

Abstract
Many types of fluorescent sensing systems have been reported for biological small molecules. Particularly, several methods have been developed for the recognition of ATP or NAD+, but they only show moderate sensitivity, and they cannot discriminate either ATP or NAD+ from their respective analogues. We have addressed these limitations and report here a dual strategy which combines split DNAzyme-based background reduction with catalytic and molecular beacon (CAMB)-based amplified detection to develop a ligation-triggered DNAzyme cascade, resulting in ultrahigh sensitivity. First, the 8–17 DNAzyme is split into two separate oligonucleotide fragments as the building blocks for the DNA ligation reaction, thereby providing a zero-background signal to improve overall sensitivity. Next, a CAMB strategy is further employed for amplified signal detection achieved through cycling and regenerating the DNAzyme to realize the true enzymatic multiple turnover (one enzyme catalyzes the cleavage of several substrates) of catalytic beacons. This combination of zero-background signal and signal amplification significantly improves the sensitivity of the sensing systems, resulting in detection limits of 100 and 50 pM for ATP and NAD+, respectively, much lower than those of previously reported biosensors. Moreover, by taking advantage of the highly specific biomolecule-dependence of the DNA ligation reaction, the developed DNAzyme cascades show significantly high selectivity toward the target cofactor (ATP or NAD+), and the target biological small molecule can be distinguished from its analogues. Therefore, as a new and universal platform for the design of DNA ligation reaction-based sensing systems, this novel ligation-triggered DNAzyme cascade method may find a broad spectrum of applications in both environmental and biomedical fields.
Graphical Abstract
INTRODUCTION
In the past few years, fluorescent sensing systems for biological small molecules have demonstrated such unique advantages as high sensitivity, rapid analysis with spatial resolution, and little proclivity to sample or cell damage.1–4 The recognition of adenosine-triphosphate (ATP) is of particular importance, because ATP is not only a universal energy source, but also an extracellular signaling mediator in many biological processes.5–7 Many fluorescent chemosensors based on organic molecules have been developed for selective detection of ATP.8–14 In addition, an anti-ATP DNA aptamer has also been isolated and converted into fluorescent biosensors using various strategies.15–19 These reported fluorescent sensors have all shown high selectivity to ATP over other nucleosides and other biological small molecules. However, most of them also exhibit only moderate sensitivity with detection limits for ATP in the micromolar range, and most cannot distinguish ATP from its analogues, such as adenosine, AMP, and ADP.
Nicotinamide adenine dinucleotide (NAD+) is another biologically important molecule. In addition to serving as a cofactor of many oxidoreductive enzymes,20 it plays a critical role in transcriptional regulation,21,22 DNA repair,23 caloric restriction-mediated lifespan extension,24 calcium homeostasis,25 and cell proliferation.26 Furthermore, several age-associated diseases, such as diabetes, cancer, and neurodegenerative diseases, have been reported to be associated with changes in NAD+ levels and/or the NAD+/NADH ratios.22,27 Although a few fluorescence methods have been developed for NAD+ by converting it into its fluoresced derivatives,28,29 they show low sensitivity, as well as an inability to discriminate NAD+ from NADH or other analogues. Therefore, a new fluorescent sensing design, having both high sensitivity and selectivity for these biological small molecules, is highly desirable.
Two different strategies have been widely employed to improve the sensitivity of a sensor: lowered background signal and amplified detection. In the first case, gold nanoparticles, carbon nanotubes, and graphene oxide have been widely applied as nanoquenchers to lower background signal and, hence, improve the sensitivity of fluorescent biosensors.30–33 While designing a sensor with zero-background signal is attractive, it remains challenging. In the second case, an excellent example of amplified detection is the use of protein enzymes, such as horseradish peroxidise (HRP) or an HRP-mimicking DNAzyme, for amplified electrochemical and colorimetric signal to achieve large signal-to-noise ratios.34–38 However, in contrast to the many amplified electrochemical and colorimetric sensors, few amplified fluorescence sensors have been reported. Moreover, to the best of our knowledge, no research efforts have yet found a way to combine these strategies into one system with high sensitivity. We recently proposed a catalytic and molecular beacon (CAMB) strategy for amplified detection of Pb2+ by employing a molecular beacon (MB) substrate to realize the true enzymatic multiple turnover (one enzyme catalyzes the cleavage of several substrates) of DNAzyme.39 Here, we propose combining CAMB with a zero-background strategy into one system to achieve a novel sensing platform with a predictably high sensitivity.
