

Abstract
This Feature overviews the basic principles of using stable organic radicals involved in reversible exchange processes as functional paramagnetic probes. We demonstrate that these probes in combination with electron paramagnetic resonance (EPR)-based spectroscopy and imaging techniques provide analytical tools for quantitative mapping of critical parameters of local chemical microenvironment. The Feature is written to be understandable to people who are laymen to the EPR field in anticipation of future progress and broad application of these tools in biological systems, especially in vivo, over the next years.
Graphical abstract
Electron paramagnetic resonance spectroscopy, EPR, is a method of choice for detection of free radicals. Specific EPR methods such as EPR spin trapping were developed to detect short-lived reactive radical intermediates in biological and chemical systems and are often considered to be a gold standard for free radical identification and quantification (for a recent review, see ref 1). The development of stable radicals, in particular nitroxyl radicals, NRs,2,3 and triarylmethyl radicals, TAMs,4,5 two main classes of soluble paramagnetic materials (see Schemes 1 and 2) revolutionized the numerous areas of EPR spectroscopy and imaging applications. Spectral sensitivity of these radicals to local physical properties of the medium such as viscosity, polarity, and temperature has been used in numerous biophysical and biomedical applications including studies of biological macromolecules and biomembranes.6,7
Scheme 1.

Chemical Structures of the Stable NRs Discussed in the Text
Scheme 2. Chemical Structures of Trityl Radicals Discussed in the Texta.

aThe peak to peak line widths in deoxygenated TAM solutions were reported being equal to 80 mG for cTAM and 32 mG for its deuterated derivative cTAMd36,48 160 mG for Ox063 and 80 mG for its deuterated derivative Ox063d24,49 and 48–52 mG for pTAM depending on the ionization state when measured at the L-band EPR frequency.28
A concept of using chemical reactions of stable radicals to learn about local chemical microenvironment via corresponding EPR spectral changes is less appreciated. In contrast to widely used NMR studies of chemical reactions based on the presence of magnetic nuclei in the most of chemical compounds, EPR detection of chemical reaction reagents or products are limited to the radical species. This significantly narrows the number of reactions that can be studied by EPR. On the other hand, overlap of the various endogenous NMR signals limits functional biomedical NMR and MRI applications while exclusive detection of specially designed paramagnetic probes provides EPR with desirable functional specificity and sensitivity.
The chemical reaction to be detectable by EPR has to comply with the two hardly compatible requirements. On the one hand, the chemical reactions should proceed in close vicinity to the radical fragment to affect significantly magnetic resonance parameters of the probe. On the other hand, the reaction should not destroy the unpaired electron of the probe to allow for its detection by EPR, with the exception of radical redox reaction measured by the decay of EPR radical signal.8 These requirements makes the design of suitable paramagnetic probes to be very challenging task. The ideal noninvasive EPR radical probe, R•, would participate in reversible exchange chemical reactions with a chemical analyte, A, namely, , that result in significant difference in magnetic resonance parameters of radical-reagent, , and radical-product, , therefore opening the possibility to measure reactivity and concentration of the analyte.9
The Heisenberg spin exchange interaction represents a physical phenomenon that mimics the simplest reversible chemical reaction affecting magnetic resonance parameters of the radicals participating in exchange.10,11 When in solution diffusion brings two free radicals so close to one another that the wave functions for the two unpaired electrons overlap, there is a probability that these electrons will exchange their spin coordinates. Thus, an increase in the exchange rate leads to a reduced lifetime for a given spin state leading to the frequency exchange and line width broadening in the EPR spectra. The rate of spin exchange is proportional to the concentration of the radical species participating in the exchange, therefore, allowing for quantification of these species from the EPR spectra. One of the most successful biomedical EPR applications, EPR oximetry,12,13 is based on spin exchange interaction between a paramagnetic probe and the oxygen molecule, the latter being diradical in its ground-state electron configuration.
Arguably the most studied chemical reactions of the stable radicals are related to the redox properties of their radical centers. In general, these reactions result in the oxidation or reduction of the radical center and therefore in loss of the EPR signal. An exception is electron self-exchange reactions which denote the one-electron transfer between the partners of a redox couple (e.g., NR and its oxoammonium cation) which results in no net chemical reaction. Electron self-exchange experimental studies were performed in a pure solvents providing useful information for the theory of electron transfer and understanding the solvent effects.14 Application of the electron self-exchange reactions of the paramagnetic probes to assess chemical microenvironment in complex biological systems seems to be hardly possible.
NRs are known to be highly succeptible toward one-electron reduction to the corresponding diamagnetic hydroxylamines that limits their stability in biological systems.15 Numerous efforts were devoted to the synthesis of the NR probes with enhanced stability toward reduction.16–18 In contrast to the NRs, TAM radicals are extraordinary stable toward reduction5,19 and major pathways of their degradation in biological systems under aerobic conditions proceed via enzymatically catalyzed TAM oxidation.20 While redox reactions of the NRs and TAMs result in loss of EPR signal and may compromise EPR sensitivity, they both were used in specific EPR applications. NRs were used as redox probes to assess reducing capacity of living tissues by MRI,21 EPR,8,22 and EPR imaging,23 in vivo. TAMs radicals have been used as superoxide-sensitive probes allowing for quantification of this biologically important radical oxygen intermediate product from the loss of TAM EPR signal.24
One of the simplest reversible chemical reactions in solution is proton exchange. Proton exchange between radical, R•, and its protonated form, RH•+, proceeds via reaction with solvated protons or proton donor, BH+, therefore allowing EPR use for measurement of [H+] (pH) and, at certain conditions, concentration of the compound B. Protonation of the N–O• radical fragment of NRs proceeds only in a very acid media25 and cannot be exploited at physiological conditions. Numerous studies of the EPR manifestation of effects of protonation of the functional groups in the vicinity to the radical center of the NRs9,26 and recently of TAMs,27,28 finally resulted in development of a set of molecular pH-meters for biological applications. The effect of H+ donors on the spectra of pH-sensitive probes has been recently utilized for quantitative EPR measurements of inorganic phosphate (H2PO4− being a H+ donor).29
Some type of the NRs, in particular imino nitroxides, form equilibrium complexes in aqueous solutions not only with protons but also with a number of metals (HgII, ZnII, CdII, AgI, etc.).30 Complexation of metals with the N-3 imino nitrogen of imino nitroxides results both in significant changes in nitrogen hyperfine splitting and appearance of the hyperfine interaction with the metal nucleus (e.g., ≈40 G hyperfine splitting for HgII bound to imino nitroxide). However, the equilibrium of the reaction of metal complexation by imino nitroxides is strongly shifted toward dissociation making its use in analytical purposes unpractical.
Among more complex chemical reactions that found applications in biological systems are the reaction of nitronyl nitroxides with nitric oxide, NO,31 and nitroxyl, HNO.32 The corresponding EPR approaches for discriminative detection of these physiologically important molecules were developed but mostly limited to application in in vitro systems due to extremely fast bioreduction of these type of the nitroxides. The reactions of nitronyl nitroxides with NO and HNO proceed via specific organic chemistry reactions, are irreversible, and are out of scope of this paper.
Finally note that specific chemical reactions that proceed comparatively far from the radical center and, therefore do not affect significantly magnetic resonance parameters of monoradicals, may significantly affect EPR spectra of biradical molecules via modification of intramolecular radical–radical interaction. Below we consider an example of the complete cancelation of intramolecular spin exchange between two monoradical fragments of the biradical that takes place upon splitting of the disulfide bond in the disulfide biradicals.33 The NR-NR33–35 and recently TAM-NR and TAM-TAM36 disulfide biradicals were developed for detection of thiols via the thiol–disulfide reaction of the disulfide biradical probes with the thiol group of the analyte.
In this feature article we reviewed the basic principles of using stable organic radicals involved in reversible exchange processes as functional EPR probes with a focus on in vivo assessment of crucial parameters of the local chemical microenvironment in living subjects, such as tissue oxygenation, acidosis, redox, glutathione, and inorganic phosphate.
SPIN EXCHANGE AND EPR OXIMETRIC PROBES
Spin exchange interaction is a consequence of the dependence of the Coulomb repulsion of electrons on their spin orientations: according to the Pauli principle two electrons with parallel spins cannot be simultaneously found at the same point of space; therefore, their electrostatic repulsion is weaker compared to that for two electrons with antiparallel spins. The energy of spin exchange interaction characterized by the exchange integral, J, becomes significant only when the radicals approach to a van der Waals distance allowing for their orbital overlap. For solution of stable radicals discussed here, electron spin exchange interactions can be described by Heisenberg pairwise exchange interactions taking place via binary collisions of the radicals:
| (1) |
The rate of the spin exchange process, We, is in complete analogy with bimolecular chemical reactions that proceed with the rate constant, ke, and can be expressed via collision rate and diffusion constant, kD, as follows:
| (2) |
where p and f are the efficiency of the spin exchange and a steric factor, correspondingly. Note that for the small radicals including most of the NRs and TAMs, it is a valid approximation of strong spin exchange (J2τc2 ≫ 1 where τc collision time). For the radicals with the spin 1/2 in the approximation of strong exchange, p is equal to 0.5 because the two electron states are approximately equally populated at room temperature.10
Figure 1 demonstrates the typical effect of the self-spin-exchange on the continuous waves (CW) EPR spectra shown for the solution of the NR1 (see Scheme 1 for the structure) at various radical concentrations. The spectrum (Figure 1a) corresponds to the diluted NR solution with a negligible effect of spin exchange and consists of three lines due to hyperfine interaction of unpaired electron with the nuclear magnetic moment of nitrogen (SN = 1) characterized by a hyperfine splitting constant, aN.
Figure 1.

EPR spectra of the di-tert-butyl nitroxide radical, NR1 (see Scheme 1) in ethanol at room temperature at various radical concentrations: (a) 0.1 mM, (b) 10 mM, (c) 100 mM, and (d) pure liquid NR. Spectra are normalized for the same peak intensity. Reproduced from ref 37 (p 323, Figure 10.8) with permission of John Wiley & Sons, Inc. Copyright 2007 John Wiley & Sons, Inc.
