

Graphical abstract

Keywords: Tuberculosis, Mycobacterium tuberculosis, Anti-tubercular activity, Triazolopyrimidines
Abstract
We identified a di-substituted triazolopyrimidine with anti-tubercular activity against Mycobacterium tuberculosis. Three segments of the scaffold were examined rationally to establish a structure-activity relationship with the goal of improving potency and maintaining good physicochemical properties. A number of compounds displayed sub-micromolar activity against Mycobacterium tuberculosis with no cytotoxicity against eukaryotic cells. Non-substituted aromatic rings at C5 and a two-carbon chain connecting a terminal aromatic at C7 were preferred features; the presence of NH at C7 and a lack of substituent at C2 were essential for potency. We identified compounds with acceptable metabolic stability in rodent and human liver microsomes. Our findings suggest that the easily-synthesized triazolopyrimidines are a promising class of potent anti-tubercular agents and warrant further investigation in our search for new drugs to fight tuberculosis.
1. Introduction
Tuberculosis (TB) and its causative agent Mycobacterium tuberculosis present a serious threat to global health. There are over 1 million deaths each year and approximately 9 million new cases.1 Taken together with the estimate that a third of the world’s population is infected with M. tuberculosis and the existence of drug-resistant strains of M. tuberculosis, it is apparent there is a pressing need for new therapies. To address these needs, there has been an increased effort directed towards TB drug discovery in recent years and a pipeline of new anti-TB drug candidates has started to emerge. The search for new molecular scaffolds with potentially novel mechanisms of action remains a priority.
Triazolopyrimidines (TZPs) are a well-known scaffold in medicinal chemistry, and their utility is exemplified by the discovery and development of novel agents to fight a wide range of diseases. For example, TZPs possess anticancer activity,2 and have been used as phosphodiesterase inhibitors for diabetes treatment.3 Recently, the first natural TZP, essramycin, was isolated and found to possess antibacterial activity.4 A considerable effort has been made to develop TZPs with antimalarial activity.5, 6, 7 Transition metal-containing TZPs have antiproliferative activity against Leishmania and Trypanosoma cruzi, the protozoa that cause leishmaniasis and Chagas disease, respectively.8 In addition TZP acylsulfonamides with anti-mycobacterial activity target acetohydroxyacid synthase.9 Similar compounds were also identified in a phenotypic screening campaign against Mycobacterium bovis BCG.10
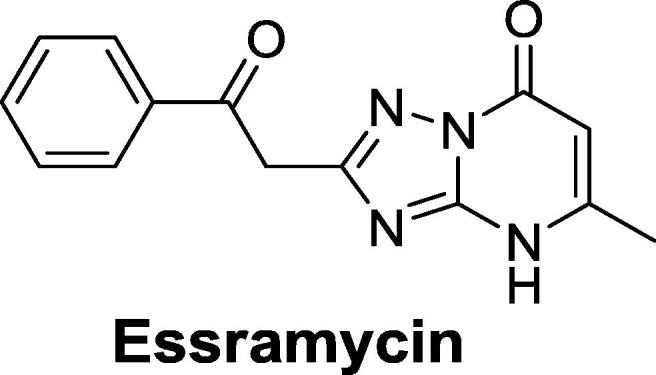
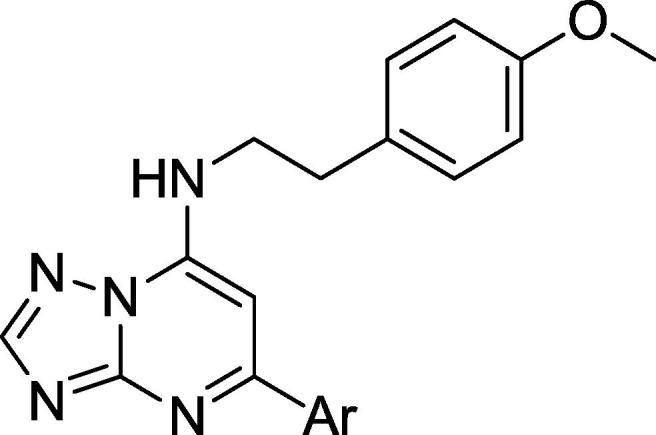
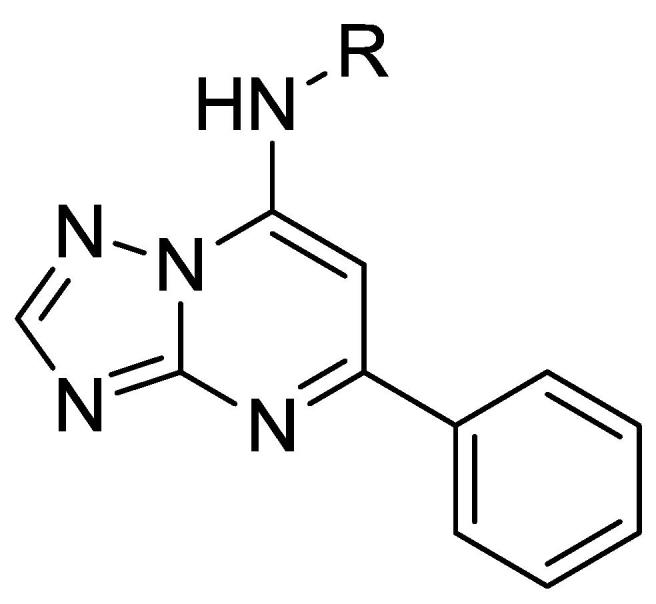
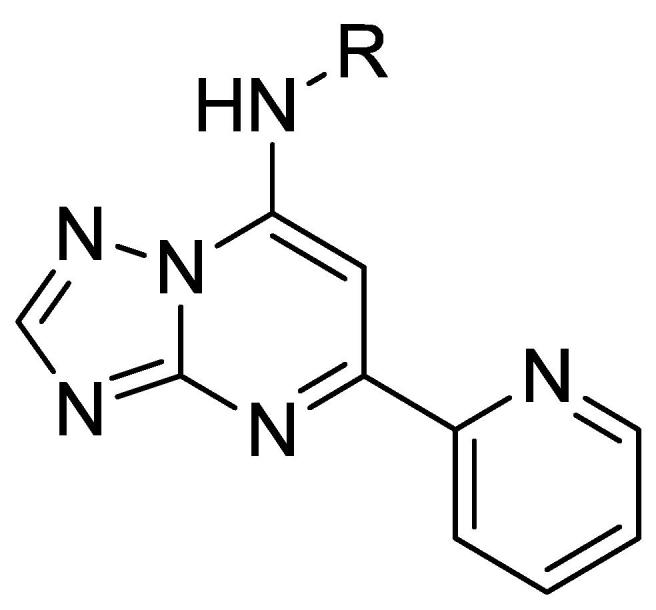
We identified a single TZP compound (1) from a whole-cell screen against M. tuberculosis which was active in liquid culture (Fig. 1). The compound had good activity against M. tuberculosis with a minimum inhibitory concentration (MIC) of 3.1 μM (Table 1). The compound was not cytotoxic, with an IC50 of >100 μM against the HepG2 cell line; the selectivity index (SI), defined as the ratio of cytotoxicity to MIC, was >32. Based on these data we initiated a structure-activity relationship (SAR) study around this singleton.
Fig. 1.

Triazolopyrimidine (TZP) 1. Three segments are illustrated with boxes and numbered.
Table 1.
| Cpd | Ar | MIC (μM) | IC50 (μM) | SI | Cpd | Ar | MIC (μM) | IC50 (μM) | SI |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 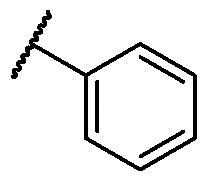 |
3.1 ± 1.3 | >100 | >32 | 13 | 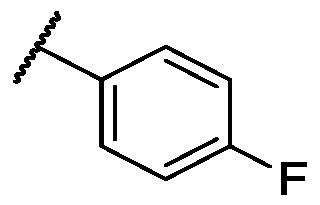 |
4.4 ± 2.0 | 89 | >21 |
| 7 | 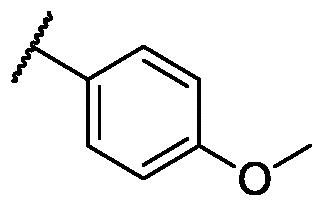 |
>20 | >100 | – | 14 | 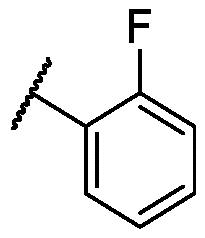 |
3.0 ± 0.21 | >100 | >30 |
| 8 | 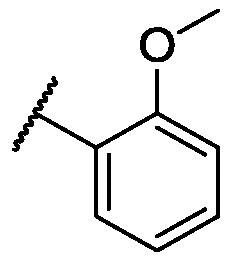 |
>20 | 87 | – | 15 | 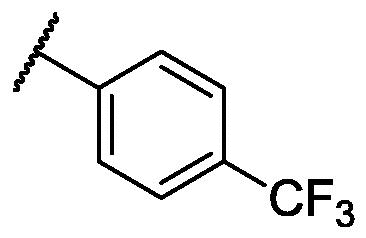 |
>20 | >100 | – |
| 9 | 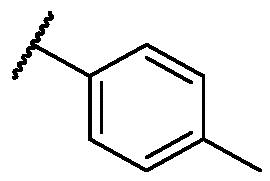 |
>20 | 37 | – | 16 | 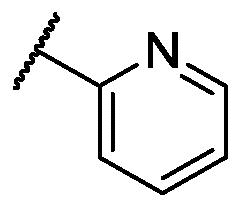 |
0.83 ± 0.26 | >100 | >130 |
| 10 | 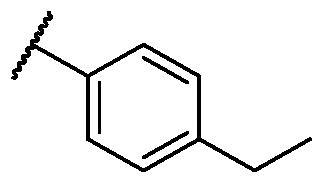 |
>20 | >100 | – | 17 | 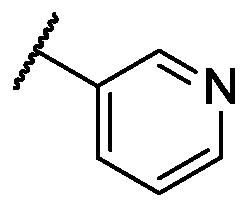 |
>20 | >100 | – |
| 11 | 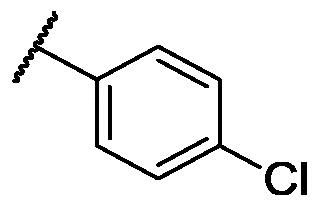 |
>20 | 65 | – | 18 | 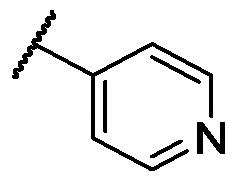 |
>20 | 69 | – |
| 12 | 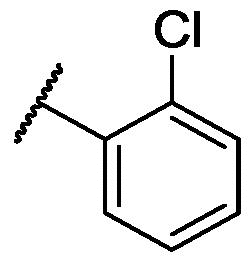 |
>20 | >100 | – | 19 | 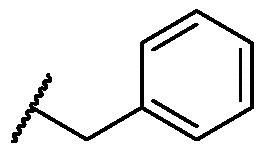 |
>20 | 57 | – |
MIC90 is the minimum concentration required to inhibit growth of M. tuberculosis by 90%. MICs are the average ± standard deviation of two independent experiments. IC50 is the concentration required to reduce the viability of HepG2 cells by 50%. Selectivity index (SI) = IC50/MIC90.
Here, we present an exploratory study to understand the SAR of the TZP series. We identified key functionalities and features necessary for anti-tubercular activity. In general, TZP compounds lack cytotoxicity and display an encouraging metabolic stability profile. In addition, we demonstrated their bactericidal activity against non-replicating bacteria.
2. Results and discussion
Our SAR investigation began with the design and synthesis of novel analogs based on modifications of the core structure of compound 1. We set out to explore modifications of the core by way of heteroatom replacement and the impact of chemical diversity at the C2, C5, and C7 positions.
2.1. Chemical synthesis
The general synthetic routes are shown in Scheme 1, Scheme 2. We initially evaluated the SAR associated with modifications at the C5 position of the TZP ring system. Synthesis of analogs 7–25 proceeded through key 7-hydroxytriazolopyrimidine intermediates 4 (Scheme 1), which were obtained via the condensation of the 1,3-di-keto compound 2 with the 5-amino-4H-1,2,4-triazole 3.9 Di-keto compounds were prepared by treating the appropriate substituted ethanone with diethyl carbonate in the presence of base. Chloro intermediates 5 were obtained upon treatment of 4 with phosphoryl chloride. Reaction of 5 with 4-methoxyphenethylamine gave C5 substituted triazolopyrimidines 7–25 in moderate to good yields. Condensation between 3-(dimethylamino)-1-phenyl-propane-1-one and 3 directly gave 27.
Scheme 1.

Synthesis of the triazolopyrimidine compounds. General conditions: (i) AcOH, 120 °C, 12–16 h; (ii) POCl3, 80 – 100 °C, 2 h; (iii) NMP, 80–100 °C or room temperature.
Scheme 2.

Synthesis of triazolopyrimidine compounds exploring core modifications. General conditions: (i) AcOH, 100–120 °C, 12–16 h; (ii) POCl3, 80–100 °C, 2 h; (iii) amine 6, NMP, 80–100 °C.
Compound 5 served as a strategic intermediate from which a diverse set of C7 analogs could be prepared. Condensation between 5 (R′ = phenyl) and a variety of amines (6) gave C7 substituted triazolopyrimidines 27–48 with good yields. Similarly, reaction of 5 (R′ = 2-pyridinyl) with a variety of amines gave C7 substituted analogs 51–60; the use of cyclic amines gave 62–64. Compound 28 was obtained after treating 7-chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine (5a) with methanolic ammonia. Synthesis of amide analogs 49 and 50 proceeded through 28 via EDCI-HOBt mediated coupling with the appropriate substituted benzeneacetic acid. The alkylation of compound 1 using iodomethane afforded analog 61.
As shown in Scheme 2, decoration of the C2 position of the TZP core proceeded through intermediate 66a-b, obtained from the condensation between ethyl 2-pyridylcarbonylacetate (2k) and a 2-substituted 5-amino-1H-1,2,4-triazole (65a-b). Commercially available triazoles 65a and 65b were treated with 26 to give 66a and 66b, respectively. Subsequent chlorination followed by treatment with 4-methoxyphenylethylamine gave C2 substituted TZP analogs 67 and 68. Heterocyclic core replacement analogs pyrazolopyrimidine 69 and imidazopyridine 70 were produced after manipulation of commercially available intermediates 65c-d.
2.2. Structure-activity relationship (SAR) studies
We determined the activity of all the compounds synthesized against both M. tuberculosis and eukaryotic cells (HepG2 cell line); we determined the MIC90 against M. tuberculosis, defined as the concentration required to inhibit 90% growth in liquid medium, and the IC50 against HepG2 cells, defined as the concentration required to reduce HepG2 viability by 50%. Selectivity index (SI) was calculated as IC50/MIC90. We completed a systematic evaluation by modification of the TZP ring at the C5, C7, C2 and core positions.
We first explored substitution of C5 phenyl with ortho- or para-electron-donating groups, which resulted in a loss of activity (Table 1, compounds 7–10). The incorporation of fluorine on the para- or ortho-positions (compounds 13 and 14) of the C5 phenyl ring was tolerated and maintained good separation from cytotoxicity. This was not seen with chloride analogs 11 and 12 which had no activity. The strongly deactivating para-CF3 phenyl moiety (compound 15) abrogated activity. Replacing the C5 phenyl with a polar 2-pyridyl gave compound 16 with slightly increased potency while having no effect on cytotoxicity, and provided a valuable point for further optimization. It is interesting to note that 3- or 4-pyridyl isomers at C5 (17 and 18) resulted in loss of activity (Table 1).
No advantage was gained by replacing the aromatic group at C5 with either a linear alkyl (20, 21) or a cyclohexyl group (25) and only small cyclic alkyl moieties such as the cyclopropyl (22), cyclopentyl 24 and cyclobutyl (23) analogs had good anti-tubercular activity (Table 2).
Table 2.
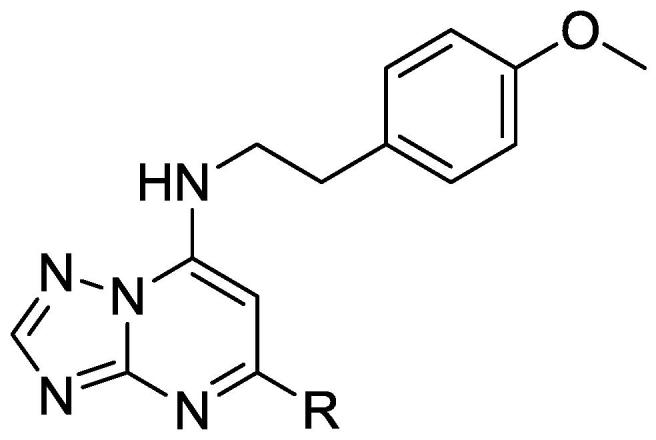
| Cpd | R | MIC (μM) | IC50 (μM) | SI |
|---|---|---|---|---|
| 1 | 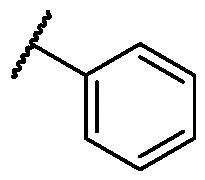 |
3.1 ± 1.3 | >100 | >32 |
| 20 |  |
>20 | >100 | – |
| 21 | 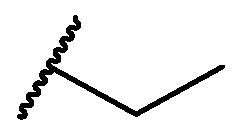 |
>20 | >100 | – |
| 22 | 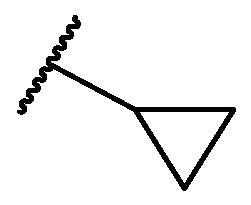 |
5.8 ± 1.6 | >100 | >17 |
| 23 | 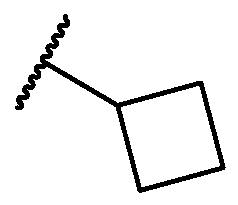 |
2.9 ± 0.71 | >100 | >34 |
| 24 | 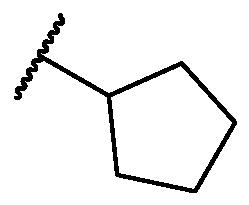 |
5.5 ± 0.07 | 95 | 17 |
| 25 | 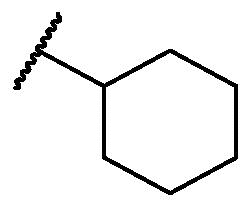 |
>20 | 47 | – |
MIC90 is the minimum concentration required to inhibit growth of M. tuberculosis by 90%. MICs are the average ± standard deviation of two independent experiments. IC50 is the concentration required to reduce the viability of HepG2 cells by 50%. Selectivity index (SI) = IC50/MIC90.
