

Abstract
A variety of oligomeric backbones with compositions deviating from biomacromolecules can fold in defined ways. Termed “foldamers,” these agents have diverse potential applications. A number of protein-inspired secondary structures (e.g., helices, sheets) have been produced from unnatural backbones, yet examples of tertiary folds combining several secondary structural elements in a single entity are rare. One promising strategy to address this challenge is the systematic backbone alteration of natural protein sequences, through which a subset of the side chains is displayed on an unnatural building block to generate a heterogeneous backbone. A drawback to this approach is that substitution at more than one or two sites often comes at a significant energetic cost to fold stability. Here we report heterogeneous-backbone foldamers that mimic the zinc finger domain, a ubiquitous and biologically important metal-binding tertiary motif, and do so with a folded stability that is superior to the natural protein on which their design is based. A combination of UV-vis spectroscopy, isothermal titration calorimetry, and multidimensional NMR reveals that suitably designed oligomers with >20% modified backbones can form native-like tertiary folds with metal-binding environments identical to the prototype sequence (the third finger of specificity factor 1) and enhanced thermodynamic stability. These results expand the scope of heterogeneous-backbone foldamer design to a new tertiary structure class and show that judiciously applied backbone modification can be accompanied by improvement to fold stability.
Graphical Abstract
INTRODUCTION
The characteristic of proteins that enables their varied and sophisticated biological roles is the ability to fold into compact, well-ordered conformations encoded by amino acid sequence. Biomacromolecules are not unique in having the capacity for sequence-encoded folding, and diverse oligomeric backbones of alternate chemical compositions can show discrete folding propensities. The bulk of research on this class of molecules, termed “foldamers,”1 has focused on secondary structures (helices, sheets, and turns) that are either inspired by motifs found in nature or completely abiotic in origin.2 More recent work has sought to create unnatural backbones mimic target folds with increased intricacy, such as multi-helix quaternary assemblies,3 multi-stranded sheets,4 and unimolecular tertiary structures.5 The design of such molecules remains a significant challenge, the magnitude of which scales with the complexity of the desired fold.
While tertiary structure is crucial to protein function, so too in many cases is the ability to bind a cognate ligand. It has been estimated that a third of all proteins bind to some metal ion and half of enzymes require a specific metal to function.6 Despite this ubiquity in nature, examples of metal binding in foldamers represent only a small fraction of the many unnatural folded units reported to date.7 Most examples have been based on oligomers with metal-coordinating backbones,8 while some have shown metal binding is possible in designed oligomers with peptide-based backbones closer to those found in nature.9
A powerful strategy for designing molecules that mimic sophisticated protein folds is the systematic backbone alteration of biologically derived sequences – inducing a natural arrangement of side chains to manifest a native-like fold on a backbone consisting of a blend of natural and unnatural building blocks.10 Such heterogeneous-backbone oligomers can resist degradation by protease enzymes, depending on the nature of the modification,11 and have potential applications in areas ranging from therapeutic12 and diagnostic13 to entirely novel functions.14 Foundational work led to robust design principles for mimicry of α-helix and β-sheet secondary structures by this approach. More recent efforts have expanded to tertiary structure contexts, specifically a disulfide-cyclized helix-turn-helix motif derived from Staphylococcal protein A13,15 and the α/β tertiary fold of the B1 domain of Streptococcal protein G (GB1).4c,5d,16
As noted above, protein backbone alteration can be accompanied by functional benefits; however, these benefits are almost always offset by compensating penalties to folded stability when more than one or two residues are altered.17 The degree to which these observations may prove general vs. system dependent is not clear from precedent. Is it the case that natural side-chain sequences, finely honed by evolution to encode folding in an α-peptide context, will always function best on a natural backbone? Alternatively, is it possible that certain significantly modified backbones may surpass nature in the capacity to manifest complex folds encoded by biologically-derived sequences? The best way to answer these questions is to expand heterogeneous-backbone foldamer design to a wider array of tertiary folds. Here, we report efforts to target the zinc finger, a ubiquitous motif in nature where folding and metal binding are intimately coupled. Examination of backbone-modified variants of a biologically derived domain found oligomers with >20% unnatural content that are identical to the native in folded structure and superior in folded stability.
Zinc fingers make up the largest class of eukaryotic proteins18 and are estimated to be encoded by 3% of human genes.19 The folding of these motifs is driven by and entirely dependent upon binding to a Zn2+ ion. Zinc finger domains are typically arrayed in tandem as part of larger DNA-binding proteins, giving rise to exquisite sequence-specific recognition of DNA through docking into the major groove of the double helix. As a modular protein domain, there has been a great deal of progress in designing and linking individual zinc fingers to engineer larger DNA-binding proteins able to modulate expression of specific genes.20 Notable examples include designer zinc fingers that inhibit the replication of HIV21 and others that stimulate angiogenesis.22 Moreover, the inherent cell permeability of zinc fingers has paved the way for their use as a delivery system.23
Zinc fingers are categorized by the tertiary folding motif and identity of residues coordinating the metal ion. The third finger of specificity factor 1 (Sp1–3) is a classical example (Figure 1). Sp1–3 is a 29-residue sequence that, in the presence of Zn2+, adopts a ββα tertiary fold supporting a metal coordination site consisting of two Cys residues and two His residues (Cys2His2, Figure 1B).24 The fold is further stabilized by a conserved hydrophobic core that includes two Phe and one Leu residue. We chose Sp1–3 as the prototype zinc finger in which to investigate sequence-guided backbone modification, detailed below.