An increase in the rate of spin exchange at larger NR concentrations results first in the line broadening and a small shift of the outer lines toward the center of the spectrum37,38 (Figure 1b). The line broadening, ΔH, occurs as a result of shortening the lifetime of the electron spin states due to spin exchange in agreement with the uncertainty principle and is proportional to the exchange frequency, ωe = ke[NR]:
| (3) |
where γ is the gyromagnetic ratio of the electron (1.76 × 107 s−1 Gauss−1, in angular frequency units); nuclear statistical factor, 2/3, arises because of the spin exchange between NRs with the same projections of nitrogen nuclear spin (for 14N, SN = 1) produces no observable line broadening; and the factor 1/√3 is included to convert the derivative line width to the absorption line width. Further increase in NR concentration results in the spectrum coalescence in a single broad line (Figure 1c) when exchange frequency is close to the difference in the EPR frequencies of the lines participating in exchange ωe ≈ γaN. At even higher radical concentrations, the width of this single line becomes exchange-narrowed (Figure 1d) proportionally to the term aN2/ωe since the electron spins are exchanging so fast that the time average of the hyperfine splitting is approaching zero.10
Spin exchange in solution of the radicals R1 with comparatively long relaxation time and radicals R2 with short relaxation time, such as oxygen diradical molecule, O2, or paramagnetic ions of Fe2+, Co2+, and Ni2+, will manifest itself only in shortening the relaxation times of the radical R1 since the EPR signal for the R2 is not detectable. In the case of CW EPR, this will result only in broadening of the lines of the R1 spectrum while practically no line shift will occur.10,39 In the particular case of the spin exchange of the stable radicals such as NRs and TAMs with oxygen, line broadening is described by eq 4 which differs from eq 3 by the absence of the nuclear statistical factor (2/3) since for the spin exchange between different particles it is always equal to unity:
| (4) |
Figure 2 shows the typical effect of oxygen on the individual line of the EPR spectrum of the nitroxides (NR2, Scheme 1). In deoxygenated solution, each component of the NR2 hyperfine triplet spectrum shows a partially resolved superhyperfine pattern arise from interaction with 12 protons of the four methyl groups and the proton at carbon C-4 of the heterocycle. An increase in oxygen concentration results first in the broadening of the components of superhyperfine structure and then in the broadening of the enveloped EPR line.
Figure 2.

EPR spectra of 50 μM aqueous solution of the NR2 measured at 37 °C at various oxygen concentrations. Spectral parameters: microwave power, 2.4 mW; time constant, 5.12 ms; sweep time, 81.92 s; number of points, 4096; modulation amplitude, 0.1 G; (0.8 G for spectrum at 100% oxygen); sweep width, 4 G (6 G for spectrum at 100% oxygen). Line width at 100% oxygen is 1.60 ± 0.03 G. Reproduced from ref 40 with permission of John Wiley & Sons, Inc. Copyright 2010 John Wiley & Sons, Inc.
Spin exchange represents pure physical interaction between the molecules of paramagnetic probe and oxygen which does not interfere with the oxygen metabolism therefore providing basis for noninvasive EPR oxygen measurements in biological systems, including that in vivo. The bimolecular collision rate of the probe with oxygen needs to be comparable either to the inverse of longitudinal (T1) or transverse (T2) relaxation time for the optimal sensitivity of T1- or T2-sensitive techniques, respectively.41 NRs were the first paramagnetic probes used for EPR oximetry.42–45 Backer et al.42 were first to apply the T2 oximetry method based on oxygen-induced line broadening of the NR to follow mitochondrial respiration in samples containing about 100 liver cells. The T1-sensitive EPR oximetry methods were introduced by Hyde et al. and include pulsed saturation-recovery, continuous wave saturation and rapid passage display modalities.41,46,47 T1-sensitive methods might have an advantage for highly viscous environments or spin-labeled macromolecules because for the nitroxides the T2 ≪ T1.
The values of the line-broadening effect of oxygen vary with the radical structure and for the aqueous solutions of the NRs and TAMs on average are about 0.5 G/mM of oxygen (≈ 0.6 mG/mmHg of pO2). Taking into account that for the rare exceptions the line width of the nitroxides (typically ≈ 0.5–1.5 G) is about 1 order of magnitude larger than that of TAMs (≈ 50–150 mG, see Scheme 2), TAMs were found to be preferable EPR oximetric soluble probes with oxygen sensitivity approaching 1 mmHg of pO2.
Self-spin exchange-induced line-broadening may interfere with accurate oxygen measurements at high probe concentrations. EPR spectra of TAMs show about 10–30 mG/mM self-spin exchange-induced concentration broadening5,50 being significantly lower compared with 100–200 mG/mM concentration broadening for the NRs.40 In the presence of hyperfine spectral structure, spectral manifestation of spin exchange with oxygen and self-spin exchange of the probe (concentration broadening) can be discriminated. As we mentioned above, spin exchange with oxygen results only in the EPR line broadening while self-spin exchange additionally narrows the distances between spectral components participating in the exchange. On the basis of this principle, Halpern et al.51 proposed the EPR oximetric application of a selectively deuterated nitroxide, NR3 (Scheme 1). The NR3 shows only one hydrogen super hyperfine doublet splitting therefore allowing for discrimination of the contributions of oxygen and probe concentrations into EPR line-shape based on their distinctive effects on line broadening and distance between the components of the doublet. Recently similar principle has been used to discriminate concentration- and oxygen-induced line broadenings for the multifunctional trityl probe, pTAM (Scheme 2)29 as it will be discussed below.
SPIN EXCHANGE IN BIRADICALS: THIOL-SENSITIVE EPR PROBES
Biradicals with unpaired electrons separated by a large number of chemical bonds simulate the situation of spin exchange in magnetically diluted solutions. In this case, the dynamics of spins in a biradical resembles that of spins in a pair of monoradicals colliding in solution when the anisotropic exchange integral is randomly modulated by rotational diffusion. The manifestation of spin exchange in the EPR spectra of biradicals depends on total number of biradical conformations, their individual values of exchange integral, and the rates of transition between conformations with a majority of studies done for the nitroxide biradicals (for reviews see ref 52). In general, a complexity of the EPR spectra results in a significantly small number of the biradical applications as functional probes compared to the monoradicals. Nevertheless some spectral features of the biradicals result in unique specific functional sensitivity unavailable in the monoradical counterpart.
Figure 3 (left) shows the EPR spectrum of R2SSR2 nitroxide biradical, which is significantly affected by intramolecular spin exchange between two radical fragments resulting in appearance of “biradical” spectral components in addition to the conventional triplet spectral pattern of the mononitroxide. An appearance of the biradical components can be qualitatively explained by the presence of the conformation with intermediate or strong spin exchange characterized by the exchange integral, J ≥ aN. The EPR spectrum of the subensemble of biradicals with both 14N nuclei of NO fragments having the same spin projections (m1, m2, +1,+1; 0,0; and −1, −1) is not affected by spin exchange, therefore providing conventional triplet pattern with relative spectral intensities, 1:1:1. Fast spin exchange in subensembles with different nuclei spin orientations results in line coalescence and appearance of one line in the center for subensembles (+1,−1 and −1,+1) and two lines shifted by aN/2 in both directions from the center for subensembles (+1,0; 0,+1; 0,−1; and −1,0), each of these three lines having relative intensity, 2. Therefore, the total superimposed spectrum is represented by quintet, 1:2:3:2:1, with the splitting equal to aN/2. In the general case, the spectrum of the nitroxide biradical is represented by the superposition of the conformations with the different values of exchange integral and dynamics of conformational transitions. The spectrum of R2SSR2 biradical (Figure 3A, top) is characteristic for the superposition of several biradical conformations with J ≈ aN.34 As seen in Figure 3, the reaction with glutathione, GSH, results in complete cancelation of intramolecular spin exchange between two monoradical fragments due to splitting of the disulfide bond via thiol–disulfide exchange reaction and formation of two monoradicals.33
Figure 3.

(A) X-band EPR spectra of 100 μM R2S-SR2 measured at various time points after incubation with 2.5 mM GSH in 0.1 M Na-phosphate buffer, pH 7.2, and 1 mM DTPA at 34 °C. The kinetics analysis provides the observed rate constant value of the reaction between GSH and R2S-SR2, kobs(pH 7.2, 34 °C) = 2.8 ± 0.2 M−1 s−1. (B) Kinetics of the monoradical spectral peak intensity change measured in mammary tumor (●) and normal mammary gland (○) using L-band EPR. The solid lines are the fits of the initial part of the kinetics by the monoexponent supposing kobs (pH 7.2, 34 °C) = 2.8 M−1 s−1 and yielding [GSH] = 10.7 mM and 3.3 mM for the tumor and normal mammary gland, correspondingly. Reproduced from ref 8 with permission of John Wiley & Sons, Inc. Copyright 2012 John Wiley & Sons, Inc.
| (5) |
Nitroxide biradical disulfides represent paramagnetic analogs of Ellman’s reagent and have found application as thiol-sensitive probes. Biradicals R1SSR1 and R2SSR2 show more than 3 orders of magnitude difference in their reactivity toward low-molecular-weight thiols such as cysteine and GSH which determines their different area of applications.
Fast reacting R1SSR1 (kGSH ≈ 5 × 103 M−1 s−1 at neutral pH and room temperature) has found application for in vitro EPR measurement of thiols, in particular GSH presented in cells in millimolar concentrations and considered to be a major intracellular redox buffer.53 Note that GSH does not penetrate the cellular membrane, and its extracellular concentrations in living tissues are at the low micromolar level. The method relies on the application of the access of the biradical over GSH and measures the fraction of the biradical split by the reaction with GSH immediately after probe addition to the sample (the R1SSR1 probe easily penetrates the cellular membrane and react with 1 mM GSH with characteristic time of about 0.2 s). GSH-induced changes in the ratio of mono- and biradical components are calibrated and used afterward for [GSH] calculation in the experimental sample. The sensitivity of the method is sufficiently high to perform the measurements of GSH in very few (≈100) cells. The described “static” EPR approach of GSH assessment has the advantage in its simplicity and represents a fast, highly sensitive, convenient, and reliable approach for in vitro measurements in optically nontransparent samples. On the other hand, the approach is based on complete consumption of the vital thiols, therefore its application in vivo is limited due to corresponding damage to cellular redox functions.
The reasonable time window for the reaction of the slow-reacting R2S-SR2 probe with glutathione at physiological intracellular tissue GSH concentrations and neutral pH (kGSH ≈ 2.8 M−1 s−1, see Figure 3A) allows for application of “kinetic” EPR approach for in vivo GSH detection as illustrated in Figure 3B. The kinetics measurements allow for using low probe concentration compared to [GSH], therefore minimizing possible probe toxicity. Noninvasive kinetics-based in vivo EPR measurement of intracellular GSH using the R2SSR2 probe is based on the dominant contribution of GSH in the reaction with biradicals, fast diffusion of the probe across the cellular membrane and their comparatively low reduction rates.8,34 Signal intensity and functional sensitivity of the probe can be increased by isotopic 15N and 2H substitutions (see R3SSR3, Scheme 3)54 which results in the decrease of the number of spectral lines and their line width, correspondingly.