We next explored modifications to the C7 position, while keeping a phenyl group at C5 (Table 3). Non-aromatic moieties at C7 (e.g. compounds 27–35) did not show any anti-tubercular activity, except for the ethyl tethered cyclohexyl analog (36).
Table 3.
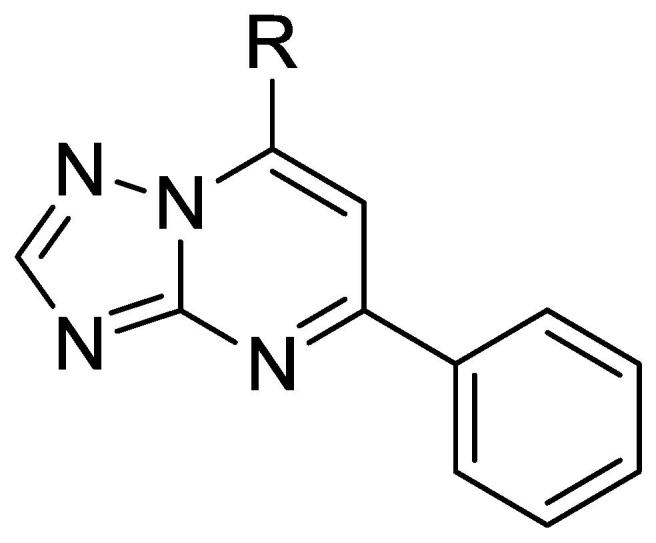
| Cpd | R | MIC (μM) | IC50 (μM) | SI | Cpd | R | MIC (μM) | IC50 (μM) | SI |
|---|---|---|---|---|---|---|---|---|---|
| 27 | 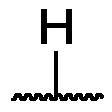 |
>20 | >20 | – | 32 | 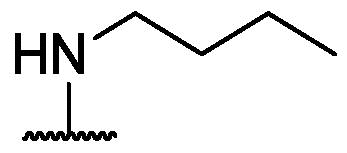 |
>20 | 98 | – |
| 28 | 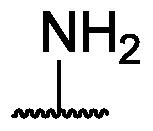 |
>20 | >20 | – | 33 | 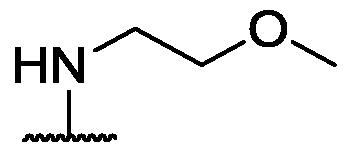 |
>20 | >100 | – |
| 29 | 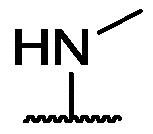 |
>20 | >20 | – | 34 | 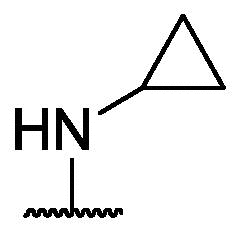 |
>20 | >100 | – |
| 30 | 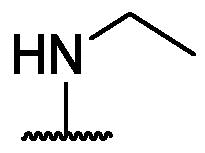 |
>20 | 37 | – | 35 | 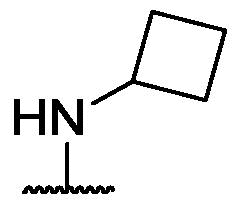 |
>20 | 57 | – |
| 31 | 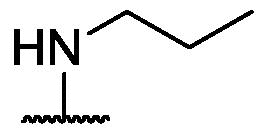 |
>20 | 94 | – | 36 | 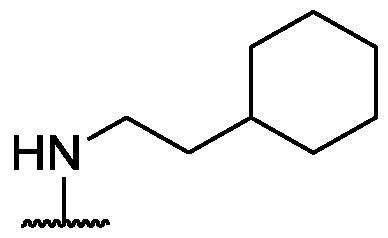 |
17 ± 2.1 | >100 | >6 |
MIC90 is the minimum concentration required to inhibit growth of M. tuberculosis by 90%. MICs are the average ± standard deviation of two independent experiments. IC50 is the concentration required to reduce the viability of HepG2 cells by 50%. Selectivity index (SI) = IC50/MIC90.
We next evaluated the effect of chain length at C7 (Table 4). Compounds 37 (with no tether), 38 (with a one carbon tether), and 40 (with a three carbon tether) were all inactive. A reduction in potency was observed for the unsubstituted terminal phenyl group as in compound 39 compared to the para-methoxy analog 1. This pointed to the importance of para-substitution for activity and selectivity. Polar aromatics at the terminal end of the C7 alkyl chain were not tolerated as demonstrated by the inactive pyridyl analogs 41–43. para-substituted analogs 44–48 were prepared, and the overall potency was rescued in all cases except for the para-fluoro analog 46. C7 amide analogs (49 and 50) retained activity and were not cytotoxic indicating that properties of the aniline nitrogen at C7 can be modified with little penalty to activity.
Table 4.
| Cpd | R | MIC (μM) | IC50 (μM) | SI | Cpd | R | MIC (μM) | IC50 (μM) | SI |
|---|---|---|---|---|---|---|---|---|---|
| 37 | 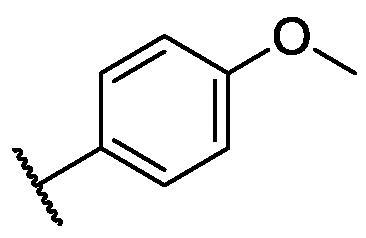 |
>20 | 49 | – | 44 | 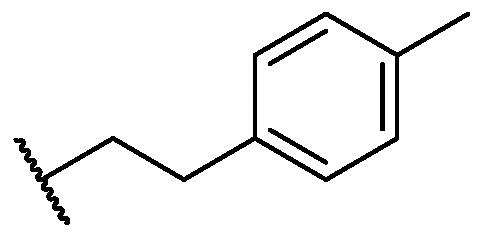 |
1.4 ± 0.5 | 53 | 43 |
| 38 | 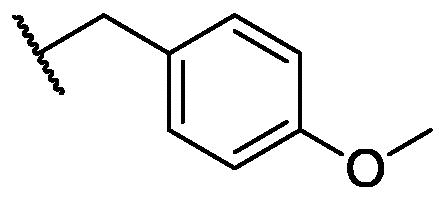 |
>20 | 43 | – | 45 | 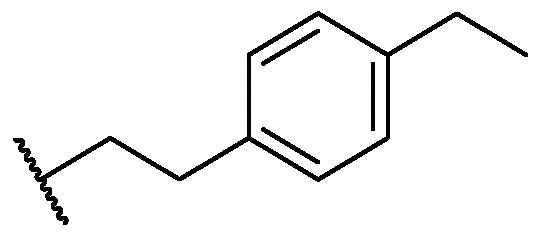 |
0.74 ± 0.1 | >100 | >138 |
| 39 | 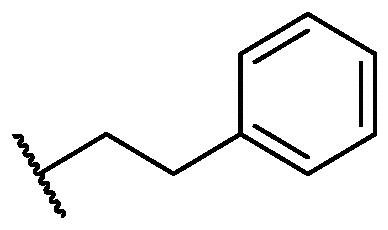 |
7.7 ± 0.7 | 27 | 3.5 | 46 | 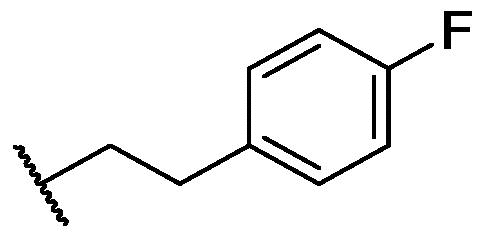 |
>20 | 50 | – |
| 40 | 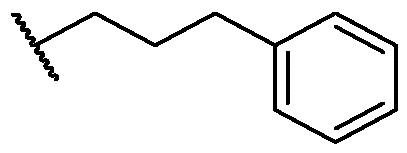 |
>20 | 8.7 | – | 47 | 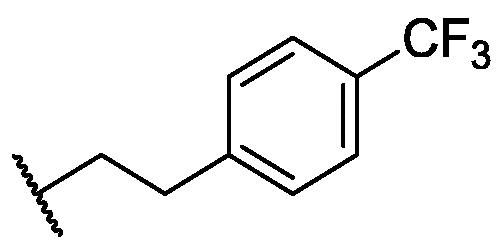 |
2.7 ± 0.7 | >100 | >38 |
| 41 | 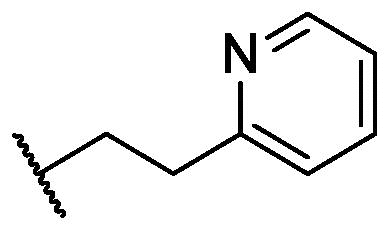 |
>20 | >100 | – | 48 | 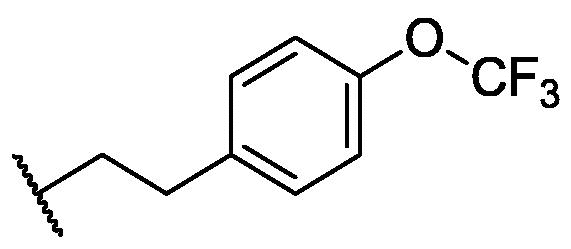 |
3.9 ± 1.1 | >100 | >27 |
| 42 | 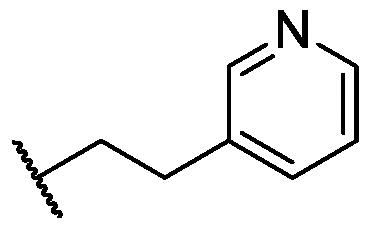 |
>20 | >100 | – | 49 | 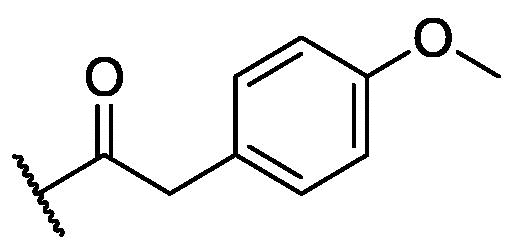 |
1.5 ± 0.7 | >100 | >73 |
| 43 | 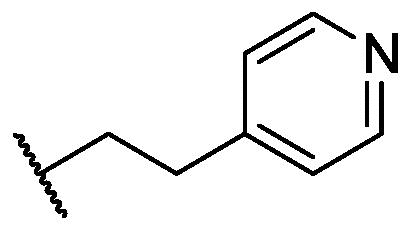 |
>20 | >100 | – | 50 |  |
10 ± 1.1 | >100 | >10 |
MIC90 is the minimum concentration required to inhibit growth of M. tuberculosis by 90%. MICs are the average of two independent experiments. IC50 is the concentration required to reduce the viability of HepG2 cells by 50%. Selectivity index (SI) = IC50/MIC90.
Thus far we demonstrated that optimal anti-tubercular activity was found in the 2-pyridyl analog 16. As with the C5 phenyl derivatives, we examined modifications to the terminal aromatic ring of the C7 side chain while keeping the 2-pyridyl at C5 constant (Table 5). A variety of para-substituted aromatic groups were tolerated at the terminal end of the C7 side chain resulting in analogs (55–60) with comparable or improved potency and good separation from cytotoxicity. It is worthy to note that analogs containing terminal pyridyl moieties on the C7 side chain (52–54) lost activity, as observed in the C5 phenyl series, demonstrating that polar residues are not tolerated in this region for either series.
Table 5.
| Cpd | R | MIC (μM) | IC50 (μM) | SI | Cpd | R | MIC (μM) | IC50 (μM) | SI |
|---|---|---|---|---|---|---|---|---|---|
| 51 |  |
2.4 ± 1.3 | >100 | >46 | 56 |  |
0.2 ± 0.02 | 44 | 220 |
| 52 | 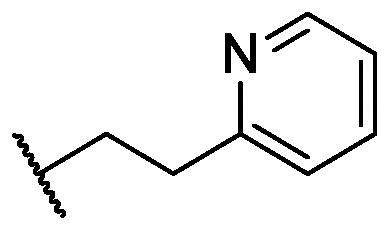 |
>20 | >100 | – | 57 | 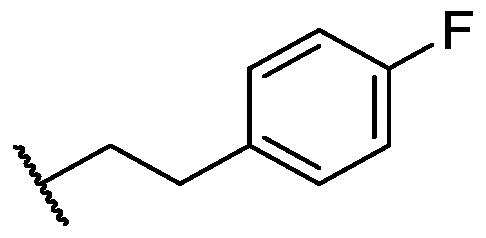 |
4.8 ± 1.3 | 81 | 17 |
| 53 | 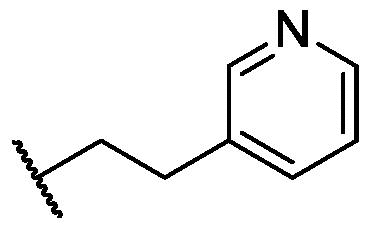 |
>20 | >100 | – | 58 | 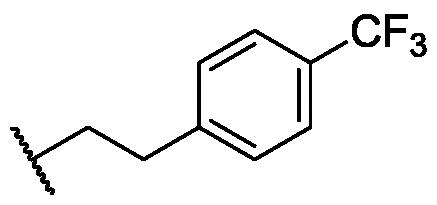 |
0.98 ± 0.4 | >100 | >119 |
| 54 | 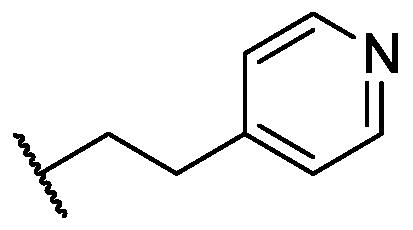 |
>20 | >100 | – | 59 | 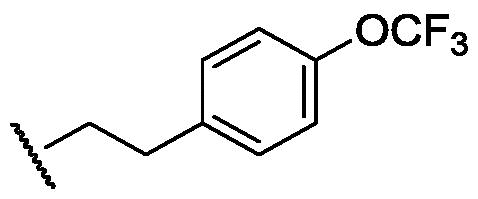 |
1.6 ± 1.1 | >100 | >72 |
| 55 | 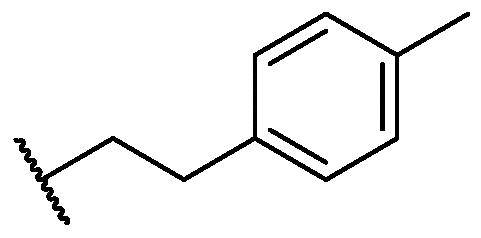 |
0.52 ± 0.1 | >100 | >196 | 60 | 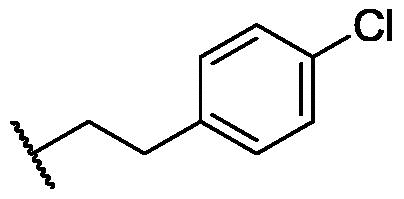 |
0.76 ± 0.25 | >100 | >135 |
MIC90 is the minimum concentration required to inhibit growth of M. tuberculosis by 90%. MICs are the average ± standard deviation of two independent experiments. IC50 is the concentration required to reduce the viability of HepG2 cells by 50%. Selectivity index (SI) = IC50/MIC90.
We investigated other SAR elements of the spacer at C7 (Table 6). The N-methylated analog 61 had a loss of activity, an indication that H-bonding may be important for the TZP compounds binding to their target (this could also be due to unfavorable steric interactions). Introducing rigidity on the amine tether as in pyrrolidine 62, piperidine 63 and morpholino 64 was not tolerated. It has been previously reported that substitution on the C2 position of the TZP scaffold gave compounds with potent antimalarial activity and with good metabolic stability.7 Based on these findings and with the goal of identifying active and metabolically stable compounds, we prepared and tested the 2-methyl (67) and 2-phenyl (68) analogs. Analog 67 had comparable activity to our original hit, but analog 68 showed an MIC > 20 μM. Pyrazolopyrimidine11 and imidazopyridine12, 13 compounds have also been reported to possess potent anti-tubercular activity. Analog 69 with a pyrazolopyrimidine core demonstrated comparable activity to original hit 1, but with increased cytotoxicity, whereas the imidazopyrimidine-based analog 70 had excellent anti-tubercular activity as well as good separation from cytotoxicity.
Table 6.