Figure 1.

(A) Primary sequence and secondary structure map of the Sp1–3 zinc finger, with metal-coordinating residues underlined. (B) NMR structure of Sp1–3 (PDB 1SP1) with close-up views of the Cys2His2 metal-binding environment (C) and β-turn between the two strands (D).
RESULTS AND DISCUSSION
Design of Backbone-Modified Sp1–3 Analogues, Synthesis, and Assessment of Metal Binding
Peptide 1 (Figure 2) served as the starting point for backbone modification and as a control for experiments aimed at characterization of folding and metal binding in the zinc finger mimics. Based on Sp1–3, it differs from the native sequence in a Phe2→Tyr substitution made to facilitate spectroscopic concentration determination. To explore the relationship between backbone composition and folding, we prepared eight analogues (2–9) of the parent Sp1–3 sequence (Figure 2). Collectively, these oligomers incorporate a variety unnatural building blocks in place of a subset of the α-residues in 1: D-α-, N-Me-α-, Cα-Me-α-, β-, and δ-residues. In terms of composition, 21–25% of the backbone in each sequence is altered. Design rationales behind the patterns of backbone substitution applied in each oligomer (Figure 3) are further detailed below. Relative to peptide 1, an additional Met12→Nle mutation was made to reduce complications arising from oxidation of this residue during synthesis, purification, and characterization. Peptides 1–9 were synthesized by microwave-assisted solid-phase methods, purified by reverse-phase HPLC, with identity and purity of final products confirmed by MALDI mass spectrometry and analytical HPLC (see Supporting Information [SI] for details).
Figure 2.

Primary sequence of peptide 1 and backbone-modified analogues 2–9 (B = norleucine). Bold residues indicate positions where the backbone is altered. For residue types with “R” groups, the identity of the side chain is that of the corresponding α-residue denoted by the single letter code found in the sequence.
Figure 3.

Summary of backbone substitutions applied in the design of oligomers 2–9. Dotted lines indicate hydrogen bonds expected in the canonical secondary structure indicated.
As a primary assay for folding and metal binding, we carried out UV-vis spectroscopy in the presence of Co2+. Substitution of the native Zn2+ ligand in this way gives rise to spectroscopic features highly sensitive to metal-binding environment and is a common tool in the study of zinc-binding proteins.25 Thus, we acquired the spectrum of a 100 μM solution of peptide in 20 mM HEPES, pH 7.0, with a slight excess of Co2+ (Figure 4). The data obtained for 1 are consistent with prior published results for Sp1–3, indicating the small sequence change did not alter the fold.26 Bands in the 300–350 nm range result from thiolate-to-Co2+ ligand-to-metal charge-transfer transitions with intensities correlating to the number of coordinating Cys residues.27 Diagnostic d-d absorption bands in the 500–700 nm region arise from a tetrahedral cobalt(II) species, and the shape, intensity, and position of these peaks are diagnostic of coordination residue type and number.28 Thus, the observation of peaks at 640 nm and 570 nm in a 3:1 ratio for 1 is consistent with the expected Cys2His2 tetrahedral metal binding site.
Figure 4.

Interaction of 1–9 with Co2+ as determined by UV-vis spectroscopy. Measurements were carried out in a 1 cm path length cell at 100 μM peptide concentration in 20 mM HEPES pH 7.0 with 1.3 equiv of Co2+ (2 equiv Co2+ for peptide 5).
The design of analogues 2 and 3 was based on our prior published observations for backbone modification strategies most effective at mimicry of the GB1 tertiary structure.4c,5d,16 A single α→N-Me-α substitution was made in each strand of the hairpin segment at sites where the newly introduced methyl group should project toward solvent. α→β3 substitutions were incorporated in each turn of the α-helix, such that added CH2 groups in the backbone reside opposite the hydrophobic core of the tertiary fold. For both the N-Me-α- and β3-residues, the native Sp1–3 side chain was retained at each point of modification. In place of the central residues of the β-turn (Pro5Glu6), we incorporated one of two unnatural turn inducers in the form of an XxxGly sequence, where Xxx is either D-Pro or the Cα-Me-α-residue Aib.
The UV signal around 300 nm was retained for 2 and 3 in the presence of Co2+, suggesting interaction between Cys residues and the metal. However, the absence of peaks in the visible region indicated the well-defined metal coordination environment of native Sp1–3, and thus the tertiary fold, was abolished upon backbone modification.
In considering the inability of 2 and 3 to mimic the parent zinc finger fold, several considerations drew our attention to the turn region. First, it is located directly between two metal-binding residues (Cys4 and Cys7); small changes in local conformation could alter alignment of these side chains and their propensity to bind metal in the folded state. Second, analysis of the NMR structure of Sp1–3 indicates the backbone reversal is best classified as a “double turn,”29 where a type-I turn encompassing Cys4–Cys7 overlaps with a type-VIII turn encompassing Pro5–Pro8 (Figure S2). The unnatural turn replacements employed in 2 and 3 are proven substitutions for canonical type-I and type-II turns;30 however, they tend to promote mirror image type-I’ and II’ turn conformations that may be incompatible with the overlapping type-VIII turn directly following in Sp1–3. Finally, the side chain of Glu6 is lost in the two analogues. This residue is found near the zinc ion in the NMR structure,25 where it may contribute as a second sphere ligand and/or stabilize the metal binding site through interaction with His24.
