

Abstract
Rhesus macaques are an important pre-clinical model of human disease. To advance our understanding of genomic variation that may influence disease, we surveyed genome-wide variation in 21 rhesus macaques. We employed best-practice variant calling, validated with Mendelian inheritance. Next, we used alignment data from our cohort to detect genomic regions likely to produce inaccurate genotypes, potentially due to either gene duplication or structural variation between individuals. We generated a final dataset of >16 million high confidence variants, including 13 million in Chinese-origin rhesus macaques, an increasingly important disease model. We detected an average of 131 mutations predicted to severely alter protein coding per animal, and identified 45 such variants that coincide with known pathogenic human variants. These data suggest that expanded screening of existing breeding colonies will identify novel models of human disease, and that increased genomic characterization can help inform research studies in macaques.
Keywords: SNP, Macaca mulatta, nonhuman primate, variant discovery, SIV, Indian-origin, Chinese-origin, genome
Introduction
The advent of next generation sequencing has made it practical to sequence the entire genome of an individual in a single experiment. The decreased cost of DNA sequencing permits the characterization of larger cohorts, and the high resolution genome-wide data from individuals will allow more precise understanding of genetic risk, ultimately informing risk, diagnostics and increasingly personalized treatments. To fully realize the benefits of new technologies, it is critical that our understanding of genetics in animal models keeps pace with humans. Animal models serve not only as a platform to experimentally test hypotheses derived from human data, but the highly controlled lifestyle experienced by many models aids in the detection and study of complex genetic traits. Due to their high level of physiological, anatomical and behavioral similarity to humans, Rhesus macaques (Macaca mulatta) are often indispensable to the study of complex human disease, and are well established as a genetic model for many diseases. There are many examples of similar disease-associated polymorphisms identified in humans and macaques, including the LOC387715/ARMS2 and HTRA1 genes in age-related macular degeneration [1], and corticotrophin-releasing hormone (CRH) in anxiety [2]. One of the most studied aspects of macaque genetics is MHC Class I, where the close similarity between macaques and humans has been particularly important as a model for the cytotoxic T-cell response in HIV/SIV, and allowed studies not possible in more evolutionarily distant organisms such as rodents [3].
Despite a long history as a genetic model, macaques have been slow to realize the full benefits of the genomic age. One of the main promises of genome sequencing is the ability to capture the entire genomic sequence of an individual in a single experiment. While it is growing ever cheaper to generate sequence data, analyzing and interpreting these data from a given organism typically requires many additional resources, including a reference genome, reference genome annotations, and population-level variation and allele frequency data. These resources are typically generated by substantial, coordinated efforts, such as the 1000Genomes and HapMap projects [4, 5]. In macaques, most attention to date has focused on the reference genome itself. A reference genome can be evaluated both in terms of the completeness of the DNA sequence, and also in the quality and breadth of annotations available. The first draft of the macaque genome was published in 2007 [6]. This draft, termed rheMac2 (also Mmul_051212), was derived from a single female Indian-origin macaque using shotgun Sanger sequencing. While it was a major step forward, as would be expected from a draft genome, there were sequence errors and gaps in the assembly and incomplete gene annotations [7, 8]. In 2014, a new rhesus genome based on the same reference animal was published, termed MacaM, which utilized new Illumina genomic and exomic sequence data to augment the original Sanger data and provided a more complete assembly [9]. MacaM also utilized macaque RNA-Seq data to better identify and annotate transcripts, resulting in a much more accurate annotation of more than 16,000 genes, a significant increase over rheMac2 that is especially important for functional studies and gene expression analyses. In late 2015, yet another version of the macaque genome was released on NCBI, Mmul_8.0.1, which uses PacBio reads to fill gaps in the assembly (accession GCA_000772875.3). However, the PacBio reads used in Mmul_8.0.1 were not error corrected, and indels in these novel regions may interfere with accurate SNP detection, as previously described [7, 8]. It should also be noted that multiple sources of Mmul_8.0.1 gene annotations exist, including a version generated by NCBI and a newer one available through Ensembl. In addition to the Indian-origin macaque assemblies, an assembly based on a Chinese-origin animal is available, termed CR_1.0 (also rheMac3) [10]; however, this assembly is less complete than any of the Indian-origin macaque genomes.
