB with leptomeningeal invasion, and tumors in the spinal cord and brain (Figure 3, C and D; Supplemental Figure 6, B and C; Table 1). In contrast to MAP mice, Cre expression is undetectable in tumor from LAP mice (Figure 3E; Supplemental Figure 6D), suggesting that transient Cre expression in cerebellar cells derived from Lmx1a+ progenitors is sufficient to trigger ATOH1-driven MB development.
ATOH1-driven tumor development recapitulates MB metastasis in humans
We analyzed tumors from MAP and Ptch1+/− mice using RNA-seq and ChIP-seq with antibody against acetylated H3K27 (H3K27Ac). Comparison of gene profiles of tumor and wild type adult cerebellum uncovered 4760 (MAP) and 6320 (Ptch1+/−) differential transcripts that show enrichment for the SHH pathway (Figure 4A; Supplemental Table 2a–2e). Compared to the cerebellum, strong correlation of H3K27Ac binding sites was observed in primary and metastatic tumor, with consistent enrichment in association with the SHH pathway (Figure 4B; Supplemental Table 3, a and b). The expression of Atoh1, Gli1, Gli2, Mycn, Ccnd1, Ccnd2, Ptch2, and Sfrp1, was markedly increased in tumor cells, whereas Ptch1 expression was lost (Figure 4C). Therefore, similar to MB in humans, the characteristic SHH signature is maintained in ATOH1-driven metastatic disease (4–6).
Figure 4. ATOH1-driven tumor development models MB metastasis in humans.

(A) Non-hierarchical clustering of differential genes between MBs from MAP, Ptch1+/− mice, and adult wild type (WT) cerebellum (n=3; one-way ANOVA, FDR < 0.05). (B) Genome-wide H3K27Ac peaks were compared among primary and metastatic tumor from MAP mice and wild type cerebellum. Scatterplots reveal the distribution of enrichment scores across the genome separated into 10-kb segments. Numbers indicate Spearman correlation coefficients. (C) RT-qPCR analysis of gene expression in primary tumor (square), spinal metastatic tumor from MAP mice (x mark), tumor from Ptch1+/− mice (circle), and adult wild type cerebellum (triangle; mean, one-way ANOVA, *P <0.05; **P <0.01; ***P <0.001). (D) Representative H&E staining images of primary tumor (arrow) in the cerebellum (*), metastatic tumor (arrowheads) in the brain and spinal cord (*) derived from SHH PDX. The expression of ATOH1, Ki-67, PAX6, and CDKN1B is shown. Scale bar, 25 μm. (E) H&E staining and ATOH1 mRNA expression are shown for representative matched primary and recurrent (distant metastasis) SHH MB from a patient. DAPI staining (blue) labels nuclei. Scale bar, 5 μm. (F) Median FKPM (fragments per kilobase of exon per million reads mapped) values of ATOH1 transcript in SHH MB in humans (primary tumor: n=4; recurrent tumor: n=15; mean ± s.e.m., Wilcoxon rank sum test, ns, non-significant).
Conversely, patient-derived xenograft (PDX) with robust ATOH1 expression spread to the brain and spinal cord following orthotopic transplantation of SHH MB into the cerebellum of recipient (Figure 4D; Supplemental Figure 7A). Though biopsy of metastatic MB is rarely performed, analysis of published data revealed similar levels of ATOH1 transcripts in one primary-metastatic pair of SHH MB (4). ATOH1 expression was also detected in primary and recurrent (local or distant metastasis) SHH MB in humans (Figure 4, E and F; Supplemental Figure 7B). Taken together, our data support a role for ATOH1 in SHH MB metastasis.
Metastatic tumor exhibits distinct molecular properties and dependence on SHH signaling
Though previous studies using the RCAS system identified several genes involved in MB metastasis, their expression was unaltered in metastatic tumor from MAP animals (Supplemental Figure 8A) (31, 32). To dissect ATOH1-driven metastasis, we searched for ATOH1-associated genes in tumor from MAP mice using ChIP-seq with HA and H3K27Ac antibodies. Analysis of pooled data uncovered 4354 and 5209 ATOH1 peaks across the genome in primary and metastatic tumor, respectively (Supplemental Table 3, c and d). Using 6 kb genomic region around the transcription start site (TSS) as regulatory domain of a gene, we identified 1664 ATOH1-associated genes in primary tumor, most of which (1446 of 1664) and an additional 1383 genes exhibit ATOH1 binding in metastatic tumor (Figure 5A; Supplemental Table 3e–3g). Consistent with characteristics of a transcription activator, ATOH1 binding was frequently associated with H3K27Ac peaks in promoter/enhancer regions and enriched in untranslated, intronic and exon regions through consensus E-box motif (Figure 5, B–D) (33).