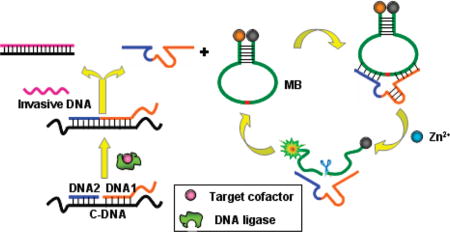
Some enzymatic reactions show specific dependence on certain biomolecules (cofactors), which, in turn, provides an efficient platform for constructing highly selective sensing systems for these cofactors. To develop sensing systems for ATP and NAD+ with high selectivity, we turned our attention to DNA ligases, as there are two kinds of DNA ligases which specifically employ ATP and NAD+ as cofactors, respectively, and their catalytic activities are cofactor-dependent.40–42 In this work, we employed a dual strategy, which combines split DNAzyme-based background reduction with a CAMB-based amplified detection to develop a ligation-triggered DNAzyme cascade (Scheme 1), for amplified sensing of small molecules such as ATP and NAD+ with sensitivity superior to that of previously reported sensors. First, the 8–17 DNAzyme is split into two separate oligonucleotide fragments for the first time as the building blocks for the DNA ligation reaction, thereby providing a zero-background signal for improved sensitivity to target biological small molecules. 8–17 DNAzyme has been previously employed for amplified detection of various targets through an allosteric strategy.43,44 However, in these sensing systems, the enzyme stand of the 8–17 DNAzyme existed in a whole molecule before and after the introduction of the target analytes, which make it possible for some enzyme stands to hybridize with substrate strands in the absence of target, and therefore result in a background signal to some extent. A CAMB strategy is further employed for amplified signal detection, achieved through the cycling and regenerating of DNAzyme to realize true enzymatic multiple turnover of catalytic beacons.
Scheme 1.

Design Strategy of the DNAzyme Cascade for Amplified Fluorescence Detection of Biological Small Moleculesa
a (a) The process for the production of activated DNAzyme; (b) amplifying fluorescence signal by the catalytic and molecular beacon. The enzyme strand of the 8–17 DNAzyme is split into two separate fragments, DNA 1 and 2 (blue and orange), which are inactive to MB substrate in the absence of DNA ligase and target cofactor. However, in the presence of DNA ligase and its cofactor, DNA 1 and 2 can be ligated to produce an activated 8–17 DNAzyme. Then, with the introduction of invasive DNA after the ligation completed, 8–17 DNAzyme can be liberated from template C-DNA and hybridized with a hairpin-structured MB substrate to form a catalytic and molecular beacon system, thus, achieving an amplified fluorescence signal for the target cofactor. The red in the molecular beacon is a cleavable ribonucleotide.
EXPERIMENTAL SECTION
General Procedures
T4 DNA ligase, Escherichia coli DNA ligase, and all synthetic oligonucleotides were purchased from Takara Biotechnology Co., Ltd. (Dalian, China). Adenosine triphosphate (ATP), adenosine, uridine triphosphate (UTP), guanosine triphosphate (GTP), and cytidine triphosphate (CTP) were obtained from Sigma-Aldrich. Nicotinamide adenine dinucleotide (NAD+), the reduced formof nicotinamide adenine dinucleotide (NADH), nicotinamide adenine dinucleotide phosphate (NADP), nicotinamide adenine dinucleotide phosphate hydride (NADPH), adenosine diphosphate (ADP), and adenosine monophosphate (AMP) were obtained from Amresco. All other chemicals were obtained from Shanghai Chemical Reagents (Shanghai) and used without further purification. All solutions were prepared in Milli-Q water (resistance >18 MΩ • cm) from a Millipore system. The pH measurements were carried out on a Mettler-Toledo Delta 320 pH meter. All fluorescence measurements were carried out on an F-7000 spectrometer (Hitachi, Japan). The instrument settings were chosen as follows: λex = 494 nm (bandpass 5 nm), λem = 518 nm (bandpass 5 nm), PMT detector voltage = 950 V. The sequences of oligonucleotides used in this work were given in Table 1.
