

Abstract
Objective
Characteristic pathologic changes define the progression of steatosis to nonalcoholic steatohepatitis (NASH), and are correlated to metabolic pathways. A common rodent model of NASH is the methionine and choline deficient (MCD) diet. The objective of this study was to perform full metabolomic analyses on liver samples to determine which pathways are altered most pronouncedly in the human condition, and to compare these changes to rodent models of nonalcoholic fatty liver disease (NAFLD).
Methods
A principal components analysis for all 91 metabolites measured indicates that metabolome perturbation is greater and less varied for humans than for rodents.
Results
Metabolome changes in human and rat NAFLD were greatest for the amino acid and bile acid metabolite families (e.g. asparagine, citrulline, GABA, lysine); although, in many cases, the trends were reversed when compared between species (cholic acid, betaine).
Conclusions
Overall, these results indicate that metabolites of specific pathways may be useful biomarkers for NASH progression, although these markers may not correspond to rodent NASH models. The MCD model may be useful when studying certain endpoints of NASH; however, the metabolomics results indicate important differences between humans and rodents in the biochemical pathogenesis of the disease.
Keywords: metabolomics, liver, nonalcoholic fatty liver disease (NAFLD), methionine and choline deficient diet (MCD), nonalcoholic steatohepatatis (NASH), models
Introduction
Nonalcoholic fatty liver disease (NAFLD) is characterized by pathological changes, starting with simple fatty liver (steatosis) and progressing to severe hepatocellular damage and inflammation in a state termed nonalcoholic steatohepatitis (NASH), which can ultimately develop into cirrhosis or liver cancer (1). The worldwide prevalence of NAFLD is estimated to be 25%, with the North American prevalence at 24% (2). Prevalence varies with age, and the presence of comorbidities such as obesity, diabetes and hypertension (2). Multiple histopathologic changes occur within the liver during disease progression, including excess fat accumulation, necroinflammation, and fibrosis (3). These hallmarks can take years to develop in humans, and are therefore difficult to model accurately in animal models. Nevertheless, because of the difficulty of obtaining human clinical samples, animal models provide critical information for understanding the molecular mechanisms responsible for histopathologic changes (4, 5).
Rodent dietary models can imitate the important histological features of human NAFLD and are powerful tools in determining the effect of disease progression on human cellular functions such as drug metabolism and disposition (6, 7). An ideal animal model should reflect both the pathologic etiologies of human NAFLD, as well as the histologic changes during disease progression.
Methionine and choline deficiency (MCD) in rodents substantially disturbs the formation of phosphatidylcholine, and the formation and export of very low density lipoprotein (VLDL) (4, 8). The MCD diet also impairs fatty acid beta-oxidation, further contributing to the development of hepatic steatosis (9). The stepwise progression of liver damage in the MCD model has been well-characterized in Wistar rats (10). Over the course of five weeks, fatty liver worsens from isolated microvesicular to diffuse macrovesicular steatosis with spotty necrosis (4). Finally, after seven weeks, clusters of inflammatory cells accumulate, and fibrosis develops in the perivenular and centrilobular regions (4). Liver injury induced by MCD is accompanied by elevated serum transaminases and proinflammatory cytokines, such as IL-1β, IL-6 and TNF-α; therefore, the model is consistent with respect to inflammatory responses (11). While the MCD diet exhibits similar hepatic characteristics to human NASH, it does not reflect the development of obesity, or insulin resistance (IR), which are recognized comorbidities of human NASH (6). The high-fat diet, while increasing free fatty acids and contributing to the development of steatosis, diabetes, and obesity, fails to produce an adequate amount of inflammation and hepatic fibrosis to be considered NASH after only eight weeks on the diet (6).
Research has demonstrated that MCD rats have similar changes in mRNA and protein expression profiles of the xenobiotic transporters to those seen in human NASH (7, 12, 13). It is unknown whether the rat MCD model also reflects changes in the metabolome seen in human NAFLD progression. Detailed knowledge of the biochemical changes that occur during NAFLD progression in humans and the prominent animal models is needed to aid in early diagnosis and development of intervention strategies.
The purpose of the present study was to compare the metabolomic response to disease progression between human steatosis and NASH and rodent models of these liver pathologies. Elucidating the similarities and differences of the metabolome in rodent models compared to human NASH will help to determine the limitations of these models. While the high-fat and MCD diets accurately reflect pathologic changes along the NAFLD spectrum, differences in the biochemical events leading to the pathology of NASH may limit their use as models in therapeutic studies.
Methods
Rodent Liver Samples
Male Sprague-Dawley rats (eight weeks old) weighing 200~250 g were purchased from Harlan Laboratories (Indianapolis, Indiana). All animals were acclimated in 12 hour light and dark cycles in a University of Arizona Association for Assessment and Accreditation of Laboratory Animal Care-certified animal facility for a week before initiation of experiments and allowed standard chow and water ad libitum. Housing and experimental procedures were in accordance with NIH guidelines for the care and use of laboratory animals and approved by the University of Arizona Institutional Animal Care and Use Committee. Rats were fed with methionine and choline sufficient diet (D#518754) as control as previously described (7, 14). Alternatively, rats were fed high fat diet to recapitulate steatosis (high cholesterol, 18% butter fat, D#112280) (n=7) or methionine-choline deficient (MCD, D#518810) (n=9) diet (all diets from Dyets, Inc., Bethlehem, PA) to recapitulate NASH. After eight weeks on diet, unfasted rats were euthanized by CO2 asphyxiation. The liver was immediately harvested, weighed, and frozen in liquid nitrogen.