To test the hypothesis that the modified turns were responsible for the inability of 2 and 3 to fold, we synthesized peptide 4. This oligomer retains most of the backbone alterations present in 2 and 3 but restores the turn to its native composition. In the UV-vis assay with Co2+, peptide 4 showed spectral features virtually identical to the parent domain 1. This result, along with further experiments detailed below, provides compelling evidence for a native-like tertiary structure.
With evidence that the Aib-Gly and D-Pro-Gly segments in 2 and 3 were incompatible with the Sp1–3 fold, we explored other modifications to the turn region (5–8). Peptides 5 and 6 were designed to examine whether the turn conformation might be the leading determinant against folding. Each incorporates a δ-residue as a dipeptide replacement in the center of the type-I turn encompassing Pro5–Pro8. Peptide 5 employs ornithine connected via its side chain to the preceding residue (δO), a well-established turn inducer.31 Peptide 6 is an analogue of 5 with a less constrained, achiral δ-residue (δX). We reasoned the enhanced flexibility of δX relative to δO might allow the two flanking Cys residues to adopt an appropriate metal binding geometry. Peptides 7 and 8 restore the native Glu6 side chain to 2 and 3, respectively, and were designed to test the hypothesis that the loss of this side chain accompanying backbone modification was responsible for the inability to fold.
Analysis of the Co2+ binding characteristics of peptide 5–8 by UV-vis did not show any evidence of a defined metal coordination environment and, thus, argues against any of these oligomers adopting a native-like tertiary fold. As with 2 and 3, ligand-to-metal-charge-transfer transitions are observed in all cases; however, none of the modified backbones show peaks characteristic of Cys2His2 tetrahedral coordination. Peptide 8 with the Aib-Glu turn did show some signal ~640 nm, but the low intensity of this peak and broad background across the visible spectrum suggest significant heterogeneity in any interaction of the peptide with Co2+.
Collectively, the inability of any of the turn modifications examined to be tolerated in Sp1–3 highlights the challenges of chemical protein backbone alteration. Nevertheless, we were heartened by the promising results obtained for peptide 4 with modifications throughout the helix and hairpin. To further explore relationships between backbone composition and folding in the zinc finger motif, we prepared peptide 9, an analogue of 4 where two of the β3-residues in the helix are replaced by the Cα-Me-α-residue Aib. We recently reported a stabilizing effect from β3→Cα-Me-α substitution in the helix of the GB1 tertiary fold and were motivated to see to what degree those observations might hold in a different structural context.16b The data obtained in the Co2+ binding assay showed that peptide 9 supports a metal coordination environment indistinguishable from both analogue 4 and the native backbone (1). Thus, we advanced peptides 1, 4, and 9 to experiments aimed at a more thorough characterization of folded structure and stability.
Thermodynamics of Folding and Metal Binding
Zinc finger domain folding and metal binding are closely coupled in a single overall process. For a series of related sequences with consistent metal binding sites, contributions from peptide-metal interactions and accompanying proton transfer to buffer are similar; differences in observed energetics within the series are then dominated by differences in folding energetics.32 Thus, comparison of metal-binding thermodynamics between wild-type peptide 1 and analogues 4 and 9 has the capacity to shed light specifically on the energetic consequences of backbone modification to folding of the zinc finger domain.
We first examined the binding affinity of each peptide for Co2+ by monitoring the intensity of the UV-vis peak at 620 nm as a function of added metal. The resulting binding isotherms (Figure 5) revealed dissociation constants (Kd) ~2 μM for wild-type sequence 1 as well as backbone-modified 4 and 9 (Table 1). These values are comparable to literature precedent for Sp1–3,26,33 indistinguishable within the uncertainty of the measurement, and at the limit of the affinity detectable under the conditions of the experiment. Some deviation from the expected curve for 1:1 binding stoichiometry was observed in the fitting. This behavior has been previously reported for other Cys2His2 zinc fingers and is attributed to a process in which multiple peptide chains bind Co2+ and compete with the formation of the 1:1 complex.25,34 Based on the relative dissociation constants for this competing pathway (Kd2), the expected 1:1 complex dominates at all but the lowest Co2+:peptide ratios.
Figure 5.
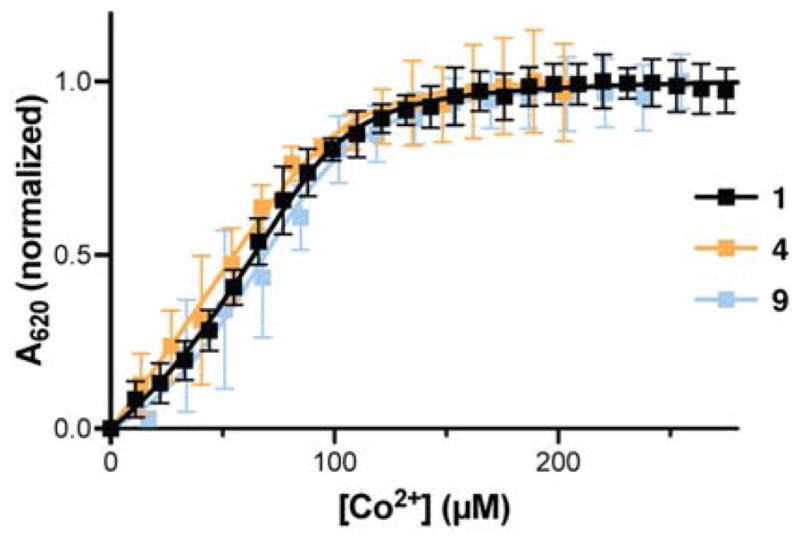
Co2+ binding isotherms for peptide 1 and backbone-modified analogues 4 and 9 determined by UV-Vis spectroscopy. Samples consisted of 100 μM peptide in 20 mM HEPES, pH 7.0. Error bars represent standard deviation from 3 independent measurements. Lines are the result of fitting to a 1:1 binding model with competing formation of a 2:1 peptide:metal complex (see SI for details).