Beyond the reference genome, which typically represents a single animal, a genomic model requires characterization of the variation that exists in the population. It has long been known that macaques are genetically diverse, and that multiple sub-populations exist within Rhesus macaques [11]. In biomedical research, the majority of Rhesus macaques are either Indian-origin or Chinese-origin. Early genome-wide SNV discovery efforts included a study of three animals that identified 3 million SNVs [12]. A later study used transcriptome and promoter regions of 14 macaques (primarily Indian-origin), detecting approximately 463,000 SNPs, to demonstrate that macaques have at least three times as much intra-species diversity as humans [13]. Similar genome-wide SNP detection has been performed using exome data of two Indian-origin and two Chinese-origin macaques [14], or with a single Indian-origin rhesus macaque [15]. Cornish et al. highlight the importance of both the completeness of the reference assembly and accuracy of gene annotation for variant detection and functional prediction, demonstrating considerable improvement in the second generation MacaM genome over the earlier rheMac2 build [14]. Most recently, a study of 133 Rhesus macaques found approximately 2.5-fold higher overall nucleotide diversity and slightly elevated putative functional variation compared with humans [16]. Each of these studies identified examples of mutations predicted to adversely affect key proteins, which suggests that many naturally occurring disease models may be present in the population. It should be noted that published efforts have been primarily focused on Indian-origin animals, although Chinese-origin macaques are genetically distinct and are of growing importance in research [17, 18]. While there is no high-density SNP array designed for macaques, a panel of 91 macaque SNPs has been designed to infer genetic ancestry [19], and the Affymetrix Human SNP 6.0 Array, which is designed using 906,000 human SNPs, has been used on macaque samples to compare the 59,691 shared markers [20]. While the efforts to date are essential, many challenges remain. The current number of macaques with genome-wide characterization is modest, and there is a critical need to expand this set and to include both Indian- and Chinese-origin animals. Here we present a survey of 21 rhesus macaque genomes (15 unrelated), from 13 Indian-origin, 6 Chinese-origin, and 2 hybrid animals. We performed high-depth whole genome sequencing, used pedigree information to validate genotypes, and generated a set of 16.6 million high confidence SNVs, which includes 12.8 million SNVs in Chinese macaques. Despite the improvements in the macaque genome, it has received less attention than other genomes such as human and mouse, and it has been demonstrated that an incomplete genome can lead to inaccurate SNV detection [7]. To help identify regions problematic for SNV detection, we used our alignment data to detect regions of the reference genome with systematic differences from our cohort, suggesting either undiscovered copy number differences or high intra-species diversity, identifying 173 genes likely to produce inaccurate or misleading variant data. This includes many polygeneic gene families, including many key immune genes, and represents an important caveat for genomic studies based in macaques and regions that should be prioritized for refinement. Within our set of validated SNVs, we focused on putative loss-of-function variants, identifying 613 total high impact single nucleotide variants (SNVs), with an average of 131 high impact SNVs per animal. We detected putative loss of function variants in many genes associated with human disease, including 45 variants identical to human variants identified as pathogenic, raising the potential for novel disease models. Together, these data advance our understanding of population-level variation in this important disease model, and improves our ability to perform accurate genome-wide genotyping.
Results
Whole Genome Sequencing and Alignment
We performed whole genome sequencing on 13 Indian-origin 6 Chinese-origin, and 2 hybrid macaques, obtaining an average of 558,371,583 reads per animal. These data were processed and aligned to the MacaM reference genome [9], providing an average genome-wide coverage of 25X (ranging from 14–48X). On average, 98.6% percent of reads mapped to the reference genome, and we did not observe significant differences in mapping rates between Indian and Chinese macaques, 98.5% and 98.7%, respectively. While the vast majority of reads aligned, we observed a slightly higher rate of high quality alignments (>Q20) in Chinese-origin animals, with 87.6% of reads mapping with >Q20, as compared to 86.5% in Indian-origin animals (Fig 1). We did not detect a significant difference in high-quality mapping rate by gender, with an average of 86.5% for females and 87.4% for males. Across all subjects, we obtained at least 10X coverage over an average of 92.8% of the genome.
Figure 1.
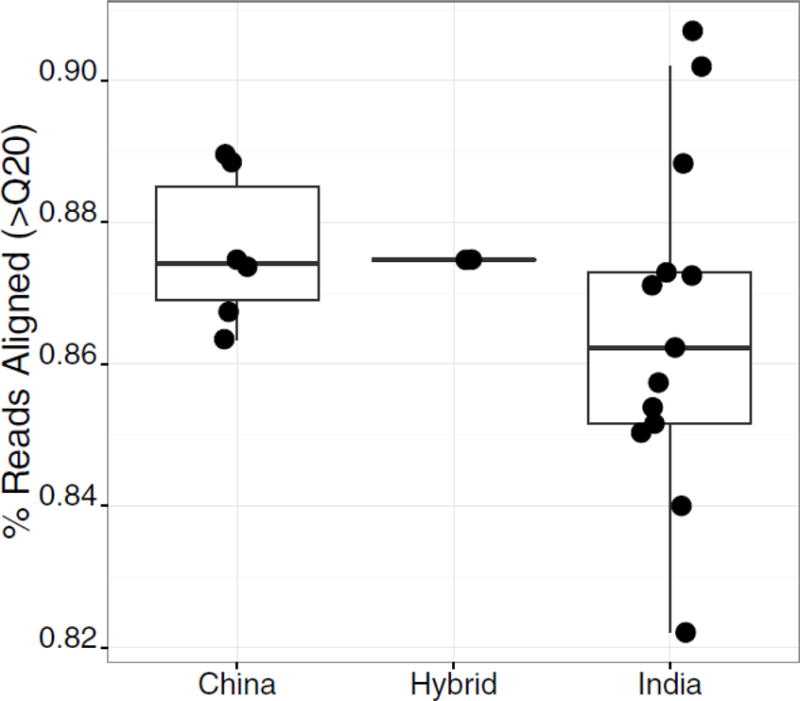
High quality mapping rates in Indian- and Chinese-origin animals. The fraction of reads aligning with high mapping quality (>Q20) is shown for each geographic origin. Dots indicate mapping rate of each individual.