Constraining C7 Side Chain and Core Modification.
| Cpd | Structure | MIC (μM) | IC50 (μM) | SI | Cpd | Structure | MIC (μM) | IC50 (μM) | SI |
|---|---|---|---|---|---|---|---|---|---|
| 61 |  |
>20 | 17 | – | 67 |  |
3.9 ± 0.21 | >100 | >26 |
| 62 |  |
>20 | 23 | – | 68 |  |
>20 | >100 | – |
| 63 | 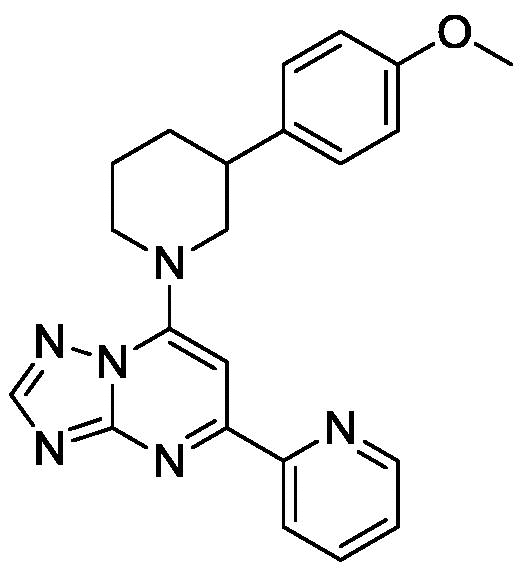 |
>20 | 16 | – | 69 | 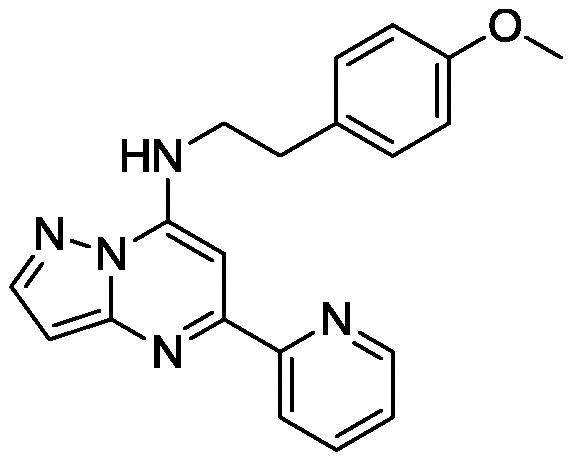 |
2.5 ± 0.15 | 23 | 9 |
| 64 | 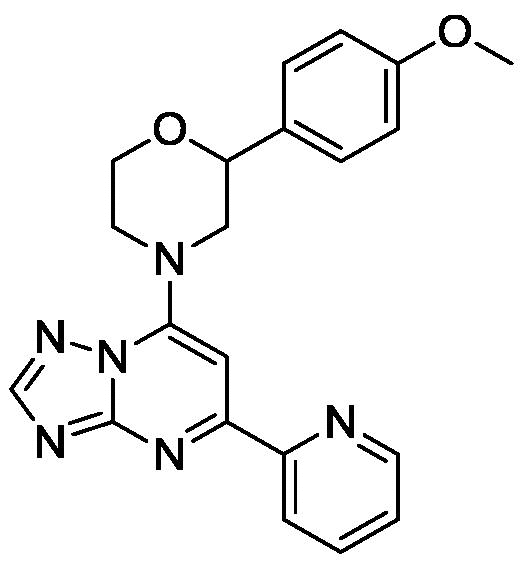 |
>20 | 50 | – | 70 | 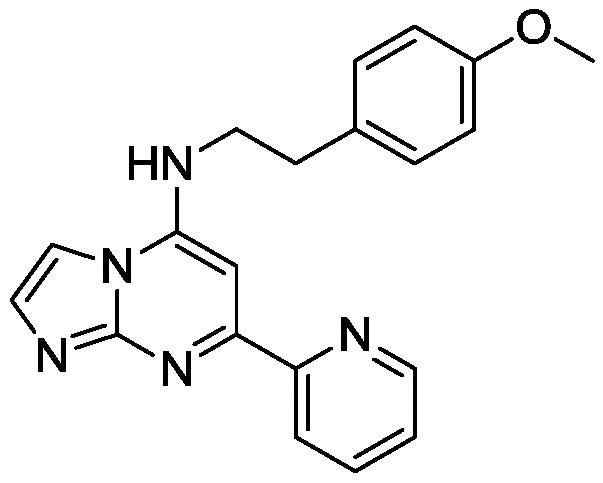 |
0.22 ± 0.07 | 30 | 142 |
MIC90 is the minimum concentration required to inhibit growth of M. tuberculosis by 90%. MICs are the average ± standard deviation of two independent experiments. IC50 is the concentration required to reduce the viability of HepG2 cells by 50%. Selectivity index (SI) = IC50/MIC90.
2.3. Microsomal stability and in vivo pharmacokinetic (PK) studies
Based on in silico ADME predictions, three compounds were chosen to cover a range of cLogP values and were evaluated for their in vitro microsomal stability (Table 7). Rapid metabolism of compound 44 was observed, in rodent and human liver microsomes. The para-OCF3 analog 48 was also rapidly metabolized. The amide (49) had improved in vitro microsomal stability, with only 12% loss after 30 min in mouse microsomes, 22% loss in rat microsomes and 31% loss in human microsomes. The difference in stability for these three compounds is probably due to presence of more than one oxidatively-labile carbon in 44 and 48 (both have a two carbon linker), while 49 has only one such soft spot, although these have not been confirmed with metabolite identification studies.
Table 7.
In vitro ADME and in vivo PK.
| Cpd | % turnover by liver microsomes in 30 min |
Mouse Fu,pla | Predicted Fu,plb | clogPc | PO AUC (nM*h) | PO Cmax (nM) | PO Tmax (h) | PO t1/2 (h) | IV clearance (mL/min/kg) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Mouse | Rat | Human | |||||||||
| 44 | 98.2 | 99.2 | 68.1 | 0.015 | 0.020 | 4.33 | 676 | 604 | 0.25 | 1.05 | 182 |
| 48 | 52.1 | 52.7 | 32.8 | 0.007 | 0.006 | 5.25 | 2750 | 958 | 0.75 | 2.49 | 33.5 |
| 49 | 12.5 | 21.8 | 30.6 | ND | 0.048 | 3.25 | 187 | 62.8 | 1.5 | ND | 248 |
Fu,pl is the fraction unbound in plasma.
Predicted value using a QSAR model built based on data generated for >3000 compounds measured internally (unpublished).
clogP was predicted by Chemaxon model (www.chemaxon.com).
The oral exposure of the three compounds was evaluated in male mice by comparing exposures following oral (PO) and intravenous (IV) administration of 10 and 1 mg/kg doses of compounds, respectively (Table 7). Overall, the three compounds showed poor to moderate in vivo mouse PK properties. Compound 44 had an AUC = 676 nM∗h, and rapid IV clearance (182 mL/min/kg) as predicted by mouse microsomes. Compound 48 had more promising mouse PK with greater oral exposure (AUC 2750 nM * h), a longer half-life, and slower IV clearance (33.5 mL/min/kg). Surprisingly, despite showing good in vitro microsomal metabolic stability, amide 49 had the lowest oral exposure and fastest IV clearance rates in vivo compared to the two other compounds.
2.4. Activity Spectrum
Three compounds (16, 48 and 49) were selected for testing against other organisms based on good activity in liquid and solid medium (MIC99 < 20 µM) against M. tuberculosis (Table 8). We tested against a non-pathogenic mycobacterial species (Mycobacterium smegmatis), Escherichia coli (Gram negative), Pseudomonas aeruginosa (Gram negative), Bacillus subtilis (Gram positive) and yeast (Saccharomyces cerevisiae) (Table 8). All three compounds had activity against M. tuberculosis, but no activity against any of the other species. Thus, the TZP compounds are selective for M. tuberculosis.
Table 8.
Spectrum of activity.
| MIC99 (μM) | ||||||
|---|---|---|---|---|---|---|
| Cpd | M. tuberculosis | M. smegmatis | E. coli | P. aeruginosa | S. cerevisiae | B. subtilis |
| 16 | 13 | >100 | >100 | >100 | >100 | >100 |
| 48 | 1.6 | >10 | >10 | >100 | >10 | >10 |
| 49 | 3.1 | >10 | >10 | >10 | >10 | >10 |
MIC99 is the lowest concentration of compound, which yielded less than 1% growth.
2.5. Activity against replicating and non-replicating M. tuberculosis
We also determined the effectiveness of the three compounds (16, 48 and 49) in killing replicating and non-replicating M. tuberculosis. Compounds exhibited static activity against replicating bacteria, preventing growth, but no kill was noted over 21 days. In contrast, we did note killing against non-replicating bacteria (starvation conditions) for all three compounds, with a 2–3 log kill over 14–21 days (see Fig. 2).
Fig. 2.

Kill kinetics against M. tuberculosis in liquid culture.
3. Conclusion
We conducted a systematic exploration of the triazolopyrimidine scaffold for activity against M. tuberculosis. Overall, the compounds in this series show good activity and selectivity. Our initial explorations suggest that non-substituted aromatic rings at C5, and a two-carbon chain connecting a terminal aromatic at C7 are preferred features for potency against M. tuberculosis and separation from cytotoxicity. The presence of NH at C7 and a lack of substituent at C2 are essential for potency of the molecule. Heteroatom replacement or homologation of the scaffold is well tolerated in the series. We have identified compounds with improved metabolic stability in rodent and human liver microsomes, however, oral exposure and clearance remains an issue for this series. We feel that these issues can be improved through further SAR exploration. Thus, there is substantial promise in developing the TZP series that warrants further investigation as novel tools in our drug arsenal to combat tuberculosis.
4. Materials and methods
4.1. Determination of minimum inhibitory concentration (MIC)
MICs were determined against M. tuberculosis H37Rv as described in.14 Briefly, M. tuberculosis was grown in Middlebrook 7H9 medium containing 10% OADC (oleic acid, albumin, dextrose, catalase) supplement (Becton Dickinson) and 0.05% w/v Tween 80 (7H9-Tw-OADC) under aerobic conditions. Bacterial growth was measured by OD590 after 5 days of incubation at 37 °C. Curves were fitted using the Levenberg–Marquardt algorithm. MIC90 was defined as the minimum concentration required to inhibit growth of M. tuberculosis by 90%.
4.2. Determination of cytotoxicity (IC50)
Cytotoxicity was assessed using the HepG2 cell line under replicating conditions. HepG2 cells were grown in DMEM, High Glucose, GlutaMAX™ (Invitrogen), 10% fetal bovine serum (FBS), and 1X penicillin-streptomycin solution (100 U/mL). Cells were incubated with compounds for 2 days at 37 °C (final DMSO concentration of 1%), 5% CO2. CellTiter-Glo® Reagent (Promega) was added and relative luminescent units (RLU) measured to assess cell viaility. Inhibition curves were fitted using the Levenberg–Marquardt algorithm. IC50 is the concentration required to reduce cell viability after 2 days by 50%. Controls were 1% DMSO only (growth control) and staurosporine (positive control).
4.3. Determination of minimum inhibitory concentration on solid medium (MIC)
The serial proportion method was used to determine MIC99 on solid medium for a variety of representative species.15 M. tuberculosis H37Rv and Mycobacterium smegmatis mc2155 were grown in Middlebrook 7H9 medium + 10% v/v OADC (oleic acid, albumin, dextrose, catalase) supplement (Becton Dickinson) and on Middlebrook 7H10 medium + 10% v/v OADC. Plates were incubated at 37 °C for 4 weeks and 3–4 days respectively; Escherichia coli DH5α and Staphylococcus aureus RN4220 were grown on LB agar and incubated at 37 °C for 1 and 2 days respectively; Pseudomonas aeruginosa HER1018 (PAO1) was grown on tryptic soy agar and incubated at 37 °C for 1 day; Bacillus subtilis Marburg was grown in on nutrient agar and incubated at 28 °C for 3–4 days; Saccharomyces cerevisiae Y187 was grown on YPD agar plus 0.003% w/v adenine hemi-sulfate and incubated at 30 °C for 3–4 days. The MIC99 was the lowest concentration of compound, which yielded less than 1% growth relative to no-compound control.
4.4. Kill kinetics
To determine kill kinetics, replicating bacteria, a late log phase culture (OD590 0.6–1.0) of M. tuberculosis was adjusted to an OD590 of 0.1 in 7H9-Tw-OADC; 50 µL was used to inoculate 5 mL 7H9-Tw-OADC containing compounds. For non-replicating conditions, bacteria were incubated at 37 °C for 14 days in PBS + 0.05% w/v Tyloxapol (PBS-Ty) at an OD590 of 0.1 and then compounds were added (final DMSO concentration of 2%). Cultures were incubated standing at 37 °C. Serial dilutions were plated on 7H10-OADC agar to determine CFU/mL.
4.5. Compound synthesis
4.5.1. General methods
1H and 13C NMR spectral data were recorded in CDCl3 or DMSO-d6 on a 300 or 400 MHz Bruker NMR spectrometer. Column chromatography was conducted on silica gel (100–300 mesh). Reactions were monitored using thin-layer chromatography (TLC) on silica gel plates. HPLC analysis was conducted on an Agilent 1100 series LC system (Agilent ChemStation Rev.A.10.02; Phenomenex-Luna-C18, 4.8 mm × 150 mm, 5 μm, 1.0 mL/min, UV 254 nm, room temperature) with MeCN/H2O (0.05% TFA or HCOOH buffer) gradient elution. HPLCMSwas performed on a Gilson 321 HPLC with detection performed by a Gilson 170 DAD and a Finnigan AQA mass spectrometer operating in electrospray ionisation mode using a Phenomenex Gemini C18 150x4.6 mm column. Purity was determined using a Waters Acquity UPLC system equipped with a BEH C18 1.7 μm 2.1 × 100 mm column.
4.5.2. General procedure for the synthesis of N-phenethyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amines (1, 27–50)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 4 (1.0 eq) was taken in NMP in a 100 mL round bottom flask under N2. To it was added amine precursor 6 (1.2 eq). The reaction mixture was heated at 90–120 °C or rt until complete by TLC analysis. The reaction mixture was then added to ice-cooled water and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using EtOAc-hexane or MeOH-DCM as eluent to afford compounds 1, 27–50.
4.5.3. General procedure for the synthesis of N-substituted-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (16, 51–60)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (1.0 eq) was taken in NMP in a 100 mL round bottom flask under N2. To it was added amine precursor (1.2 eq). The reaction mixture was heated at 90–100 °C or rt until complete by TLC analysis. The reaction mixture was then added to ice-cooled water and extracted with EtOAc. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using EtOAc-hexane or MeOH-DCM as eluent to afford compounds 16, 51–60.
4.5.4. N-(4-methoxyphenethyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (1)
7-chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 5 (500 mg, 2.2 mmol) was taken in NMP (5 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (365 mg, 2.4 mmol). The reaction mixture was heated at 100 °C and monitored by TLC analysis (EtOAc, 100%) until completion. The reaction mixture was then poured into ice water (50 g) and extracted with EtOAc (2 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 1 as a white solid (300 mg, 40%). M.P. 163–164 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.34 (bs, 1H), 8.15–8.17 (m, 2H), 7.52–7.53 (m, 3H), 7.23 (d, J = 8.3 Hz, 2H), 6.84 (d, J = 7.2 Hz, 3H), 3.73–3.76 (m, 2H), 3.66 (s, 3H), 2.92–2.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.2, 157.8, 155.5, 154.7, 147.9, 137.6, 130.7, 130.2, 129.9, 128.5, 127.4, 113.7, 84.8, 54.9, 43.2, 33.7. LCMS (ESI) m/z 346.45.
4.5.5. Ethyl 3-(2-methoxyphenyl)-3-oxopropanoate (2c)
Sodium hydride (480 mg, 20.0 mmmol) was taken in dry THF (18 mL) in a 100 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of ethyl 3-(2-methoxyphenyl)-3-oxopropanoate (1.0 g, 6.7 mmol) in THF (2 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (3.2 mL, 26.8 mmol). The reaction mixture was then stirred at rt for 14 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 75 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 10% hexane-EtOAc as eluent to afford 2c as a colourless liquid (1.3 g, 88%). LC-MS (ESI) m/z 223.32 [M+H+]; 97% (purity).
4.5.6. Ethyl 3-oxo-3-(p-tolyl)propanoate (2d)
Sodium hydride (2.68 g, 112 mmmol) was taken in dry DMF (25 mL) in a 250 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 1-(p-tolyl)ethan-1-one (5.0 g, 37.3 mmol) in DMF (5 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (18 mL, 149.2 mmol). The reaction mixture was then stirred at rt for 16 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 10% hexane-EtOAc as eluent to afford 2d as a colourless liquid (6.2 g, 81%). LCMS (ESI) m/z 207.32 [M+H+]; 84.88% (purity).
4.5.7. Ethyl 3-(4-ethylphenyl)-3-oxopropanoate (2e)
Sodium hydride (161 mg, 6.74 mmmol) was taken in dry THF (8 mL) in a 100 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 1-(4-ethylphenyl)ethan-1-one (500 mg, 3.37 mmol) in THF (2 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (1.6 mL, 13.5 mmol). The reaction mixture was then stirred at rt for 12 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with Et2O afford 2e as a brown solid (500 mg, 67%). LCMS(ESI) m/z 221.29 [M+H+]; 50% (purity).
4.5.8. Ethyl 3-(2-fluorophenyl)-3-oxopropanoate (2i)
Sodium hydride (1.74 g, 72.4 mmmol) was taken in dry THF (30 mL) in a 100 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 1-(2-fluorophenyl)ethan-1-one (5.0 g, 36.2 mmol) in THF (5 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (17.5 mL, 144.8 mmol). The reaction mixture was then stirred at rt for 12 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 75 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with Et2O afford 2i as a brown liquid (4.0 g, 52%). MS (ESI) m/z 211.17 [M+H+].
4.5.9. Ethyl 3-oxo-3-(4-(trifluoromethyl)phenyl)propanoate (2j)
Sodium hydride (2.55 g, 106.4 mmmol) was taken in dry THF (40 mL) in a 250 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 1-(4-(trifluoromethyl)phenyl)ethan-1-one (10 g, 53.2 mmol) in THF (10 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (25.8 mL, 212.8 mmol). The reaction mixture was then stirred at rt for 14 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with Et2O afford 2j as a brown solid (10.0 g, 72%). LCMS(ESI) m/z 259.14 [M−H+]; 61% (purity).