Table 1.
Thermodynamic Parameters for Metal-Binding Interactions of Peptides 1, 4, and 9 with Co2+ and Zn2+
| Parameter | Peptide
|
||
|---|---|---|---|
| 1 | 4 | 9 | |
| Co2+ | |||
| Kd (μM)a | 1.4 ± 0.2 | 2.7 ± 1.2 | 1.4 ± 0.5 |
| Kd2 (μM2)a | 150 ± 30 | 1500 ± 1100 | 70 ± 40 |
| Zn2+ | |||
| Kd (μM)b | 2.2 ± 0.3 | 0.8 ± 0.1 | 1.7 ± 0.2 |
| ΔG (kcal mol−1)b | −7.72 ± 0.08 | −8.30 ± 0.09 | −7.90 ± 0.07 |
| ΔH (kcal mol−1)b | −13.5 ± 0.2 | −9.8 ± 0.1 | −11.0 ± 0.1 |
| TΔS (kcal mol−1)b | −5.8 ± 0.2 | −1.5 ± 0.1 | −3.3 ± 0.1 |
| nb | 0.81 | 0.8 | 0.78 |
Determined by UV-vis titration with Co2+; Kd and Kd2 are the dissociation constants for formation of the 1:1 and 2:1 ligand to metal complex, respectively (see SI for details).
Determined by ITC with Zn2+.
We next assessed the interaction of peptides 1, 4, and 9 with Zn2+ by isothermal titration calorimetry (ITC). Besides confirming the ability of the modified backbones to fold in the presence of the native ligand, ITC experiments provide more accurate measurements of energetics for folding and metal binding compared to UV-vis titration as well as new information in the form of entropy/enthalpy components. ITC results (Figure 6, Table 1) show that the affinity of both backbone-modified analogues for Zn2+ is higher than that of parent sequence 1. Consistent with prior studies on zinc finger domains, binding stoichiometries (n) were found to be slightly less than 1, which we attribute to partial oxidation of Cys residues during the measurement.33a
Figure 6.

Isothermal titration calorimetry (ITC) data for the interaction of Zn2+ with peptide 1 and backbone modified analogues 4 and 9 in 50 mM HEPES, 100 mM NaCl, pH 7.4 at 25 °C.
The observation that both the metal binding affinities and the folding free energies are so similar among 1, 4, and 9 is significant. In our previous work on analogous backbone modifications in the GB1 tertiary structure, a comparable fraction of backbone alteration led to a destabilization of the tertiary fold by ~8 kcal mol−1.5d Here, backbone modification has an opposite favorable effect on folding, with a magnitude as large as 0.5 kcal mol−1 in the case of peptide 4. Because the folded state of each analogue remains identical to that of the native sequence (vide infra), we reason that the differences in fold stability originate elsewhere along the folding pathway, either in the denatured ensemble16a or folding intermediates.35
ITC data also provided insights into the thermodynamic trends underlying the observed stabilization of the zinc finger fold upon backbone modification. Like the parent sequence 1, folding and metal binding in analogues 4 and 9 are enthalpically driven and entropically opposed. Taking the energetic values for 1 as a baseline (Figure 7), the backbone modifications in 4 (2 α→N-Me-α; 4 α→β3) have a moderately unfavorable effect on folding ΔH (~0.6 kcal mol−1 per substitution) that is more than compensated for by a favorable effect on folding TΔS (~0.7 kcal mol−1 per substitution). The above qualitative trend follows that seen upon α→β3 modifications in the helix of GB1;16a however, the quantitative balance between parameters differs between the two systems.5d The magnitude of the unfavorable ΔΔH in 4 vs. 1 is comparable in GB1 and Sp1–3 yet the corresponding favorable ΔΔS is much greater in the zinc finger motif.
Figure 7.

Summary of changes to the free energy, enthalpy, and entropy for the interaction between the indicated peptide and Zn2+ as determined by ITC. Changes are reported relative to corresponding parameters for natural backbone 1.
In our prior analysis of folding thermodynamics in heterogeneous-backbone GB1 analogues, we proposed the favorable entropic effect of α→β3 substitution results from altered desolvation entropy.16a In short, each β3-residue substitution adds one freely rotatable bond to the oligomer backbone. The resulting enhanced flexibility leads to a less ordered denatured ensemble. The decrease in residual folded structure in the denatured state, in turn, results in a greater solvent-accessible surface area for the unfolded heterogeneous-backbone oligomer compared to the corresponding all-α backbone. If the tertiary folds of the natural and heterogeneous backbones are identical, folding the less ordered denatured state releases more bound water to bulk. We suggest this favorable difference in desolvation entropy is the origin of entropic stabilization of the folded state observed upon α→β3 substitution.