Single Nucleotide Variant Discovery
We next used these alignments to identify single nucleotide variants (SNVs). Accurate variant calling in macaques presents several challenges. First, unlike human and many other common models, there is substantially less characterization of population-level variation and no curated databases of canonical variants or allele frequencies, such as the resources available for humans [4, 5, 21]. Second, macaques lack a high-density commercial genotyping SNP array, which is one of the most common techniques used to validate genotypes derived from whole genome sequencing. Finally, the reference genome has undergone less revision than the human genome, and an incomplete or inaccurate genome can cause inaccurate variant calls through the misalignment of reads. To overcome these issues we approached variant discovery in two phases: first we used best-practice variant discovery pipelines and Mendelian inheritance to generate an initial set of SNVs. Next, we used our alignment data to identify genomic regions likely to produce inaccurate variant calls through systematic misalignment, either due to genome errors or high intra-species variation.
Variant Filtration and Validation
Single nucleotide variants (SNVs) were called using an adapted version of the GATK Variant Discovery Pipeline [21], using hard filters to eliminate low confidence variants, as described in the methods. In addition to these filters, our cohort design included two trios (a child with both parents), and multiple parent/child pairs, which allowed us to use Mendelian inheritance to validate genotypes of these subjects. Prior to filtering, our raw data included 42,447,532 SNVs. We detected Mendelian violations in 303,278 of these sites (0.71%). After applying filters at both the site and genotype level, we generated a set of 16,605,886 passing SNVs. Of the sites where Mendelian violations were detected, 84% were removed by filters. The transition/transversion ratio (Ti/Tv) within these SNV data is 2.21, consistent with data from previous publications [13]. It is worth noting that of the filters applied, masking sites in repetitive regions was the most significant, responsible for 55.9% of filtered sites. Many of these repetitive regions are subject to more rapid evolution, meaning that the genomes of individuals are more likely be divergent from the macaque reference genome [22, 23]. This results in both genuine variation, and artifacts due to misalignment. In addition to manual inspection of variant calls using alignment data, we designed PCR amplicons to span the regions surrounding 7 SNVs and independently verified the genotypes obtained from WGS (Supplemental Table I).
Identification of Problematic Genomic Regions Using Alignment Data
We then took a second approach to validate these variants. Short read sequencing is highly dependent on the accuracy of the reference genome. If the genome is either incomplete or if the reference differs structurally from the genome of the individual being sequenced, sequence reads can misalign, causing both false positive and false negative genotypes. In addition to the fact that the macaque genome has undergone less revision than the human genome, the macaque genome was derived from a single individual, and even a perfect sequence of that individual will not represent the population at large across all regions. For example, there are many regions of the genome known for significant structural variation and segmental duplication between individuals [24, 25]. We hypothesized that we could use our alignment data to identify regions where our empirical data systematically differ from the reference genome, indicating either an incomplete reference genome or high structural variation across individuals. Identifying these regions is important both for the interpretation of experiments based on the current macaque genome and to prioritize genomic regions for refinement in the future. To identify these regions, we scanned all alignments to identify genomic positions where at least three alleles were detected in a single animal, and the third allele was present in at least 10% of reads. While in some cases these tri-allelic positions could represent sequencing errors, many indicate regions of the genome in which there is a copy number difference relative to the reference genome. We detected 580,530 tri-allelic positions across the genome with an average of 70,794 per animal (Fig 2). When we compared these locations against predicted genes, we detected 173 distinct genes with at least one tri-allelic position in at least one animal, suggesting a duplication in those subjects. There were 43 genes with tri-allelic positions in 10 or more animals, and 18 genes with tri-allelic positions in 100% of the cohort (Supplemental Table II). Many of these genes are known to be polygenic, including the leukocyte immunoglobulin-like receptor (LILR) family, the killer cell immunoglobulin-like receptor (KIR) family and the major histocompatibility class II DRB locus. These three families are key members of the immune response, including the response to many viral pathogens [26–28]. The systematic differences observed in our cohort relative to the reference genome suggests SNVs called over these regions may be artificially high. In addition to these immune-associated genes, we also detected pregnancy specific beta-1-glycoprotein (PSG3), a soluble protein secreted at high levels by the placenta [29], and ankyrin repeat domain 18A (ANKRD18A), which has been implicated in lung cancer [30]. It should be noted that because most genotyping tools are designed to predict diploid genotypes, these tri-allelic positions frequently may only manifest as a low quality or non-called genotypes. We excluded SNVs in these regions from subsequent analyses. After this second filter step, a total of 16,440,634 high-confidence SNVs remained, with an average of 4,414,820 SNVs per animal (Table I).