Figure 5. Metastatic tumor displays distinct molecular properties.
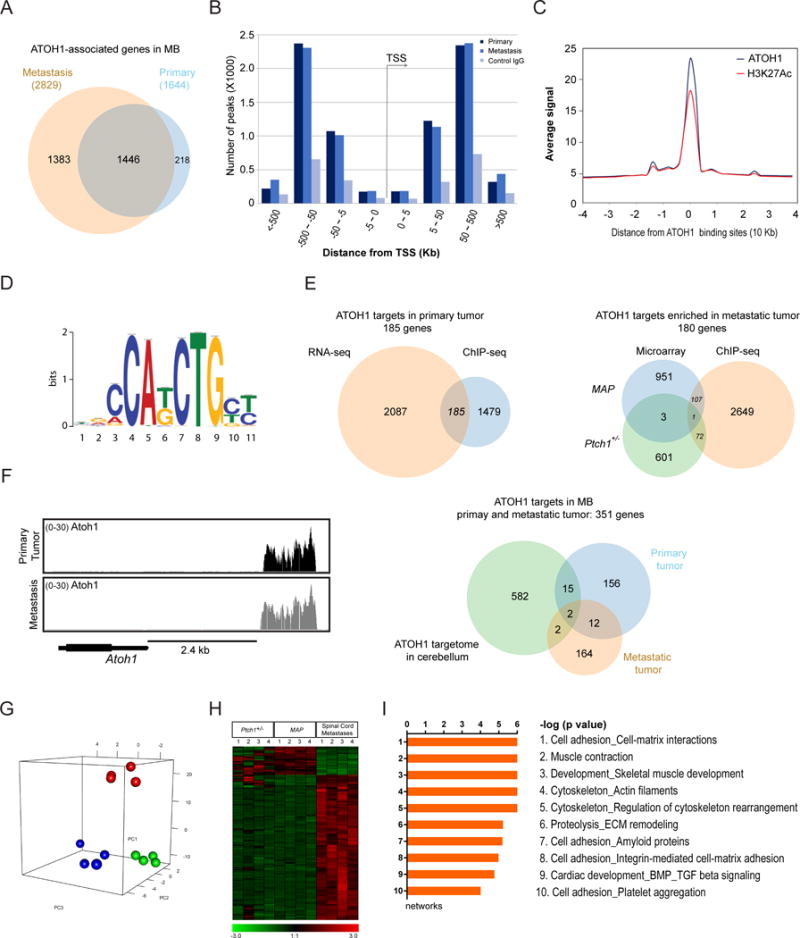
(A) Venn diagram indicates overlap of genes with ATOH1-binding peaks within 6 kb of the TSS in primary and metastatic tumor from MAP mice. (B) Distribution of ATOH1 peaks according to the distance from TSS in primary and metastatic tumor. (C) Comparison of ATOH1 and H3K27Ac signals generated from ChIP-seq fragment counts in the 40 kb genomic regions surrounding ATOH1 peaks. (D) Logos for the motif enriched in ATOH1-binding sequences identified by de novo motif analysis. (E) Venn diagrams show the overlap of ATOH1-associated genes and differentially expressed genes in primary and metastatic tumor, as well as the overlap of putative ATOH1 targets in tumor with previously identified ATOH1 targets in the cerebellum. (F) The peak density plot of fragment counts across the indicated genomic interval. Atoh1 is labeled in black with coding sequence in single exon as thick rectangle. (G) Principal component analysis of gene expression profile of primary (green dots) and spinal metastatic tumor (red dots) from MAP mice, and tumor from Ptch1+/− mice (blue dots, n=4 for each tumor group). (H) Non-hierarchical clustering of differential genes between primary and spinal metastatic tumor from MAP animals, and tumor from Ptch1+/− mice (n=4; one-way ANOVA, FDR < 0.05). (I) MetaCore analysis of primary and spinal metastatic tumor from MAP animals.