Scheme 3.

Chemical Structures of the Imidazoline, R1SSR1, Imidazolidine, R2SSR2 and R3SSR3, and Pyrrolidine Disulfide Biradicals, R4SSR4 and R5SSR5, Discussed in the Text
Currently EPR spectroscopy in combination with the optimized imidazolidine nitroxide disulfide biradicals R2SSR2 and R3SSR3 is the only direct noninvasive method that proves its capability for quantitative measurement of glutathione in living subjects.8,54 Recently new NR-NR,35 TAM-NR, and TAM-TAM36 disulfide biradicals were synthesized. Note that TAM-containing disulfides TAM-NR and TAM-TAM36 are cell-impermeable and therefore their application for intracellular GSH measurements is hardly possible. The feasibility of using pyrrolidine disulfide biradicals (see Scheme 3 for the structures) for thiols detection was first demonstrated in vitro35,55 and very recently in vivo for imaging thiol redox status in murine tumors.56 Further studies are required to determine their potential for quantitative in vivo GSH detection, which strongly depends on the comparative contributions of the various factors into the EPR spectra change, namely, the rates of probe reaction with GSH, probe penetration into the cells, and probe reduction.
PROTON EXCHANGE AND SPIN pH PROBES
In aqueous solutions chemical reactions of proton exchange between spectroscopically distinguishable free radical R• and its conjugate acid RH•+ in the presence of buffer molecules, B, can be described as follows:
| (6) |
| (7) |
The equilibrium constant of the proton exchange reaction 7 is determined by thermodynamic parameters of the compounds involved, namely, by the pK values of the radical and buffer, pK7 = pKR − pKB. The concentration ratio of [RH•+] to [R•] does not depend on buffer concentration and is determined by the values of pH and pK of the radical (pKR):
| (8) |
Equation 8 represents the standard pH titration curve providing a basis for using radicals as pH-sensitive probes. An accuracy of [RH•+]/[R•] ratio determination depends on extent of protonation effect on the EPR spectrum of the radical, and the signal-to-noise ratio (SNR) of the EPR spectra, and in many reported cases is equal or better than 10% which is equivalent to accuracy of pH measurement of 0.05 pH units. Note that pH probe has optimal sensitivity in a linear part of the titration curve (8) at pH ≈ pKR ± 1.
The kinetics of reactions 6 and 7 determine the rate of proton-exchange-induced frequency exchange in the EPR spectrum and therefore its line shape. Intermediate exchange on the EPR time scale is observed when lifetimes of the radical in the RH•+ and R• forms are close to the frequency difference between corresponding EPR lines, . In the absence of buffer, this condition is equivalent to two requirements k6[H+] ≈ k−6 and k6[H+] ≈ Δω. The first condition, k6[H+] ≈ k−6, is a rather obvious statement that the simultaneous observation of the EPR spectra of both radical forms is possible only at pH ≈ pKR in agreement with eq 8. The second condition, k6[H+] ≈ Δω, for diffusion-controlled proton exchange reactions (k6 ≈ 109 M−1 s−19 and Δω ≈ (106 – 107) s−1 (see Figure 4) yields [H+] ≈ 10−3–10−2 M or pH ≈ 2–3. In agreement with this estimate, a fast or intermediate proton exchange was observed for the number of NRs with pKR < 3 in very acidic solutions at pH < 3.9 Interestingly, the fast exchange was also reported for the radicals with pKR > 11 in alkaline solutions at pH > 11.9 The latter cases are determined by the proton exchange via the mechanism in eq 9 facilitated by a high concentration of hydroxyl anion in alkaline solutions:9
Figure 4.

(A) Second-derivative of high-field component of X-band EPR spectra of aqueous 0.3 mM solutions of the radical NR4 (pKR = 6.1) measured at various phosphate buffer concentrations, pH = 5.9 and temperature, 23 °C. At low phosphate concentration (top spectrum), the narrow lines of this deuterated probe allows for distinguishing not only R• and RH•+ and forms but also discriminating two protonated radical forms with different nuclear spin projection of attached proton, and . The fitting calculated spectra (dotted lines) to the experimental ones performed as described in ref 57 provides the values of the rate constants of proton exchange of the NR4 with phosphate, k7(NR4) = 2.6 × 108 M−1 s−1, k−7 (NR4) = 1.6 × 109 M−1 s−1. Reproduced from ref 57 with permission of Springer. Copyright 2001 Springer. (B) High-field component of the L-band EPR spectra of aqueous 0.2 mM solutions of the pTAM radical (pkR = 6.9) measured at various phosphate buffer concentrations, pH 6.8, under anoxic conditions and temperature 37 °C. The fitting calculated spectra (dotted lines) to the experimental ones performed as described in ref 29 provides the values of the rate constants of proton exchange of the pTAM with phosphate, k7(pTAM) = 2.1 × 107 M−1 s−1, k−7(pTAM) = 3.3 × 107 M−1 s−1. Reproduced from ref 28. Copyright 2013 American Chemical Society.
| (9) |
The observed cases of the proton exchange of the radicals with solvated proton in aqueous solution at neutral pH show slow frequency EPR exchange due to the low concentrations of protons or hydroxyl anions, [H+], [OH−] ≪ 1 mM.
In the presence of buffer, the condition of intermediate exchange ( ) transforms into the requirements, k7[BH+] ≈ k−7[B], k7[BH+] ≈ Δω, or pKR ≈ pH and [BH+] ≈ [B] ≈ 1–10 mM. Figure 4 shows proton exchange-induced spectral transformations for the nitroxide NR4 (Figure 4A) and trityl pTAM (Figure 4B) observed at pH close to the pKR of the radicals upon increase of phosphate buffer ( ) concentration. At a low concentration of phosphate, the frequency exchange does not affect significantly the EPR spectra. The ratio of the spectral components of the RH•+ and R• forms corresponds to the differences between the values of pH of solution and pKR according to eq 8. At higher phosphate concentrations, the proton exchange results in line broadening of the spectral components participating in the exchange due to a decrease of the life times of the corresponding ionization states of the radical proportionally to the phosphate concentration ( ). A further increase of the phosphate concentration results in the lines coalescence followed by the enveloped line narrowing in the region of fast frequency exchange ( ).
Summarizing, manifestation of the proton exchange in the EPR spectra of aqueous solutions of stable radicals such as NRs and TAMs allows for quantitative measurements of the equilibrium and kinetics parameters of this chemical reaction, and as a consequence in particular situations this allows for concurrent quantitative assessment of the concentrations of protons (pH) and buffer molecules, e.g., concentration of inorganic phosphate, Pi.
MUTIFUNCTIONAL ASSESSMENT OF CHEMICAL MICROENVIRONMENT
Noninvasive assessment of chemical parameters in living tissue such as oxygen, pO2, pH, interstitial inorganic phosphate, Pi, redox status, and glutathione may provide unique information related to normal physiological as well as pathophysiological processes. Significant efforts have been made to upgrade low-field EPR spectroscopy and imaging techniques with capabilities for functional measurements when combined with specific paramagnetic probes including pO2-, pH-, redox-, and GSH-sensitive probes. Multifunctional probes would have the advantage in the capability for assessment of several parameters simultaneously independent on probe distribution and time of probe delivery. This opens a new functional dimension allowing for correlation of the parameters between each other, therefore providing insight into the underlying biological mechanisms. Until recently there were no available techniques for the concurrent assessment of several chemical parameters in tissue microenvironment in living subjects.
In theory, NRs with ionizable groups possess multifunctional spectral sensitivity toward pH, redox, oxygen, and radical concentration. In practice, it is enormously difficult to design the NR structure that combines reasonable spectral sensitivity toward several of these parameters at physiological conditions. First, except for the rare exceptions, NRs have limited oxygen sensitivity due to a comparatively broad line width which is further compromised by concentration broadening. Second, the NRs reduction in reducing biological medium into EPR-silent products often compromises their EPR detection, and therefore synthesis of the NRs with enhanced stability toward reduction is highly desirable for in vivo use.
NR5 represents one of the successful examples of dual function pH and redox probe developed for in vivo monitoring of the tissue microenvironment. This probe combines a number of structural features that makes it a suitable probe for monitoring tumor extracellular tissue pH (pHe) and the redox status in animal models of cancer (Scheme 4). Note that extracellular probe localization is required to report on tumor tissue acidosis occurring in extracellular medium only. This has been achieved by binding the NR label to glutathione preventing the resulting NR5 probe from diffusion across the hydrophobic lipid bilayer of the biomembranes and therefore enforcing probe localization in extracellular aqueous volumes.58 The pKR value of the probe has been tuned by variation of the substitute at position 4 of the radical heterocycle resulting in the ideal range of pH sensitivity centered on slightly acidic pH in tumors (pKR = 6.6 at 37 °C). The bulky ethyl substitutes at positions 2 and 5 around the paramagnetic NO fragment of the NR5 have been introduced to enhance the probe stability toward bioreduction. The NR5 probe has been used to assess tissue acidosis in various mouse models of cancer8,59,60 and in the ischemia-reperfusion model in isolated perfused rat hearts.61,62 The sufficiently long reduction rate of the NR5 even in highly reducing tumor tissue (τ1/2 ≈ 5 min) allowed for quantitative assessment of tumor tissue reducing capacity in complement to pH measurements.8
Scheme 4.

Structure of the NR5
Trityl radicals have been found to be the most sensitive soluble EPR oximetric probes with extraordinary stability in vivo. A decade ago we published the concept of dual function oxygen-and pH-sensitive TAM probes based on introduction of ionizable groups in their structure.27 Following this concept, TAM probes containing amino-63 and phosphono-28,64,65 groups with pH sensitivity in the physiological range of pH were synthesized based on the cTAM structure (see Scheme 2). The cTAM radical bearing two thioacetonide moieties and one carboxylic acid on each aromatic group exhibits an extraordinary stability in vivo as a result of steric protection and electronic stabilization.5 Conformational studies using EPR spectroscopy, DFT calculation, and X-ray diffraction have shown that the TAMs adopt a helix conformation (Scheme 2) which protects the central carbon bearing approximately 60% of the unpaired electron spin density66 and provides helicoidally chirality to the EPR spin probe.67,68
The EPR spectra of TAMs containing amino-63 and phosphono-28,64,65 groups exhibit complex EPR spectra therefore making the spectrum simulation and extraction of both functional parameters, pO2 and pH, challenging. In particular, the EPR spectra of triphosphonated TAM radicals64,65 (Scheme 5) contain the superposition of four ionic species arising from the independent ionization of three phosphonic groups with slightly different pKa2 (6.4, 6.9, and 7.7) each having different phosphorus hyperfine splitting pattern. The first dissociation constant of phosphonic acid with pKa1 ≈ 2 keeps the molecule ionized, therefore enhancing its aqueous solubility under physiological pH and ensuring its extracellular localization by preventing transmembrane diffusion.28,64,65 The recent p3TAM noninvasive application in vivo for pH measurements based on in vitro calibration of the apparent phosphorus hyperfine splitting showed the results in agreement with the ones measured using invasive pH electrodes.69 However, an extraction of the second function, pO2, from the EPR spectrum of p3TAM is hardly possible due to the overlap of the numerous individual spectral lines. Isotopic substitution of all 36 methyl hydrogen atoms for deuterium in p3TAMd36 (Scheme 5) significantly decreases the individual line width from about 100 mG down to 40 mG65 allowing for discriminative measurements of both pH and pO2. Still complex multiline spectrum of p3TAMd36 probe compromises signal intensity and accurate line shape analysis making its application in biological systems difficult.