Human Liver Samples
Frozen postmortem human liver samples were obtained previously from the National Institutes of Health-funded Liver Tissue Cell Distribution System (LTCDS) coordinated through Virginia Commonwealth University (Richmond, VA) and the Universities of Minnesota (Minneapolis, MN), and Pittsburgh (Pittsburgh, PA), funded by NIH Contract # HHSN276201200017C. The detailed clinical information of these human liver samples was published previously, fasting status is unknown (15). Histology scoring was performed using the NAFLD activity scoring system (16). The samples used in these metabolomics analyses were diagnosed as healthy (n=17), steatosis (n=4), NASH (fatty) (n=14) or NASH (not fatty) (n=23). Samples exhibiting over 10% fat deposition in hepatocytes without inflammation and fibrosis were diagnosed as steatosis. Samples exhibiting over 5% fatty deposition accompanying inflammation and fibrosis were diagnosed as NASH (fatty). The NASH (not fatty) was diagnosed when less than 5% fatty infiltration of hepatocyte accompanied significant inflammation and fibrosis.
Analytical methods for LC-MS/MS
50 ~ 100mg rat or human liver samples were homogenized in 10 times the tissue weight of ice-cold methanol solution with 0.1% formic for 18–20 s using a polytron homogenizer over ice. Supernantants centrifuged prior to being aliquoted, dried and frozen in a 96-well plate as described previously (17). Samples were reconstituted in 90:10 (v:v) water:methanol prior to analysis. The internal standard d5-hippurate was added to the samples, which were then injected in a randomized fashion to Thermo UHPLC Accela coupled to a Thermo Exactive high resolution orbitrap mass spectrometer. The peaks were acquired in positive and negative ion mode (separate injections) with a mass accuracy within 5 ppm at 25 K resolution. Metabolite peak area under the curve (AUC) measurements for LC-MS was calculated using Component Elucidator, a software package developed by Bristol-Myers Squibb (Princeton, NJ) as described previously (17). The potential biomarkers were identified according to chromatographic retention time, molecular weight (m/z) and MS/MS fragmentation. The metabolites were matched using the databases KEGG (http://www.kegg.com/), METLIN (http://metlin.scripps.edu/), and HMDB (http://www.hmdb.ca/) and so on.
Bioinformatics and Statistical analysis
Ninety-one metabolites were analyzed by the University of Arizona Statistical and Bioinformatics Consortium. Principal Component Analysis (PCA) was used as an unsupervised statistical method to study the differences of intergroup metabolites among all experimental groups. Hierarchical clustering analysis was performed on the metabolite data to reveal any clustering effects related to the stage of the disease and species. Peak AUC values from the LC-MS data were log transformed into a normal distribution approximation. After log transformation, a one-way ANOVA with Tukey honest significant difference (HSD) testing was used for multiple comparisons of metabolites among the diagnosis groups and species to test for mean differences. The Benjamini-Hochberg procedure was used to adjust the reported p-values to control for the rate of false discoveries. Although human metabolite analyses were performed using the four diagnosis categories, due to the lack of statistical differences between human NASH (fatty) and NASH (not fatty), these two categories were combined as human NASH. Significance was defined as adjusted p values ≤ 0.05. The interpretation of the data described herein refers to the reported log-transformed values and the respective adjusted p values.
Results
The distribution of 91 rat and human metabolites is shown by PCA which demonstrates any relationship or differences between rat and human metabolic profiles. The PCA plot (Figure 1) reflects the fact that the human and rat metabolites were predominantly separated by the first component (PC1), the x-axis, completely differentiating the two species as indicated by the lack of overlap between any rat and human sample. The NASH and control samples were unable to be completely separated by the second component (PC2), the y-axis. Diagnoses were not able to be differentiated on an axis orthogonal to species in this analysis. The variance explained by PC1 was 26.71%, and that explained by PC2 was 13.64%.
Figure 1.
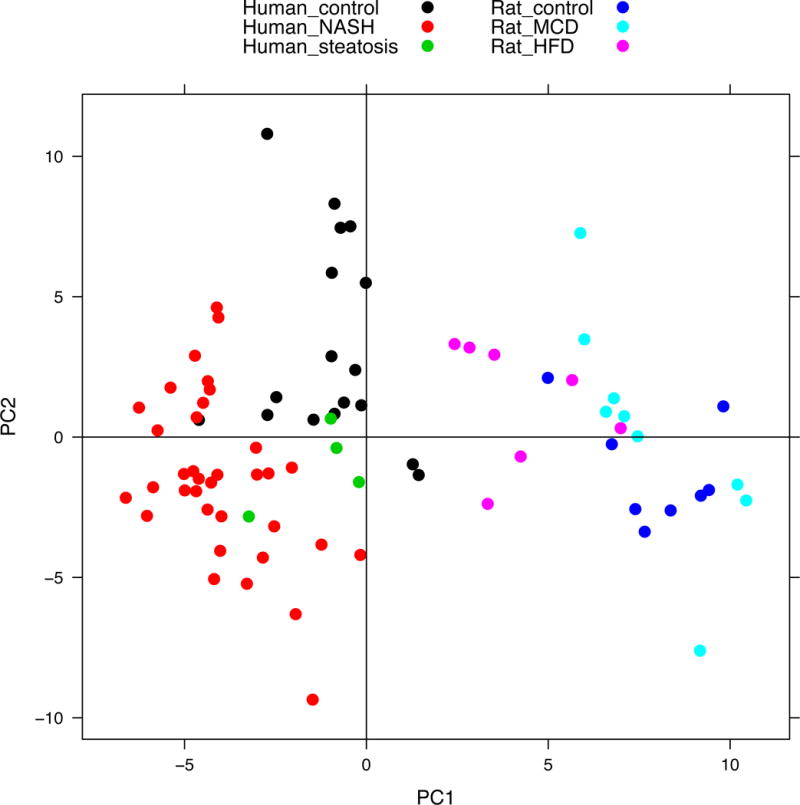
Principal Component Analysis (PCA) distinguished any correlated variables between PC1 and PC2 for the metabolite’s projections to the orthogonal coordinate transformation. The distribution of 91 metabolites in the plot indicates the relationship between rat HFD and MCD, and human nonalcoholic fatty liver disease (NAFLD) progression. The human and rat metabolites were predominantly separated by the first component (PC1) and the diagnosis/model was mostly separated by the second component (PC2).