While the qualitative trend remains the same in each system, the magnitude of the favorable effect on ΔΔS upon backbone modification is greater in the Sp1–3 context than in GB1. While the reason for this observation is not clear, two structural differences between GB1 and Sp1–3 may contribute: chain length [56 residues in GB1 vs. 29 in Sp1–3] and fraction apolar side chains [59% in GB1 vs. 28% in Sp1–3]. It is important to note that 4 has, in addition to four α→β3 replacements, two α→N-Me-α substitutions relative to 1 that complicate the above analysis. These alterations introduce additional backbone freedom in the form of tertiary amide isomerization and have been shown to destabilize the fold of isolated sheet secondary structure.4c
In considering the differences in folding energetics between 4 and 9, the two β3→Cα-Me-α replacements have offsetting effects: a reduced enthalpic penalty and a reduced entropic boost to folding. In prior work on GB1, we saw a similar effect and attributed it to the known ability of Aib to restrict backbone conformational freedom and promote helical folds.16b Interestingly and in contrast to GB1, the balance of enthalpy / entropy compensation differs in Sp1–3, and β3→Cα-Me-α substitution leads to a net destabilization of the tertiary fold.
Collectively, data obtained for the GB1 and zinc finger systems show some emerging themes in the relationship between backbone composition and tertiary folding thermodynamics. For example, β3-residues appear to be a consistent source of both an unfavorable ΔΔHfold as well as a favorable ΔΔSfold in heterogeneous backbones vs. their natural counterparts. The effect of Cα-Me-α residue incorporation is found to be system dependent, which may be a consequence of the identity of the side chain lost when Aib replaces a residue other than Ala. In prior work on GB1, Aib was introduced in place of sites bearing small side chains (Ala, Asn). In contrast, residues replaced by Aib in 1 to generate 9 were larger and more hydrophilic (Lys, Gln). Thus, side-chain changes were more dramatic in Sp1–3 and may contribute more to folding energetics as a result. It is important to emphasize that the above are working hyoptheses and that additional studies will be required to gain a more thorough understanding of the effects of backbone modification on folding pathways and the structure of the denatured ensemble.
High-Resolution Structural Characterization of the Folded State
The experiments detailed above all involve measurements related to the interaction between the zinc finger and metal. While folding and metal binding are highly interdependent, such analyses provide only indirect evidence bearing on the nature of the folded state. For a direct assessment of tertiary structure in the modified backbones and how it compares to the parent zinc finger, we subjected peptides 1, 4, and 9 to multidimensional NMR spectroscopy. We acquired 1H/1H COSY, TOCSY, and NOESY spectra for each peptide at 1.3–1.6 mM peptide concentration with 1.2 equiv. ZnCl2 in 9:1 H2O/D2O at pH 7.0 (uncorrected) and 4 °C. Comparison of backbone Hα chemical shifts for canonical α-residues present in all three sequences (Figure 8) showed significant deviations from random coil values and an identical pattern and magnitude of those deviations along the chain. Collectively, these observations argue strongly for a similar folded state.
Figure 8.

Backbone Hα chemical shift deviation from predicted random coil values for peptides 1, 4 and 9; asterisks indicate unnatural residues (no literature random coil chemical shift available for comparison) and open circles cases where Hα could not be unambiguously assigned.
To evaluate the folds of 4 and 9 at higher resolution, we assigned the majority of backbone and side-chain resonances in each and analyzed both local and long-range NOEs. Select short-range correlations confirmed the secondary structure pattern of the parent domain was retained in the modified backbones (Figure 9A). Strong sequential i→i+1 Hα-HN cross peaks were observed in each strand of the putative hairpin, along with a strong inter-strand Hα-Hα correlation between N-Me-Ala3 and N-Me-Arg10. Near-continuous i→i+1 HN-HN and i→i+3 Hα-HN NOEs were seen in the expected helical region. In addition to short-range correlations indicative of secondary structure, a number of long-range NOEs were observed between the helix and hairpin that were consistent with the expected tertiary fold (Figure S3).
Figure 9.

Analysis of the folded structure of 4 and 9 by NMR. (A) Secondary structure maps with select short- and medium-range NOE correlations supporting that assignment. (B) Overlay of the 9 lowest energy structures of peptide 4 as determined by simulated annealing with NOE distance restraints. Color code for carbons matches that in Figure 2. (C) Overlay of the NMR structure of peptide 4 (color) with that of Sp1–3 (white).
To obtain a high-resolution picture of the folded state for one of the heterogeneous backbones, we tabulated a set of unambiguous inter-residue NOEs for peptide 4 and performed simulated annealing with distance restraints derived from NOE intensities. Preliminary calculations with just the NOE restraints supported the native-like pattern of secondary structures, overall tertiary folding topology, and proximity of Cys4, Cys7, His20 and His24 side chains around the expected binding site for Zn2+. We therefore repeated the simulation with additional geometric restraints for hydrogen-bonds in the helix as well as the idealized tetrahedral coordination of the Zn2+ ion. The resulting ensemble of lowest energy structures showed excellent internal agreement and close homology to the previously reported tertiary folded structure for the native Sp1–3 domain (Figure 9B,C).24
CONCLUSIONS
In summary, we have demonstrated here that the systematic backbone alteration of a ubiquitous metal-binding tertiary structure, the zinc finger domain Sp1–3, can lead to heterogeneous-backbone mimics with native-like tertiary folded structures and superior folded stability. Beyond mimicry of this particular target fold, these findings are significant in the demonstration that a natural sequence of amino acid side chains can fold more effectively on a ~20% unnatural backbone than on the natural all-α-peptide backbone on which that sequence evolved.