Through comparison of ATOH1-bound genes with differential transcripts from RNA-seq, we identified 185 potential ATOH1 targets in primary tumor, 156 of which were maintained in metastatic tumor, including Atoh1 itself as shown previously (Figure 5, E and F; Supplemental Table 3j) (33). Principal component analysis revealed unique molecular profile of metastatic tumor characterized by 2021 and 1992 differential genes compared to primary tumors in MAP and Ptch1+/− animals, respectively (Figure 5, G and H; Supplemental Table 3, h and i). Integrative analysis of gene profiling and ChIP-seq data uncovered 180 potential ATOH1 targets in metastatic tumor that, together with ATOH1 targets in primary tumor, comprised ATOH1 targetome in MB (351 genes, Figure 5E; Supplemental Table 3j). Analysis of candidate targets revealed an enrichment for pathways involved in cerebellar development and MB formation, including CXCR4 and actin cytoskeleton signaling pathways, whereas ATOH1 targets in metastatic tumor were significantly enriched in extracellular matrix remodeling (Supplemental Figure 8, B and C; Supplemental Table 3, k and l). Consistently, metastatic tumor exhibits an enrichment of signaling pathways regulating cytoskeleton and extracellular matrix remodeling, suggesting interaction with microenvironment (Figure 5I). RT-qPCR analysis also revealed increased expression Pdgfb and Pdgfrb in metastatic tumor, supporting a role in metastasis (Supplemental Figure 8D; Supplemental Table 2, f and g) (34, 35).
To explore potential therapies for metastatic tumor, we targeted the conserved SHH signature and ATOH1 expression. Treatment of tumor cells with either BMP4, to inhibit ATOH1, or SMO inhibitor cyclopamine, effectively suppressed tumor cell proliferation (Figure 6A). When MAP animals underwent a 2-week treatment with vismodegib, a clinically-approved SMO inhibitor (12), tumor mass and cell density along the spinal cord were significantly reduced, especially at cervical and thoracic levels (Figure 6B). Vismodegib treatment significantly decreased proliferation and markedly increased differentiation of tumor cells, whereas the expression of Gli1 and Mycn in metastatic lesions was dramatically reduced or absent in vismodegib-treated animals (Figure 6, C and D), indicating that SHH signaling is required for metastatic growth.
Figure 6. Metastatic tumor growth depends on SHH signaling.

(A) Representative images of of eGFP (green) and Ki-67 (red) expression are shown in tumor cells from MAP mice treated with BMP4 (100 ng/ml), or cyclopamine (10 μM), respectively. Scale bar, 25 μm. Quantitation of the percentage of Ki-67+ cells in eGFP+ tumor cells after 72-hour treatment as indicated is shown (n=3; mean, one-way ANOVA, *P <0.05; **P <0.01). (B) H&E staining of metastatic tumor at different levels of the spinal cord in representative MAP mice treated with vismodegib or vehicle for 2 weeks. Inset pictures of spinal cord cross sections show the boxed regions magnified. Scale bar, 12.5 μm. Quantitation of metastatic tumor cell density and burden at different levels of the spinal cord is shown (vehicle: n=5, vismodegib: n=7; mean, two-tailed unpaired t-test, *P <0.05; **P <0.01; ****P <0.0001). (C) H&E staining and the expression of Ki-67 and NeuN are shown in tumor from MAP mice as shown in (B). Boxed regions in representative images shown in top row are magnified in the lower rows. Scale bars, 250 μm (top row) and 12.5 μm (lower rows). Quantitation of the percentage of Ki-67+ or NeuN+ tumor cell is shown (vehicle: n=3, vismodegib: n=7; mean, two-tailed unpaired t-test, **P <0.01). (D) Analysis of Gli1 and Mycn mRNA expression in metastatic tumor at different levels of the spinal cord shown in (B). Boxed regions in representative images are magnified in bottom row of each panel. Bracket lines mark the border of spinal metastatic tumor. Scale bars, 250 μm (top rows) and 12.5 μm (bottom rows).
Discussion
Current efforts to understand the biology of MB metastasis and recurrence are driven by an urgent need to improve patient survival while reducing long term treatment-related toxicity (7–9). Metastatic (pre- and post-therapy) and recurrent SHH MB exhibits ATOH1 expression, whereas ATOH1 overexpression enhances MB development and metastasis, consistent with a crucial role in MB (13–18). SHH pathway regulates the transcription of ATOH1 as well as the degradation of its protein product through E3 ubiquitin ligase HUWE1 (36). HUWE1-deficient clones are present in metastatic and recurrent SHH MB in humans and mice, respectively (9, 27). In addition, ATOH1 forms a heterodimer with E-protein TCF4 to regulate the differentiation of rhombic lip progenitors (37). Loss of TCF4 is commonly observed in a mouse model of SHH MB recurrence (local and metastatic relapse), and thus may enhance ATOH1 functions in metastasis (9). Therefore, the effects of ATOH1 on MB can be achieved through transcriptional and post-translational mechanisms.