To explore the potentially significant biochemical differences in NAFLD progression by metabolite class, the metabolites were divided into functional categories: bile acids, amino acids, fatty acids and carnitine metabolites, and “other” metabolites.
Bile acid metabolomics
Select bile acids present in both human and rats at detectable levels were chosen for this metabolomic comparison (18). Eleven bile acid metabolites in rat and human liver samples with NAFLD were quantified by LC-MS/MS. Further analysis revealed significant differences in the bile acid profile between rat MCD samples and human samples diagnosed as NASH (Table 1). The average log-transformed area under the curve (AUC) data for cholic acid (CA), and deoxycholic acid (DCA) was significantly increased in rat NASH. Conversely, significant decreases in CA and DCA were observed in human NASH, though there were increases in glycochenodeoxycholic acid (GCDCA), taurocholic acid (TCA) and taurodeoxycholic acid (TDCA). CA, DCA, GCDCA, glycodeoxycholic acid (GDCA), glycoursocholic acid (GUCA), and tauroursocholic acid (TUCA) exhibited significant interspecies differences (p≤0.05).
Table 1. Bile acids profiled in rat and human NAFLD liver samples.
Bile acid metabolite values represent natural log transformed AUC values relative to species specific control samples ± the standard deviation.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Bile Acid | Rat | Human | Interspecies Adj. p value | ||||
|
| |||||||
| HFD | MCD | Adj. p value | Steatosis | NASH | Adj. p value | ||
| CA | 0.73±0.51 | 1.06±0.53 | 0.003* | −0.38±0.16 | −1.01±0.47 | 0.000* | 0.000# |
| DCA | 0.76±1.74 | 2.91±1.56 | 0.008* | 0.53±2.02 | −3.22±1.79 | 0.000* | 0.000# |
| GCDCA | −0.71±0.97 | −0.82±0.93 | 0.896 | −0.46±0.81 | 0.70±0.83 | 0.028* | 0.000# |
| GCA | −0.70±1.02 | −0.31±1.24 | 0.954 | −0.34±0.74 | 0.28±0.79 | 0.716 | 0.117 |
| GDCA | −0.76±1.34 | 0.30±0.92 | 0.954 | −0.07±0.87 | −2.50±1.11 | 0.000* | 0.000# |
| GUCA | −1.05±1.96 | 0.34±1.31 | 0.954 | −1.08±1.64 | −1.09±1.38 | 0.105 | 0.015# |
| GUDCA | −2.64±1.54 | −0.71±1.13 | 0.931 | −0.67±1.60 | −0.60±1.91 | 0.708 | 0.956 |
| TCA | 0.67±1.27 | 1.15±0.84 | 0.102 | 0.03±0.51 | 1.15±0.94 | 0.001* | 0.999 |
| TDCA | 1.12±0.54 | 1.02±1.06 | 0.102 | −0.27±0.55 | 1.65±0.83 | 0.000* | 0.090 |
| TUCA | 1.44±0.65 | 0.99±0.77 | 0.094 | −0.81±2.51 | −0.15±1.21 | 0.992 | 0.018# |
| TUDCA | −1.78±1.72 | 0.16±0.62 | 0.954 | 0.35±0.99 | 0.37±1.59 | 0.827 | 0.841 |
represents significant difference across samples for that species,
represents for significant difference in human NASH to rat MCD samples. Test statistics calculated conducting the one way ANOVA with Tukey honest significant difference (HSD), with significance define as adjusted (adj.) p value ≤ 0.05.
Abbreviation: HFD: high fat diet; MCD: methionine and choline deficient; NASH: nonalcoholic steatohepatitis; CA: cholic acid; DCA: deoxycholic acid; GCDCA: glycochenodeoxycholic acid; GCA: glycocholic acid; GDCA: glycodeoxycholic acid; GUCA: glycoursocholic acid; GUDCA: glycoursodeoxycholic acid; TCA: taurocholic acid; TDCA: taurodeoxycholic acid; TUCA: tauroursocholic acid; TUDCA: tauroursodeoxycholic acid.
A heat map of a hierarchical cluster analysis in represents the clustering of bile acid metabolites in human and rat samples within different NAFLD diagnosis categories for each species (Figure 2). However, bile acid metabolome alterations were only able to differentiate the dissimilarity between species at this higher level of analysis, and showed no clustering effects for diagnosis in either humans or rats. Chenodeoxycholic acid and ursodeoxycholic acid were not able to be differentiated, as the peaks were superimposed, and were thus excluded from further analysis.
Figure 2. Hierarchical clustering analysis of bile acid metabolomics.