The present work leaves important open questions for future research on foldameric zinc finger domain mimics. Backbone modifications examined in this study were limited to residues with side chains projected toward solvent. The finding that such modifications were energetically favorable to the tertiary fold suggests that side-chain-retaining substitutions at sites involved in metal-binding or hydrophobic-core contacts may be well tolerated or even more favorable. Another important question is whether heterogeneous backbone zinc finger domains will retain the key biological activity of the prototype—sequence-specific recognition of double stranded DNA. Experiments aimed at probing the above questions are ongoing.
Given the fact that the tertiary fold of Sp1–3 is structurally representative of a wide array of natural zinc finger domains, we anticipate the design principles successful here will be generalizable to other systems. More broadly, the successful mimicry of a new tertiary structure class through sequence-guided backbone alteration broadens the potential scope of this approach and suggests it can recreate a wide array of folding motifs. The reported results on the Sp1–3 system also highlight the challenges and limitations of applying the method to complex folds through the extreme sensitivity of the metal-binding β-turn to modification. Continued expansion of the structural diversity of building blocks used for modification as well as application to new prototype tertiary structures will lead to a greater understanding of the structural and functional consequences of backbone chemical alteration and refinement of state-of-the-art design principles.
Supplementary Material
Acknowledgments
Funding for this work was provided by the National Institutes of Health (GM107161) and support for MALDI-TOF MS instrumentation by the National Science Foundation (CHE-1625002).
Footnotes
The Supporting Information is available free of charge on the ACS Publications website at DOI: ##
Figures S1–S3, Tables S1–S2, and methods (PDF). Ensemble coordinates and additional experimental data for the NMR structure of 4 are deposited in the PDB under accession code 5US3.
References
- 1.Gellman SH. Acc Chem Res. 1998;31:173–180. [Google Scholar]
- 2.(a) Bautista AD, Craig CJ, Harker EA, Schepartz A. Curr Opin Chem Biol. 2007;11:685–692. doi: 10.1016/j.cbpa.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Goodman CM, Choi S, Shandler S, DeGrado WF. Nat Chem Biol. 2007;3:252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Seebach D, Gardiner J. Acc Chem Res. 2008;41:1366–1375. doi: 10.1021/ar700263g. [DOI] [PubMed] [Google Scholar]; (d) Guichard G, Huc I. Chem Commun. 2011;47:5933–5941. doi: 10.1039/c1cc11137j. [DOI] [PubMed] [Google Scholar]; (e) Martinek TA, Fulop F. Chem Soc Rev. 2012;41:687–702. doi: 10.1039/c1cs15097a. [DOI] [PubMed] [Google Scholar]; (f) Nair RV, Vijayadas KN, Roy A, Sanjayan GJ. Eur J Org Chem. 2014;2014:7763–7780. [Google Scholar]; (g) Laursen JS, Engel-Andreasen J, Olsen CA. Acc Chem Res. 2015;48:2696–2704. doi: 10.1021/acs.accounts.5b00257. [DOI] [PubMed] [Google Scholar]; (h) Robertson EJ, Battigelli A, Proulx C, Mannige RV, Haxton TK, Yun L, Whitelam S, Zuckermann RN. Acc Chem Res. 2016;49:379–389. doi: 10.1021/acs.accounts.5b00439. [DOI] [PubMed] [Google Scholar]
- 3.(a) Qiu JX, Petersson EJ, Matthews EE, Schepartz A. J Am Chem Soc. 2006;128:11338–11339. doi: 10.1021/ja063164+. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Horne WS, Price JL, Keck JL, Gellman SH. J Am Chem Soc. 2007;129:4178–4180. doi: 10.1021/ja070396f. [DOI] [PubMed] [Google Scholar]; (c) Delsuc N, Léger JM, Massip S, Huc I. Angew Chem Int Ed. 2007;46:214–217. doi: 10.1002/anie.200603390. [DOI] [PubMed] [Google Scholar]; (d) Collie GW, Pulka-Ziach K, Lombardo CM, Fremaux J, Rosu F, Decossas M, Mauran L, Lambert O, Gabelica V, Mackereth CD, Guichard G. Nat Chem. 2015;7:871–878. doi: 10.1038/nchem.2353. [DOI] [PubMed] [Google Scholar]