Our results show that infiltration of leptomeningeal space by tumor cells occurs at early stage of tumor development, leading to metastasis at terminal stage. The similarities in morphology, gene expression, and a conserved SHH signature in metastatic tumor in murine and PDX models, all reminiscent of SHH MB metastasis in patients, indicate that ATOH1-driven tumorigenesis provides an opportunity to study metastasis of naïve SHH MB in humans (4, 7, 38–42). However, the role of ATOH1 in recurrence (local or distant relapse) remains unclear. The development of “humanized mouse model” that receives standard therapies used in patients is necessary to answer this question.
Unlike adult wild type animals, most Ptch1+/− mice display ectopic Atoh1+ cells on cerebellar surface at 3–8 weeks after birth characterized by constitutive SHH signaling that leads to MB in a subset of animals (26). MAP mice exhibit normal cerebellar development and form nascent tumor from 6 weeks of age, suggesting that Atoh1 transgene expression drives MB development from pre-neoplastic cells in LAP or MAP mice. Consistently, enforced expression of Atoh1 or Mycn in GNPs or pre-neoplastic cells of Ptch1+/− mice leads to MB (17, 26). However, the origin of spinal tumor remains unclear due to the presence of Lmx1a+ and Atoh1+ progenitors in the spinal cord during development (43, 44). We directed Atoh1 transgene to pre-neoplastic cells through tamoxifen-induced Cre expression in adult AAP mice, when Atoh1 is absent from the spinal cord in Ptch1+/− mice (26). Nascent tumor development only in the cerebellum shortly after tamoxifen administration, together with subsequent formation of tumor in the cerebellum and spinal cord in all tamoxifen-treated AAP mice, indicates that tumor in the spinal cord and brain originates from primary tumor in the cerebellum in Ptch1+/− mice.
The role of ATOH1 in MB is mediated through transcriptional regulation of downstream targets (13, 17). We identified candidate ATOH1 targets in MB that partially overlap with ATOH1 targets in the cerebellum (33). Analysis of these targets uncovered multiple signaling pathways implicated in MB (45). Our results also revealed unique ATOH1 targets in metastatic tumor, indicating a shift in epigenomic and transcriptomic landscapes during metastasis characterized by dynamic interaction with microenvironment. Genetic divergence from dominant clone in primary or untreated tumor requires optimized treatment strategies based on targets in metastatic and recurrent compartments (8, 9, 27, 46, 47). Treatment of MAP tumors with BMP4 or SHH pathway inhibitors effectively suppresses tumor cell proliferation and metastasis, respectively, suggesting that therapies aimed at subgroup molecular susceptibility might be efficacious at metastasis and recurrence. Moreover, epigenetic and transcriptional alterations in metastatic tumor may provide actionable targets and thus warrants further investigation.
Supplementary Material
Acknowledgments
Financial support: L. Bihannic was supported by a PhD fellowship from the Ministère français de l’enseignement supérieur et de la recherche and Ligue nationale contre le cancer. O. Ayrault is supported by Marie Curie Integration grant FP7-PEOPLE-2011-CIG 294010, CNRS, Inserm, and Institut Curie. M.D. Taylor is supported by The Canadian Cancer Society Research Institute. E. Zeng is supported by the University of South Dakota Faculty Startup Fund. E. Zeng and A.H. Heglin are supported by an Institutional Development Award (IDeA) from the National Institutes of Health (NIH) under grant number P20GM103443 and South Dakota BioSystems Networks & Translational Research through the National Science Foundation/EPSCoR Award #IIA-1335423. H. Zhao is supported by Sanford Research, Matthew Larson Foundation, and IDeA from the NIH under grant number 5P20GM103548 (Cancer), and 1P20GM103620-01A1 (Pediatrics). H. Zhao and R.J. Bram are supported by T. Denny Sanford Pediatric Collaborative Research Grant.