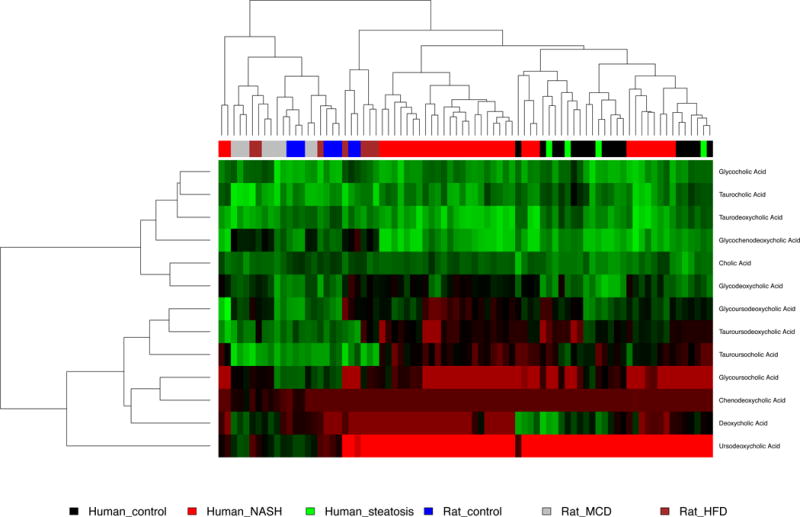
The heat map of bile acids metabolites indicates the increase or decrease of each metabolite in human hepatic samples diagnosed as control, steatosis and nonalcoholic steatohepatitis (NASH), or samples from rat models representing those disease states (control, HFD, or MCD, respectively). The metabolites were clustered based on elucidation distances. Red, black and green denote low, medium and high concentration, respectively.
Amino acid metabolomics
Analysis of 19 amino acid metabolites (Table 2) in rat and human hepatic samples measured by LC-MS revealed that asparagine, citrulline and lysine exhibit a significant increase in both human and rat NASH, while other changes in amino acid levels did not carry the trend across species. Alanine, GABA, and α-ketoglutarate were decreased, while serine and threonine were increased in MCD rats compared to control. In human NASH, histidine, methionine, and tryptophan were decreased, and isoleucine, phenylalanine, tyrosine, valine, and succinic acid were increased. Significant interspecies interaction was seen in alanine, asparagine, citrulline, histidine, α-ketoglutarate, methionine, phenylalanine, proline, serine, succinic acid, threonine, tryptophan, and valine.
Table 2. Amino acid metabolomics in hepatic samples of rat and human.
Amino acid metabolite values represent natural log transformed AUC values relative to species specific control samples ± the standard deviation.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Amino Acid | Rat | Human | Interspecies Adj. p value | ||||
|
| |||||||
| HFD | MCD | Adj. p value | Steatosis | NASH | Adj. p value | ||
| Alanine | −0.34±0.18 | −0.46±0.38 | 0.024* | −0.07±0.25 | −0.19±0.28 | 0.117 | 0.035# |
| Asparagine | −0.12±0.50 | 0.72±0.32 | 0.010* | −0.75±0.40 | 0.28±0.26 | 0.006* | 0.000# |
| Citrulline | 1.05±0.69 | 2.19±0.60 | 0.000* | −1.46±2.32 | 2.86±1.11 | 0.000* | 0.121# |
| GABA | −0.16±0.24 | −0.68±0.47 | 0.022* | −0.58±0.67 | −0.74±0.52 | 0.000* | 0.755 |
| Glutamine | −0.48±0.22 | −0.10±0.63 | 0.964 | 0.01±0.42 | 0.09±0.29 | 0.657 | 0.211 |
| Histidine | −0.05±0.13 | −0.11±0.27 | 0.752 | −2.25±0.46 | −2.6±0.31 | 0.000* | 0.000# |
| Isoleucine | 0.34±0.34 | 0.09±0.45 | 0.964 | −0.49±0.34 | 0.32±0.38 | 0.022* | 0.165 |
| α-ketoglutarate | 0.90±0.58 | −2.68±2.45 | 0.094 | −0.89±3.37 | −0.50±1.85 | 0.862 | |
| Leucine | 0.32±0.39 | 0.10±0.45 | 0.964 | −0.52±0.42 | 0.25±0.35 | 0.076 | 0.007# |
| Lysine | 0.82±0.64 | 0.73±0.52 | 0.036* | −0.53±0.37 | 0.44±0.37 | 0.001* | 0.303 |
| Methionine | 0.47±0.82 | 0.21±0.85 | 0.964 | −0.44±0.61 | −0.74±0.80 | 0.016* | 0.092 |
| Phenylalanine | 0.08±0.28 | −0.14±0.40 | 0.848 | −0.47±0.43 | 0.46±0.38 | 0.001* | 0.005# |
| Proline | 0.35±0.35 | 0.35±0.49 | 0.436 | −0.19±0.10 | −0.20±0.35 | 0.308 | 0.000# |
| Serine | −1.12±0.72 | 1.40±0.44 | 0.004* | −0.30±0.64 | −0.25±0.34 | 0.099 | 0.001# |
| Succinic acid | −0.3±0.3 | −0.48±0.47 | 0.199 | −0.15±0.29 | 0.43±0.40 | 0.033* | 0.000# |
| Threonine | −0.58±0.30 | 0.98±0.45 | 0.001* | −1.05±0.28 | 0.14±0.40 | 0.571 | 0.000# |
| Tryptophan | 0.01±0.29 | −0.05±0.49 | 0.998 | −1.65±0.07 | −0.69±0.35 | 0.038* | 0.000# |
| Tyrosine | 0.23±0.52 | 0.23±0.53 | 0.796 | −0.55±0.57 | 0.36±0.38 | 0.021* | 0.413 |
| Valine | 0.16±0.25 | −0.35±0.44 | 0.283 | −0.50±0.35 | 0.40±0.28 | 0.000* | 0.000# |
represents significant difference across samples for that species,
represents for significant difference in human NASH to rat MCD samples. Test statistics calculated conducting the one way ANOVA with Tukey honest significant difference (HSD), with significance define as adjusted p value ≤ 0.05.