- 4.(a) Cheng PN, Pham JD, Nowick JS. J Am Chem Soc. 2013;135:5477–5492. doi: 10.1021/ja3088407. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Hegedüs Z, Wéber E, Kriston-Pál É, Makra I, Czibula Á, Monostori É, Martinek TA. J Am Chem Soc. 2013;135:16578–16584. doi: 10.1021/ja408054f. [DOI] [PubMed] [Google Scholar]; (c) Lengyel GA, Reinert ZE, Griffith BD, Horne WS. Org Biomol Chem. 2014;12:5375–5381. doi: 10.1039/c4ob00886c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Cheng RP, DeGrado WF. J Am Chem Soc. 2002;124:11564–11565. doi: 10.1021/ja020728a. [DOI] [PubMed] [Google Scholar]; (b) Price JL, Hadley EB, Steinkruger JD, Gellman SH. Angew Chem Int Ed. 2010;49:368–371. doi: 10.1002/anie.200904714. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Delsuc N, Massip S, Léger JM, Kauffmann B, Huc I. J Am Chem Soc. 2011;133:3165–3172. doi: 10.1021/ja110677a. [DOI] [PubMed] [Google Scholar]; (d) Reinert ZE, Lengyel GA, Horne WS. J Am Chem Soc. 2013;135:12528–12531. doi: 10.1021/ja405422v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bou-Abdallah F, Giffune TR. Biochim Biophys Acta. 2016;1860:879–891. doi: 10.1016/j.bbagen.2015.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maayan G. Eur J Org Chem. 2009;2009:5699–5710. [Google Scholar]
- 8.(a) Prince RB, Okada T, Moore JS. Angew Chem Int Ed. 1999;38:233–236. [Google Scholar]; (b) Zhang F, Bai S, Yap GPA, Tarwade V, Fox JM. J Am Chem Soc. 2005;127:10590–10599. doi: 10.1021/ja050886c. [DOI] [PubMed] [Google Scholar]; (c) Zhao Y, Zhong Z. J Am Chem Soc. 2006;128:9988–9989. doi: 10.1021/ja062001i. [DOI] [PubMed] [Google Scholar]; (d) Tashiro S, Matsuoka K, Minoda A, Shionoya M. Angew Chem Int Ed. 2012;51:13123–13127. doi: 10.1002/anie.201206968. [DOI] [PubMed] [Google Scholar]
- 9.(a) Viles JH, Patel SU, Mitchell JBO, Moody CM, Justice DE, Uppenbrink J, Doyle PM, Harris CJ, Sadler PJ, Thornton JM. J Mol Biol. 1998;279:973–986. doi: 10.1006/jmbi.1998.1764. [DOI] [PubMed] [Google Scholar]; (b) Lelais G, Seebach D, Jaun B, Mathad RI, Flögel O, Rossi F, Campo M, Wortmann A. Helv Chim Acta. 2006;89:361–403. [Google Scholar]; (c) Delsuc N, Hutin M, Campbell VE, Kauffmann B, Nitschke JR, Huc I. Chem Eur J. 2008;14:7140–7143. doi: 10.1002/chem.200800988. [DOI] [PubMed] [Google Scholar]; (d) Lee BC, Chu TK, Dill KA, Zuckermann RN. J Am Chem Soc. 2008;130:8847–8855. doi: 10.1021/ja802125x. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Schafmeister CE, Belasco LG, Brown PH. Chem Eur J. 2008;14:6406–6412. doi: 10.1002/chem.200701942. [DOI] [PubMed] [Google Scholar]; (f) Sénèque O, Bourlès E, Lebrun V, Bonnet E, Dumy P, Latour JM. Angew Chem Int Ed. 2008;47:6888–6891. doi: 10.1002/anie.200800677. [DOI] [PubMed] [Google Scholar]; (g) Sénèque O, Bonnet E, Joumas FL, Latour JM. Chem Eur J. 2009;15:4798–4810. doi: 10.1002/chem.200900147. [DOI] [PubMed] [Google Scholar]; (h) Ma Z, Olechnowicz F, Skorik YA, Achim C. Inorg Chem. 2011;50:6083–6092. doi: 10.1021/ic200138b. [DOI] [PubMed] [Google Scholar]; (i) Miller JP, Melicher MS, Schepartz A. J Am Chem Soc. 2014;136:14726–14729. doi: 10.1021/ja508872q. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Baskin M, Maayan G. Chem Sci. 2016;7:2809–2820. doi: 10.1039/c5sc04358a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reinert ZE, Horne WS. Org Biomol Chem. 2014;12:8796–8802. doi: 10.1039/c4ob01769b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Werner HM, Cabalteja CC, Horne WS. ChemBioChem. 2016;17:712–718. doi: 10.1002/cbic.201500312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheloha RW, Maeda A, Dean T, Gardella TJ, Gellman SH. Nat Biotechnol. 2014;32:653–655. doi: 10.1038/nbt.2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Checco JW, Kreitler DF, Thomas NC, Belair DG, Rettko NJ, Murphy WL, Forest KT, Gellman SH. Proc Natl Acad Sci USA. 2015;112:4552–4557. doi: 10.1073/pnas.1420380112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Werner HM, Horne WS. Curr Opin Chem Biol. 2015;28:75–82. doi: 10.1016/j.cbpa.2015.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Gopalakrishnan R, Frolov AI, Knerr L, Drury WJ, Valeur E. J Med Chem. 2016;59:9599–9621. doi: 10.1021/acs.jmedchem.6b00376. [DOI] [PubMed] [Google Scholar]; (c) Checco JW, Gellman SH. Curr Opin Struct Biol. 2016;39:96–105. doi: 10.1016/j.sbi.2016.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Checco JW, Gellman SH. ChemBioChem. 2017;18:291–299. doi: 10.1002/cbic.201600545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.