Scheme 5.
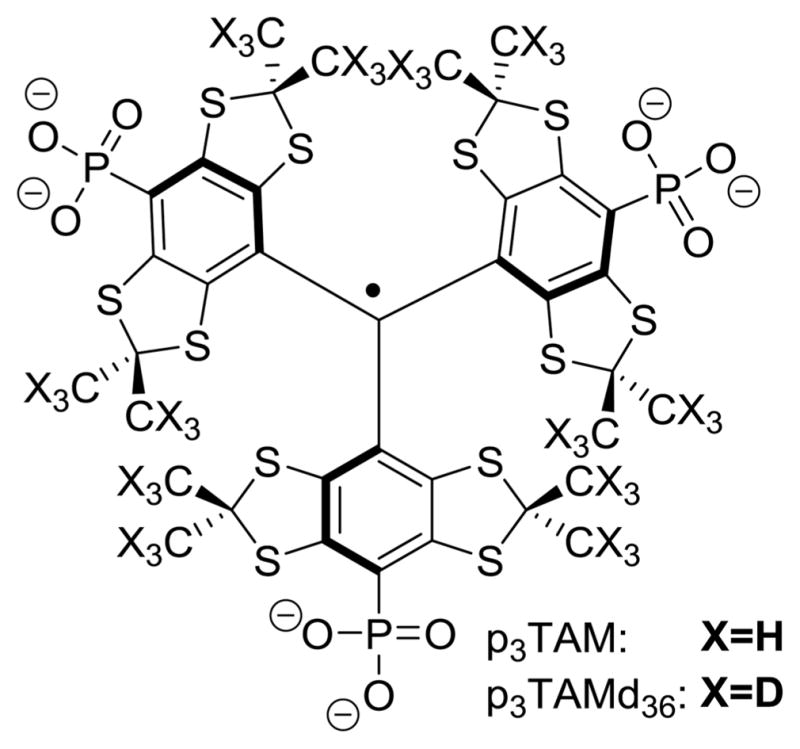
Chemical Structure of p3TAM and Its Deuterated Derivative, p3TAMd36
The synthesis of the monophosphonated TAM derivative decreases the number of phosphonic acids from three in p3TAM and p3TAMd36 (Scheme 5) to one in pTAM (Scheme 2) and provides the simplest doublet EPR hyperfine splitting pattern sufficient for multifunctional sensitivity.28,29 The doublet of very narrow EPR lines of the pTAM probe arises from phosphorus hyperfine splitting (SP = 1/2) allows for the quantitative line shape analysis and discriminative concurrent extraction not only of pH and pO2 values but also concentrations of inorganic phosphate, Pi, and concentration of the probe.29 Because of unsurpassed multiple functional sensitivity to pH, pO2, and Pi in the extracellular tissue microenvironment, we further abbreviated this type of probes as a HOPE probes.
Figure 5 illustrates the multifunctional assessment of the chemical microenvironment using the pTAM HOPE probe.
Figure 5.

Multifunctional assessment of chemical microenvironment using pTAM HOPE probe. (a) Setup for in vivo L-band EPR measurements of the tissue microenvironment parameters in living mice. Photograph shows the anesthetized mouse between the magnets of the EPR spectrometer with the inset on the right showing placement and positioning of the loop resonator on top of the measured tissue. (b) The scheme of pH-dependent equilibrium between two ionization states of the probe. (c) L-band EPR spectrum of pTAM. (d) The EPR line width of the pTAM is a pO2 marker (accuracy, ≈ 1 mmHg; pO2 range, 1–100 mmHg). (e) The fraction of protonated pTAM is a pH marker in the range from 6 to 8.0 (accuracy, ±0.05). (f) Dependence of proton exchange rate (expressed in mG) of the pTAM with inorganic phosphate on Pi concentration extracted by spectra simulation (accuracy, ±0.1 mM; range, 0.1–20 mM).28,29 Note interstitial extracellular localization of the pTAM probe: it does not penetrate into the cells due to bulky charged structures and the pTAM signal from the blood is not detected by EPR due to signal broadening by pTAM complexation with plasma albumin.70 Reproduced from ref 71 with permission of Nature Publishing Group. Copyright 2017 Nature Publishing Group
The pTAM doublet hyperfine splitting on phosphorus nucleus differs for two ionization states shown in Figure 5b (ap(pTAM3−) = 3.63G; ap(pTAM4−) = 3.37G) resulting in additional doublet splitting of the EPR lines of about 130 mG (Figure 5c). A fraction of one of the ionization states measured from the integral intensities of the individual lines of this doublet is a pH marker (Figure 5e) while the line width of the individual components is a pO2 marker (Figure 5d). In contrast to oxygen-induced line-broadening, phosphate-facilitated exchange between the EPR lines of two ionization states of pTAM results both in line broadening and in narrowing the distance between the lines of the smaller doublet (Figure 5c). In its turn, spin self-exchange between trityl radicals in addition to line broadening results in a shift of the positions of all the EPR lines of pTAM (see ref 29 for details). Therefore, in general, the contributions of all four factors, pH, pO2, [Pi], and [pTAM] in the EPR spectrum of the probe are qualitatively different and can be discriminated by advanced spectra analysis using theory of exchange between several sites in noncoupled systems.72 Note that this spectra simulation in complex multicomponent system are preceded by the analysis of spectra changes separately for each of the ionization states of pTAM upon variation of one of the factors only: (a) pO2 variations in the absence of phosphate and low [pTAM] to exclude concentration broadening; (b) [Pi] variations in deoxygenated solutions at low [pTAM] as shown in Figure 4b; and (c) [pTAM] variation in deoxygenated solutions in the absence of phosphate.29 These measurements allow for calibration of probe reactivity yielding the values of the bimolecular rate constants of the corresponding exchange processes.29 The obtained values of the rate constants about 109 M−1 s−1 for the spin exchange of pTAM with a small oxygen diradical molecule; almost 2 orders of magnitude lower, in the range (2.1–3.3) × 107 M−1 s−1, for the proton exchange reactions of pTAM with phosphate anions; and the lowest values, (0.74–1.43) × 107 M−1 s−1, for the spin self-exchange between bulky pTAM radicals support significant contribution of steric hindrance in the reactivity of trityl radicals. The observed decrease of the values of the rate constants for exchange reactions between different ionization states of pTAM and between pTAM and phosphate upon increase of the negative charge of the anions participating in the exchange supports electrostatic repulsion contribution in the pTAM reactivity.29
Tissue pO2 and pHe are well recognized hallmarks in solid tumors while extracellular Pi has been recently identified as a new signaling molecule of importance in tumorigenesis. An extraordinary high sensitivity of pTAM to pO2 allows one to detect an oxygen tension as low as the threshold of anoxia, about 1 mmHg. pTAM possesses a pH sensitivity in a physiologically important pH range that makes it possible to monitor the acidity both in normal tissues and in acidic tumors with an accuracy of 0.05 pH units. The range of Pi measured by pTAM from 0.1 to 20 mM covers the physiological range of the Pi values previously reported in the literature. This unique multifunctional sensitivity of the pTAM probe decreases the method invasiveness and allows for a better correlation of the parameters independent of the distribution of the probe. pTAM sensitivity to the probe concentration can be used in imaging modalities to map the perfusion areas. Note that long relaxation times of the pTAM probe (depending on ionization state, T1 ≈ 18–21 μs and T2 ≈ 6–11 μs in deoxygenated solutions29) favors its application using pulsed and rapid scan EPR techniques and Overhauser-enhanced MRI.
Recently pTAM probe has been used for in vivo multifunctional assessment of tumor microenvironment (TME) in various animal models of cancer using L-band EPR spectroscopy. The measurements in PyMT transgenic mice which spontaneously develop breast cancer showed existence of hypoxic and acidic areas in TME and significantly (up to 3-fold) higher concentrations of interstitial [Pi] in tumors vs normal mammary glands. Figure 6 shows correlation analysis between measured parameters.
Figure 6.
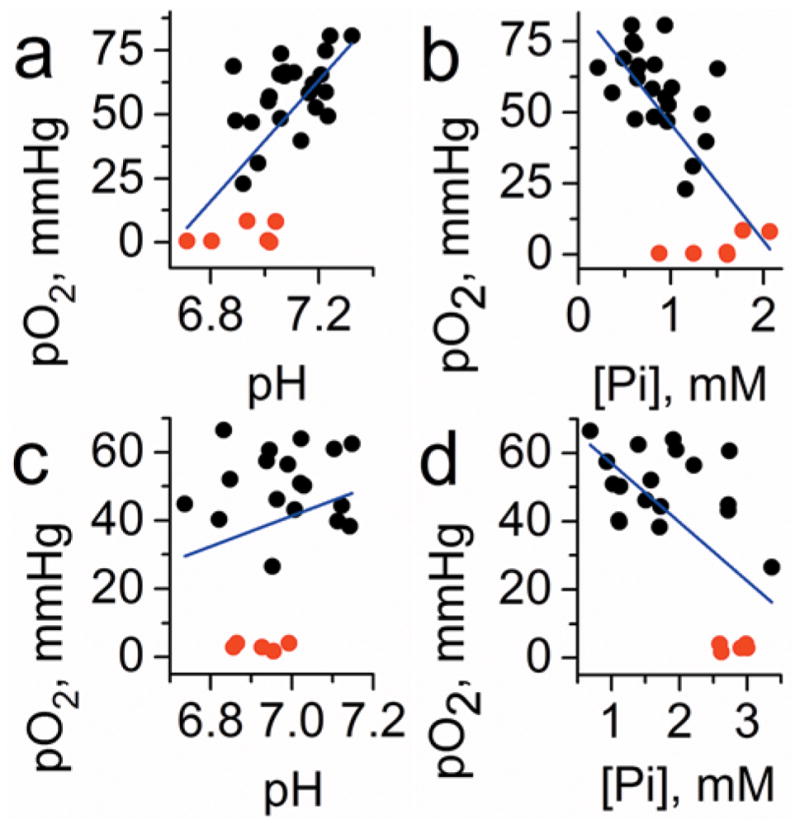
Correlation between interstitial pO2, pHe, and Pi values measured in normal mammary glands of FVB/N wild type mice and in the TME of breast cancer in MMTV-PyMT transgenic mice (n = 23). To extend the range of oxygen variations, anoxic conditions in interstitial space were established by i.t. injection of oxygen-consuming enzymatic system of glucose/glucose oxidase (red symbols). Blue lines represent linear fit for the total data sets. (a) A positive correlation between pO2 and pHe in normal tissue (r = 0.5, p = 0.014 for black symbols; r = 0.64, p = 1.8 × 10−4 for total data set) vs (c) no significant correlation between pO2 and pHe in TME (r = 0.01, p = 0.97 for black symbols; r = 0.23, p = 0.3 for total data set) were found. A negative correlations between pO2 and Pi both in (b) normal tissue (r = −0.51, p = 0.013 for black symbols; r = −0.7, p = 2.3 × 10−5 for total data set), and (d) in TME (b, bottom: r = −0.4, p = 0.079 for black symbols; r = −0.62, p = 0.001 for total data set) were found. Adapted from ref 71 with permission. Copyright 2017 Springer Nature.