Abbreviation: HFD: high fat diet; MCD: methionine and choline deficient; NASH: nonalcoholic steatohepatitis
The metabolism pathways of selected amino acid metabolites analyzed illustrates the important role in the citric acid and urea cycle pathways (Figure 3). All amino acids ultimately enter the citrate cycle or undergo glycolysis/gluconeogenesis for degradation or metabolism. The concurrent changes and contrasts observed in these metabolites in rat and human NAFLD are highlighted (Figure 3).
Figure 3. Amino acid metabolism pathway analysis.
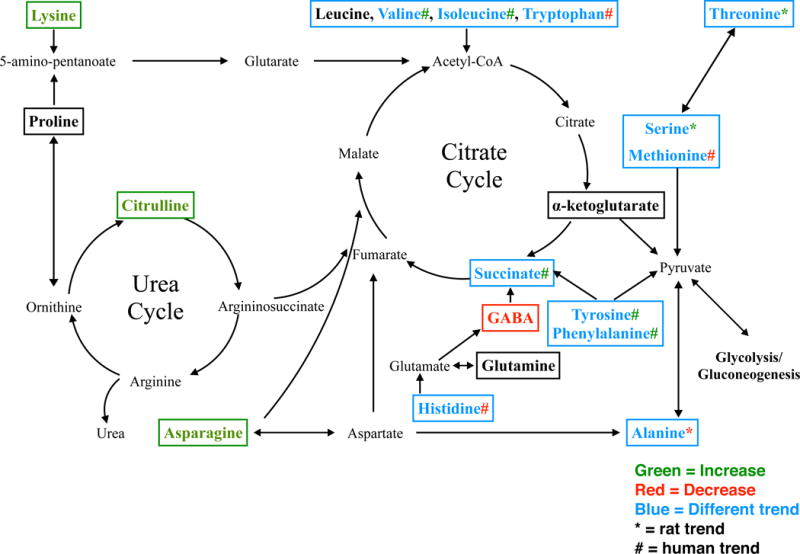
Amino acid analysis was performed to show the change of amino acid metabolites between different species and diagnosis/model. The boxed amino acids were analyzed and matched by LC-MS and metabolomics software. Green means the log transformed area under the curve (AUC) significantly increased in rat MCD and human NASH. Red stands for significantly decreased in rat MCD and human NASH. Black shows no change regardless of species. Blue shows that the change trend comparing control to NASH was different between human and rat. The asterisk sign (*) represents the trend in rat and the pound sign (#) represents the trend in human.
Fatty acids, carnitine and lysophosphatidyl choline metabolomics
Metabolomics analysis identified 15 fatty acids (including carnitine) and 17-lysophosphatidyl choline. Fatty acid metabolites significantly differed between humans and rats for controls and NASH (Table 3). All highlighted metabolites in table 3 were significantly altered in human NASH, and revealed significant interspecies interaction. Arachidonic acid, choline, linoleic acid and stearoyl-lyso-PE (18:0) were significantly decreased in human NASH, whereas stearoyl-lyso-PE was increased in MCD rats. Additionally, significant increases in the levels of arachidonoyl-lyso-PC (20:4), butyryl carnitine (C4), lauryl carnitine (C12), stearoyl carnitine (C18:0), and tetradecanoyl carnitine (C14) were observed in human samples during NAFLD progression. Rat MCD rats also exhibited increased stearoyl carnitine (C18:0) levels.
Table 3. Fatty acids, carnitine and lysophosphatidyl choline metabolomics in hepatic samples of rat and human.
Fatty acid metabolite values represent natural log transformed AUC values relative to species specific control samples ± the standard deviation.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Fatty acids | Rat | Human | Interspecies Adj. p value | ||||
|
| |||||||
| HFD | MCD | Adj. p value | Steatosis | NASH | Adj. p value | ||
| Arachidonic Acid | −0.12±0.57 | 1.02±0.82 | 0.056 | −1.00±0.51 | −0.92±0.60 | 0.000* | 0.000# |
| Arachidonoyl-lyso-PE (20:4) | 0.1±0.31 | 0.19±0.38 | 0.589 | −0.36±0.13 | 0.59±0.39 | 0.000* | 0.011# |
| Butyryl Carnitine (C4) | 0.13±0.84 | −2.00±2.24 | 0.116 | 1.76±0.92 | 1.75±0.69 | 0.000* | 0.000# |
| Choline | 0.79±0.58 | 0.20±0.70 | 0.878 | −0.11±0.23 | 0.57±0.27 | 0.000* | 0.000# |
| Lauryl Carnitine (C12) | 1.62±0.78 | −0.17±1.10 | 1.000 | 2.25±1.51 | 2.50±0.81 | 0.000* | 0.000# |
| Linoleic Acid | 0.29±1.43 | 2.20±1.88 | 0.110 | −1.43±2.25 | −2.12±1.45 | 0.001* | 0.000# |
| Stearoyl Carnitine (C18:0) | 1.3±0.48 | 1.37±0.81 | 0.016* | 0.36±0.35 | 0.54±0.65 | 0.020* | 0.004# |
| Stearoyl-lyso-PE (18:0) | 0.95±0.52 | 1.39±0.65 | 0.001* | −0.54±0.25 | −0.86±0.45 | 0.000* | 0.000# |
| Tetradecanoyl Carnitine (C14) | 2.17±0.55 | −0.32±1.56 | 1.000 | 2.30±0.80 | 1.48±0.77 | 0.000* | 0.000# |
represents significant difference across samples for that species,
represents for significant difference in human NASH to rat MCD samples. Test statistics calculated conducting the one way ANOVA with Tukey honest significant difference (HSD), with significance define as adjusted p value ≤ 0.05.