(a) Reinert ZE, Horne WS. Chem Sci. 2014;5:3325–3330. doi: 10.1039/C4SC01094A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tavenor NA, Reinert ZE, Lengyel GA, Griffith BD, Horne WS. Chem Commun. 2016;52:3789–3792. doi: 10.1039/c6cc00273k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.(a) Newberry RW, VanVeller B, Raines RT. Chem Commun. 2015;51:9624–9627. doi: 10.1039/c5cc02685g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zhang Y, Malamakal RM, Chenoweth DM. J Am Chem Soc. 2015;137:12422–12425. doi: 10.1021/jacs.5b04590. [DOI] [PubMed] [Google Scholar]; (c) Kreitler DF, Mortenson DE, Forest KT, Gellman SH. J Am Chem Soc. 2016;138:6498–6505. doi: 10.1021/jacs.6b01454. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Simon MD, Maki Y, Vinogradov AA, Zhang C, Yu H, Lin YS, Kajihara Y, Pentelute BL. J Am Chem Soc. 2016;138:12099–12111. doi: 10.1021/jacs.6b03765. [DOI] [PubMed] [Google Scholar]; (e) Walters CR, Szantai-Kis DM, Zhang Y, Reinert ZE, Horne WS, Chenoweth DM, Petersson EJ. Chem Sci. 2017:2868–2877. doi: 10.1039/c6sc05580j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanas JS, Larabee JL, Hocker JR. Zinc Finger Proteins: From Atomic Contact to Cellular Function. Springer US; 2005. [Google Scholar]
- 19.Klug A. Annu Rev Biochem. 2010;79:213–231. doi: 10.1146/annurev-biochem-010909-095056. [DOI] [PubMed] [Google Scholar]
- 20.Gersbach CA, Gaj T, Barbas CF. Acc Chem Res. 2014;47:2309–2318. doi: 10.1021/ar500039w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reynolds L, Ullman C, Moore M, Isalan M, West MJ, Clapham P, Klug A, Choo Y. Proc Natl Acad Sci USA. 2003;100:1615–1620. doi: 10.1073/pnas.252770699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rebar EJ, Huang Y, Hickey R, Nath AK, Meoli D, Nath S, Chen B, Xu L, Liang Y, Jamieson AC, Zhang L, Spratt SK, Case CC, Wolffe A, Giordano FJ. Nat Med. 2002;8:1427–1432. doi: 10.1038/nm1202-795. [DOI] [PubMed] [Google Scholar]
- 23.Gaj T, Liu J, Anderson KE, Sirk SJ, Barbas CF. ACS Chem Biol. 2014;9:1662–1667. doi: 10.1021/cb500282g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Narayan VA, Kriwacki RW, Caradonna JP. J Biol Chem. 1997;272:7801–7809. doi: 10.1074/jbc.272.12.7801. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, Beger RD, Berg JM. Biophys J. 1993;64:749–753. doi: 10.1016/S0006-3495(93)81435-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Posewitz MC, Wilcox DE. Chem Res Toxicol. 1995;8:1020–1028. doi: 10.1021/tx00050a005. [DOI] [PubMed] [Google Scholar]
- 27.(a) Giedroc DP, Keating KM, Williams KR, Konigsberg WH, Coleman JE. Proc Natl Acad Sci USA. 1986;83:8452–8456. doi: 10.1073/pnas.83.22.8452. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen X, Chu M, Giedroc DP. J Biol Inorg Chem. 2000;5:93–101. doi: 10.1007/s007750050012. [DOI] [PubMed] [Google Scholar]
- 28.(a) May SW, Kuo JY. Biochemistry. 1978;17:3333–3338. doi: 10.1021/bi00609a025. [DOI] [PubMed] [Google Scholar]; (b) Vasak M, Kaegi JHR, Holmquist B, Vallee BL. Biochemistry. 1981;20:6659–6664. doi: 10.1021/bi00526a021. [DOI] [PubMed] [Google Scholar]
- 29.Hutchinson EG, Thornton JM. Protein Sci. 1994;3:2207–2216. doi: 10.1002/pro.5560031206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.(a) Haque TS, Little JC, Gellman SH. J Am Chem Soc. 1996;118:6975–6985. [Google Scholar]; (b) Masterson LR, Etienne MA, Porcelli F, Barany G, Hammer RP, Veglia G. Pept Sci. 2007;88:746–753. doi: 10.1002/bip.20738. [DOI] [PubMed] [Google Scholar]
- 31.Nowick JS, Brower JO. J Am Chem Soc. 2003;125:876–877. doi: 10.1021/ja028938a. [DOI] [PubMed] [Google Scholar]
- 32.Berg JM, Godwin HA. Annu Rev Biophys Biomol Struct. 1997;26:357–371. doi: 10.1146/annurev.biophys.26.1.357. [DOI] [PubMed] [Google Scholar]
- 33.(a) Rich AM, Bombarda E, Schenk AD, Lee PE, Cox EH, Spuches AM, Hudson LD, Kieffer B, Wilcox DE. J Am Chem Soc. 2012;134:10405–10418. doi: 10.1021/ja211417g. [DOI] [PubMed] [Google Scholar]; (b) Miloch A, Krezel A. Metallomics. 2014;6:2015–2024. doi: 10.1039/c4mt00149d. [DOI] [PubMed] [Google Scholar]
- 34.Michael SF, Kilfoil VJ, Schmidt MH, Amann BT, Berg JM. Proc Natl Acad Sci USA. 1992;89:4796–4800. doi: 10.1073/pnas.89.11.4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miura T, Satoh T, Takeuchi H. Biochim Biophys Acta. 1998;1384:171–179. doi: 10.1016/s0167-4838(98)00015-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.