Table 1.
Sequences of Oligonucleotides Used in This Worka
| oligonucleotide | sequences (from 5′ to 3′) |
|---|---|
| MB2 | /FAM/-CCACCACATTCAAATTCACCAACTATrAGGAAG-AGATGTTACGAGGCGGTGGTGG-/BHQ/ |
| DNA 1 | CATCTCTTCTCCGAGCCGGTCG |
| DNA 2 | P-AAATAGTGGGTG |
| C-DNA | CCCCCCCCCCACCCACTATTTCGACCGGCTCGGCCCCCCC |
| Invasive DNA | GGGGGGGCCGAGCCGGTCGAAATAGTGGGTGGGGGGGGGG |
| 8–17 DNAzyme | CATCTCTTCTCCGAGCCGGTCG AAATAGTGGGTG |
The underlined sequences in MB2 represent the stem of MB, and rA denotes adenosine ribonucleotide at that position, while all others are deoxyribonucleotides.
Ligation Procedure
The buffer containing 66 mM Tris-HCl (pH = 7.6), 6.6 mM MgCl2, and 10 mM dithiothreitol was used for both ligation reaction and fluorescence detection. To detect ATP, a mixture containing 0.15 µM of DNA 1, 0.15 µM of DNA 2, and 0.1 µM of template C-DNA was prepared in a ligation buffer of 42 µL volume in the presence of varying concentrations of ATP. The mixture was heated to 90 °C for 5 min and then cooled to room temperature. T4 DNA ligase at a final concentration of 0.04 U/µL was then introduced into the mixture to induce the ligation. The reaction was allowed to proceed at 37 °C for 30 min. Subsequently, the invasive DNA at a final concentration of 0.3 µM was added to the resulting mixture to inhibit the ligation reaction by forming a more stable invasive C-DNA hybrid and releasing the active 8–17 DNAzyme to perform its catalytic function. The produced mixture was directly introduced into the next kinetic fluorescence assay step. For the detection of NAD+, a procedure similar to that used for ATP detection was conducted, except for the use of E. coli DNA ligase at a final concentration of 0.06 U/µL instead of T4 DNA ligase to trigger the ligation and the use of NAD+ at varying concentrations instead of ATP as cofactor. Different ligation conditions, including varying concentrations of DNA ligase, different molar ratios of DNA segments (1 and 2) to the template C-DNA (fixed at 0.1 µM), and different incubation times, were also employed in the above ligation procedures for optimization.
Fluorescence Assay Procedures
To conduct the fluorescence assay for target cofactor, 0.2 µM of MB2 and 100 µM Zn2+ were then added to the as-prepared free 8–17 DNAzyme-containing mixture in a 100 µL quartz cuvette. After thoroughly mixing the components, the cuvette was quickly placed in a fluorometer thermostatted at 25 °C, and the rate of the fluorescence increase was recorded. The excitation and emission wavelengths were set at 494 and 518 nm, respectively. To study the effect of the sequence of substrate strand, 0.2 µM of MB substrate with different sequences (MB1 and MB2) was incubated with 0.1 µM of purchased 8–17 DNAzyme at room temperature for 20 min in a 100 µL quartz cuvette in the fluorometer. After the initial reading, the quartz cuvette was removed, and 100 µM Zn2+ was added to induce the cleavage reaction. After vortexing, the cuvette was quickly placed back into the fluorometer to continue the kinetics assay. The effect of the concentration of cofactor Zn2+ was also investigated using a similar procedure by incubating 0.2 µM of MB2 with 0.1 µM purchased 8–17 DNAzyme in the presence of varying concentrations of Zn2+.
To investigate the effect of ligation conditions on the sensing performance of the system, MB2 at 0.2 and 100 µM of Zn2+ was mixed with the crude products obtained using different ligation conditions, by varying the concentration of T4 DNA ligase, the molar ratio of DNA segments (1 and 2) to the template C-DNA (fixed at 0.1 µM), and the incubation time. The catalytic reaction was allowed to proceed for 25 min, and a fluorometer was then used to record the fluorescence intensity change at 518 nm upon excitation at 494 nm. A similar procedure was also used to investigate the effect of molar ratio of the MB2 to template C-DNA by fixing the concentration of template DNA C at 0.1 µM and changing the concentration of MB2.