Abbreviation: HFD: high fat diet; MCD: methionine and choline deficient; NASH: nonalcoholic steatohepatitis
Other metabolomics
The remaining metabolites analyzed from the liver samples were classified as other metabolites and includes miscellaneous metabolites involved in energetics pathway processes and by-products as well as organic acids that were significantly different in subjects with NASH or MCD rats. These include 4-hydroxyproline, betaine, creatine, creatinine, glucose, and taurine (Table 4). Among these metabolites, all but creatinine exhibited an interspecies interaction. 4-hyddroxyproline was increased in human NASH, but unchanged in MCD rats. Betaine was decreased in MCD rats, but increased in NASH, while creatine was increased in MCD rats, but decreased in human NASH. Glucose levels decreased in MCD rats, and trended toward increase in NASH. Taurine was increased significantly in human NASH, and trended toward an increase in MCD rats.
Table 4. Other metabolomics in hepatic samples of rat and human.
Metabolite values represent natural log transformed AUC values relative to species-specific control samples ± the standard deviation.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Metabolite | Rat | Human | Interspecies Adj. p value | ||||
|
| |||||||
| HFD | MCD | Adj. p value | Steatosis | NASH | Adj. p value | ||
| 4-hydroxyproline | 0.62±1.55 | 1.03±0.56 | 0.209 | −0.52±1.29 | 2.99±0.70 | 0.000* | 0.000# |
| Betaine | −0.02±0.18 | −1.03±0.62 | 0.024* | −0.29±0.29 | 1.09±0.55 | 0.000* | 0.000# |
| Creatine | 0.30±0.46 | 0.90±0.32 | 0.001* | −0.08±0.08 | −0.56±0.32 | 0.000* | 0.000# |
| Creatinine | 1.42±0.76 | 0.83±0.86 | 0.115 | −0.35±0.39 | 1.03±0.70 | 0.000* | 0.502 |
| Glucose | −0.51±0.53 | −1.32±0.71 | 0.002* | 0.41±0.27 | 0.41±0.54 | 0.196 | 0.000# |
| Taurine | 1.03±0.74 | 0.57±0.55 | 0.385 | 0.05±0.05 | 1.14±0.27 | 0.000* | 0.000# |
represents significant difference across samples for that species
represents for significant difference in human NASH to rat MCD samples. Test statistics calculated conducting the one way ANOVA with Tukey honest significant difference (HSD), with significance define as adjusted p value ≤ 0.05.
Abbreviation: HFD: high fat diet; MCD: methionine and choline deficient; NASH: nonalcoholic steatohepatitis
Discussion
The utility of NAFLD rodent models in the future is both extensive and selective: extensive in the need for appropriate mechanistic studies in animal models, but selective due to characteristics needed to be accurately reflected in the model. Rodent NAFLD models vary dramatically in their ability to reproduce both the clinical and histopathological features of the disease (7). Both rats and mice fed an MCD diet develop histopathological features associated with NASH, but fail to represent the full spectrum of the clinical features of NASH such as insulin resistance (19).
Changes in metabolic pathways that occur from NAFLD progression can be ascertained by studying the metabolome. Moreover, perturbed levels of metabolites can reflect the degree of liver injury. The current study describes a metabolomic analysis of liver samples to compare the biochemical changes in both human NAFLD progression and in rodent models designed to reflect stages of the NAFLD spectrum: HFD representing steatosis, and MCD representing NASH. Importantly, many significant changes were found in metabolic pathways in NAFLD that were more pronounced with progression of the disease, and we determined whether these trends are reflected in the rodent models.
A more detailed look at bile acid metabolomics in human NAFLD has been published previously, highlighting elevated levels of TCA and TDCA in NASH, and decreased levels of CA and GDCA (20). In this study, those changes are compared to the rodent HF and MCD diet models, which conversely exhibit increased CA, and no changes in the other bile acids measured. A major contributor of this discrepancy is the interspecies difference in bile acid composition. Targeted profiling research of hepatic bile acids in humans and rats demonstrates that glycine-conjugated bile acids were the most abundant in the human liver, whereas tauro-bile acids were the predominant conjugated forms in MCD rats (18). This is coincident with our finding that the metabolic profile of hepatic bile acids is dominated by conjugated bile acids (Table 1). The interspecies differences in the hepatic bile acid pool composition may be a determinant in potential liver damage induced by the accumulation of specific bile acids in humans, which may be missing in rodent models (21, 22). Among bile acids, hydrophobic bile acids are recognized as the main nuclear receptor signaling ligands and can also behave as toxic precursors that can lead to the direct activation of cell apoptosis and necrosis (23, 24). Changes in bile acid composition in humans and rodent models of disease must be interpreted cautiously and include analysis of nuclear receptor activation and regulation of rate-limiting enzymes due to well-defined differences in hydrophilic and hydrophobic bile acid pools and signaling between rodents and humans (25).
The liver is the main site for amino acid catabolism, and would be a prime target in metabolomic remodeling in NAFLD. In the present study, almost all amino acids exhibited a significant change in NASH, and had a significant interspecies interaction. A detailed investigation into branched chain amino acid regulation in NASH has been published previously (26); however, it is worth noting that none of the altered branched chain amino acids were changed in either HFD or MCD. Only asparagine, citrulline, GABA, and lysine exhibited the same trend across both species. Although each amino acid has its own unique catabolic pathway, nitrogen metabolism is mainly via urea formation and carbon conservation via gluconeogenesis and the citrate cycle (Figure 3). Asparagine, citrulline, and lysine contribute directly to the formation of acetyl-CoA and disruption of these levels can have a direct impact on the metabolic and antioxidant function of the mitochrondria (27).