4.5.10. Ethyl 3-oxo-3-(pyridin-3-yl)propanoate (2l)
Sodium hydride (2.9 g, 124 mmmol) was taken in dry THF (45 mL) in a 250 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 1-(pyridin-3-yl)ethan-1-one (5.0 g, 41.2 mmol) in THF (5 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (20 mL, 165.0 mmol). The reaction mixture was then stirred at rt for 14 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 10% hexane-EtOAc as eluent to afford 2l as a colourless liquid (4.1 g, 52%). LCMS(ESI) m/z 194.25 [M+H+]; 88% (purity).
4.5.11. Ethyl 3-cyclobutyl-3-oxopropanoate (2r)
Sodium hydride (2.44 g, 101.9 mmmol) was taken in dry THF (55 mL) in a 250 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 1-cyclobutylethan-1-one (5.0 g, 50.9 mmol) in THF (5 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (24.7 mL, 203.6 mmol). The reaction mixture was then stirred at rt for 14 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 200 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 10% hexane-EtOAc as eluent to afford 2r as a colourless semi-solid (2.6 g, 30%). LCMS(ESI) m/z 169.07 [M−H+]; 80% (purity).
4.5.12. Synthesis of 5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4)
Ethyl 3-oxo-3-phenylpropanoate 2 (10.0 g, 52.1 mmol) was taken in AcOH (50 mL) in a 250 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine 3 (0.4 mL, 62.5 mmol). The reaction mixture was heated at 120 °C for 14 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vaccum which affored a white solid (10.5 g, 95%). This was then used in the next step without any further purification. LCMS(ESI) m/z 213.09 [M+H+]; 59.54% (purity).
4.5.13. 5-Phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4a)
Ethyl 3-oxo-3-phenylpropanoate 2 (10.0 g, 52.1 mmol) was taken in AcOH (50 mL) in a 250 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (0.4 mL, 62.5 mmol). The reaction mixture was heated at 120 °C for 14 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded a white solid (10.5 g, 95%). This was then used in the next step without any further purification. LCMS(ESI) m/z 213.09 [M+H+]; 59.54% (purity).
4.5.14. 5-(4-Methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4b)
Ethyl 3-(4-methoxyphenyl)-3-oxopropanoate (1.0 g, 4.5 mmol) was taken in AcOH (5 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (416 mg, 4.9 mmol). The reaction mixture was heated at 120 °C for 12 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4b as a brown solid (510 mg, 47%). This was then used in the next step without any further purification. LCMS(ESI) m/z 243.10 [M+H]+; 80.54% (purity).
4.5.15. 5-(2-Methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4c)
Ethyl 3-(2-methoxyphenyl)-3-oxopropanoate 2c (1.5 g, 6.7 mmol) was taken in AcOH (15 mL) in a 100 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine 2 (681 mg, 8.1 mmol). The reaction mixture was heated at 120 °C for 14 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4c as an off-white solid (1.2 g, crude). This was then used in the next step without any further purification. LCMS(ESI) m/z 243.11 [M+H+]; 95.10% (purity).
4.5.16. 5-(p-Tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4d)
Ethyl 3-oxo-3-(p-tolyl)propanoate 2d (6.0 g, 29.1 mmol) was taken in AcOH (40 mL) in a 250 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (2.9 g, 34.9 mmol). The reaction mixture was heated at 120 °C for 14 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4d as a white solid (1.8 g, 27%). This was then used in the next step without any further purification. LCMS(ESI) m/z 227.22 [M+H+]; 75.24% (purity).
4.5.17. 5-(4-Ethylphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4e)
Ethyl 3-(4-ethylphenyl)-3-oxopropanoate 2e (500 mg, 2.27 mmol) was taken in AcOH (6 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (286 mg, 3.4 mmol). The reaction mixture was heated at 115 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4e as a brown solid (400 mg, 73%). This was then used in the next step without any further purification. LCMS(ESI) m/z 239.20 [M−H+]; 45% (purity).
4.5.18. 5-(4-Chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4f)
Ethyl 3-(4-chlorophenyl)-3-oxopropanoate (1.0 g, 4.4 mmol) was taken in AcOH (5 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (406 mg, 4.8 mmol). The reaction mixture was heated at 120 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4f as a white solid (560 mg, 51%). This was then used in the next step without any further purification. LCMS(ESI) m/z 247.15 [M+H+]; 98.87% (purity).
4.5.19. 5-(4-Fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4h)
Ethyl 3-(4-fluorophenyl)-3-oxopropanoate (2.0 g, 9.5 mmol) was taken in AcOH (10 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine 2 (958 mg, 11.4 mmol). The reaction mixture was heated at 120 °C for 12 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4h as a yellow solid (1.1 g, 50%). This was then used in the next step without any further purification. LCMS(ESI) m/z 231.25 [M+H+]; 79.73% (purity).
4.5.20. 5-(2-Fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4i)
Ethyl 3-(2-fluorophenyl)-3-oxopropanoate 2i (4.0 g, 19.0 mmol) was taken in AcOH (20 mL) in a 100 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (1.92 g, 22.8 mmol). The reaction mixture was heated at 115 °C for 12 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4i as a yellow solid (800 mg, 18%). This was then used in the next step without any further purification. LCMS(ESI) m/z 231.06 [M+H+]; 54% (purity).
4.5.21. 5-(4-(Trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4j)
Ethyl 3-oxo-3-(4-(trifluoromethyl)phenyl)propanoate 2j (10.0 g, 38.5 mmol) was taken in AcOH (50 mL) in a 250 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (4.85 g, 57.7 mmol). The reaction mixture was heated at 115 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4j as a yellow solid (6.0 g, 56%). This was then used in the next step without any further purification. LCMS(ESI) m/z 279.43 [M−H+]; 78% (purity).
4.5.22. 5-(Pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4k)
Ethyl 3-oxo-3-(pyridin-2-yl)propanoate 2k (8.0 g, 41.4 mmol) was taken in AcOH (50 mL) in a 250 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (4.2 g, 49.7 mmol). The reaction mixture was heated at 120 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4k as a deep brown solid (4.5 g, 51%). This was then used in the next step without any further purification. MS (ESI) m/z 212.2 [M−H+].
4.5.23. 5-(Pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4l)
Ethyl 3-oxo-3-(pyridin-3-yl)propanoate 2l (4.0 g, 20.7 mmol) was taken in AcOH (40 mL) in a 250 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (2.08 g, 24.8 mmol). The reaction mixture was heated at 120 °C for 14 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4l as a white solid (2.1 g, 48%). This was then used in the next step without any further purification. LCMS(ESI) m/z 214.13 [M+H+]; 62% (purity).
4.5.24. 5-(Pyridin-4-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4m)
Ethyl 3-oxo-3-(pyridin-4-yl)propanoate (2.0 g, 10.4 mmol) was taken in AcOH (10 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (1.04 g, 12.4 mmol). The reaction mixture was heated at 110 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4m as a yellow solid (1.2 g, 54%). This was then used in the next step without any further purification. LCMS(ESI) m/z 214.19 [M+H+]; 56% (purity).
4.5.25. 5-Benzyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4n)
Ethyl 3-oxo-4-phenylbutanoate (1.5 g, 7.2 mmol) was taken in AcOH (8 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (733 mg, 8.7 mmol). The reaction mixture was heated at 115 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4n as a white solid (910 mg, 55%). This was then used in the next step without any further purification.
4.5.26. 5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4o)
Ethyl 3-oxobutanoate (1.0 g, 7.7 mmol) was taken in AcOH (5 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (646 mg, 7.7 mmol). The reaction mixture was heated at 120 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4o as a white solid (610 mg, 53%). This was then used in the next step without any further purification. LCMS (ESI) m/z 151.06 [M+H+]; 78% (purity).
4.5.27. 5-Ethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4p)
Ethyl 3-oxopentanoate (200 mg, 1.4 mmol) was taken in AcOH (1.0 mL) in a 25 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (139 mg, 1.6 mmol). The reaction mixture was heated at 120 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4p as an off-white solid (110 mg, 48%). This was then used in the next step without any further purification. LCMS(ESI) m/z 165.43 [M+H+]; 90% (purity).
4.5.28. 5-Cyclobutyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4r)
Ethyl 3-cyclobutyl-3-oxopropanoate 2r (5.0 g, 29.4 mmol) was taken in AcOH (25 mL) in a 100 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (2.9 g, 35.2 mmol). The reaction mixture was heated at 110 °C for 14 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4r as a yellow semi-solid (3.8 g, crude). This was then used in the next step without any further purification. LCMS(ESI) m/z 191.30 [M+H+]; 84.60% (purity).
4.5.29. 5-Cyclopentyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4s)
Ethyl 3-cyclopentyl-3-oxopropanoate (2.0 g, 10.8 mmol) was taken in AcOH (10 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (1.08 g, 13.0 mmol). The reaction mixture was heated at 110 °C for 12 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4s as a brown solid (1.1 g, 50%). This was then used in the next step without any further purification. LCMS(ESI) m/z 205.05 [M+H+]; 66.67% (purity).
4.5.30. 5-Cyclohexyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol (4t)
Ethyl 3-cyclohexyl-3-oxopropanoate (800 mg, 4.04 mmol) was taken in AcOH (4 mL) in a 50 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine (407 mg, 4.84 mmol). The reaction mixture was heated at 115 °C for 16 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 4t as an off-white solid (410 mg, 46%). This was then used in the next step without any further purification. LCMS(ESI) m/z 217.30 [M−H+]; 52.65% (purity).
4.5.31. 7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine (5a)
To a solution of 5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4a (10.0 g, 47.2 mmol) was added POCl3 (44 mL, 472.0 mmol) at 0 °C. The reaction mixture was then heated at 120 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 15% hexane-EtOAc as eluent to afford 5a as a pale yellow solid (4.2 g, 39%). 1H NMR (400 MHz, DMSO-d6): δ 8.77 (s, 1H), 8.38 (s, 1H), 8.32–8.34 (m, 2H), 7.60–7.61 (m, 3H). LCMS(ESI) m/z 231.03 [M+H+]; 92.15% (purity).
4.5.32. 7-Chloro-5-(4-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5b)
To a solution of 5-(4-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4b (500 mg, 2.1 mmol) was added POCl3 (1.6 mL, 16.8 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 5% hexane-EtOAc as eluent to afford 5b as a brown solid (210 mg, 39%). LCMS(ESI) m/z 261.15 [M+H+]; 85.81% (purity).
4.5.33. 7-Chloro-5-(2-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5c)
To a solution of 5-(2-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4c (500 mg, 2.1 mmol) was added POCl3 (5 mL, 52.5 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure to afford 5c. This was then used in the next step without any further purification. LCMS(ESI) m/z 261.0 [M+H+]; 53.5% (purity).
4.5.34. 7-Chloro-5-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5d)
To a solution of 5-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4d (1.5 g, 6.6 mmol) was added POCl3 (20 mL, 211 mmol) at 0 °C. The reaction mixture was then heated at 80 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 20% hexane-EtOAc as eluent to afford 5d as a yellow solid (800 mg, 49%). LCMS(ESI) m/z 245.08 [M+H+]; 76.82% (purity).
4.5.35. 7-Chloro-5-(4-ethylphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5e)
To a solution of 5-(4-ethylphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4e (400 mg, 1.7 mmol) was added POCl3 (5 mL, 52.7 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (30%, EtOAc-hexane). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 25 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 12% EtOAc-hexane as eluent to afford 5e as a pale yellow solid (250 mg, 58%). LCMS(ESI) m/z 259.44 [M+H+]; 91.33% (purity).
4.5.36. 7-Chloro-5-(4-chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5f)
To a solution of 5-(4-chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4f (550 mg, 2.2 mmol) was added POCl3 (2.5 mL, 26.4 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (100% EtOAc). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with diethyl ether to afford 5f as an off-white solid (310 mg, 52%). 1H NMR (400 MHz, DMSO-d6): δ 8.78 (s, 1H), 8.42 (s, 1H), 8.35 (d, J = 8.4 Hz, 2H), 7.67 (d, J = 8.0 Hz, 2H).
4.5.37. 7-Chloro-5-(4-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5h)
To a solution of 5-(4-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4h (1.1 g, 4.8 mmol) was added POCl3 (6.0 mL, 62.4 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (100% EtOAc). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with diethyl ether to afford 5h as a yellow solid (635 mg, 53%). LCMS(ESI) m/z 249.07 [M+H+]; 75.08% (purity).
4.5.38. 7-Chloro-5-(2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5i)
To a solution of 5-(2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4i (800 mg, 3.5 mmol) was added POCl3 (10 mL, 112 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (30%, EtOAc-hexane). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (30 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 14% EtOAc-hexane as eluent to afford 5i as a pale yellow solid (100 mg, 11%). LCMS(ESI) m/z 248.90 [M+H+]; 50% (purity).
4.5.39. 7-Chloro-5-(4-(trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (5j)
To a solution of 5-(4-(trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4j (3.0 g, 10.7 mmol) was added POCl3 (20 mL, 214 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (50%, EtOAc-hexane). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 25 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 13% EtOAc-hexane as eluent to afford 5j as a pale yellow solid (1.75 g, 55%). LCMS(ESI) m/z 299.08 [M+H+]; 93.67% (purity).
4.5.40. 7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine (5k)
To a solution of 5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4k (4.0 g, 18.8 mmol) was added POCl3 (18 mL, 188 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 20% hexane-EtOAc as eluent to afford 5k as a yellow solid (1.5 g, 35%). LCMS(ESI) m/z 232.04 [M+H+]; 98.9% (purity).
4.5.41. 7-Chloro-5-(pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidine (5l)
To a solution of 5-(pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4l (900 mg, 4.2 mmol) was added POCl3 (10 mL, 105 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 20% hexane-EtOAc as eluent to afford 5l as a yellow solid (480 mg, 49%). LCMS(ESI) m/z 232.19 [M+H+]; 74.46% (purity).
4.5.42. 7-Chloro-5-(pyridin-4-yl)-[1,2,4]triazolo[1,5-a]pyrimidine (5m)
To a solution of 5-(pyridin-4-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4m (500 mg, 2.3 mmol) was added POCl3 (3.0 mL, 34.0 mmol) at 0 °C. The reaction mixture was then heated at 85 °C and monitored by TLC analysis (100% EtOAc). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with diethyl ether to afford 5m as a yellow solid (260 mg, 48%). LCMS(ESI) m/z 232.18 [M+H+]; 72% (purity).
4.5.43. 5-Benzyl-7-chloro-[1,2,4]triazolo[1,5-a]pyrimidine (5n)
To a solution of 5-benzyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4n (900 mg, 3.9 mmol) was added POCl3 (4.5 mL, 47.8 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (100% EtOAc). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with diethyl ether to afford 5n as an off-white solid (610 mg, 62%). LCMS(ESI) m/z 245.22 [M+H+]; 94.26% (purity).
4.5.44. 7-Chloro-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidine (5o)
To a solution of 5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4o (600 mg, 4.0 mmol) was added POCl3 (2.0 mL, 22.0 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (50% EtOAc-hexane). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 20% EtOAc-hexane as eluent to afford 5o as a white solid (340 mg, 50%). LCMS(ESI) m/z 169.03 [M+H+]; 92.15% (purity).
4.5.45. 7-Chloro-5-ethyl-[1,2,4]triazolo[1,5-a]pyrimidine (5p)
To a solution of 5-ethyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4p (100 mg, 0.6 mmol) was added POCl3 (1.0 mL, 10.8 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (50% EtOAc-hexane). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 15 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 12% EtOAc-hexane as eluent to afford 5p as a white solid (85 mg, 76%).1H NMR (400 MHz, CDCl3): δ 8.51 (s, 1H), 7.11 (s, 1H), 2.98 (q, J = 7.6 Hz, 2H), 1.41 (t, J = 7.6 Hz, 3H).
4.5.46. 7-Chloro-5-cyclobutyl-[1,2,4]triazolo[1,5-a]pyrimidine (5r)
To a solution of 5-cyclobutyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4r (3.8 g, 20 mmol) was added POCl3 (30 mL, 320 mmol) at 0 °C. The reaction mixture was then heated at 80 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 25% hexane-EtOAc as eluent to afford 5r as a yellow semi-solid (1.8 g, 43%). LCMS(ESI) m/z 209.06 [M+H+]; 86.29% (purity).
4.5.47. 7-Chloro-5-cyclopentyl-[1,2,4]triazolo[1,5-a]pyrimidine (5s)
To a solution of 5-cyclopentyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4s (1.0 g, 4.9 mmol) was added POCl3 (6.0 mL, 63.7 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (100% EtOAc). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with diethyl ether to afford 5s as an off-white solid (270 mg, 25%). LCMS(ESI) m/z 223.09 [M+H+]; 69.45% (purity).
4.5.48. 7-Chloro-5-cyclohexyl-[1,2,4]triazolo[1,5-a]pyrimidine (5t)
To a solution of 5-cyclohexyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 4t (400 mg, 1.8 mmol) was added POCl3 (2.0 mL, 21.6 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (100% EtOAc). The reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was triturated with diethyl ether to afford 5t as a yellow solid (110 mg, 25%). LCMS(ESI) m/z 237.0 [M+H+]; 61.42% (purity).
4.5.49. N-(4-Methoxyphenethyl)-5-(4-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (7)
7-Chloro-5-(4-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine 5b (200 mg, 0.77 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (127 mg, 0.84 mmol). The reaction mixture was heated at 100 °C and monitored by TLC analysis (EtOAc, 100%) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 2% MeOH-DCM as eluent to afford 7 as a white solid (75 mg, 26%). M.P. 130–131 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.52 (s, 1H), 8.37–8.40 (m, 1H), 8.15 (d, J = 8.8 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H), 7.07 (d, J = 8.9 Hz, 2H), 6.82–6.85 (m, 3H), 3.85 (s, 3H), 3.73–3.78 (m, 2H), 3.67 (s, 3H), 2.95 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 161.3, 160.2, 157.8, 154.1, 152.8, 147.9, 130.6, 129.9, 129.3, 129.1, 113.9, 113.7, 84.9, 55.3, 54.9, 43.3, 33.7. LCMS (ESI) m/z 376.37.