RESULTS AND DISCUSSION
Design and Optimization of the DNAzyme Cascade-Based Sensing System
The design of this new DNAzyme cascade is shown in Scheme 1. The 8–17 DNAzyme was chosen as the catalytic unit for the amplified sensing system based on its highly catalytic activity and the ability to expand its functionality by adopting either Pb2+ or Zn2+ as cofactors. The enzyme strand of the 8–17 DNAzyme was split into two separate fragments, DNA 1 and 2, which are inactive to MB substrate in the absence of DNA ligase and target cofactor Zn2+, thus, providing a zero-background for the sensing system. However, both oligonucleotide fragments can be separately hybridized to the template C-DNA to form a ligatable nick. Therefore, in the presence of T4 DNA ligase, together with the cofactor ATP, or E. coli DNA ligase, together with the cofactor NAD+, the two oligonucleotides can be ligated to produce activated 8–17 DNAzyme in an amount that is positively related to the concentration of the cofactor ATP or NAD+. With the introduction of invasive DNA, 8–17 DNAzyme can be liberated from C-DNA by the formation of a more stable duplex of invasive DNA and C-DNA. The liberated 8–17 DNAzyme can then be hybridized with a hairpin-structured MB substrate to form the CAMB system.29 Upon the addition of the cofactor metal ions, the DNAzyme catalyzes the cleavage of the MB substrate, causing the quenched MB fluorophore/quencher pair to be separated, thereby producing a dramatic increase of fluorescent signal. Eventually, each released 8–17 DNAzyme can undergo many cycles to trigger the cleavage of many MB substrates, providing an amplified detection signal for the target ATP or NAD+.
To achieve the best sensing performance, the sequence of the MB substrate, the concentration of cofactor Zn2+, and the ligation conditions were optimized. Similar to previously reported findings,39 an MB substrate called MB2 with a 7 base pairs stem could provide satisfactory performance with a 12.3-fold fluorescence enhancement in the presence of 100 µM Zn2+ (see Supporting Information, Figure S1); therefore, this optimized MB2 substrate was chosen for further investigation. Experimental results showed that the following conditions could provide maximum S/N ratio for the sensing system: 100 µM Zn2+, 0.04 U/µL of T4 DNA ligase, a molar ratio of 1.5:1 for DNA segments (1 and 2) to the template C-DNA (fixed at 0.1 µM) with incubation time of 30 min for the ligation reaction, and a molar ratio of 2:1 for the MB substrate to the template C-DNA (see Supporting Information Figures S2–S4).
Application of the DNAzyme Cascade toward ATP Detection
T4 DNA ligase was first chosen to construct the sensing system for its cofactor ATP. As shown in Figure 1a, no fluorescence enhancement was observed in the absence of ATP, indicating that the DNA ligase was fully inactive and could not catalyze the ligation reaction of DNA 1 and 2. Under these conditions, the separated DNA 1 and 2 segments could neither form an active DNAzyme to catalyze the cleavage of the hairpin-structured substrate in the presence of Zn2+ nor efficiently hybridize with the MB substrate. The result is a zero-background signal in the absence of ATP. However, in the presence of ATP, the two oligonucleotides could be ligated to produce an activated 8–17 DNAzyme, and the introduction of invasive DNA could then release 8–17 DNAzyme to catalyze the cleavage of the hairpin-structured substrate to induce a fluorescence enhancement. In addition, the initial rate of fluorescence enhancement increased with the increase of ATP concentration (Figure 1a).
Figure 1.

Sensitivity of the DNAzyme cascade for ATP detection. (a) Time-dependent fluorescence response over background fluorescence with varying concentrations of ATP. The concentrations for template C-DNA, DNA 1, 2 and MB2 are 0.1, 0.15, 0.15, and 0.2 µM, respectively, using 0.04 U/µL of T4 DNA ligase. The buffer contained 66 mM Tris-HCl (pH = 7.6), 6.6 mM MgCl2, and 10 mM dithiothreitol. Inset: Responses at low ATP concentrations. (b) Calibration curve of the DNAzyme cascade for ATP. The curve was plotted with the initial rate of fluorescence enhancement vs ATP concentration. Inset shows the linear responses at low ATP concentrations.