The observed positive correlation between pO2 and pHe in normal mammary gland vs absence of correlation in tumors support tumor reliance on glycolysis independent of oxygen concentration; in our opinion, this is an exemplified in vivo demonstration of the Warburg effect. In its turn, the observed negative correlation between interstitial [Pi] and pO2 both in normal and tumor tissues is in agreement with association of high [Pi] (and low ATP/Pi ratio) with changes in bioenergetics status upon lower oxygen supply. Of particular note, these first pTAM applications in animal models of cancer reveal a potential role of interstitial Pi as TME marker for tumor progression and aggressiveness. The interest to Pi role in cancer has been periodically revisited. The selective uptake of phosphorus 32P by tumors upon intravenous injection of radioactive phosphate has been known since the 1940 report by Marshak et al.73 supported by further observations.74–76 In 1980s, the endogenous inorganic phosphate and/or NTP/Pi ratio were considered as candidates to be used as 31P magnetic resonance spectroscopy (MRS)-measured markers to help differentiate hypoxic tumors from normal tissues and optimize and monitor ongoing radiation therapies.77,78 Recently, a high phosphorus content requirement for the malignant cells has been proposed based on the “growth rate hypothesis” (GRH).79–81 The GRH hypothesis being applied to cancer predicts that tumors are richer in phosphorus than the surrounding tissue due to the requirement of a high amount of ribosomes and other P-rich RNA components that are necessary to manufacture proteins in rapidly proliferating cancer cells. This was also suggested experimentally by significantly higher intracellular concentration of phosphorus in some types of tumors compared to somatic tissues.79 Future studies are warranted to evaluate whether high interstitial [Pi] levels in TME observed using pTAM probe have physiological significance and may provide additional avenues for therapy.
FUTURE DIRECTION IN DESIGN OF MULTIFUNCTIONAL TAM PROBES
With the exception of the polar carboxylic acid and phosphonic acid, the aryl core of cTAM and pTAM HOPE probes has a relatively lipophilic nature which facilitates probe hydrophobic interaction with biomacromolecules such as albumin.70 The latter results in a drastic decrease of the EPR signal intensity and therefore preventing systemic administration of these probes. Further development of HOPE probes may proceed via structure modification and/or formulation to allow for a systemic delivery of the probe and to optimize the circulation time as well as to achieve targeting to specific tissue.
A first strategy based on conjugation of the probe with biocompatible and hydrophilic polyethylene glycol (PEG) chains is illustrated in Figure 7. This modification will prevent hydrophobic interactions with biomacromolecules such as albumin82 while the size of the PEG chains will determine the circulation time. A tissue selectivity can be achieved by conjugation of the probe to a ligand of a particular receptor. For example, adhesion integrin receptors αvβ3 has been shown to be overexpressed on tumoral endothelium during angiogenesis and numerous cancer cells but they are poorly expressed in mature endothelium which make the integrin αvβ3 a popular target for tumor imaging and treatment. This receptor recognized the peptide RGD sequence of appropriate conformation.83,84 The use of folic acid is another popular ligand to target the tumor.
Figure 7.

Structure rationale for the development of PEGylated and targeted HOPE probes. Blue zone encompasses the chemical structural core which has a direct influence on the EPR signal pattern and line width, the latter determines oxygen sensitivity. Red zone encompasses the phosphonic acid moiety that provides pH and [Pi] sensitivities. Green zone encircles a deuterated linker that prevent line broadening of the EPR signal. Yellow zone shows PEGylating which provides high aqueous solubility and prevents hydrophobic interactions with biomacromolecules. Finally, the purple arrow indicates the introduction of a particular ligand that enables a targeting effect of the HOPE probes.
An alternative strategy for development of HOPE advanced structures illustrated in Figure 8 that schematically shows the covalent conjugation of the radical with a water-soluble biocompatible carriers such as dextran, chitosan, and others. Similar to the first strategy of the synthesis of PEGylated HOPEs, targeting can be achieved using a specific ligand.
Figure 8.

Loading of a water-soluble biocompatible polymer with HOPE probe. Active targeting could be achieved by conjugation of a particular ligand.
In theory, such formulation would allow for the injection of smaller probe doses compared to PEGylated HOPE as a higher spin density can be obtained for the same molecular weight. A size between 10 kDa and 40 kDa would represent the optimal zone of investigation in order to increase the circulation time of the HOPE probes and favor probe clearance by the kidney. The possible variations in probe mobility upon binding to polymer and intramolecular spin exchange interaction between pTAM radicals may contribute to the EPR spectrum and has to be minimized to avoid spectrum complication, therefore implementing some structural restrictions in the probe design (e.g., limiting the number of radicals per polymer).
MULTIFUNCTIONAL IMAGING OF PARAMAGNETIC PROBES
In functional EPR imaging (EPRI), spatial variation of EPR line shape and intensity are measured. The former reports local chemical environment, while the latter is proportional to the number of spins in a given voxel. If only one functional parameter, such as pO2 or pH, needs to be measured in the experiment, it is sufficient to image a single EPR line. Its line width can be translated into oxygen concentration and its position would report local pH. Monofunctional EPRI has been performed with single-line trityl probes and the nitroxides.40,85 In the case of the nitroxides because of a large nitrogen hyperfine splitting, aN, between the lines, it is possible to use selected line for imaging.86 However, the maximum gradient is limited to aN/L, where L is the characteristic object linear size, therefore limiting spatial resolution. Multifunctional EPRI requires use of the spin probes discussed above with multiline spectra. In the presence of gradient the lines overlap to give a spectrum that may exceed 10 G in line width. In this situation, the standard filtered backprojection algorithm fails to reconstruct spectral-spatial images.87 In addition, pulsed EPR may not be used because of the excitation bandwidth limitations. CW EPR is not limited with respect to the spectral width but is known to suffer from low sensitivity, especially when it comes to imaging because the signal intensity drops as gradient squared. To our opinion, the future of multifunctional EPRI lays in further development of (i) relatively new methodology, rapid scan EPR and (ii) new algorithms for spectral-spatial image reconstruction.
Alternatively, Overhauser-enhanced MRI (OMRI, also termed proton–electron double-resonance imaging, PEDRI) represents a MRI-based approach for imaging of paramagnetic probes based on the enhancement of the proton MRI image after EPR irradiation due to transfer of polarization from electron spins to nucleus spins by the Overhauser effect.88 It inherently offers high spatial resolution, plane selectivity, and rapid image data collection. Recently developed variable field (VF)89 and variable radio frequency (VRF) OMRI90 approaches allow for functional mapping using NR59 or TAM91 probes while additionally keeping the capability to provide an anatomical image.
A brief review describing recent developments in RS EPRI and functional OMRI and future directions for their use for multifunctional imaging are given below.
Rapid Scan EPR Spectroscopy and Imaging
Since its beginning, the magnetic resonance methodology has been developing along two mainstream directions: CW and pulsed EPR. Both methods have their advantages and limitations. CW EPR does not require high powers and is unrestricted with respect to the spectral width. Pulsed EPR can be only applied to a limited set of samples with relatively long relaxations times, often at low temperatures. In the early days of NMR there were attempts to explore the rapid passage effect92,93 to improve sensitivity. However, neither rapid scan NMR nor CW NMR have never become mainstream methods because of the Fourier transform (FT) NMR success. Long relaxation times of nuclear spins permit using very sophisticated pulse sequences that generate rich data no other method can provide. It is different for EPR. The electron spin time scale is much shorter, so that CW EPR has been and continues to be a mainstream method.
A new development, sinusoidal RS EPR has been demonstrated that CW methodology has not reached its sensitivity limit. A 1–2 orders of magnitude signal enhancement has been achieved compared to the standard modality.94,95 The gain is achieved due to overcoming two major limiting factors of slow scan CW EPR: (i) only a very small portion of the spins contribute to the signal as illustrated in Figure 9; (ii) power saturation of the spin system limits signal intensity. By employing rapid passage, the spins are not readily saturated and more of them contribute to the signal. In fact, in RS EPR all spins are excited and detected twice the scan period as demonstrated in Figure 9. The gain is even more profound for imaging. Gradients of the external magnetic field used to encode spatial information broaden the spectrum. As a result, even smaller portions of the spins contribute to the signal in CW vs RS experiments.
Figure 9.
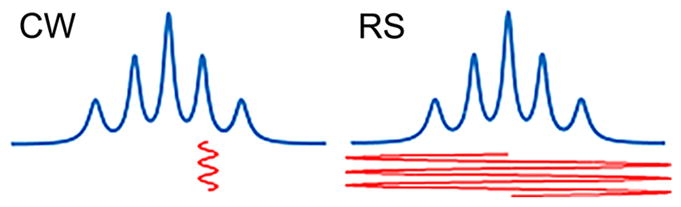
In CW EPR, a small percentage of the spins contribute to the signal. In RS, the entire EPR spectrum is measured twice in the scan period with the SNR gain of 1–2 orders of magnitude. RS signal needs to be postprocessed into the EPR spectrum.