We thank members of the laboratory for helpful discussions. We are grateful to Dr. Kathleen Millen (Seattle Children’s Hospital Research Institute, USA) and Dr. Ryoichiro Kageyama (Institute for Virus Research Kyoto University, Japan) for providing Lmx1a-Cre mouse strain and pCI-RAGE-Math1-IRES-LacZ vector, respectively. We are indebted to Claire Evans, Jessica Zylla, Sara Maria Cigna, Sophie Leboucher, Coralie Lefevre, Christophe Roulin, Hamasseh Shirvani, and Hua Yu for excellent technical assistance.
References
- 1.Northcott PA, Korshunov A, Pfister SM, Taylor MD. The clinical implications of medulloblastoma subgroups. Nature reviews Neurology. 2012;8(6):340–51. doi: 10.1038/nrneurol.2012.78. [DOI] [PubMed] [Google Scholar]
- 2.Gilbertson RJ, Ellison DW. The origins of medulloblastoma subtypes. Annual review of pathology. 2008;3:341–65. doi: 10.1146/annurev.pathmechdis.3.121806.151518. [DOI] [PubMed] [Google Scholar]
- 3.Aref D, Croul S. Medulloblastoma: recurrence and metastasis. CNS oncology. 2013;2(4):377–85. doi: 10.2217/cns.13.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang X, Dubuc AM, Ramaswamy V, Mack S, Gendoo DM, Remke M, et al. Medulloblastoma subgroups remain stable across primary and metastatic compartments. Acta neuropathologica. 2015;129(3):449–57. doi: 10.1007/s00401-015-1389-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ramaswamy V, Remke M, Bouffet E, Faria CC, Perreault S, Cho YJ, et al. Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. The lancet oncology. 2013;14(12):1200–7. doi: 10.1016/S1470-2045(13)70449-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zollo M. Genetics of recurrent medulloblastoma. The lancet oncology. 2013;14(12):1147–8. doi: 10.1016/S1470-2045(13)70482-0. [DOI] [PubMed] [Google Scholar]
- 7.Hill RM, Kuijper S, Lindsey JC, Petrie K, Schwalbe EC, Barker K, et al. Combined MYC and P53 defects emerge at medulloblastoma relapse and define rapidly progressive, therapeutically targetable disease. Cancer cell. 2015;27(1):72–84. doi: 10.1016/j.ccell.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Ramaswamy V, Remke M, Mack SC, Dubuc AM, Northcott PA, et al. Intertumoral and intratumoral heterogeneity as a barrier for effective treatment of medulloblastoma. Neurosurgery. 2013;60(Suppl 1):57–63. doi: 10.1227/01.neu.0000430318.01821.6f. [DOI] [PubMed] [Google Scholar]
- 9.Morrissy AS, Garzia L, Shih DJ, Zuyderduyn S, Huang X, Skowron P, et al. Divergent clonal selection dominates medulloblastoma at recurrence. Nature. 2016;529(7586):351–7. doi: 10.1038/nature16478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Blanpain C. Tracing the cellular origin of cancer. Nature cell biology. 2013;15(2):126–34. doi: 10.1038/ncb2657. [DOI] [PubMed] [Google Scholar]
- 11.Rohatgi R, Scott MP. Patching the gaps in Hedgehog signalling. Nature cell biology. 2007;9(9):1005–9. doi: 10.1038/ncb435. [DOI] [PubMed] [Google Scholar]
- 12.Amakye D, Jagani Z, Dorsch M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nature medicine. 2013;19(11):1410–22. doi: 10.1038/nm.3389. [DOI] [PubMed] [Google Scholar]
- 13.Flora A, Klisch TJ, Schuster G, Zoghbi HY. Deletion of Atoh1 disrupts Sonic Hedgehog signaling in the developing cerebellum and prevents medulloblastoma. Science. 2009;326(5958):1424–7. doi: 10.1126/science.1181453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao H, Ayrault O, Zindy F, Kim JH, Roussel MF. Post-transcriptional down-regulation of Atoh1/Math1 by bone morphogenic proteins suppresses medulloblastoma development. Genes & development. 2008;22(6):722–7. doi: 10.1101/gad.1636408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grimmer MR, Weiss WA. BMPs oppose Math1 in cerebellar development and in medulloblastoma. Genes & development. 2008;22(6):693–9. doi: 10.1101/gad.1657808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dubuc AM, Northcott PA, Kenney AM, Taylor MD. Calculating a cure for cancer: managing medulloblastoma MATH1-ematically. Expert review of neurotherapeutics. 2010;10(10):1489–92. doi: 10.1586/ern.10.126. [DOI] [PubMed] [Google Scholar]