4.5.50. N-(4-Methoxyphenethyl)-5-(2-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (8)
7-Chloro-5-(2-methoxyphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine 5c (100 mg, 0.38 mmol) was taken in NMP (2 mL) in a 25 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (69 mg, 0.46 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 25 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 8 as an off-white solid (54 mg, 37%). M.P. 148–149 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.45 (s, 1H), 8.33 (bs, 1H), 7.78 (d, J = 7.4 Hz, 1H), 7.45–7.49 (m, 1H), 7.20 (d, J = 8.2 Hz, 3H), 7.09 (t, J = 7.5 Hz, 1H), 6.84 (d, J = 8.9 Hz, 3H), 3.87 (s, 3H), 3.69 (s, 3H), 3.61–3.62 (m, 2H), 2.91–2.94 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.6, 157.8, 157.0, 155.3, 154.5, 146.9, 131.1, 130.6, 130.5, 129.7, 127.5, 120.5, 113.8, 112.1, 89.3, 55.7, 54.9, 43.3, 33.4. LCMS (ESI) m/z 376.24.
4.5.51. N-(4-Methoxyphenethyl)-5-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (9)
7-Chloro-5-(p-tolyl)-[1,2,4]triazolo[1,5-a]pyrimidine 5d (250 mg, 1.02 mmol) was taken in NMP (5 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (185 mg, 1.22 mmol) and the reaction mixture was stirred at rt for 20 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 25 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 9 as an off-white solid (100 mg, 27%). M.P. 155–156 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.44 (s, 1H), 8.28 (m, 1H), 8.07 (d, J = 7.8 Hz, 2H), 7.33 (d, J = 7.8 Hz, 2H), 7.23 (d, J = 8.1 Hz, 2H), 6.80–6.85 (m, 3H), 3.72–3.75 (m, 2H), 3.67 (s, 3H), 2.92–2.95 (m, 2H), 2.39 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.2, 157.8, 155.5, 154.6, 147.8, 140.0, 134.8, 130.7, 129.9, 129.2, 127.3, 113.7, 84.4, 54.9, 43.2, 33.7, 20.9. LCMS (ESI) m/z 360.40.
4.5.52. 5-(4-Ethylphenyl)-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (10)
7-Chloro-5-(4-ethylphenyl)-[1,2,4]triazolo[1,5-a]pyrimidine 5e (100 mg, 0.38 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (70 mg, 0.46 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 25 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 26% EtOAc-hexane as eluent to afford 10 as a pale yellow solid (78 mg, 54%). M.P. 151–12 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.44 (s, 1H), 8.29 (t, J = 5.6 Hz, 1H), 8.08 (d, J = 8.2 Hz, 2H), 7.36 (d, J = 8.2 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 6.79 (s, 1H), 3.72–3.77 (m, 2H), 3.66 (s, 3H), 2.91–2.95 (m, 2H), 2.68 (q, J = 7.6 Hz, 2H), 1.23 (t, J = 7.6 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.2, 157.8, 155.5, 154.6, 147.8, 146.2, 135.1, 130.7, 129.8, 127.9, 127.4, 113.7, 84.5, 54.9, 43.2, 33.7, 27.9, 15.4. LCMS (ESI) m/z 373.99.
4.5.53. 5-(4-Chlorophenyl)-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (11)
7-Chloro-5-(4-chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine 60 (200 mg, 0.77 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (127 mg, 0.84 mmol). The reaction mixture was heated at 100 °C and monitored by TLC analysis (EtOAc, 100%) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 45% EtOAc-hexane as eluent to afford 11 as a white solid (110 mg, 30%). M.P. 178–180 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.47 (s, 1H), 8.40 (bs, 1H), 8.20 (d, J = 8.5 Hz, 2H), 7.59 (d, J = 8.5 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 6.82–6.86 (m, 3H), 3.76–3.78 (m, 2H), 3.66 (s, 3H), 2.92–2.95 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 158.8, 157.8, 155.4, 154.8, 147.9, 136.4, 135.1, 130.6, 129.9, 129.2, 128.6, 113.7, 84.8, 54.9, 43.3, 33.7. LCMS (ESI) m/z 380.34.
4.5.54. 5-(4-Fluorophenyl)-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (13)
7-Chloro-5-(4-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine 5h (125 mg, 0.5 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (91 mg, 0.6 mmol). The reaction mixture was stirred at rt and monitored by TLC analysis (70% EtOAc-hexane) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 25% EtOAc-hexane as eluent to afford 13 as a white solid (40 mg, 22%). M.P. 153–155 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.45 (s, 1H), 8.35–8.38 (m, 1H), 8.22–8.25 (m, 2H), 7.35 (t, J = 8.8 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 6.8 Hz, 3H), 3.73–3.78 (m, 2H), 3.66 (s, 3H), 2.92–2.95 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 163.5 (d, J = 246 Hz), 159.8, 159.1, 157.8, 155.4, 154.7, 147.9, 134.1 (d, J = 3 Hz), 130.7, 129.8 (d, J = 8.5 Hz), 115.4 (d, J = 21 Hz), 113.7, 84.7, 54.9, 43.2, 33.8. LCMS (ESI) m/z 364.15.
4.5.55. 5-(2-Chlorophenyl)-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (12)
7-Chloro-5-(2-chlorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine (106 mg, 0.4 mmol) was taken in NMP (2 mL) in a 25 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (72 mg, 0.48 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 25 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 27% EtOAc-hexane as eluent to afford 12 as a white solid (68 mg, 45%). 1H NMR (400 MHz, DMSO-d6): δ 8.52 (s, 2H), 7.60–7.51 (m, 2H), 7.50–7.46 (m, 2H), 7.18–7.16 (m, 2H), 6.82–6.80 (m, 2H), 6.55 (s, 1H), 3.68–3.64 (m, 5H), 2.91–2.88 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.7, 157.8, 155.1, 154.7, 147.3, 138.4, 131.1, 131.1, 130.5, 130.5, 129.9, 129.8, 127.2, 113.8, 89.3, 54.9, 43.3, 33.7. LCMS (ESI) m/z 380.34.
4.5.56. 5-(2-Fluorophenyl)-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (14)
7-Chloro-5-(2-fluorophenyl)-[1,2,4]triazolo[1,5-a]pyrimidine 5i (100 mg, 0.4 mmol) was taken in NMP (2 mL) in a 25 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (72 mg, 0.48 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 25 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 27% EtOAc-hexane as eluent to afford 14 as a white solid (58 mg, 40%). M.P. 153–155 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.50 (s, 2H), 7.93 (t, J = 6.7 Hz, 1H), 7.53–7.58 (m, 1H), 7.34–7.40 (m, 2H), 7.19 (d, J = 8.3 Hz, 2H), 6.82 (d, J = 8.4 Hz, 2H), 6.68 (s, 1H), 3.65–3.67 (m, 5H), 2.90–2.93 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.8 (d, J = 248 Hz), 157.8, 156.9, 155.3, 154.8, 147.5, 131.8 (d, J = 8.7 Hz), 131.0, 130.4, 129.8, 126.4 (d, J = 11.2 Hz), 124.7 (d, J = 3.0 Hz), 116.4 (d, J = 22 Hz), 113.8, 88.5 (d, J = 8.0 Hz), 54.9, 43.4, 33.5. LCMS (ESI) m/z 364.33.
4.5.57. N-(4-Methoxyphenethyl)-5-(4-(trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (15)
7-Chloro-5-(4-(trifluoromethyl)phenyl)-[1,2,4]triazolo[1,5-a]pyrimidine 5j (100 mg, 0.33 mmol) was taken in NMP (2 mL) in a 25 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (62 mg, 0.4 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 25 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 27% EtOAc-hexane as eluent to afford 15 as a pale yellow solid (75 mg, 54%). M.P. 191–193 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.48–8.50 (m, 2H), 8.37 (d, J = 8.0 Hz, 2H), 7.89 (d, J = 8.3 Hz, 2H), 7.23 (d, J = 8.4 Hz, 2H), 6.92 (s, 1H), 6.83 (d, J = 8.4 Hz, 2H), 3.75–3.80 (m, 2H), 3.65 (s, 3H), 2.92–2.96 (m, 2H). LCMS (ESI) m/z 414.39.
4.5.58. N-(4-Methoxyphenethyl)-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (16)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (200 mg, 0.86 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (156 mg, 1.0 mmol) and the reaction mixture was stirred at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 45% EtOAc-hexane as eluent to afford 16 as a white solid (80 mg, 27%). M.P. 172–173 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.0 Hz, 1H), 8.50–8.51 (m, 2H), 8.45 (d, J = 8.0 Hz, 1H), 8.01 (t, J = 7.8 Hz, 1H), 7.53–7.56 (m, 1H), 7.34 (s, 1H), 7.21 (d, J = 8.5 Hz, 2H), 6.84 (d, J = 8.5 Hz, 2H), 3.69 (m, 2H), 3.67 (s, 3H), 2.93–2.97 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.0, 157.8, 155.5, 154.9, 153.8, 149.2, 148.0, 137.4, 130.5, 129.7, 125.3, 121.4, 113.9, 84.5, 54.9, 43.5, 33.4. LCMS (ESI) m/z 347.39.
4.5.59. N-(4-Methoxyphenethyl)-5-(pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (17)
7-Chloro-5-(pyridin-3-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5l (200 mg, 0.86 mmol) was taken in NMP (5 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (156 mg, 1.04 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 25 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 25% EtOAc-hexane as eluent to afford 17 as an off-white solid (60 mg, 17%). M.P. 152–153 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.33 (s, 1H), 8.69–8.70 (m, 1H), 8.47–8.52 (m, 3H), 7.55–7.58 (m, 1H), 7.23 (d, J = 8.2 Hz, 2H), 6.94 (s, 1H), 6.82 (d, J = 8.2 Hz, 2H), 3.77 (m, 2H), 3.65 (s, 3H), 2.92–2.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 157.9, 157.8, 155.4, 154.8, 150.9, 148.5, 148.1, 134.8, 133.1, 130.6, 129.9, 123.6, 113.7, 85.1, 54.9, 43.2, 33.8. LCMS (ESI) m/z 347.37.
4.5.60. N-(4-Methoxyphenethyl)-5-(pyridin-4-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (18)
7-Chloro-5-(pyridin-4-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5m (180 mg, 0.77 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (141 mg, 0.92 mmol). The reaction mixture was stirred at rt and monitored by TLC analysis (100% EtOAc) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 3% MeOH-DCM as eluent to afford 18 as a white solid (70 mg, 26%). M.P. 217–219 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 6.0 Hz, 2H), 8.52–8.56 (m, 2H), 8.11 (d, J = 6.0 Hz, 2H), 7.23 (d, J = 8.5 Hz, 2H), 6.96 (s, 1H), 6.82 (d, J = 8.5 Hz, 2H), 3.75–3.80 (m, 2H), 3.64 (s, 3H), 2.92–2.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 157.8, 157.6, 155.4, 155.0, 150.2, 148.2, 144.6, 130.6, 129.9, 121.4, 113.7, 85.5, 54.9, 43.3, 33.8. LCMS (ESI) m/z 347.44.
4.5.61. 5-Benzyl-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (19)
5-Benzyl-7-chloro-[1,2,4]triazolo[1,5-a]pyrimidine 5n (250 mg, 1.0 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (166 mg, 1.1 mmol). The reaction mixture was heated at 100 °C and monitored by TLC analysis (EtOAc, 100%) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 19 as an off-white solid (80 mg, 21%). M.P. 119–121 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.38 (s, 1H), 8.27–8.31 (m, 1H), 7.28–7.34 (m, 4H), 7.19–7.23 (m, 1H), 7.15 (d, J = 8.1 Hz, 2H), 6.83 (d, J = 8.1 Hz, 2H), 6.36 (s, 1H), 3.99 (s, 2H), 3.70 (s, 3H), 3.52–3.57 (m, 2H), 2.83–2.86 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 165.9, 157.8, 155.3, 154.2, 147.4, 138.9, 130.4, 129.8, 129.0, 128.3, 126.3, 113.8, 87.7, 54.9, 44.0, 43.3, 33.5. LCMS (ESI) m/z 360.48.
4.5.62. N-(4-Methoxyphenethyl)-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (20)
7-Chloro-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidine 5o (280 mg, 1.7 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (301 mg, 2.0 mmol). The reaction mixture was heated at 100 °C and monitored by TLC analysis (EtOAc, 100%) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 25 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 45% EtOAc-hexane as eluent to afford 20 as a white solid (130 mg, 27%). M.P. 129–130 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.35 (s, 1H), 8.16 (bs, 1H), 7.20 (d, J = 8.2 Hz, 2H), 6.85 (d, J = 7.9 Hz, 2H), 6.31 (s, 1H), 3.71 (s, 3H), 3.53–3.56 (m, 2H), 2.86–2.90 (m, 2H), 2.41 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 163.7, 157.8, 155.2, 154.0, 147.1, 130.5, 129.8, 113.7, 87.7, 54.9, 43.2, 33.5, 24.7. LCMS (ESI) m/z 284.44.
4.5.63. Synthesis of 5-ethyl-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (21)
7-Chloro-5-ethyl-[1,2,4]triazolo[1,5-a]pyrimidine 5p (60 mg, 0.32 mmol) was taken in NMP (1 mL) in a 25 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (59 mg, 0.39 mmol). The reaction mixture was stirred at rt and monitored by TLC analysis (70% EtOAc-hexane) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 25 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 45% EtOAc-hexane as eluent to afford 21 as a white solid (34 mg, 35%). M.P. 70–72 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.36 (s, 1H), 8.16 (bs, 1H), 7.19 (d, J = 7.7 Hz, 2H), 6.83 (d, J = 7.7 Hz, 2H), 6.24 (s, 1H), 3.69 (s, 3H), 3.56–3.58 (m, 2H), 2.86–2.88 (m, 2H), 2.63–2.67 (m, 2H), 1.21 (t, J = 7.5 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 168.4, 157.8, 155.3, 154.1, 147.3, 130.6, 129.8, 113.7, 86.8, 54.9, 43.2, 33.6, 31.1, 13.1. LCMS (ESI) m/z 298.15.
4.5.64. 5-Cyclobutyl-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (23)
7-Chloro-5-cyclobutyl-[1,2,4]triazolo[1,5-a]pyrimidine 5r (200 mg, 0.96 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (174 mg, 1.1 mmol) and the reaction mixture was stirred at rt for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 50% EtOAc-hexane as eluent to afford 23 as an off-white solid (22 mg, 7%). M.P. 99–100 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.37 (s, 1H), 8.16 (bs, 1H), 7.18 (d, J = 8.1 Hz, 2H), 6.83 (d, J = 8.1 Hz, 2H), 6.12 (s, 1H), 3.69 (s, 3H), 3.54–3.59 (m, 3H), 2.85–2.89 (m, 2H), 2.20–2.32 (m, 4H), 1.94–2.01 (m, 1H), 1.82–1.83 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 169.2, 157.8, 155.4, 154.1, 147.3, 130.6, 129.8, 113.7, 85.7, 54.9, 43.2, 41.6, 33.7, 27.3, 17.5. LCMS (ESI) m/z 324.36.
4.5.65. 5-Cyclopentyl-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (24)
7-Chloro-5-cyclopentyl-[1,2,4]triazolo[1,5-a]pyrimidine 5s (260 mg, 1.2 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine 5a (212 mg, 1.4 mmol). The reaction mixture was stirred at rt and monitored by TLC analysis (50% EtOAc-hexane) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 28% EtOAc-hexane as eluent to afford 24 as a white solid (90 mg, 23%). M.P. 99–100 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.36 (s, 1H), 8.13 (t, J = 6.0 Hz, 1H), 7.19 (d, J = 8.5 Hz, 2H), 6.82 (d, J = 8.5 Hz, 2H), 6.18 (s, 1H), 3.69 (s, 3H), 3.56–3.61 (m, 2H), 3.06–3.09 (m, 1H), 2.85–2.89 (m, 2H), 1.91–1.95 (m, 2H), 1.75–1.76 (m, 4H), 1.61–1.62 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 170.7, 157.8, 155.4, 154.1, 147.2, 130.6, 129.8, 113.7, 86.6, 54.9, 47.4, 43.3, 33.7, 32.5, 25.5. LCMS (ESI) m/z 338.36.
4.5.66. 5-Cyclohexyl-N-(4-methoxyphenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (25)
7-Chloro-5-cyclohexyl-[1,2,4]triazolo[1,5-a]pyrimidine 5t (100 mg, 0.42 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (76 mg, 0.51 mmol). The reaction mixture was stirred at rt and monitored by TLC analysis (100% EtOAc) until completion. The reaction mixture was then poured into ice water (30 g) and extracted with EtOAc (3 × 30 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 25 as a white solid (40 mg, 27%). M.P. 193–194 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.36 (s, 1H), 8.13 (t, J = 6.0 Hz, 1H), 7.19 (d, J = 8.4 Hz, 2H), 6.83 (d, J = 8.5 Hz, 2H), 6.14 (s, 1H), 3.69 (s, 3H), 3.57–3.62 (m, 2H), 2.85–2.89 (m, 2H), 2.58–2.59 (m, 1H), 1.77–1.80 (m, 4H), 1.69–1.72 (m, 1H), 1.45–1.54 (m, 2H), 1.18–1.38 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 171.2, 157.8, 155.3, 154.1, 147.4, 130.7, 129.8, 113.7, 86.0, 54.9, 46.1, 43.3, 33.8, 31.7, 25.8, 25.5. LCMS (ESI) m/z 352.48.