Our CAMB strategy could further improve the sensitive detection of ATP by realizing the true enzymatic multiple turnover of DNAzyme. A linear relationship is observed between the fluorescence enhancement and target DNA concentration up to 300 nM (Figure 1b), with a detection limit of 100 pM (3σ/slope). This detection limit is much lower than that of the previously reported anti-ATP DNA aptamer-based fluorescence sensing systems (detection limit in the µM range).15–19 The ultrahigh sensitivity indicates the success of our dual strategy design.
Achieving highly selective response to the analyte of interest over other potentially competing species is required for biosensors with applications to practical samples. Although several anti-ATP DNA aptamer-based fluorescent biosensors have been reported with satisfactory sensitivity, almost none of them can distinguish ATP from its analogues, such as adenosine, adenosine-monophosphate (AMP), and adenosine-diphosphate (ADP). Because an enzymatic reaction cannot occur in the absence of target biomolecules, our system achieves improved selectivity by using the T4 DNA ligase-based ligation reaction. The selectivity of this method has been tested by comparing the fluorescence signal change rates of samples containing ATP with those of its analogues, including adenosine (A), AMP and ADP, uridine triphosphate (UTP), cytidine triphosphate (CTP), and guanosine triphosphate (GTP), as shown in Figure 2. When 0.5 µM of ATP, A, AMP, ADP, UTP, CTP, and GTP were added, respectively, to the reaction mixture, only ATP caused a marked fluorescence increase. This result obviously indicated that the proposed strategy had sufficient selectivity in ATP detection and was able to detect ATP among its analogues. The selectivity of the sensing system is obviously better than that of previously reported anti-ATP DNA apatmer-based fluorescent biosensors.15–19
Figure 2.

Selectivity of the DNAzyme cascade for ATP compared to its analogues. The initial rate of fluorescence enhancement of the sensing system induced by different compounds at a concentration of 0.5 µM is shown. The concentrations for DNA 1, 2, and MB2 are 0.15, 0.15, and 0.2 µM, respectively, using 0.04 U/µL of T4 DNA ligase. The buffer contained 66 mM Tris-HCl (pH = 7.6), 6.6 mM MgCl2, and 10 mM dithiothreitol.
Application of the DNAzyme Cascade to NAD+ Detection
To demonstrate the generality of this design, a dual-strategy ligation reaction-based sensing system was also designed for NAD+ using E. coli DNA ligase instead of T4 DNA ligase. To obtain the best performance of the sensing system, the ligation conditions were first optimized. Experimental results indicated that a concentration of 0.06 U/µL of ligase and a molar ratio of 1.5:1 for DNA segments (1 and 2) to the template C-DNA with incubation time of 30 min for the ligation reaction could provide maximum S/N ratio for the sensing system (see Supporting Information, Figure S5). Similar to the ATP sensing system, E. coli DNA ligase is inactive in the absence of NAD+ and cannot catalyze the ligation reaction of A and B, again providing a zero-background signal for the sensing system (Figure 3a). In the presence of NAD+, DNA 1 and 2 can be ligated to produce an activated 8–17 DNAzyme, which can then be liberated by the introduction of invasive DNA and can subsequently catalyze the cleavage of the hairpin-structured substrate to induce a fluorescence enhancement. Similar to ATP results, the fluorescence enhancement rate increased with increasing concentration of NAD+ (Figure 3a). The initial rate of fluorescence increased proportionally to the concentration of NAD+ within the range of 0.1 to 100 nM, and a detection limit of 50 pM (3σ/slope) was estimated for NAD+ (Figure 3b). The detection limit of the newly designed NAD+ sensing system is 2–3 orders of magnitude lower than that of previously reported fluorescent methods.28,29 Taken together, these experimental results also indicate successful fluorescence amplification for NAD+ recognition, further demonstrating the feasibility of our novel dual-strategy, ligation reaction-based sensing system in the context of overall improved sensitivity.
Figure 3.

Sensitivity of the DNAzyme cascade for NAD+ detection. (a) Time-dependent fluorescence response over background fluorescence with varying concentrations of NAD+. The concentrations for DNA 1, 2, and MB2 are 0.15, 0.15, and 0.2 µM, respectively, using 0.06 U/µL of E. coli DNA ligase. The buffer contained 30 mM Tris-HCl (pH = 8.0), 4 mM MgCl2, and 10 mM (NH4)2SO4. Inset: Responses at low NAD+ concentrations. (b) Calibration curve of the DNAzyme cascade for NAD+. The curve was plotted with the initial rate of fluorescence enhancement vs NAD+ concentration. Inset shows the linear responses at low NAD+ concentrations.