The theoretical sensitivity limit for RS EPR approaches that of FT EPR. To reach the limit, a number of hardware and mathematical problems need to be solved. On the hardware side, further development of bimodal resonators96 together with fast sinusoidal scan system is required. RS EPR enhancement comes with the price of increasing the excitation power. More power delivers stronger signal but also may saturate the detection system. That is why bimodal resonators with good isolation are required. On the side of data processing, the current algorithm97–99 is limited by the scan rate. If free induction decay (FID) time in the rapid scan experiment becomes comparable to the scan period, FIDs produced by up- and down-scans through the resonance overlap. Interference patterns formed by the two oscillating signals is quite difficult but not impossible to detangle.
Multifunctional imaging, simultaneous detection of several physiological parameters, utilizes spin probes with complex spectra. Collected data in the form of projections is transformed into four-dimensional spectral-spatial images. For each 3D voxel in the image there is an EPR spectrum. Spectrum shape analysis provides information on local concentration of oxygen, pH, and other important parameters as discussed above. As a result, 4D image is turned into a set of 3D maps for each parameter. The standard reconstruction method, called filtered backprojection, which was developed for pure spatial imaging, limits the use of multiline spin probes. A new approach to image reconstruction is being evaluated that is based on the concept of solving the inverse problem in a form of large system of linear equations using Tikhonov regularization.87 It was demonstrated that the size of the problem can be reduce by one in the Fourier domain, which makes it possible to handle with a modern desktop computer. The new method has been shown to work in a less computationally demanding two-dimensional case. It is now being optimized for 4D reconstruction.
Overhauser-Enhanced Magnetic Resonance Imaging
OMRI represents double resonance imaging modality in which the polarization of the EPR transition of the paramagnetic probe induced by rf irradiation is transferred into polarization of the NMR transition by the Overhauser effect,88 e.g., resulting in enhancement of NMR signal of water protons. An NMR signal enhancement upon EPR irradiation, nuclear Overhauser effect (NOE) is proportional to several factors, among them ratio of electron and nuclear gyromagnetic ratios ( ) and saturation factor, s, of irradiated EPR transition which varies from 0 to 1 depending on power, time, and frequency of irradiation, probe concentration, relaxation times, and its EPR spectrum shape. The maximum theoretical value of NOE, 329, requires long high power saturation irradiation,100 which is not compatible with living subjects due to overheating problems. Therefore, in in vivo studies short (under 1 s) and weak rf saturation pulses are used providing NOE values around 10.59
The dependence of NOE on the EPR spectral parameters and therefore on the frequency of EPR irradiation provides opportunity to acquire OMRI at several selected spectral points or several power values of rf irradiation, therefore enabling extraction of specific spectral-related parameters and conversion of OMRI images to a functional map. OMRI acquisition in the absence of EPR irradiations and upon rf irradiations at selected EPR parameters allows for coimaging of the anatomical structure and functional mapping of the chemical microenvironment.
The simplest OMRI experiments that measure only spatially resolved changes of NMR signal intensity upon irradiation of the paramagnetic probe at a fixed EPR frequency do not reveal any spectral information on the probe but still may provide functional information on probe distribution and kinetics of the EPR signal change. This type of experiment has been used to assess tissue redox properties in living animals. Both nitroxide reduction rate101–103 and hydroxylamine oxidation rate102 measured by OMRI experiments can be used to map reducing and oxidizing capacities in living subjects, correspondingly. In addition to redox measurements, spatially resolved kinetics measurements of EPR signal intensity provided by OMRI can be used to map the rates of the biradical disulfide, e.g., R2SSR2, reaction with GSH. Taking into account the convenient time-window of the reaction of R2SSR2 with GSH of a few minutes in living tissue and a short time of acquisition of OMRI images of about 10–20 s (see Figure 10), the OMRI mapping of tissue GSH looks feasible and worthy to explore.
Figure 10.

(a) Illustration of the variable radio frequency (VRF) OMRI approach for the case of pH-mapping. Two OMRI images of the phantom were acquired during EPR irradiation (8.4 s) at two preselected EPR frequencies, rf1 = 559.3 MHz and rf2 = 562.1 MHz, which correspond to EPR resonances of RH+ and R forms of the deuterated NR5 probe. pH values of 1 mM solutions of NR5 are indicated on the phantom picture. The calibration curve was calculated as pH dependence of the ratio of signal amplitudes, I(rf1)/I(rf2). (b) pHe mapping of living mouse by OMRI. pH map (in color) was superimposed with low-field MRI (gray scale) showing the transverse view of the mouse. The pH probe was injected into the number 4 mammary gland containing tumor (left) and in the number 9 normal mammary gland (right) before OMRI/MRI acquisitions. The pH map was calculated from two OMRI images, acquired during EPR irradiation at two EPR frequencies, rf1 and rf2. Total acquisition, 24.8 s. From ref 8 with permission from John Wiley & Sons, Ltd. Copyright 2012 John Wiley & Sons, Ltd.
The most known application of functional OMRI that utilizes NOE dependence on electron relaxation times T1e and T2e of paramagnetic probe is represented by the imaging of tissue oxygen concentrations.104–107 The Heisenberg spin exchange of probe with oxygen results in shortening electron relaxation times of the probe, which can be elucidated by comparison of OMRI images taken at different EPR powers.104
VF89 and VRF OMRI90 approaches allow for mapping chemical microenvironment using the probes with a functionally dependent ratiometric spectral parameter. Figure 10 demonstrates application of VRF OMRI for pH mapping using a deuterated analogue of the NR5 radical. The narrow EPR signal of the deuterated probe is easily saturated by rf irradiation which is of critical importance for obtaining higher NOE in OMRI and allowing for shorter acquisition time. Two OMRI images are acquired during 8.4 s EPR irradiation at two preselected EPR frequencies which correspond to EPR resonances of RH+ and R forms of deuterated NR5 probe. The ratio of the signal intensities of these two images correspond to the [RH+]/[R] ratio and therefore allows for pH calculation according to eq 8. Figure 10a illustrates the principles of VRF OMRI and method calibration using a phantom sample. Figure 10b demonstrates OMRI pH mapping of a tumor-bearing mouse after NR5 injection in the tumor and mammary gland. Two areas of probe localization correspond to tumor (Figure 10b,left) and mammary gland (Figure 10b,right) with higher pH heterogeneity in the tumor. Mean pHe values in the tumor and mammary gland were found to be in agreement with EPR spectroscopy and microelectrode data measured in the same mice being about 0.4 pH values lower for TME.59 Note that decay of OMRI signal at each pixel can provide spatially resolved information on the tissue redox status. Therefore, the same pH probe can be used for simultaneous mapping of pH and redox status. The OMRI functional approach may find applications for pH mapping of other living tissues and has potential for applications to humans.
In summary, the OMRI technique offers a comparatively fast and easy to build system (only rf EPR coils have to be integrated with the MRI scanner) to follow specific values of the tissue microenvironment using functional paramagnetic probes. In addition, several paramagnetic probes could be used simultaneously extending OMRI multifunctionality.107 Application of multifunctional radicals such as HOPE probes can further extend OMRI capability for concurrent mapping of several chemical parameters of the local microenvironment. Note that optimization of the OMRI imaging procedure for the pTAM HOPE probe requires consideration of contribution of all exchange processes in the EPR spectrum and NOE, namely, exchange of the probe with oxygen, self-spin exchange, and proton exchange with inorganic phosphate.
CONCLUSIONS
EPR-based spectroscopy and imaging of stable organic radicals involved in reversible exchange processes may allow for quantitative measurements of the kinetic and thermodynamic parameters of these processes. This provides noninvasive analytical tool for assessment of local chemical microenvironment. A need for quantitative mapping of critical parameters of the local chemical microenvironment in biological systems, especially in vivo, with spatial and temporal resolution is emerging. In particular, tissue oxygenation, acidosis, redox, and GSH content are physiologically important markers of the processes related to tumorigenesis characterizing cancer progression, aggressiveness, and efficacy of tumor microenvironment (TME)-targeted anticancer therapy.108 Among the other chemical parameters of TME recently identified as important for a signaling molecule in tumorigenesis is inorganic phosphate, Pi.109
Here we demonstrate that currently available NR and TAM probes can be efficiently used for multifunctional assessment of chemical microenvironment in living tissues providing new insights in the underlying biochemical and physiological processes. The proposed future directions include advances in the synthesis of multifunctional paramagnetic probes and progress in EPR-based imaging techniques such as RS EPR imaging87 and Overhauser-enhanced MRI.59,91
Acknowledgments
This work was partially supported by NIH Grants CA194013, CA192064, EB022775, and U54GM104942. The WVCTSI is acknowledged for start-up to V.V.K., A.B., and M.T. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Biographies
Valery V. Khramtsov is a Professor in the Department of Biochemistry at West Virginia University (WVU) and Director of the In Vivo Multifunctional Magnetic Resonance (IMMR) center (http://www.hsc.wvu.edu/immr/), Health Sciences Center, WVU. Current research includes the development of advanced EPR-based magnetic resonance techniques for multifunctional mapping of the chemical microenvironment and their application for in vivo tumor tissue analysis for obtaining knowledge on the role of tumor microenvironment in tumor progression and therapy.
Andrey A. Bobko is a Research Assistant Professor in the Department of Biochemistry at West Virginia University. He was awarded a Ph.D. in chemistry from the Russian Academy of Sciences in 2005. His research is focused on the development and application of novel paramagnetic probes for multifunctional assessment of tumor microenvironment using low-field EPR and Overhauser-enhanced MRI techniques.
Mark Tseytlin is an Assistant Professor in the Department of Biochemistry at West Virginia University. He was awarded a Ph.D. in physical-mathematical sciences from the Russian Academy of Sciences in 2002 under Kev Salikhov. His research is focused on the development of novel spectroscopy and imaging methods and instruments in the field of electron paramagnetic resonance.
Benoit Driesschaert is a Research Scholar in the Khramtsov laboratory in the Department of Biochemisty at West Virginia University. He was awarded a Ph.D in Chemistry in 2013 from University of Louvain, Belgium, under Bernard Gallez and Jacqueline Marchand-Brynaert. His current research includes the synthesis and characterization of biocompatible magnetic resonance probes for the in vivo profiling of important markers of the tumor microenvironment.
Footnotes
Notes
The authors declare no competing financial interest.