4.5.67. Ethyl 3-oxo-3-(pyridin-2-yl)propanoate (26)
Sodium hydride (2.9 g, 124 mmmol) was taken in dry THF (50 mL) in a 100 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 1-(pyridin-2-yl)ethan-1-one (5.0 g, 41.3 mmol) in THF (5 mL). The reaction mixture was stirred at rt for 30 min followed by the addition of diethyl carbonate (20 mL, 165 mmol). The reaction mixture was then stirred at rt for 12 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further triturated with diethyl ether which afforded 26 as a pale yellow semi-solid (4.0 g, 50%). LCMS(ESI) m/z 194.06 [M+H+]; 40% (purity).
4.5.68. 5-Phenyl-[1,2,4]triazolo[1,5-a]pyrimidine (27)
3-(Dimethylamino)-1-phenylpropan-1-one (300 mg, 1.7 mmol) was taken in AcOH (1 mL) in a 25 mL round bottom flask under N2. To it was added 1H-1,2,4-triazol-5-amine 2 (171 mg, 2.0 mmol). The reaction mixture was heated at 110 °C for 10 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 15% EtOAc-hexane as eluent to afford 27 as a white solid (35 mg, 10%). M.P. 179–181 °C. 1H NMR (400 MHz, DMSO-d6): δ 9.47 (d, J = 7.2 Hz, 1H), 8.68 (s, 1H), 8.29–8.32 (m, 2H), 7.99 (d, J = 7.2 Hz, 1H), 7.57–7.61 (m, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.9, 156.4, 154.7, 137.6, 135.8, 131.5, 129.1, 127.7, 107.9. LCMS (ESI) m/z 197.28.
4.5.69. 5-Phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (28)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (500 mg, 2.2 mmol) was taken sealed tube under N2. To it was added methanolic NH3 (20 mL) and the reaction mixture was heated at 120 °C for 16 h. The reaction mixture concentrated in vacuo and the crude was purified by flash chromatography on silica gel (100–200 mesh) using 5% MeOH-DCM as eluent to afford 28 as a yellow solid (410 mg, 63%). M.P. 302–304 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.45 (s, 1H), 8.17 (bs, 2H), 8.05–8.07 (m, 2H), 7.49–7.54 (m, 3H), 6.78 (s, 1H); 13C NMR (100 MHz, DMSO-d6): δ 159.7, 155.9, 155.0, 149.3, 137.5, 130.2, 128.8, 126.9, 87.0. LCMS (ESI) m/z 212.14.
4.5.70. N-Methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (29)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) in NMP (3 mL) was taken sealed tube under N2. To it was added MeNH2 (2.0 M in THF) (0.6 mL, 1.3 mmol) and the reaction mixture was heated at 100 °C for 6 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 29 as a white solid (84 mg, 34%). M.P. 200–202 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.36–8.37 (m, 1H), 8.23 (d, J = 4.4 Hz, 2H), 7.53–7.54 (m, 3H), 6.87 (s, 1H), 3.10 (d, J = 4.4 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.5, 154.7, 148.6, 137.6, 130.3, 128.6, 127.4, 84.6, 28.6. LCMS (ESI) m/z 224.28.
4.5.71. N-Ethyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (30)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) in NMP (5 mL) was taken sealed tube under N2. To it was added EtNH2 (59 mg, 1.3 mmol) and the reaction mixture was heated at 100 °C for 6 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 30 as a white solid (87 mg, 34%). M.P. 170–172 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.38 (bs, 1H), 8.22–8.23 (m, 2H), 7.53 (m, 3H), 6.94 (s, 1H), 3.56–3.60 (m, 2H), 1.27 (t, J = 6.9 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.6, 154.7, 147.8, 137.6, 130.2, 128.6, 127.4, 84.4, 36.5, 14.1. LCMS (ESI) m/z 240.40.
4.5.72. 5-Phenyl-N-propyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (31)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) in NMP (3 mL) was taken sealed tube under N2. To it was added 1-propylamine (77 mg, 1.3 mmol) and the reaction mixture was heated at 90 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 32% EtOAc-hexane as eluent to afford 31 as a white solid (100 mg, 36%). M.P. 139–140 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.40 (bs, 1H), 8.21–8.22 (m, 2H), 7.52 (m, 3H), 6.94 (s, 1H), 3.48–3.53 (m, 2H), 1.67–1.72 (m, 2H), 0.95 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.6, 154.6, 148.0, 137.7, 130.2, 128.6, 127.4, 84.5, 43.2, 21.8, 11.2. LCMS (ESI) m/z found 252.30.
4.5.73. N-Butyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (32)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (120 mg, 0.5 mmol) in NMP (2 mL) was taken sealed tube under N2. To it was added n-butylamine (45 mg, 0.6 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 27% EtOAc-hexane as eluent to afford 32 as a white solid (40 mg, 28%). M.P. 125–126 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.38 (t, J = 6.0 Hz, 1H), 8.21–8.23 (m, 2H), 7.52–7.54 (m, 3H), 6.93 (s, 1H), 3.51–3.56 (m, 2H), 1.62–1.70 (m, 2H), 1.35–1.44 (m, 2H), 0.93 (t, J = 7.3 Hz, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.6, 154.7, 147.9, 137.6, 130.2, 128.6, 127.4, 84.5, 41.3, 30.5, 19.4, 13.7. LCMS (ESI) m/z 268.33.
4.5.74. N-(2-Methoxyethyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (33)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) in NMP (3 mL) was taken sealed tube under N2. To it was added 2-methoxyethan-1-amine (99 mg, 1.3 mmol) and the reaction mixture was heated at 90 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 45% EtOAc-hexane as eluent to afford 33 as a white solid (95 mg, 32%). M.P. 116–118 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.47 (s, 1H), 8.27–8.28 (m, 1H), 8.21–8.23 (m, 2H), 7.53–7.54 (m, 3H), 7.02 (s, 1H), 3.71–3.74 (m, 2H), 3.61–3.63 (m, 2H), 3.29 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.5, 154.8, 148.2, 137.6, 130.3, 128.6, 127.4, 84.9, 70.2, 58.1, 41.4. LCMS (ESI) m/z 268.27.
4.5.75. N-Cyclopropyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (34)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (120 mg, 0.5 mmol) in NMP (2 mL) was taken sealed tube under N2. To it was added cyclopropanamine (35 mg, 0.6 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 25% EtOAc-hexane as eluent to afford 34 as a white solid (95 mg, 72%). M.P. 193–195 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.77 (s, 1H), 8.47 (s, 1H), 8.17–8.20 (m, 2H), 7.54–7.58 (m, 3H), 7.03 (s, 1H), 2.84–2.89 (m, 1H), 0.88–0.95 (m, 2H), 0.76–0.80 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.2, 155.6, 154.8, 147.3, 137.6, 130.4, 128.7, 127.3, 85.8, 23.8, 6.6. LCMS (ESI) m/z 252.19.
4.5.76. N-Cyclobutyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (35)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (150 mg, 0.65 mmol) in NMP (5 mL) was taken sealed tube under N2. To it was added cyclobutanamine (55.4 mg, 0.78 mmol) and the reaction mixture was stirred at rt for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 25% EtOAc-hexane as eluent to afford 35 as an off-white solid (50 mg, 29%). M.P. 193–194 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.62 (d, J = 7.2 Hz, 1H), 8.48 (s, 1H), 8.21–8.23 (m, 2H), 7.53–7.54 (m, 3H), 6.87 (s, 1H), 4.43–4.53 (m, 1H), 2.38–2.44 (m, 2H), 2.23–2.32 (m, 2H), 1.71–1.78 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.7, 154.6, 146.8, 137.5, 130.3, 128.6, 127.4, 85.0, 46.6, 29.4, 14.6. LCMS (ESI) m/z 266.24.
4.5.77. N-(2-Cyclohexylethyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (36)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) in NMP (3 mL) was taken sealed tube under N2. To it was added 2-cyclohexylethan-1-amine (99 mg, 1.3 mmol) and the reaction mixture was heated at 90 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 45% EtOAc-hexane as eluent to afford 36 as an off-white solid (60 mg, 29%). M.P. 168–169 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.32–8.35 (m, 1H), 8.20–8.22 (m, 2H), 7.53–7.54 (m, 3H), 6.89 (s, 1H), 3.52–3.57 (m, 2H), 1.77–1.80 (m, 2H), 1.55–1.68 (m, 5H), 1.38 (m, 1H), 1.12–1.13 (m, 3H), 0.90–0.98 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 154.8, 147.9, 137.7, 130.2, 128.6, 127.4, 84.5, 40.1, 35.6, 34.6, 32.6, 26.0, 25.7. LCMS (ESI) m/z 322.28.
4.5.78. N-(4-Methoxyphenyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (37)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) in NMP (5 mL) was taken sealed tube under N2. To it was added 4-methoxyaniline (140 mg, 1.2 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 37 as a white solid (135 mg, 39%). M.P. 200-202 °C. 1H NMR (400 MHz, DMSO-d6): δ 10.16 (s, 1H), 8.58 (s, 1H), 7.98 (bs, 2H), 7.44–7.50 (m, 5H), 7.08 (d, J = 8.0 Hz, 2H), 6.69 (s, 1H), 3.81 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.5, 157.7, 155.9, 155.0, 147.3, 137.4, 130.3, 129.2, 128.8, 127.1, 126.6, 114.8, 85.6, 55.3. LCMS (ESI) m/z 318.38.
4.5.79. N-(4-Methoxybenzyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (38)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) in NMP (3 mL) was taken sealed tube under N2. To it was added (4-methoxyphenyl)methanamine (178 mg, 1.3 mmol) and the reaction mixture was heated at 100 °C for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 38 as a white solid (185 mg, 51%). M.P. 162-163 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.98 (bs, 1H), 8.49 (s, 1H), 8.13–8.14 (m, 2H), 7.51 (m, 3H), 7.42 (d, J = 8.0 Hz, 2H), 6.89–6.92 (m, 3H), 4.71 (d, J = 6.2 Hz, 2H), 3.69 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.1, 158.5, 155.6, 154.8, 147.9, 137.5, 130.3, 129.8, 128.7, 128.6, 127.3, 113.9, 85.1, 54.9, 44.0. LCMS (ESI) m/z 332.43.
4.5.80. N-Phenethyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (39)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) was taken in NMP (5 mL) in a 50 mL round bottom flask under N2. To it was added 2-phenylethan-1-amine (140 mg, 1.2 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 39 as an off-white solid (210 mg, 61%). M.P. 164–165 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.37–8.39 (m, 1H), 8.17–8.18 (m, 2H), 7.52–7.53 (m, 3H), 7.27–7.34 (m, 4H), 7.16–7.20 (m, 1H), 6.89 (s, 1H), 3.78–3.83 (m, 2H), 2.99–3.03 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.7, 155.5, 154.7, 147.9, 138.9, 137.6, 130.2, 128.9, 128.6, 128.3, 127.4, 126.3, 84.8, 42.9, 34.6. LCMS (ESI) m/z 316.20.
4.5.81. 5-Phenyl-N-(3-phenylpropyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (40)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (120 mg, 0.5 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 3-phenylpropan-1-amine (84 mg, 0.6 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 40 as a white solid (80 mg, 46%). M.P. 143–145 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46–8.49 (m, 2H), 8.15–8.17 (m, 2H), 7.52–7.57 (m, 3H), 7.25–7.30 (m, 4H), 7.17–7.21 (m, 1H), 6.81 (s, 1H), 3.53–3.58 (m, 2H), 2.69–2.73 (m, 2H), 1.95–2.03 (m, 2H). LCMS (ESI) m/z 330.20.
4.5.82. 5-Phenyl-N-(2-(pyridin-2-yl)ethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (41)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (200 mg, 0.8 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(pyridin-2-yl)ethan-1-amine (127 mg, 1.0 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 41 as an off-white solid (135 mg, 49%). M.P. 111–114 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.52 (d, J = 4.1 Hz, 1H), 8.46 (bs, 2H), 8.17–8.19 (m, 2H), 7.70 (t, J = 7.6 Hz, 1H), 7.53–7.56 (m, 3H), 7.21 (d, J = 7.8 Hz, 1H), 7.20–7.23 (m, 1H), 6.92 (s, 1H), 3.91–3.96 (m, 2H), 3.15–3.19 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.2, 158.7, 155.5, 154.7, 149.1, 147.9, 137.6, 136.5, 130.3, 128.6, 127.4, 123.5, 121.7, 84.8, 41.3, 36.5. LCMS (ESI) m/z 317.19.
4.5.83. 5-Phenyl-N-(2-(pyridin-3-yl)ethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (42)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(pyridin-3-yl)ethan-1-amine (159 mg, 1.3 mmol) and the reaction mixture was heated at 100 °C for 8 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 45% EtOAc-hexane as eluent to afford 42 as an off-white solid (146 mg, 42%). M.P. 169–171 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.53 (d, J = 1.6 Hz, 1H), 8.43–8.47 (m, 2H), 8.38–8.40 (m, 1H), 8.17–8.20 (m, 2H), 7.74 (d, J = 7.9 Hz, 1H), 7.52–7.55 (m, 3H), 7.28–7.31 (m, 1H), 6.93 (s, 1H), 3.81–3.86 (m, 2H), 3.02–3.05 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.5, 154.7, 150.1, 147.9, 147.5, 137.6, 136.5, 134.3, 130.3, 128.6, 127.4, 123.3, 84.8, 42.4, 31.6. LCMS (ESI) m/z 317.48.
4.5.84. 5-Phenyl-N-(2-(pyridin-4-yl)ethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (43)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (400 mg, 1.7 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(pyridin-4-yl)ethan-1-amine (256 mg, 2.1 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 50% EtOAc-hexane as eluent to afford 43 as an off-white solid (120 mg, 22%). M.P. 231-233 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.41–8.46 (m, 4H), 8.19–8.21 (m, 2H), 7.52–7.54 (m, 3H), 7.36 (d, J = 5.7 Hz, 2H), 6.96 (s, 1H), 3.83–3.88 (m, 2H), 3.03–3.06 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.9, 160.3, 155.5, 154.7, 149.4, 147.9, 147.8, 137.6, 130.3, 128.6, 127.4, 124.4, 84.8, 41.7, 33.6. LCMS (ESI) m/z 317.38.
4.5.85. N-(4-Methylphenethyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (44)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(p-tolyl)ethan-1-amine (176 mg, 1.3 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 44 as a white solid (85 mg, 23%). M.P. 167–168 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.35 (t, J = 6.0 Hz, 1H), 8.14–8.16 (m, 2H), 7.51–7.53 (m, 3H), 7.20 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 7.8 Hz, 2H), 6.82 (s, 1H), 3.74–3.79 (m, 2H), 2.94–2.97 (m, 2H), 2.21 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 160.2, 155.5, 154.7, 147.9, 137.6, 135.8, 135.2, 130.2, 128.9, 128.8, 128.5, 127.4, 84.8, 43.1, 34.3, 20.5. LCMS (ESI) m/z 330.39.
4.5.86. N-(4-Ethylphenethyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (45)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-ethylphenyl)ethan-1-amine (78 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 45 as a pale yellow solid (107 mg, 72%). M.P. 152–154 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.45 (s, 1H), 8.34 (bs, 1H), 8.15 (s, 2H), 7.31 (m, 3H), 7.21–7.23 (m, 2H), 7.09–7.11 (m, 2H), 6.80 (s, 1H), 3.77 (m, 2H), 2.96 (m, 2H), 2.49 (m, 2H), 1.08 (t, J = 7.5 Hz, 3H). LCMS (ESI) m/z 344.37.
4.5.87. N-(4-Fluorophenethyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (46)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (250 mg, 1.1 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-fluorophenyl)ethan-1-amine (181 mg, 1.3 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 25% EtOAc-hexane as eluent to afford 46 as an off-white solid (120 mg, 33%). M.P. 169–170 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.38 (t, J = 6.0 Hz, 1H), 8.16–8.19 (m, 2H), 7.52–7.53 (m, 3H), 7.34–7.38 (m, 2H), 7.01 (t, J = 8.8 Hz, 2H), 6.89 (s, 1H), 3.76–3.81 (m, 2H), 2.98–3.02 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.9 (d, J = 240 Hz), 160.3, 155.5, 154.7, 147.9, 137.6, 135.0 (d, J = 3.0 Hz), 130.7 (d, J = 8.0 Hz), 130.2, 128.6, 127.4, 114.9 (d, J = 21 Hz), 84.8, 42.9, 33.7. LCMS (ESI) m/z 334.37.
4.5.88. 5-Phenyl-N-(4-(trifluoromethyl)phenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (47)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (150 mg, 0.65 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-(trifluoromethyl)phenyl)ethan-1-amine (147 mg, 0.78 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 50% EtOAc-hexane as eluent to afford 47 as a light colour solid (120 mg, 20%). M.P. 183–184 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.43–8.46 (m, 2H), 8.16–8.17 (m, 2H), 7.63–7.65 (m, 2H), 7.52–7.56 (m, 5H), 6.89 (s, 1H), 3.84–3.85 (m, 2H), 3.09–3.13 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.4, 154.7, 147.8, 143.9, 137.5, 130.2, 129.7, 128.5, 127.4, 127.2, 125.0 (q, J = 4.0 Hz), 124.3 (q, J = 270 Hz), 84.8, 42.5, 34.3. LCMS(ESI) m/z 384.38.