Previously reported fluorescence methods for NAD+ necessitated the conversion of NAD+ into its fluoresced derivatives, such as NADH, thus, limiting the ability of these methods to discriminate NAD+ from NADH or other analogues. An inherent advantage of our ligation reaction-based sensing system is its high specificity. To test the selectivity of the biosensor toward NAD+, the fluorescence responses of several potential interferents were also recorded separately with excitation fixed at 497 nm and emission at 518 nm (Figure 4). Experimental results showed that the addition of 1 µM of NADH, nicotinamide adenine dinucleotide phosphate (NADP), nicotinamide adenine dinucleotide phosphate hydride (NADPH), ATP, ADP, or AMP yielded only negligible fluorescence changes when compared to the yield of NAD+, indicating that the proposed ligation reaction-based sensing system achieved high selectivity for the detection of NAD+ and was able to discriminate NAD+ from its analogues.
Figure 4.

Selectivity of the DNAzyme cascade for NAD+ compared its analogues. The initial rate of fluorescence enhancement of the sensing system induced by different compounds at a concentration of 1 µM is shown. The concentrations for DNA 1, 2, and MB2 are 0.15, 0.15, and 0.2 µM, respectively, using 0.06 U/µL E. coli DNA ligase. The buffer contained 30 mM Tris-HCl (pH = 8.0), 4 mM MgCl2, and 10 mM (NH4)2SO4.
CONCLUSION
In summary, we have developed a ligation-triggered DNAzyme cascade by combining a split DNAzyme-based background reduction strategy with CAMB-based amplification in one system, resulting in ultrahigh sensitivity. The 8–17 DNAzyme was split into two separate oligonucleotide fragments as the building blocks for the DNA ligation reaction, which could then provide a zero-background signal in the absence of target for the sensing system, thus, affording high sensitivity for the detection of target biological small molecules. A CAMB strategy was further employed to amplify the detection signal by cycling and regenerating the DNAzyme to realize true enzymatic multiple turnover of catalytic beacons. Combining zero-background signal and CAMB-based signal amplification significantly improves the sensitivity of the sensing systems, resulting in a detection limit of 100 and 50 pM for ATP and NAD+, respectively, much lower than those of previously reported biosensors. By taking advantage of the highly specific biomolecule-dependence of the DNA ligation reaction, the proposed DNAzyme cascades showed significantly high selectivity toward target cofactor (ATP or NAD+) and could distinguish the target biological small molecules from their analogues. Thus, the ligation-triggered DNAzyme cascade provides a new and universal platform for the design of novel DNA ligation reaction-based sensing systems for detection of cofactors, monitoring activity of DNA ligases,45 and real-time investigation of nucleic acids phosphorylation process,46 and, as such, may find wide applications in environmental and biomedical fields.
Supplementary Material
Acknowledgments
This work was supported by the National Natural Science Foundation of China (20975034), the National Key Scientific Program of China (2011CB911001, 2011CB911003), “973” National Key Basic Research Program of China (2007CB310500), and Program for Changjiang Scholars and Innovative Research Team in University. This work is also suported by grants awarded by the National Institutes of Health (GM066137, GM079359 and CA133086).