The pathogenesis of NAFLD involves the accumulation of excess triglyceride (TG) and free fatty acids (FFAs) in the hepatocytes, which contributes to the hallmark inflammatory response (28). TG accumulation in hepatocytes arises from an imbalance between lipid uptake and removal (28). TG synthesis is stimulated to dispose of excess of FFAs, which come from peripheral adipose tissue to the liver via the lipogenic pathway (29). Animal models such as the MCD diet exhibit TG and lipoperoxide accumulation as early as 2-5 days after starting the diet (30), and these changes in TG composition and levels are reflected in the results of the analysis described in Table 3. The absence of choline, an essential nutrient, induces steatosis and liver cell death in humans, and these data support decreased choline levels in human NASH (31). The current studies show the short-chain carnitines were significantly higher in individuals with NASH, consistent with our previous study (26), while long-chain fatty acid and phospholipids were lower in NASH. This is similar to previous studies that performed target lipidomics analyses of hepatic tissue in subjects with NAFLD (32, 33).
In the comparison of human and MCD rat model metabolomes, several endogenous metabolites in the “energetics metabolite” category in Table 4 were assessed. 4-hydroxyproline, betaine, glucose, creatinine, creatine and taurine were all significantly changed in human NASH samples. Taurine is an organic acid that conjugates bile acids, and was significantly increased in human NASH. Recent clinical studies support the use of taurine administration to alleviate inflammation and lipid accumulation in NAFLD (34). Taurine increases in the rat models did not reach statistical significance. Betaine and creatine exhibited opposite changes between rat models, and the human NAFLD samples. Overall, some of the similarities between human NASH and rodent MCD diet model metabolites may be developed as useful biomarkers, while others may not.
In summary, this comparison profiling analysis of human and rodent NAFLD provides a foundation for further investigation into diagnosis and monitoring. This analysis did have some limitations, including the inability to differentiate diagnoses in a PCA, and that the human liver samples were obtained by LTCDS, most likely from deceased donors, thus over-representing the more severe pre-cirrhotic end of the NAFLD spectrum. Nevertheless, select essential amino acids could be potential biomarkers for NAFLD progression. The combination of metabolomic changes of bile acids, amino acids, and fatty acids lends insight into the physiological and biochemical response integral to NAFLD pathogenesis in both species. Furthermore, the dissimilarities noted between human NAFLD progression and rodent models suggest a limited utility of these models for comparing biochemical biomarkers. While they are appropriate for certain endpoints, such as pharmacokinetics or enzyme regulation, therapeutic intervention or the biochemical aspects of pathogenesis of NASH may prove to have important differences.
Importance Questions.
What is already known about this subject?
Metabolic pathways are altered in human nonalcoholic fatty liver disease (NAFLD) progression.
Rodent models of nonalcoholic steatohepatitis (NASH) reflect many pathologic and histologic changes that occur in humans.
What does your study add?
A liver-specific profile of metabolomic pathways that are altered in NAFLD progression to NASH.
A comparison of metabolomics profile between human NASH and the rodent model of NASH, the MCD diet.
Acknowledgments
Funding Sources: This work was supported by the National Institute of Environmental Health Sciences Toxicology Training Grant (ES007091) and National Institutes of Health grants (HD062489, AI083927, and ES019487). PN supported by CAS (grant RVO:60077344).
Footnotes
Author Contributions:
Participated in research design: Lake, Cherrington, Lehman-McKeeman
Conducted experiments: Lake, Shipkova, Aranibar, Robertson, Reily, Lehman-McKeeman
Contributed new reagents or analytical tools: Lehman-McKeeman, Novak
Performed data analysis: Han, Lu, Lake, Novak, Torabzadehkhorasani, Billheimer
Wrote or contributed to the writing of the manuscript: Han, Dzierlenga, Li, Cherrington
Disclosure: The authors declared no conflict of interest.