4.5.89. 5-Phenyl-N-(4-(trifluoromethoxy)phenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (48)
7-Chloro-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidine 6 (150 mg, 0.65 mmol) was taken in NMP (5 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-(trifluoromethoxy)phenyl)ethan-1-amine (267 mg, 0.78 mmol) and the reaction mixture was heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 25% EtOAc-hexane as eluent to afford 48 as an off-white solid (170 mg, 39%). M.P. 179–180 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.46 (s, 1H), 8.39–8.42 (m, 1H), 8.17–8.18 (m, 2H), 7.52–7.53 (m, 3H), 7.45 (d, J = 8.3 Hz, 2H), 7.27 (d, J = 8.0 Hz, 2H), 6.90 (s, 1H), 3.79–3.84 (m, 2H), 3.03–3.06 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.3, 155.5, 154.7, 147.9, 146.9, 138.4, 137.6, 130.7, 130.2, 128.5, 127.4, 120.8, 120.0 (q, J = 256 Hz), 84.8, 42.7, 33.8. LCMS (ESI) m/z 400.43.
4.5.90. 2-(4-Methoxyphenyl)-N-(5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)acetamide (49)
5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine 28 (150 mg, 0.71 mmol) was taken in DMF (5 mL) in a 50 mL round bottom flask under N2 and cooled it to 0 °C. To it were sequentially added 2-(4-methoxyphenyl)acetic acid (141 mg, 0.85 mmol), EDCI.HCl (216 mg, 1.1 mmol), HOBt (148 mg, 1.1 mmol) and DIPEA (181 mg, 1.4 mmol). The reaction mixture was then heated at 90 °C for 24 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 49 as an off-white solid (70 mg, 27%). M.P. 220–222 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.59 (s, 1H), 8.72 (s, 1H), 8.41 (s, 1H), 8.11–8.12 (m, 2H), 7.57–7.58 (m, 3H), 7.31 (d, J = 8.4 Hz, 2H), 6.91 (d, J = 8.5 Hz, 2H), 4.02 (s, 2H), 3.73 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 172.1, 161.5, 158.2, 155.2, 142.1, 136.7, 130.9, 130.5, 129.0, 127.3, 126.4, 113.8, 94.1, 55.0, 41.9. LCMS (ESI) m/z 360.35.
4.5.91. 2-(3,4-Dimethoxyphenyl)-N-(5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)acetamide (50)
5-Phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine 28 (120 mg, 0.57 mmol) was taken in THF:CHCl3 (1:2, 6 mL) in a 50 mL round bottom flask under N2 and cooled it to 0 °C. To it were sequentially added 2-(3,4-dimethoxyphenyl)acetic acid (334 mg, 1.7 mmol), EDCI.HCl (163 mg, 0.85 mmol), HOBt (116 mg, 0.85 mmol) and DIPEA (181 mg, 1.4 mmol). The reaction mixture was then heated at 100 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 30 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 22% EtOAc-hexane as eluent to afford 50 as a white solid (38 mg, 17%). M.P. 202–204 °C. 1H NMR (400 MHz, DMSO-d6): δ 11.56 (s, 1H), 8.72 (s, 1H), 8.41 (s, 1H), 8.12 (m, 2H), 7.58 (m, 3H), 7.01 (s, 1H), 6.91 (m, 2H), 4.00 (s, 2H), 3.75 (s, 3H), 3.73 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 171.9, 161.5, 155.3, 155.2, 148.6, 147.8, 142.1, 136.7, 131.1, 129.1, 127.3, 126.9, 121.6, 113.4, 111.9, 94.2, 55.5, 55.4, 42.4. LCMS (ESI) m/z 390.08.
4.5.92. N-Phenethyl-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (51)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-phenylethan-1-amine (63 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 50% EtOAc-hexane as eluent to afford 51 as an off-white solid (60 mg, 44%). M.P. 214–215 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.0 Hz, 1H), 8.52 (bs, 2H), 8.46 (d, J = 7.8 Hz, 1H), 8.01 (t, J = 7.8 Hz, 1H), 7.53–7.56 (m, 1H), 7.37 (s, 1H), 7.27–7.30 (m, 4H), 7.19–7.20 (m, 1H), 3.73–3.75 (m, 2H), 3.01–3.05 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.0, 155.5, 154.9, 153.8, 149.2, 148.0, 138.6, 137.4, 128.7, 128.4, 126.3, 125.2, 121.4, 84.4, 43.2, 34.2. LCMS (ESI) m/z 317.22.
4.5.93. 5-(Pyridin-2-yl)-N-(2-(pyridin-2-yl)ethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (52)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(pyridin-2-yl)ethan-1-amine (63 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 52 as a pale yellow solid (52 mg, 38%). M.P. 156–157 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75–8.76 (m, 1H), 8.58 (bs, 1H), 8.51–8.53 (m, 2H), 8.45 (d, J = 7.9 Hz, 1H), 7.98–8.02 (m, 1H), 7.67–7.71 (m, 1H), 7.53–7.56 (m, 1H), 7.37 (s, 1H), 7.33 (d, J = 7.8 Hz, 1H), 7.19–7.22 (m, 1H), 3.89–3.91 (m, 2H), 3.16–3.20 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.0, 158.4, 155.5, 154.9, 153.8, 149.2, 149.1, 148.0, 137.4, 136.6, 125.2, 123.4, 121.7, 121.3, 84.4, 41.5, 36.3. LCMS (ESI) m/z 318.27.
4.5.94. 5-(Pyridin-2-yl)-N-(2-(pyridin-3-yl)ethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (53)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(pyridin-3-yl)ethan-1-amine 5i (63 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 15 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 53 as a pale yellow solid (104 mg, 76%). M.P. 212–213 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.74–8.75 (m, 1H), 8.56–8.59 (m, 1H), 8.50–8.52 (m, 2H), 8.46 (d, J = 7.9 Hz, 1H), 8.39–8.40 (m, 1H), 7.99–8.03 (m, 1H), 7.73 (d, J = 7.8 Hz, 1H), 7.54–7.56 (m, 1H), 7.37 (s, 1H), 7.29–7.32 (m, 1H), 3.77–3.82 (m, 2H), 3.03–3.07 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.0, 155.5, 154.9, 153.8, 149.9, 149.2, 148.0, 147.6, 137.4, 136.3, 134.2, 125.2, 123.4, 121.4, 84.5, 42.7, 31.3. LCMS (ESI) m/z 318.37.
4.5.95. 5-(Pyridin-2-yl)-N-(2-(pyridin-4-yl)ethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (54)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(pyridin-4-yl)ethan-1-amine (63 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 15 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 54 as a pale yellow solid (70 mg, 51%). M.P. 201–202 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.3 Hz, 1H), 8.57 (bs, 1H), 8.51 (s, 1H), 8.45–8.46 (m, 3H), 7.99–8.03 (m, 1H), 7.54–7.57 (m, 1H), 7.38 (s, 1H), 7.34 (d, J = 7.6 Hz, 2H), 3.81–3.82 (m, 2H), 3.04–3.08 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.1, 155.5, 155.0, 153.8, 149.5, 149.2, 148.0, 147.7, 137.4, 125.3, 124.3, 121.4, 84.5, 42.0, 33.3. LCMS (ESI) m/z 318.27.
4.5.96. N-(4-Methylphenethyl)-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (55)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(p-tolyl)ethan-1-amine (70 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 10 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 10 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 55 as a pale yellow solid (60 mg, 42%). M.P. 194–196 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.0 Hz, 1H), 8.51 (bs, 2H), 8.45 (d, J = 7.9 Hz, 1H), 7.98–8.02 (m, 1H), 7.53–7.56 (m, 1H), 7.34 (s, 1H), 7.18 (d, J = 7.8 Hz, 2H), 7.08 (d, J = 7.7 Hz, 2H), 3.71 (m, 2H), 2.96–2.99 (m, 2H), 2.21 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 159.0, 155.5, 154.9, 153.8, 149.2, 148.0, 137.4, 135.5, 135.3, 128.9, 128.6, 125.2, 121.3, 84.5, 43.3, 33.8, 20.5. LCMS (ESI) m/z 331.16.
4.5.97. N-(4-Ethylphenethyl)-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (56)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-ethylphenyl)ethan-1-amine (70 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 10 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 10 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 56 as a pale yellow solid (75 mg, 50%). M.P. 154–155 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.1 Hz, 1H), 8.50 (d, J = 5.0 Hz, 2H), 8.45 (d, J = 8.0 Hz, 1H), 8.00 (t, J = 7.7 Hz, 1H), 7.53–7.55 (m, 1H), 7.33 (s, 1H), 7.20 (d, J = 7.9 Hz, 2H), 7.11 (d, J = 7.8 Hz, 2H), 3.72–3.73 (m, 2H), 2.96–3.00 (m, 2H), 2.37 (m, 2H), 1.09 (t, J = 7.6 Hz, 3H). LCMS (ESI) m/z 345.39.
4.5.98. N-(4-Fluorophenethyl)-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (57)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-fluorophenyl)ethan-1-amine (72 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 10 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 57 as a white solid (69 mg, 48%). M.P. 190-192 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.0 Hz, 1H), 8.51 (bs, 2H), 8.45 (d, J = 7.9 Hz, 1H), 7.98–8.02 (m, 1H), 7.53–7.56 (m, 1H), 7.32–7.35 (m, 3H), 7.10 (t, J = 8.9 Hz, 2H), 3.72–3.76 (m, 2H), 3.00–3.03 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 160.4 (d, J = 240 Hz), 159.0, 155.5, 154.9, 153.8, 149.1, 148.0, 137.4, 134.8 (d, J = 2.9 Hz), 130.5 (d, J = 7.9 Hz), 125.2, 121.3, 115.0 (d, J = 21 Hz), 84.4, 43.2, 33.3. LCMS (ESI) m/z 335.26.
4.5.99. 5-(Pyridin-2-yl)-N-(4-(trifluoromethyl)phenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (58)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-(trifluoromethyl)phenyl)ethan-1-amine (98 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 50% EtOAc-hexane as eluent to afford 58 as a pale yellow solid (80 mg, 48%). M.P. 186-188 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.74 (d, J = 4.2 Hz, 1H), 8.56 (bs, 1H), 8.51 (s, 1H), 8.44 (d, J = 8.0 Hz, 1H), 8.00 (t, J = 7.4 Hz, 1H), 7.62–7.64 (m, 2H), 7.52–7.56 (m, 3H), 7.31 (s, 1H), 3.80–3.81 (m, 2H), 3.11–3.14 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.0, 155.5, 154.9, 153.8, 149.1, 148.0, 143.8, 137.4, 129.6, 127.1 (q, J = 32 Hz), 125.1 (q, J = 5 Hz), 124.3 (q, J = 270 Hz), 121.3, 115.6, 84.5, 42.8, 34.0. LCMS (ESI) m/z 385.21.
4.5.100. 5-(Pyridin-2-yl)-N-(4-(trifluoromethoxy)phenethyl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (59)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (4 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-(trifluoromethoxy)phenyl)ethan-1-amine (107 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 0.5 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 32% EtOAc-hexane as eluent to afford 59 as a white solid (45 mg, 48%). M.P. 163-165 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.74 (d, J = 4.3 Hz, 1H), 8.55 (bs, 1H), 8.54 (s, 1H), 8.45 (d, J = 8.0 Hz, 1H), 8.01 (t, J = 7.6 Hz, 1H), 7.53–7.56 (m, 1H), 7.43 (d, J = 8.5 Hz, 2H), 7.33 (s, 1H), 7.26 (d, J = 7.9 Hz, 2H), 3.75–3.80 (m, 2H), 3.04–3.08 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.0, 155.5, 154.9, 153.8, 149.2, 148.1, 146.9, 138.3, 137.4, 130.6, 125.2, 121.4, 120.9, 120.0 (q, J = 260 Hz), 84.5, 43.0, 33.5. LCMS (ESI) m/z 401.10.
4.5.101. N-(4-Chlorophenethyl)-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (60)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (120 mg, 0.52 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-chlorophenyl)ethan-1-amine (96 mg, 0.62 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 30% EtOAc-hexane as eluent to afford 60 as a white solid (60 mg, 33%). M.P. 194–195 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 3.8 Hz, 1H), 8.51 (m, 2H), 8.45 (d, J = 7.5 Hz, 1H), 8.08 (t, J = 7.2 Hz, 1H), 7.53–7.56 (m,1H), 7.33 (m, 5H), 3.74–3.76 (m, 2H), 3.00–3.04 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.5, 156.0, 155.4, 154.3, 149.6, 148.5, 138.3, 137.9, 131.5, 131.2, 128.7, 125.7, 121.8, 84.9, 43.5, 33.9. LCMS (ESI) m/z 351.10.
4.5.102. N-(4-Methoxyphenethyl)-N-methyl-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (61)
Sodium hydride (25 mg, 1.1 mmmol) was taken in anhydrous DMF (5 mL) in a 50 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of N-(4-methoxyphenethyl)-5-phenyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine 1 (250 g, 0.72 mmol) in DMF (1 mL). It was kept stirred at rt for 30 min. Methyl iodide (120 mg, 0.86 mmol) was then added to it and the reaction mixture was stirred at rt for 2 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 20 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 50% hexane-EtOAc as eluent to afford 61 as a pale yellow solid (180 mg, 69%). M.P. 170-171 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.47 (s, 1H), 8.16–8.18 (m, 2H), 7.53–7.54 (m, 3H), 7.16 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.6 Hz, 3H), 4.32–4.35 (m, 2H), 3.65 (s, 3H), 3.34 (s, 3H), 2.92–2.96 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 159.7, 157.8, 157.2, 154.2, 149.9, 137.4, 130.3, 130.2, 129.8, 128.6, 127.3, 113.6, 89.8, 54.9, 54.1, 40.1, 33.3. LCMS (ESI) m/z 360.22.
4.5.103. 7-(3-(4-Methoxyphenyl)pyrrolidin-1-yl)-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine (62)
7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 5k (100 mg, 0.43 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 3-(4-methoxyphenyl)pyrrolidine (92 mg, 0.52 mmol) and the reaction mixture was heated at 75 °C for 2 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 3% MeOH-DCM as eluent to afford 62 as a light yellow solid (46 mg, 29%). M.P. 222–223 °C. 1H NMR (400 MHz, CDCl3): δ 8.64–8.66 (m, 2H), 8.26 (s, 1H), 7.86 (t, J = 7.8 Hz, 1H), 7.36–7.39 (m, 1H), 7.23–7.26 (m, 3H), 6.91 (d, J = 8.5 Hz, 2H), 4.60 (m, 1H), 4.39 (m, 1H), 4.16 (m, 1H), 4.04 (m, 1H), 3.81 (s, 3H), 3.48–3.56 (m, 1H), 2.45–2.50 (m, 1H), 2.16–2.26 (m, 1H); 13C NMR (100 MHz, CDCl3): δ 160.0, 158.9, 158.0, 154.6, 154.5, 148.9, 148.3, 137.2, 132.0, 128.2, 125.0, 122.4, 114.3, 88.2, 55.5, 51.0, 43.3, 32.1, 22.8. LCMS (ESI) m/z 373.19.
4.5.104. 7-(3-(4-Methoxyphenyl)piperidin-1-yl)-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine (63)
(a) 1-Bromo-4-methoxybenzene (500 mg, 2.7 mmol) was taken in DMF (8 mL) in a 50 mL round bottom flask under N2. To it were sequentially added pyridin-3-ylboronic acid (394 mg, 3.2 mmol), Pd(OAc)2 (60 mg, 0.27 mmol), dppf (149 mg, 0.27 mmol), CuCl (27 mg, 0.27 mmol) and Cs2CO3 (1.7 g, 5.4 mmol). The reaction mixture was heated at 110 °C for 2 h. It was then filtered through sintered funnel with a pad of Celite, washed with EtOAc (40 mL). The filtrate was then poured into ice water (40 g) and extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 15% EtOAc-hexane as eluent to afford 3-(4-methoxyphenyl)pyridine as a light brown liquid (325 mg, 66%). 1H NMR (400 MHz, DMSO-d6): δ 8.85 (s, 1H), 8.50–8.51 (m, 1H), 8.02 (d, J = 7.9 Hz, 1H), 7.68 (d, J = 8.6 Hz, 2H), 7.43–7.46 (m, 1H), 7.06 (d, J = 8.7 Hz, 2H), 3.81 (s, 3H). (b) A par flask was charged with 3-(4-methoxyphenyl)pyridine 49 (140 mg, 0.75 mmole) and MeOH (5 mL) followed by addition of HCl () and PtO2 (205 mg, 0.9 mmol). The flask was evacuated under vacuum and then purged with hydrogen. The reaction was stirred under hydrogen atmosphere (20 psi) for 6 h. It was then filtered through sintered funnel with a pad of Celite, washed with MeOH (20 mL) and concentrated under reduced pressure to afford 3-(4-methoxyphenyl)piperidine as a white solid (100 mg, 69%) that was used as such for the next step without any further purification. LCMS(ESI) m/z 192.10 (M+H)+, 97.34% (purity). (c) 7-Chloro-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine 35 (100 mg, 0.43 mmol) was taken in NMP (3 mL) in a 50 mL round bottom flask under N2. To it was added 3-(4-methoxyphenyl)piperidine (99 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 34% EtOAc-hexane as eluent to afford 63 as a semi-solid (50 mg, 30%). 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.0 Hz, 1H), 8.54 (s, 1H), 8.48 (d, J = 7.9 Hz, 1H), 8.01–8.05 (m, 1H), 7.55–7.58 (m, 2H), 7.28 (d, J = 8.6 Hz, 2H), 6.92 (d, J = 8.6 Hz, 2H), 4.73–4.76 (m, 1H), 4.64–4.68 (m, 1H), 3.74 (s, 3H), 3.25–3.28 (m, 2H), 2.92–2.94 (m, 1H), 1.95–1.98 (m, 2H), 1.81–1.89 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 158.8, 158.0, 157.1, 154.6, 153.4, 150.4, 149.3, 137.6, 134.9, 128.1, 125.5, 121.4, 113.9, 90.8, 55.0, 54.4, 48.4, 31.2, 24.8. LCMS (ESI) m/z 387.23.