- 17.Ayrault O, Zhao H, Zindy F, Qu C, Sherr CJ, Roussel MF. Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer research. 2010;70(13):5618–27. doi: 10.1158/0008-5472.CAN-09-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Briggs KJ, Eberhart CG, Watkins DN. Just say no to ATOH: how HIC1 methylation might predispose medulloblastoma to lineage addiction. Cancer research. 2008;68(21):8654–6. doi: 10.1158/0008-5472.CAN-08-1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Safran M, Kim WY, Kung AL, Horner JW, DePinho RA, Kaelin WG., Jr Mouse reporter strain for noninvasive bioluminescent imaging of cells that have undergone Cre-mediated recombination. Molecular imaging. 2003;2(4):297–302. doi: 10.1162/15353500200303154. [DOI] [PubMed] [Google Scholar]
- 20.Zhao H, Yang Y, Rizo CM, Overbeek PA, Robinson ML. Insertion of a Pax6 consensus binding site into the alphaA-crystallin promoter acts as a lens epithelial cell enhancer in transgenic mice. Investigative ophthalmology & visual science. 2004;45(6):1930–9. doi: 10.1167/iovs.03-0856. [DOI] [PubMed] [Google Scholar]
- 21.Li L, Grausam KB, Wang J, Lun MP, Ohli J, Lidov HG, et al. Sonic Hedgehog promotes proliferation of Notch-dependent monociliated choroid plexus tumour cells. Nature cell biology. 2016;18(4):418–30. doi: 10.1038/ncb3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z, Merkurjev D, Yang F, Li W, Oh S, Friedman MJ, et al. Enhancer activation requires trans-recruitment of a mega transcription factor complex. Cell. 2014;159(2):358–73. doi: 10.1016/j.cell.2014.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell. 2007;131(1):146–59. doi: 10.1016/j.cell.2007.07.021. [DOI] [PubMed] [Google Scholar]
- 24.Matei V, Pauley S, Kaing S, Rowitch D, Beisel KW, Morris K, et al. Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Developmental dynamics : an official publication of the American Association of Anatomists. 2005;234(3):633–50. doi: 10.1002/dvdy.20551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Machold R, Fishell G. Math1 is expressed in temporally discrete pools of cerebellar rhombic-lip neural progenitors. Neuron. 2005;48(1):17–24. doi: 10.1016/j.neuron.2005.08.028. [DOI] [PubMed] [Google Scholar]
- 26.Kessler JD, Hasegawa H, Brun SN, Emmenegger BA, Yang ZJ, Dutton JW, et al. N-myc alters the fate of preneoplastic cells in a mouse model of medulloblastoma. Genes & development. 2009;23(2):157–70. doi: 10.1101/gad.1759909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Northcott PA, Dubuc A, Dupuy AJ, Shih DJ, Witt H, et al. Clonal selection drives genetic divergence of metastatic medulloblastoma. Nature. 2012;482(7386):529–33. doi: 10.1038/nature10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer research. 2008;68(6):1768–76. doi: 10.1158/0008-5472.CAN-07-5092. [DOI] [PubMed] [Google Scholar]
- 29.Chizhikov VV, Lindgren AG, Mishima Y, Roberts RW, Aldinger KA, Miesegaes GR, et al. Lmx1a regulates fates and location of cells originating from the cerebellar rhombic lip and telencephalic cortical hem. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(23):10725–30. doi: 10.1073/pnas.0910786107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng FY, Huang X, Sarangi A, Ketova T, Cooper MK, Litingtung Y, et al. Widespread contribution of Gdf7 lineage to cerebellar cell types and implications for hedgehog-driven medulloblastoma formation. PloS one. 2012;7(4):e35541. doi: 10.1371/journal.pone.0035541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mumert M, Dubuc A, Wu X, Northcott PA, Chin SS, Pedone CA, et al. Functional genomics identifies drivers of medulloblastoma dissemination. Cancer research. 2012;72(19):4944–53. doi: 10.1158/0008-5472.CAN-12-1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jenkins NC, Kalra RR, Dubuc A, Sivakumar W, Pedone CA, Wu X, et al. Genetic drivers of metastatic dissemination in sonic hedgehog medulloblastoma. Acta neuropathologica communications. 