References
- 1.Barriga-Gonzalez G, Olea-Azar C, Zuniga-Lopez MC, Folch-Cano C, Aguilera-Venegas B, Porcal W, Gonzalez M, Cerecetto H. Curr Top Med Chem. 2015;15:484–495. doi: 10.2174/1568026615666150206155108. [DOI] [PubMed] [Google Scholar]
- 2.Neiman MB, Rozantzev EG, Mamedova YG. Nature. 1962;196:472–474. [Google Scholar]
- 3.Rozantsev EG. Free Nitroxyl Radicals. Springer Science + Business Media, LLC; New York: 1970. p. 249. [Google Scholar]
- 4.Anderson S, Radner F, Rydbeek A, Servin R, Wistrand L-G. Free radicals. 5,530,140. US Patent. 1996 Jun 25;
- 5.Ardenkjaer-Larsen JH, Laursen I, Leunbach I, Ehnholm G, Wistrand LG, Petersson JS, Golman K. J Magn Reson. 1998;133:1–12. doi: 10.1006/jmre.1998.1438. [DOI] [PubMed] [Google Scholar]
- 6.Berliner LJ. In: Nitroxides - Theory, Experiment and Applications. Kokorin AI, editor. InTech; Rijeka, Croatia: 2012. pp. 3–24. [Google Scholar]
- 7.Bagryanskaya EG, Krumkacheva OA, Fedin MV, Marque SRA. Methods Enzymol. 2015;563:365–396. doi: 10.1016/bs.mie.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Bobko AA, Eubank TD, Voorhees JL, Efimova OV, Kirilyuk IA, Petryakov S, Trofimiov DG, Marsh CB, Zweier JL, Grigor’ev IA, Samouilov A, Khramtsov VV. Magn Reson Med. 2012;67:1827–1836. doi: 10.1002/mrm.23196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Khramtsov VV, Volodarsky LB. In: In Spin Labeling: The Next Millennium. Berliner LJ, editor. Plenum Press; New York: 1998. pp. 109–180. [Google Scholar]
- 10.Molin YN, Salikhov KM, Zamaraev KI. Spin Exchange: Principles and Applications in Chemistry and Biology. Springer-Verlag; New York: 1980. p. 242. [Google Scholar]
- 11.Eaton SS, Eaton GR. Coord Chem Rev. 1978;26:207–262. [Google Scholar]
- 12.Epel B, Redler G, Tormyshev V, Halpern HJ. Adv Exp Med Biol. 2016;876:363–369. doi: 10.1007/978-1-4939-3023-4_45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Swartz HM, Hou HG, Khan N, Jarvis LA, Chen EY, Williams BB, Kuppusamy P. Adv Exp Med Biol. 2014;812:73–79. doi: 10.1007/978-1-4939-0620-8_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grampp G, Rasmussen K. In: Nitroxides - Theory, Experiment and Applications. Kokorin AI, editor. InTech; Rijeka, Croatia: 2012. pp. 25–56. [Google Scholar]
- 15.Kocherginsky N, Swartz HM. Nitroxide Spin Labels: Reactions in Biology and Chemistry. CRC Press; Boca Raton, FL: 1995. p. 270. [Google Scholar]
- 16.Kinoshita Y, Yamada K, Yamasaki T, Mito F, Yamato M, Kosem N, Deguchi H, Shirahama C, Ito Y, Kitagawa K, Okukado N, Sakai K, Utsumi H. Free Radical Biol Med. 2010;49:1703–1709. doi: 10.1016/j.freeradbiomed.2010.08.027. [DOI] [PubMed] [Google Scholar]
- 17.Kirilyuk IA, Bobko AA, Grigor’ev IA, Khramtsov VV. Org Biomol Chem. 2004;2:1025–1030. doi: 10.1039/b400252k. [DOI] [PubMed] [Google Scholar]
- 18.Marx L, Chiarelli R, Guiberteau T, Rassat A. J Chem Soc Perk T. 2000;1:1181–1182. [Google Scholar]
- 19.Decroos C, Boucher JL, Mansuy D, Yun XL. Chem Res Toxicol. 2014;27:627–636. doi: 10.1021/tx400467p. [DOI] [PubMed] [Google Scholar]
- 20.Decroos C, Li Y, Bertho G, Frapart Y, Mansuy D, Boucher JL. Chem Res Toxicol. 2009;22:1342–1350. doi: 10.1021/tx9001379. [DOI] [PubMed] [Google Scholar]
- 21.Davis RM, Sowers AL, DeGraff W, Bernardo M, Thetford A, Krishna MC, Mitchell JB. Free Radical Biol Med. 2011;51:780–790. doi: 10.1016/j.freeradbiomed.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Swartz HM, Khan N, Khramtsov VV. Antioxid Redox Signaling. 2007;9:1757–1771. doi: 10.1089/ars.2007.1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuppusamy P, Krishna MC. Curr Top Biophys. 2002;26:29–34. [Google Scholar]
- 24.Rizzi C, Samouilov A, Kutala VK, Parinandi NL, Zweier JL, Kuppusamy P. Free Radical Biol Med. 2003;35:1608–1618. doi: 10.1016/j.freeradbiomed.2003.09.014. [DOI] [PubMed] [Google Scholar]
- 25.Malatesta V, Ingold KU. J Am Chem Soc. 1973;95:6404–6407. [Google Scholar]
- 26.Khramtsov VV. Curr Org Chem. 2005;9:909–923. [Google Scholar]
- 27.Bobko AA, Dhimitruka I, Zweier JL, Khramtsov VV. J Am Chem Soc. 2007;129:7240–7241. doi: 10.1021/ja071515u. [DOI] [PubMed] [Google Scholar]
- 28.Dhimitruka I, Bobko AA, Eubank TD, Komarov DA, Khramtsov VV. J Am Chem Soc. 2013;135:5904–5910. doi: 10.1021/ja401572r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bobko AA, Dhimitruka I, Zweier JL, Khramtsov VV. Angew Chem, Int Ed. 2014;53:2735–2738. doi: 10.1002/anie.201310841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner BE, Helbert JN, Bates RD, Poindexter Eh. J Chem Soc, Chem Commun. 1973:748–749. [Google Scholar]
- 31.Akaike T, Yoshida M, Miyamoto Y, Sato K, Kohno M, Sasamoto K, Miyazaki K, Ueda S, Maeda H. Biochemistry. 1993;32:827–832. doi: 10.1021/bi00054a013. [DOI] [PubMed] [Google Scholar]
- 32.Bobko AA, Ivanov A, Khramtsov VV. Free Radical Res. 2013;47:74–81. doi: 10.3109/10715762.2012.746460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Khramtsov VV, Yelinova VI, Weiner LM, Berezina TA, Martin VV, Volodarsky LB. Anal Biochem. 1989;182:58–63. doi: 10.1016/0003-2697(89)90718-5. [DOI] [PubMed] [Google Scholar]
- 34.Khramtsov VV, Yelinova VI, Glazachev YuI, Reznikov VA, Zimmer G. J Biochem Biophys Methods. 1997;35:115–128. doi: 10.1016/s0165-022x(97)00035-3. [DOI] [PubMed] [Google Scholar]
- 35.Legenzov EA, Sims SJ, Dirda NDA, Rosen GM, Kao JPY. Biochemistry. 2015;54:6973–6982. doi: 10.1021/acs.biochem.5b00531. [DOI] [PubMed] [Google Scholar]
- 36.Liu YP, Song YG, Rockenbauer A, Sun J, Hemann C, Villamena FA, Zweier JL. J Org Chem. 2011;76:3853–3860. doi: 10.1021/jo200265u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weil JA, Bolton JR. Electron Paramagnetic Resonance: Elementary Theory and Practical Applications. John Wiley & Sons; Hoboken, NJ: 2007. p. 664. [Google Scholar]
- 38.Bales B, Peric M. J Phys Chem B. 1997;101:8707–8716. [Google Scholar]
- 39.Eaton SS, Eaton GR. Coord Chem Rev. 1988;83:29–72. [Google Scholar]
- 40.Khramtsov VV, Zweier JL. In: Stable Radicals: Fundamentals and Applied Aspects of Odd-Electron Compounds. Hicks R, editor. John Wiley & Sons, Ltd; Chichester, U.K: 2010. pp. 537–566. [Google Scholar]
- 41.Hyde JS, Yin JJ, Feix JB, Hubbell WL. Pure Appl Chem. 1990;62:255–260. [Google Scholar]
- 42.Backer JM, Budker VG, Eremenko SI, Molin YN. Biochim Biophys Acta, Bioenerg. 1977;460:152–156. doi: 10.1016/0005-2728(77)90161-x. [DOI] [PubMed] [Google Scholar]
- 43.Lai CS, Hopwood LE, Hyde JS, Lukiewicz S. Proc Natl Acad Sci U S A. 1982;79:1166–1170. doi: 10.1073/pnas.79.4.1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bacic G, Nilges MJ, Magin RL, Walczak T, Swartz HM. Magn Reson Med. 1989;10:266–272. doi: 10.1002/mrm.1910100211. [DOI] [PubMed] [Google Scholar]
- 45.Halpern HJ, Yu C, Peric M, Barth E, Grdina DJ, Teicher BA. Proc Natl Acad Sci U S A. 1994;91:13047–13051. doi: 10.1073/pnas.91.26.13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Froncisz W, Lai CS, Hyde JS. Proc Natl Acad Sci U S A. 1985;82:411–415. doi: 10.1073/pnas.82.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hyde JS, Subszynski WK. In: Spin Labeling: Theory and Application. Berliner LJ, Reubens J, editors. Plenum Press; New York: 1989. pp. 399–425. [Google Scholar]
- 48.Talmon Y, Shtirberg L, Harneit W, Rogozhnikova OYu, Tormyshev V, Blank A. Phys Chem Chem Phys. 2010;12:5998–6007. doi: 10.1039/b922060g. [DOI] [PubMed] [Google Scholar]
- 49.Epel B, Redler G, Halpern HJ. Oxygen Transport to Tissue XXXVI. In: Swartz HM, Harrison DK, Bruley DF, editors. Advances in Experimental Medicine and Biology. Vol. 812. Springer; Berlin, Germany: 2014. pp. 113–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Epel B, Halpern H. J Methods Enzymol. 2015;564:501–527. doi: 10.1016/bs.mie.2015.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lin YJ, Teicher BA, Halpern HJ. J Labelled Compd Radiopharm. 1990;28:621–631. [Google Scholar]
- 52.Luckhurst GR. In: Spin Labeling: Theory and Application. Berliner LJ, editor. Academic Press; New York: 1976. pp. 133–181. [Google Scholar]
- 53.Schafer FQ, Buettner GR. Free Radical Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 54.Roshchupkina GI, Bobko AA, Bratasz A, Reznikov VA, Kuppusamy P, Khramtsov VV. Free Radical Biol Med. 2008;45:312–320. doi: 10.1016/j.freeradbiomed.2008.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elajaili H, Biller JR, Rosen GM, Kao JP, Tseytlin M, Buchanan LA, Rinard GA, Quine RW, McPeak J, Shi Y, Eaton SS, Eaton GR. J Magn Reson. 2015;260:77–82. doi: 10.1016/j.jmr.2015.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Epel B, Sundramoorthy SV, Krzykawska-Serda M, Maggio MC, Tseytlin M, Eaton GR, Eaton SS, Rosen GM, Kao JPY, Halpern HJ. J Magn Reson. 2017;276:31–36. doi: 10.1016/j.jmr.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Glazachev YI, Grigor’ev IA, Reijerse EJ, Khramtsov VV. Appl Magn Reson. 2001;20:489–505. [Google Scholar]