Footnotes
ASSOCIATED CONTENT
Supporting Information. Supplementary spectral data. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Martinez-Manez R, Sancenon F. Chem. Rev. 2003;103:4419–4476. doi: 10.1021/cr010421e. [DOI] [PubMed] [Google Scholar]
- 2.Borisov SM, Wolfbeis OS. Chem. Rev. 2008;108:423–461. doi: 10.1021/cr068105t. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Cao Z, Lu Y. Chem. Rev. 2009;109:1948–1998. doi: 10.1021/cr030183i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gale PA. Chem. Soc. Rev. 2010;39:3746–3771. doi: 10.1039/c001871f. [DOI] [PubMed] [Google Scholar]
- 5.Lipscomb WN, Strater N. Chem. Rev. 1996;96:2375–2433. doi: 10.1021/cr950042j. [DOI] [PubMed] [Google Scholar]
- 6.Abraham EH, Okunieff P, Scala S, M Oosterveld JS, Chen AY, Shrivastav B, Guidotti G. Science. 1997;275:1324–1325. doi: 10.1126/science.275.5304.1324. [DOI] [PubMed] [Google Scholar]
- 7.Gourine AV, Llaudet E, Dale N, Spyer KM. Nature. 2005;436:108–111. doi: 10.1038/nature03690. [DOI] [PubMed] [Google Scholar]
- 8.Ojida A, Mito-oka Y, Sada K, Hamachi I. J. Am. Chem. Soc. 2004;126:2454–2463. doi: 10.1021/ja038277x. [DOI] [PubMed] [Google Scholar]
- 9.Yamaguchi S, Yoshimura I, Kohira T, Tamaru S, Hamachi I. J. Am. Chem. Soc. 2005;127:11835–11841. doi: 10.1021/ja052838y. [DOI] [PubMed] [Google Scholar]
- 10.Numata C, Li M, Takeuchi M, Shinkai S. Angew. Chem. Int. Ed. 2005;44:6371–6374. doi: 10.1002/anie.200501823. [DOI] [PubMed] [Google Scholar]
- 11.Ojida A, Nonaka H, Miyahara Y, Tamaru S, Sada K, Hamachi I. Angew. Chem. Int. Ed. 2006;45:5518–5521. doi: 10.1002/anie.200601315. [DOI] [PubMed] [Google Scholar]
- 12.Zyryanov GV, Palacios MA, Anzenbacher P. Angew. Chem. Int. Ed. 2007;46:7849–7852. doi: 10.1002/anie.200702611. [DOI] [PubMed] [Google Scholar]
- 13.Xu Z, Jiten Singh N, Lim J, Pan J, Kim HN, Park S, Kim KS, Yoon J. J. Am. Chem. Soc. 2009;131:15528–15533. doi: 10.1021/ja906855a. [DOI] [PubMed] [Google Scholar]
- 14.Kurishita Y, Kohira T, Ojida A, Hamachi I. J. Am. Chem. Soc. 2010;132:13290–13299. doi: 10.1021/ja103615z. [DOI] [PubMed] [Google Scholar]
- 15.Mei SHJ, Liu Z, Brennan JD, Li Y. J. Am. Chem. Soc. 2003;125:412–420. doi: 10.1021/ja0281232. [DOI] [PubMed] [Google Scholar]
- 16.Nutiu R, Li Y. J. Am. Chem. Soc. 2003;125:4771–4778. doi: 10.1021/ja028962o. [DOI] [PubMed] [Google Scholar]
- 17.Wang J, Jiang YX, Zhou CS, Fang XH. Anal. Chem. 2005;77:3542–3546. doi: 10.1021/ac050165w. [DOI] [PubMed] [Google Scholar]
- 18.Li N, Ho CM. J. Am. Chem. Soc. 2008;130:2380–2381. doi: 10.1021/ja076787b. [DOI] [PubMed] [Google Scholar]
- 19.Li TH, Fu RZ, Park HG. Chem. Commun. 2010;46:3271–3273. doi: 10.1039/b923462d. [DOI] [PubMed] [Google Scholar]
- 20.Matthew CK, Van Holde KE, Ahern KG. Biochemistry. 3. Addison-Wesley; Boston, MA: 2000. [Google Scholar]
- 21.Rutter J, Reick M, Wu LC, McKnight SL. Science. 2001;293:510–514. doi: 10.1126/science.1060698. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Q, Piston DW. Science. 2002;295:1895–1897. doi: 10.1126/science.1069300. [DOI] [PubMed] [Google Scholar]
- 23.Wilkinson A, Day J, Bowater R. Mol. Microbiol. 2001;40:1241–1248. doi: 10.1046/j.1365-2958.2001.02479.x. [DOI] [PubMed] [Google Scholar]