2014;2:85. doi: 10.1186/s40478-014-0085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klisch TJ, Xi Y, Flora A, Wang L, Li W, Zoghbi HY. In vivo Atoh1 targetome reveals how a proneural transcription factor regulates cerebellar development. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(8):3288–93. doi: 10.1073/pnas.1100230108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacDonald TJ, Brown KM, LaFleur B, Peterson K, Lawlor C, Chen Y, et al. Expression profiling of medulloblastoma: PDGFRA and the RAS/MAPK pathway as therapeutic targets for metastatic disease. Nature genetics. 2001;29(2):143–52. doi: 10.1038/ng731. [DOI] [PubMed] [Google Scholar]
- 35.Gilbertson RJ, Langdon JA, Hollander A, Hernan R, Hogg TL, Gajjar A, et al. Mutational analysis of PDGFR-RAS/MAPK pathway activation in childhood medulloblastoma. Eur J Cancer. 2006;42(5):646–9. doi: 10.1016/j.ejca.2005.11.023. [DOI] [PubMed] [Google Scholar]
- 36.Forget A, Bihannic L, Cigna SM, Lefevre C, Remke M, Barnat M, et al. Shh signaling protects Atoh1 from degradation mediated by the E3 ubiquitin ligase Huwe1 in neural precursors. Developmental cell. 2014;29(6):649–61. doi: 10.1016/j.devcel.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 37.Flora A, Garcia JJ, Thaller C, Zoghbi HY. The E-protein Tcf4 interacts with Math1 to regulate differentiation of a specific subset of neuronal progenitors. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(39):15382–7. doi: 10.1073/pnas.0707456104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kumar S, Handa A, Jha DK, Choudhary A. Supratentorial metastasis of medulloblastoma in adults. Asian journal of neurosurgery. 2016;11(3):320. doi: 10.4103/1793-5482.149993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Garrity JA, Herman DC, Dinapoli RP, Waller RR, Campbell RJ. Isolated metastasis to optic nerve from medulloblastoma. Ophthalmology. 1989;96(2):207–10. doi: 10.1016/s0161-6420(89)32912-5. [DOI] [PubMed] [Google Scholar]
- 40.Gerlach R, Kieslich M, van de Nes J, Galow W, Seifert V. Supratentorial leptomeningeal metastasis of a medulloblastoma without cerebellar tumor recurrence. Acta neurochirurgica. 2002;144(2):201–4. doi: 10.1007/s007010200025. discussion 4. [DOI] [PubMed] [Google Scholar]
- 41.Garcia Escrig M, Diaz Guzman J, Soto Tellez O, Simon de las Heras R, Mateos Beato F, Munoz A, et al. Cerebellar medulloblastoma in childhood: supratentorial metastasis. Neurologia. 1993;8(4):131–4. [PubMed] [Google Scholar]
- 42.George RE, Laurent JP, McCluggage CW, Cheek WR. Spinal metastasis in primitive neuroectodermal tumors (medulloblastoma) of the posterior fossa: evaluation with CT myelography and correlation with patient age and tumor differentiation. Pediatric neuroscience. 1985;12(3):157–60. doi: 10.1159/000120239. [DOI] [PubMed] [Google Scholar]
- 43.Helms AW, Johnson JE. Progenitors of dorsal commissural interneurons are defined by MATH1 expression. Development. 1998;125(5):919–28. doi: 10.1242/dev.125.5.919. [DOI] [PubMed] [Google Scholar]
- 44.Chizhikov VV, Millen KJ. Control of roof plate formation by Lmx1a in the developing spinal cord. Development. 2004;131(11):2693–705. doi: 10.1242/dev.01139. [DOI] [PubMed] [Google Scholar]
- 45.Ward SA, Rubin JB. Not so Fast: Co-Requirements for Sonic Hedgehog Induced Brain Tumorigenesis. Cancers (Basel) 2015;7(3):1484–98. doi: 10.3390/cancers7030848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Huang X, He Y, Dubuc AM, Hashizume R, Zhang W, Reimand J, et al. EAG2 potassium channel with evolutionarily conserved function as a brain tumor target. Nature neuroscience. 2015;18(9):1236–46. doi: 10.1038/nn.4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faria CC, Golbourn BJ, Dubuc AM, Remke M, Diaz RJ, Agnihotri S, et al. Foretinib is effective therapy for metastatic sonic hedgehog medulloblastoma. Cancer research. 2015;75(1):134–46. doi: 10.1158/0008-5472.CAN-13-3629. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.