4.5.105. 2-(4-Methoxyphenyl)-4-(5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)morpholine (64)
(a) 2-Bromo-1-(4-methoxyphenyl)ethan-1-one (4.0 g, 17.5 mmmol) was taken in dry THF (45 mL) in a 100 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added a solution of 2-(benzylamino)ethan-1-ol (3.7 g, 24.4 mmol) in THF (5 mL). The reaction mixture was heated at 50 °C for 12 h. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 75 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 15% hexane-EtOAc as eluent to afford 2-(benzyl(2-hydroxyethyl)amino)-1-(4-methoxyphenyl)ethan-1-one as a brown solid (2.5 g, 48%). LCMS(ESI) m/z 300.09 [M+H+]; 52% (purity). (b) 2-(Benzyl(2-hydroxyethyl)amino)-1-(4-methoxyphenyl)ethan-1-one (2.5 g, 8.3 mmmol) was taken in MeOH (20 mL) in a 100 mL round bottom flask under N2 and cooled it down to 0 °C. To it was added sodiumboro hydride (3.7 g, 24.4 mmol) in portion-wise. The reaction mixture was stirred at rt for 3 h. The reaction mixture was evaporated to dryness. Ice-cooled water was added dropwise to quench the reaction. It was extracted with EtOAc (3 × 100 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. This afforded 2-(benzyl(2-hydroxyethyl)amino)-1-(4-methoxyphenyl)ethan-1-ol as a brown solid (1.2 g, 48%). This was then used in the next step without any further purification. LCMS(ESI) m/z 302.21 [M+H+]; 42% (purity). (c) 2-(Benzyl(2-hydroxyethyl)amino)-1-(4-methoxyphenyl)ethan-1-ol (1.2 g, 3.9 mmmol) was taken in HCl:H2O mixture (10 mL, 60% HCl in H2O) in a 50 mL round bottom flask fitted with a condenser. The reaction mixture was heated at 110 °C for 12 h. The reaction mixture was evaporated to dryness under reduced pressure. This was triturated with Et2O which afforded 4-benzyl-2-(4-methoxyphenyl)morpholine as a brown semi-solid (800 mg, 71%). This was then used in the next step without any further purification. LCMS(ESI) m/z 284.10 [M+H+]; 98.71% (purity). (d) A par flask was charged with 4-benzyl-2-(4-methoxyphenyl)morpholine (800 mg, 2.8 mmmol) and MeOH (20 mL) followed by the addition of Pd(OH)2 (393 mg, 2.8 mmol). The flask was evacuated under vacuum and then purged with hydrogen. The reaction was stirred under hydrogen atmosphere (20 psi) for 4 h. It was then filtered through sintered funnel with a pad of Celite, washed with MeOH (20 mL) and concentrated under reduced pressure to afford 2-(4-methoxyphenyl)morpholine as a white solid (500 mg, 92%) that was used as such for the next step without any further purification. LCMS(ESI) m/z 194.28 (M+H)+; 85% (purity). (e) 7-Chloro-2-methyl-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine (200 mg, 0.86 mmol) was taken in NMP (6 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)morpholine (200 mg, 1.04 mmol) and the reaction mixture was stirred at rt for 1 h. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 40% EtOAc-hexane as eluent to afford 64 as a white solid (130 mg, 39%). 1H NMR (400 MHz, DMSO-d6): δ 8.75 (d, J = 4.4 Hz, 1H), 8.59 (s, 1H), 8.49 (d, J = 7.9 Hz, 1H), 8.01–8.06 (m, 1H), 7.56–7.60 (m, 2H), 7.40 (d, J = 8.6 Hz, 2H), 6.97 (d, J = 8.6 Hz, 2H), 4.70–4.75 (m, 2H), 4.58–4.70 (m, 1H), 4.17–4.19 (m, 1H), 3.92–3.97 (m, 1H), 3.76 (s, 3H), 3.39–3.46 (m, 1H), 3.21–3.27 (m, 1H); 13C NMR (100 MHz, DMSO-d6): δ 159.1, 159.0, 156.9, 154.8, 153.3, 150.5, 149.3, 137.6, 131.2, 127.6, 125.6, 121.4, 113.7, 90.9, 76.3, 65.6, 55.1, 53.3, 47.2. LCMS (ESI) m/z 389.16.
4.5.106. N-(4-Methoxyphenethyl)-2-methyl-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (67)
(a) Ethyl 3-oxo-3-(pyridin-2-yl)propanoate (400 mg, 2.07 mmol) was taken in AcOH (10 mL) in a 50 mL round bottom flask under N2. To it was added 5-methyl-4H-1,2,4-triazol-3-amine (334 mg, 2.49 mmol). The reaction mixture was heated at 115 °C for 12 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vaccum which affored 66a as a brown solid (350 mg, 75%). This was then used in the next step without any further purification. LCMS(ESI) m/z 226.03 [M−H+]; 56% (purity). (b) To a solution of 2-methyl-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidin-7-ol 66a (350 mg, 1.5 mmol) was added POCl3 (6 mL, 60 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 1:1). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 15% EtOAc-hexane as eluent to afford 7-chloro-2-methyl-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine as a brown solid (125 mg, 33%). LCMS(ESI) m/z 246.20 [M+H+]; 49% (purity). (c) 7-Chloro-2-methyl-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine (100 mg, 0.41 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine 5a (74 mg, 0.49 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 67 as a white solid (80 mg, 54%). M.P. 139-141 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.73–8.74 (m, 1H), 8.39–8.42 (m, 2H), 7.97–8.01 (m, 1H), 7.51–7.55 (m, 1H), 7.27 (s, 1H), 7.20 (d, J = 8.6 Hz, 2H), 6.84 (d, J = 8.6 Hz, 2H), 3.65–3.70 (m, 5H), 2.92–2.95 (m, 2H), 2.50 (s, 3H); 13C NMR (100 MHz, DMSO-d6): δ 164.1, 158.4, 157.8, 155.9, 153.9, 149.1, 147.5, 137.3, 130.5, 129.7, 125.1, 121.2, 113.8, 84.2, 54.9, 43.4, 33.4, 14.8. LCMS (ESI) m/z 359.06.
4.5.107. N-(4-Methoxyphenethyl)-2-phenyl-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-amine (68)
(a) Ethyl 3-oxo-3-(pyridin-2-yl)propanoate (500 mg, 2.6 mmol) was taken in AcOH (50 mL) in a 250 mL round bottom flask under N2. To it was added 3-phenyl-1H-1,2,4-triazol-5-amine (497 mg, 3.1 mmol). The reaction mixture was heated at 115 °C for 12 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 66b as a deep brown solid (300 mg, 40%). This was then used in the next step without any further purification. LCMS(ESI) m/z 288.02 [M−H+]; 57% (purity). (b) To a solution of 2-phenyl-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidin-7-ol 66b (300 mg, 1.04 mmol) was added POCl3 (2 mL, 21 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 3:7). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 17% hexane-EtOAc as eluent to afford 7-chloro-2-phenyl-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine as a brown solid (150 mg, 47%).LCMS(ESI) m/z 307.99 [M+H+]; 87.88% (purity). (c) 7-Chloro-2-phenyl-5-(pyridin-2-yl)-[1,2,4]triazolo[1,5-a]pyrimidine (125 mg, 0.41 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (74 mg, 0.49 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 50% EtOAc-hexane as eluent to afford 68 as a white solid (65 mg, 38%). M.P. 146–148 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.76 (d, J = 4.0 Hz, 1H), 8.47 (d, J = 7.8 Hz, 2H), 8.25–8.27 (m, 2H), 8.00–8.05 (m, 1H), 7.54–7.59 (m, 4H), 7.36 (s, 1H), 7.24 (d, J = 8.5 Hz, 2H), 6.86 (d, J = 8.5 Hz, 2H), 3.73–3.75 (m, 2H), 3.67 (s, 3H), 2.97–3.01 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 163.6, 158.8, 157.8, 156.3, 153.9, 149.2, 147.8, 137.4, 130.8, 130.5, 130.3, 129.7, 128.8, 126.8, 125.3, 121.3, 113.9, 84.8, 54.9, 43.6, 33.5. LCMS (ESI) m/z 423.20.
4.5.108. N-(4-Methoxyphenethyl)-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidin-7-amine (69)
(a) Ethyl 3-oxo-3-(pyridin-2-yl)propanoate (500 mg, 2.6 mmol) was taken in AcOH (6 mL) in a 50 mL round bottom flask under N2. To it was added 1H-pyrazol-3-amine (258 mg, 3.1 mmol). The reaction mixture was heated at 120 °C for 14 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vaccum which affored 66c as a colourless solid (420 mg, 77%). This was then used in the next step without any further purification. MS (ESI) m/z 213.08 [M+H+]. (b) To a solution of 5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidin-7-ol 66c (400 mg, 1.8 mmol) was added POCl3 (6 mL, 66.5 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 3:7). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 5% MeOH-DCM as eluent to afford 7-chloro-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine as a brown liquid (320 mg, 74%). LCMS(ESI) m/z 231.06 [M+H+]; 75.59% (purity). (c) 7-Chloro-5-(pyridin-2-yl)pyrazolo[1,5-a]pyrimidine (100 mg, 0.43 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine (79 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 35% EtOAc-hexane as eluent to afford 69 as a light yellow solid (30 mg, 20%). M.P. 116–117 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.72–8.73 (m, 1H), 8.42 (d, J = 7.9 Hz, 1H), 8.13 (d, J = 2.2 Hz, 1H), 8.01–8.05 (m, 1H), 7.93–7.98 (m, 1H), 7.48–7.51 (m, 1H), 7.23 (d, J = 8.6 Hz, 2H), 7.15 (s, 1H), 6.86 (d, J = 8.6 Hz, 2H), 6.53 (d, J = 2.2 Hz, 1H), 3.65–3.71 (m, 5H), 2.95–2.99 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 157.8, 154.8, 154.6, 149.0, 148.6, 147.0, 143.9, 137.1, 130.6, 129.6, 124.6, 120.9, 113.8, 95.3, 81.2, 54.9, 43.2, 33.4. LCMS (ESI) m/z 346.18.
4.5.109. N-(4-Methoxyphenethyl)-7-(pyridin-2-yl)imidazo[1,2-a]pyrimidin-5-amine (70)
(a) Ethyl 3-oxo-3-(pyridin-2-yl)propanoate (2.0 g, 10.3 mmol) was taken in AcOH (20 mL) in a 100 mL round bottom flask under N2. To it was added 1H-imidazol-2-amine (1.03 g, 12.4 mmol). The reaction mixture was heated at 120 °C for 12 h. The reaction mixture was then evaporated to dryness using toluene as an azeotropic solvent and triturated with diethyl ether. This was finally dried under high vacuum which afforded 66d as a deep brown solid (1.0 g, 45%). This was then used in the next step without any further purification. LCMS(ESI) m/z 212.98 [M+H+]; 52% (purity). (b) To a solution of 7-(pyridin-2-yl)imidazo[1,2-a]pyrimidin-5-ol 66d (700 mg, 3.3 mmol) was added POCl3 (10 mL, 105.6 mmol) at 0 °C. The reaction mixture was then heated at 100 °C and monitored by TLC analysis (Hexane/EtOAc = 3:7). Upon completion, the reaction mixture was concentrated using toluene as an azeotropic solvent and quenched with ice cooled saturated NaHCO3 solution (20 mL) to pH 8. It was extracted with EtOAc (3 × 50 mL). The combined organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. The crude was further purified by flash chromatography on silica gel (60–120 mesh) using 5% MeOH-DCM as eluent to afford 5-chloro-7-(pyridin-2-yl)imidazo[1,2-a]pyrimidine as a brown liquid (350 mg, 46%). LCMS(ESI) m/z 231.03 [M+H+]; 92.15% (purity). (c) 5-Chloro-7-(pyridin-2-yl)imidazo[1,2-a]pyrimidine (100 mg, 0.43 mmol) was taken in NMP (2 mL) in a 50 mL round bottom flask under N2. To it was added 2-(4-methoxyphenyl)ethan-1-amine 5a (79 mg, 0.52 mmol) and the reaction mixture was stirred at rt for 30 min. The reaction mixture concentrated in vacuo and the resulting solid taken up in EtOAc and H2O, the organic layer was separated and the aqueous layer extracted with EtOAc (3 × 20 mL). The organic layers combined, washed with brine, dried over Na2SO4 and the solvent removed under reduced pressure. The crude was purified by flash chromatography on silica gel (100–200 mesh) using 2% MeOH-DCM as eluent to afford 70 as a light yellow solid (32 mg, 21%). M.P. 234–235 °C. 1H NMR (400 MHz, DMSO-d6): δ 8.71–8.72 (m, 1H), 8.44 (d, J = 7.9 Hz, 1H), 7.95–7.99 (m, 2H), 7.90–7.92 (m, 1H), 7.62 (d, J = 1.4 Hz, 1H), 7.48–7.51 (m, 1H), 7.23 (d, J = 8.5 Hz, 2H), 7.15 (d, J = 6.5 Hz, 1H), 6.87 (d, J = 8.6 Hz, 2H), 3.67 (s, 3H), 3.62–3.64 (m, 2H), 2.93–2.99 (m, 2H); 13C NMR (100 MHz, DMSO-d6): δ 157.9, 155.6, 154.7, 149.8, 149.0, 147.4, 137.2, 133.9, 130.7, 129.7, 124.7, 120.8, 113.8, 106.2, 81.4, 54.9, 43.9, 33.1. LCMS (ESI) m/z 346.19.
Acknowledgements
Funding: This work was supported by the Bill & Melinda Gates Foundation, Seattle, WA [grant number OPP1024038]; and Eli Lilly and Company [Lilly TB Drug Discovery Initiative].
We thank Dean Thompson, Lindsay Flint, Olena Anoshchenko, James Vela and David Roberts for technical assistance.
References
- 1.WHO, Global Tuberculosis Report 2015, Geneva, 2015.
- 2.Zhang N., Ayral-Kaloustian S., Nguyen T. Synthesis and SAR of [1,2,4]triazolo[1,5-a]pyrimidines, a class of anticancer agents with a unique mechanism of tubulin inhibition. J Med Chem. 2007;50:319–327. doi: 10.1021/jm060717i. [DOI] [PubMed] [Google Scholar]
- 3.DeNinno M.P., Wright S.W., Etienne J.B. Discovery of triazolopyrimidine-based PDE8B inhibitors: exceptionally ligand-efficient and lipophilic ligand-efficient compounds for the treatment of diabetes. Bioorg Med Chem Lett. 2012;22:5721–5726. doi: 10.1016/j.bmcl.2012.06.079. [DOI] [PubMed] [Google Scholar]
- 4.El-Gendy M.M., Shaaban M., Shaaban K.A., El-Bondkly A.M., Laatsch H. Essramycin: a first triazolopyrimidine antibiotic isolated from nature. J Antibiot (Tokyo) 2008;61:149–157. doi: 10.1038/ja.2008.124. [DOI] [PubMed] [Google Scholar]
- 5.Gujjar R., Marwaha A., El Mazouni F. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J Med Chem. 2009;52:1864–1872. doi: 10.1021/jm801343r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gujjar R., El Mazouni F., White K.L. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem. 2011;54:3935–3949. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coteron J.M., Marco M., Esquivias J. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J Med Chem. 2011;54:5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caballero A.B., Rodríguez-Diéguez A., Quirós M. Triazolopyrimidine compounds containing first-row transition metals and their activity against the neglected infectious Chagas disease and leishmaniasis. Eur J Med Chem. 2014;85:526–534. doi: 10.1016/j.ejmech.2014.08.026. [DOI] [PubMed] [Google Scholar]
- 9.Patil V., Kale M., Raichurkar A. Design and synthesis of triazolopyrimidine acylsulfonamides as novel anti-mycobacterial leads acting through inhibition of acetohydroxyacid synthase. Bioorg Med Chem Lett. 2014;24:2222–2225. doi: 10.1016/j.bmcl.2014.02.054. [DOI] [PubMed] [Google Scholar]
- 10.Ballell L., Bates R.H., Young R.J. Fueling open-source drug discovery: 177 small-molecule leads against tuberculosis. ChemMedChem. 2013;8:313–321. doi: 10.1002/cmdc.201200428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Candice Sde M., Feng T.S., van der Westhuyzen R. Aminopyrazolo[1,5-a]pyrimidines as potential inhibitors of Mycobacterium tuberculosis: structure activity relationships and ADME characterization. Bioorg Med Chem. 2015;23:7240–7250. doi: 10.1016/j.bmc.2015.10.021. [DOI] [PubMed] [Google Scholar]
- 12.Moraski G.C., Markley L.D., Hipskind P.A. Advent of imidazo[1,2-a]pyridine-3-carboxamides with potent multi- and extended drug resistant antituberculosis activity. ACS Med Chem. 2011;2:466–470. doi: 10.1021/ml200036r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abrahams K.A., Cox J.A., Spivey V.L. Identification of novel imidazo[1,2-a]pyridine inhibitors targeting M. tuberculosis QcrB. PLoS ONE. 2012;7:e52951. doi: 10.1371/journal.pone.0052951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ollinger J., Bailey M.A., Moraski G.C. A dual read-out assay to evaluate the potency of compounds active against Mycobacterium tuberculosis. PLoS ONE. 2013;8:e60531. doi: 10.1371/journal.pone.0060531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sirgel F.A., Wiid I.J., van Helden P.D. Measuring minimum inhibitory concentrations in mycobacteria. In: Parish T., Amanda Claire B., editors. Mycobacteria Protocols. Humana Pres; Totowa, NJ: 2009. pp. 173–186. [DOI] [PubMed] [Google Scholar]