- 58.Woldman YY, Semenov SV, Bobko AA, Kirilyuk IA, Polienko JF, Voinov MA, Bagryanskaya EG, Khramtsov VV. Analyst. 2009;134:904–910. doi: 10.1039/b818184e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Samouilov A, Efimova OV, Bobko AA, Sun Z, Petryakov S, Eubank TD, Trofimov DG, Kirilyuk IA, Grigor’ev IA, Takahashi W, Zweier JL, Khramtsov VV. Anal Chem. 2014;86:1045–1052. doi: 10.1021/ac402230h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Goodwin J, Yachi K, Nagane M, Yasui H, Miyake Y, Inanami O, Bobko AA, Khramtsov VV, Hirata H. NMR Biomed. 2014;27:453–458. doi: 10.1002/nbm.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Komarov DA, Dhimitruka I, Kirilyuk IA, Trofimiov DG, Grigor’ev IA, Zweier JL, Khramtsov VV. Magn Reson Med. 2012;68:649–655. doi: 10.1002/mrm.23251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gorodetsky AA, Kirilyuk IA, Khramtsov VV, Komarov DA. Magn Reson Med. 2016;76:350–358. doi: 10.1002/mrm.25867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dhimitruka I, Bobko AA, Hadad CM, Zweier JL, Khramtsov VV. J Am Chem Soc. 2008;130:10780–10787. doi: 10.1021/ja803083z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Driesschaert B, Marchand V, Leveque P, Gallez B, Marchand-Brynaert J. Chem Commun. 2012;48:4049–4051. doi: 10.1039/c2cc00025c. [DOI] [PubMed] [Google Scholar]
- 65.Bobko AA, Dhimitruka I, Komarov DA, Khramtsov VV. Anal Chem. 2012;84:6054–6060. doi: 10.1021/ac3008994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xia S, Villamena FA, Hadad CM, Kuppusamy P, Li Y, Zhu H, Zweier JL. J Org Chem. 2006;71:7268–7279. doi: 10.1021/jo0610560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Driesschaert B, Robiette R, Lucaccioni F, Gallez B, Marchand-Brynaert J. Chem Commun. 2011;47:4793–4795. doi: 10.1039/c1cc10988j. [DOI] [PubMed] [Google Scholar]
- 68.Driesschaert B, Robiette R, Le Duff CS, Collard L, Robeyns K, Gallez B, Marchand-Brynaert J. Eur J Org Chem. 2012;2012:6517–6525. [Google Scholar]
- 69.Marchand V, Levêque P, Driesschaert B, Marchand-Brynaert J, Gallez B. Magn Reson Med. 2016 doi: 10.1002/mrm.26316. [DOI] [PubMed] [Google Scholar]
- 70.Song YG, Liu YP, Liu WB, Villamena FA, Zweier JL. RSC Adv. 2014;4:47649–47656. doi: 10.1039/C4RA04616A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bobko AA, Eubank TD, Driesschaert B, Dhimitruka I, Evans J, Mohammad R, Tchekneva EE, Dikov MM, Khramtsov VV. Sci Rep. 2017;7:41233. doi: 10.1038/srep41233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Martin ML, Martin GJ, Delpuech J-J. Practical NMR Spectroscopy. xxxi. Heyden; London: 1980. p. 460. [Google Scholar]
- 73.Marshak A. Science. 1940;92:460–461. doi: 10.1126/science.92.2394.460. [DOI] [PubMed] [Google Scholar]
- 74.Papaloucas AC. Nucl Med (Stuttg) 1959;1:62–70. [PubMed] [Google Scholar]
- 75.Kouloulias V, Tolia M, Tsoukalas N, Papaloucas C, Pistevou-Gombaki K, Zygogianni A, Mystakidou K, Kouvaris J, Papaloucas M, Psyrri A, Kyrgias G, Gennimata V, Leventakos K, Panayiotides I, Liakouli Z, Kelekis N, Papaloucas A. Asian Pacific journal of cancer prevention: APJCP. 2015;16:77–81. doi: 10.7314/apjcp.2015.16.1.77. [DOI] [PubMed] [Google Scholar]
- 76.Low-Beer BV. Science. 1946;104:399. [PubMed] [Google Scholar]
- 77.Rofstad EK, DeMuth P, Sutherland RM. Radiother Oncol. 1988;12:315–326. doi: 10.1016/0167-8140(88)90021-7. [DOI] [PubMed] [Google Scholar]
- 78.Stubbs M, Rodrigues LM, Griffiths JR. Br J Cancer. 1989;60:701–707. doi: 10.1038/bjc.1989.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Elser JJ, Kyle MM, Smith MS, Nagy JD. PLoS One. 2007;2:e1028. doi: 10.1371/journal.pone.0001028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.ELSER JJ, NAGY JD, KUANG Y. BioScience. 2003;53:1112–1120. [Google Scholar]
- 81.Kareva I. PLoS One. 2013;8:e51844. doi: 10.1371/journal.pone.0051844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Song Y, Liu Y, Hemann C, Villamena FA, Zweier JL. J Org Chem. 2013;78:1371–1376. doi: 10.1021/jo301849k. [DOI] [PubMed] [Google Scholar]
- 83.Driesschaert B, Levêque P, Gallez B, Marchand-Brynaert J. Eur J Org Chem. 2014;2014:8077–8084. [Google Scholar]
- 84.Driesschaert B, Levêque P, Gallez B, Marchand-Brynaert J. Tetrahedron Lett. 2013;54:5924–5926. [Google Scholar]
- 85.Subramanian S, Matsumoto KI, Mitchell JB, Krishna MC. NMR Biomed. 2004;17:263–294. doi: 10.1002/nbm.897. [DOI] [PubMed] [Google Scholar]
- 86.Koda S, Goodwin J, Khramtsov VV, Fujii H, Hirata H. Anal Chem. 2012;84:3833–3837. doi: 10.1021/ac203415w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tseitlin M, Biller JR, Elajaili H, Khramtsov VV, Dhimitruka I, Eaton GR, Eaton SS. J Magn Reson. 2014;245:150–155. doi: 10.1016/j.jmr.2014.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Overhauser AW. Phys Rev. 1953;92:411–415. [Google Scholar]
- 89.Khramtsov VV, Caia GL, Shet K, Kesselring E, Petryakov S, Zweier JL, Samouilov A. J Magn Reson. 2010;202:267–273. doi: 10.1016/j.jmr.2009.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Efimova OV, Sun Z, Petryakov S, Kesselring E, Caia GL, Johnson D, Zweier JL, Khramtsov VV, Samouilov A. J Magn Reson. 2011;209:227–232. doi: 10.1016/j.jmr.2011.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Takahashi W, Bobko AA, Dhimitruka I, Hirata H, Zweier JL, Samouilov A, Khramtsov VV. Appl Magn Reson. 2014;45:817–826. doi: 10.1007/s00723-014-0570-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dadok J, Sprecher RF. J Magn Reson. 1974;13:243–248. [Google Scholar]
- 93.Gupta RK, Ferretti JA, Becker ED. J Magn Reson. 1974;13:275–290. [Google Scholar]
- 94.Mitchell DG, Quine RW, Tseitlin M, Meyer V, Eaton SS, Eaton GR. Radiat Meas. 2011;46:993–996. doi: 10.1016/j.radmeas.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Mitchell DG, Tseitlin M, Quine RW, Meyer V, Newton ME, Schnegg A, George B, Eaton SS, Eaton GR. Mol Phys. 2013;111:2664–2673. [Google Scholar]
- 96.Rinard GA, Quine RW, Biller JR, Eaton GR. Concepts Magn Reson, Part B. 2010;37B:86–91. doi: 10.1002/cmr.b.20161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tseitlin M, Rinard GA, Quine RW, Eaton SS, Eaton GR. J Magn Reson. 2011;208:279–283. doi: 10.1016/j.jmr.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tseitlin M, Quine RW, Rinard GA, Eaton SS, Eaton GR. J Magn Reson. 2010;203:305–310. doi: 10.1016/j.jmr.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Tseitlin M, Mitchell DG, Eaton SS, Eaton GR. J Magn Reson. 2012;223:80–84. doi: 10.1016/j.jmr.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Türke MT, Bennati M. Appl Magn Reson. 2012;43:129–138. doi: 10.1007/s00723-012-0362-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bačić G, Pavićević A, Peyrot F. Redox Biol. 2016;8:226–242. doi: 10.1016/j.redox.2015.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hyodo F, Soule BP, Matsumoto K-i, Matusmoto S, Cook JA, Hyodo E, Sowers AL, Krishna MC, Mitchell JB. J Pharm Pharmacol. 2008;60:1049–1060. doi: 10.1211/jpp.60.8.0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Utsumi H, Yamada K-i, Ichikawa K, Sakai K, Kinoshita Y, Matsumoto S, Nagai M. Proc Natl Acad Sci U S A. 2006;103:1463–1468. doi: 10.1073/pnas.0510670103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Golman K, Petersson JS, Ardenkjær-Larsen JH, Leunbach I, Wistrand LG, Ehnholm G, Liu K. J Magn Reson Imaging. 2000;12:929–938. doi: 10.1002/1522-2586(200012)12:6<929::aid-jmri17>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 105.Krishna MC, English S, Yamada K, Yoo J, Murugesan R, Devasahayam N, Cook JA, Golman K, Ardenkjaer-Larsen JH, Subramanian S, Mitchell JB. Proc Natl Acad Sci U S A. 2002;99:2216–2221. doi: 10.1073/pnas.042671399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Efimova OV, Caia GL, Sun Z, Petryakov S, Kesselring E, Samouilov A, Zweier JL. J Magn Reson. 2011;212:197–203. doi: 10.1016/j.jmr.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ahn KH, Scott G, Stang P, Conolly S, Hristov D. Magn Reson Med. 2011;65:1416–1422. doi: 10.1002/mrm.22732. [DOI] [PubMed] [Google Scholar]
- 108.Khramtsov VV, Gillies RJ. Antioxid Redox Signaling. 2014;21:723–729. doi: 10.1089/ars.2014.5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lin YM, McKinnon KE, Ha SW, Beck GR. Mol Carcinog. 2015;54:926–934. doi: 10.1002/mc.22153. [DOI] [PMC free article] [PubMed] [Google Scholar]