References
- 1.Ali R, Cusi K. New diagnostic and treatment approaches in non-alcoholic fatty liver disease (NAFLD) Ann Med. 2009;41:265–78. doi: 10.1080/07853890802552437. [DOI] [PubMed] [Google Scholar]
- 2.Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global Epidemiology of Non-Alcoholic Fatty Liver Disease-Meta-Analytic Assessment of Prevalence, Incidence and Outcomes. Hepatology. 2015 doi: 10.1002/hep.28431. [DOI] [PubMed] [Google Scholar]
- 3.Ratziu V, Bellentani S, Cortez-Pinto H, Day C, Marchesini G. A position statement on NAFLD/NASH based on the EASL 2009 special conference. J Hepatol. 2010;53:372–84. doi: 10.1016/j.jhep.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 4.Kucera O, Cervinkova Z. Experimental models of non-alcoholic fatty liver disease in rats. World J Gastroenterol. 2014;20:8364–8376. doi: 10.3748/wjg.v20.i26.8364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hebbard L, George J. Animal models of nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2011;8:35–44. doi: 10.1038/nrgastro.2010.191. [DOI] [PubMed] [Google Scholar]
- 6.Imajo K, Yoneda M, Kessoku T, et al. Rodent models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Int J Mol Sci. 2013;14:21833–57. doi: 10.3390/ijms141121833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Canet MJ, Hardwick RN, Lake AD, Dzierlenga AL, Clarke JD, Cherrington NJ. Modeling Human Nonalcoholic Steatohepatitis-Associated Changes in Drug Transporter Expression Using Experimental Rodent Models. Drug Metab Dispos. 2014;42:586–595. doi: 10.1124/dmd.113.055996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ghoshal AK. New insight into the biochemical pathology of liver in choline deficiency. Crit Rev Biochem Mol Biol. 1995;30:263–73. doi: 10.3109/10409239509083487. [DOI] [PubMed] [Google Scholar]
- 9.Serviddio G, Giudetti AM, Bellanti F, et al. Oxidation of hepatic carnitine palmitoyl transferase-I (CPT-I) impairs fatty acid beta-oxidation in rats fed a methionine-choline deficient diet. PLoS One. 2011;6:e24084. doi: 10.1371/journal.pone.0024084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Veteläinen R, van Vliet A, van Gulik TM. Essential pathogenic and metabolic differences in steatosis induced by choline or methione-choline deficient diets in a rat model. J Gastroenterol Hepatol. 2007;22:1526–33. doi: 10.1111/j.1440-1746.2006.04701.x. [DOI] [PubMed] [Google Scholar]
- 11.Tahan V, Eren F, Avsar E, et al. Rosiglitazone attenuates liver inflammation in a rat model of nonalcoholic steatohepatitis. Dig Dis Sci. 2007;52:3465–72. doi: 10.1007/s10620-007-9756-x. [DOI] [PubMed] [Google Scholar]
- 12.Hardwick RN, Fisher CD, Canet MJ, Scheffer GL, Cherrington NJ. Variations in ATP-binding cassette transporter regulation during the progression of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:2395–2402. doi: 10.1124/dmd.111.041012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lake A, Novak P, Fisher C, et al. Analysis of global and absorption, distribution, metabolism, and elimination gene expression in the progressive stages of human nonalcoholic fatty liver disease. Drug Metab Dispos. 2011;39:1954–1960. doi: 10.1124/dmd.111.040592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stärkel P, Sempoux C, Leclercq I, et al. Oxidative stress, KLF6 and transforming growth factor-beta up-regulation differentiate non-alcoholic steatohepatitis progressing to fibrosis from uncomplicated steatosis in rats. J Hepatol. 2003;39:538–46. doi: 10.1016/s0168-8278(03)00360-x. [DOI] [PubMed] [Google Scholar]
- 15.Fisher CDC, Lickteig AJA, Augustine LML, et al. Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos. 2009;37:2087–2094. doi: 10.1124/dmd.109.027466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kleiner DE, Brunt EM, Van Natta M, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–21. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- 17.Lake AD, Novak P, Shipkova P, et al. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol Appl Pharmacol. 2013;268:132–40. doi: 10.1016/j.taap.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.García-Cañaveras JC, Donato MT, Castell JV, Lahoz A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J Lipid Res. 2012;53:2231–41. doi: 10.1194/jlr.D028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fisher CD, Lickteig AJ, Augustine LM, et al. Experimental non-alcoholic fatty liver disease results in decreased hepatic uptake transporter expression and function in rats. Eur J Pharmacol. 2009;613:119–127. doi: 10.1016/j.ejphar.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lake AD, Novak P, Shipkova P, et al. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol Appl Pharmacol. 2013;268:132–40. doi: 10.1016/j.taap.2013.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile-acid signalling for metabolic diseases. Nat Rev Drug Discov. 2008;7:678–93. doi: 10.1038/nrd2619. [DOI] [PubMed] [Google Scholar]
- 22.Palmeira CM, Rolo AP. Mitochondrially-mediated toxicity of bile acids. Toxicology. 2004;203:1–15. doi: 10.1016/j.tox.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 23.Perez M-J, Briz O. Bile-acid-induced cell injury and protection. World J Gastroenterol. 2009;15:1677–89. doi: 10.3748/wjg.15.1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sharma R, Majer F, Peta VK, et al. Bile acid toxicity structure-activity relationships: correlations between cell viability and lipophilicity in a panel of new and known bile acids using an oesophageal cell line (HET-1A) Bioorg Med Chem. 2010;18:6886–95. doi: 10.1016/j.bmc.2010.07.030. [DOI] [PubMed] [Google Scholar]
- 25.Chiang JYL. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–66. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lake AD, Novak P, Shipkova P, et al. Branched chain amino acid metabolism profiles in progressive human nonalcoholic fatty liver disease. Amino Acids. 2015;47:603–15. doi: 10.1007/s00726-014-1894-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids. 2009;37:1–17. doi: 10.1007/s00726-009-0269-0. [DOI] [PubMed] [Google Scholar]
- 28.Berlanga A, Guiu-Jurado E, Porras JA, Auguet T. Molecular pathways in non-alcoholic fatty liver disease. Clin Exp Gastroenterol. 2014;7:221–39. doi: 10.2147/CEG.S62831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23:201–29. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 30.Larter CZ, Yeh MM. Animal models of NASH: getting both pathology and metabolic context right. J Gastroenterol Hepatol. 2008;23:1635–48. doi: 10.1111/j.1440-1746.2008.05543.x. [DOI] [PubMed] [Google Scholar]
- 31.Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol. 2012;28:159–65. doi: 10.1097/MOG.0b013e32834e7b4b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kalhan SC, Guo L, Edmison J, et al. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism. 2011;60:404–13. doi: 10.1016/j.metabol.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Puri P, Baillie RA, Wiest MM, et al. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–90. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 34.Gentile CL, Nivala AM, Gonzales JC, et al. Experimental evidence for therapeutic potential of taurine in the treatment of nonalcoholic fatty liver disease. Am J Physiol Regul Integr Comp Physiol. 2011;301:R1710–22. doi: 10.1152/ajpregu.00677.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]