- 24.Lin SJ, Defossez PA, Guarente L. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 25.Guse AH, Gu XF, Zhang LR, Weber K, Krämer E, Yang ZJ, Jin HW, Li Q, Carrier L, Zhang LH. J. Biol. Chem. 2005;280:15952–15959. doi: 10.1074/jbc.M414032200. [DOI] [PubMed] [Google Scholar]
- 26.Bruzzone S, Flora AD, Usai C, Graeff R, Lee HC. Biochem. J. 2003;375:395–403. doi: 10.1042/BJ20030556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charron MJ, Bonner-Weir S. Nat. Med. 1999;5:269–270. doi: 10.1038/6479. [DOI] [PubMed] [Google Scholar]
- 28.Putt KS, Hergenrother PJ. Anal. Biochem. 2004;326:78–86. doi: 10.1016/j.ab.2003.11.015. [DOI] [PubMed] [Google Scholar]
- 29.Hausler RE, Fischer KL, Flugge UI. Anal. Biochem. 2000;281:1–8. doi: 10.1006/abio.2000.4556. [DOI] [PubMed] [Google Scholar]
- 30.Dubertret B, Calame M, Libchaber AJ. Nat. Biotechnol. 2001;19:365–370. doi: 10.1038/86762. [DOI] [PubMed] [Google Scholar]
- 31.Yang RH, Jin JY, Chen Y, Shao N, Tang ZW, Wu YR, Zhu Z, Tan WH. J. Am. Chem. Soc. 2008;130:8351–8358. doi: 10.1021/ja800604z. [DOI] [PubMed] [Google Scholar]
- 32.Lu CH, Yang HH, Zhu CL, Chen X, Chen GN. Angew. Chem. Int. Ed. 2009;48:4785–4787. doi: 10.1002/anie.200901479. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Li Z, Hu D, Lin CT, Li J, Lin Y. J. Am. Chem. Soc. 2010;132:9274–9276. doi: 10.1021/ja103169v. [DOI] [PubMed] [Google Scholar]
- 34.Liu G, Wan Y, Gau V, Zhang J, Wang L, Song S, Fan C. J. Am. Chem. Soc. 2008;130:6820–6825. doi: 10.1021/ja800554t. [DOI] [PubMed] [Google Scholar]
- 35.Rodenko B, Toebes M, Celie PHN, Perrakis A, Schumacher TNM, Ovaa H. J. Am. Chem. Soc. 2009;131:12305–12313. doi: 10.1021/ja9037565. [DOI] [PubMed] [Google Scholar]
- 36.Kolpashchikov DM. J. Am. Chem. Soc. 2008;130:2934–2935. doi: 10.1021/ja711192e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deng M, Zhang D, Zhou Y, Zhou X. J. Am. Chem. Soc. 2008;130:13095–13102. doi: 10.1021/ja803507d. [DOI] [PubMed] [Google Scholar]
- 38.Nakayama S, Sintim HOJ. J. Am. Chem. Soc. 2009;131:10320–10333. doi: 10.1021/ja902951b. [DOI] [PubMed] [Google Scholar]
- 39.Zhang XB, Wang ZD, Xing H, Lu Y. Anal. Chem. 2010;82:5005–5011. doi: 10.1021/ac1009047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lehman IR. Sciences. 1974;186:790–797. doi: 10.1126/science.186.4166.790. [DOI] [PubMed] [Google Scholar]
- 41.Sriskanda V, Shuman S. Nucleic Acids Res. 2001;29:4930–4934. doi: 10.1093/nar/29.24.4930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kuhn H, Frank-Kamenetskii MD. FEBS J. 2005;272:5991–6000. doi: 10.1111/j.1742-4658.2005.04954.x. [DOI] [PubMed] [Google Scholar]
- 43.Liu J, Lu Y. Angew. Chem. Int. Ed. 2007;46:7587–7590. doi: 10.1002/anie.200702006. [DOI] [PubMed] [Google Scholar]
- 44.Ali MM, Li Y. Angew. Chem. Int. Ed. 2009;48:3512–3515. doi: 10.1002/anie.200805966. [DOI] [PubMed] [Google Scholar]
- 45.Liu L, Tang Z, Wang K, Tan W, Li J, Guo Q, Meng X, Ma C. Analyst. 2005;130:350–357. doi: 10.1039/b413959c. [DOI] [PubMed] [Google Scholar]
- 46.Tang Z, Wang K, Tan W, Ma C, Li J, Liu L, Guo Q, Meng X. Nucleic Acids Res. 2005;33:e97. doi: 10.1093/nar/gni096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.