Figure 2.
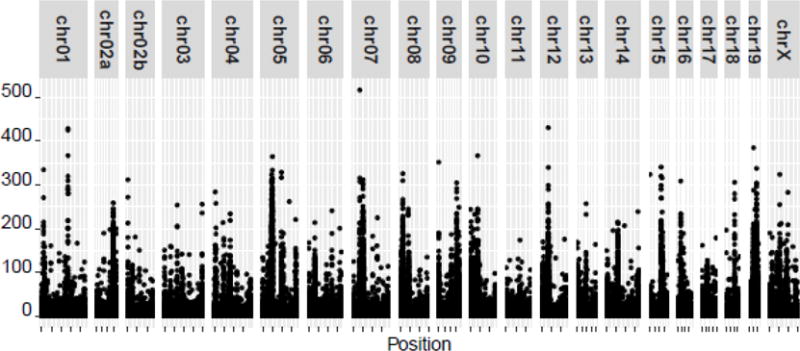
Discrepancies between reference genome and alignment data. All alignments against the MacaM reference genome were analyzed, and genomic positions where more than two alleles were detected in a single individual were identified. The graph shows the number of times these tri-allelic positions were detected in the genome.
Table I.
Summary of SNVs detected by geographic origin.
| Avg. Reads | High Confidence SNVs | Predicted High Impact SNVs | Predicted High Impact SNVs Per Animal | |
|---|---|---|---|---|
| All | 558,371,583 | 16,440,634 | 612 | 130.8 (94–169) |
| Indian-Origin | 560,852,139 | 10,067,896 | 371 | 120.1 (95–137) |
| Chinese-Origin | 552,170,192 | 12,846,731 | 427 | 147.8 (137–155) |
Comparison of Indian-origin and Chinese-origin SNVs
We next compared polymorphisms between Indian- and Chinese-origin macaques using these validated SNVs. The two hybrid macaques were excluded from these analyses. We detected significantly more SNVs in Chinese-origin macaques, with an average of 5,185,500 per animal, as compared to an average of 3,992,244 per Indian-origin macaque. A greater number of SNVs among Chinese macaques is not unexpected, given the macaque reference genome was generated from an Indian rhesus macaque and that others have reported higher sequence diversity in Chinese macaques [6, 13, 31]. This is mirrored by the observation that the Chinese origin animals have an average of 2.1 million homozygous non-reference sites per animal, as compared to 1.5 million per animal in Indian rhesus macaques. We also detected 753,143 SNVs with an allele frequency of 1.0 in Chinese-origin animals, as compared to only 116,648 in Indian-origin macaques. This suggests these are positions that are systematically different in Chinese-origin macaques; however, it should be noted that as more animals are sequenced it is possible shared minor alleles could be detected.
The majority of SNVs were unique to one population, with 38.3% detected only in Chinese macaques and 21.6% unique to Indian rhesus macaques (Fig 3A). A significant number of variants were rare, detected in only a single animal, representing 37.9% in the unrelated Indian-origin animals (n=15) and 40.6% in Chinese-origin animals. It should also be noted that large scale human studies have reported a slightly higher error rate in rare variants (97.1% concordance compared with >99% overall) [32], and similarly, there may be a slightly higher error rate in the singleton macaque variants compared to the broader dataset. The distribution of allele frequencies by geographic origin is shown in Figure 3B. To further evaluate the similarity of Indian-origin versus Chinese-origin animals, we performed identity-by-state (IBS) clustering on the cohort, including the 2 hybrid animals, using all 16.6 million SNVs. Animals from each geographic origin clustered together (Fig 3C), consistent with previous studies suggesting there is significant genetic separation between these two populations [19]. As expected, the two hybrid macaques are clustered between both Indian and Chinese animals (Fig 3C, red dots). A scree plot supporting Figure 3C in available in the supplemental material (Supplemental Figure S1).
Figure 3.
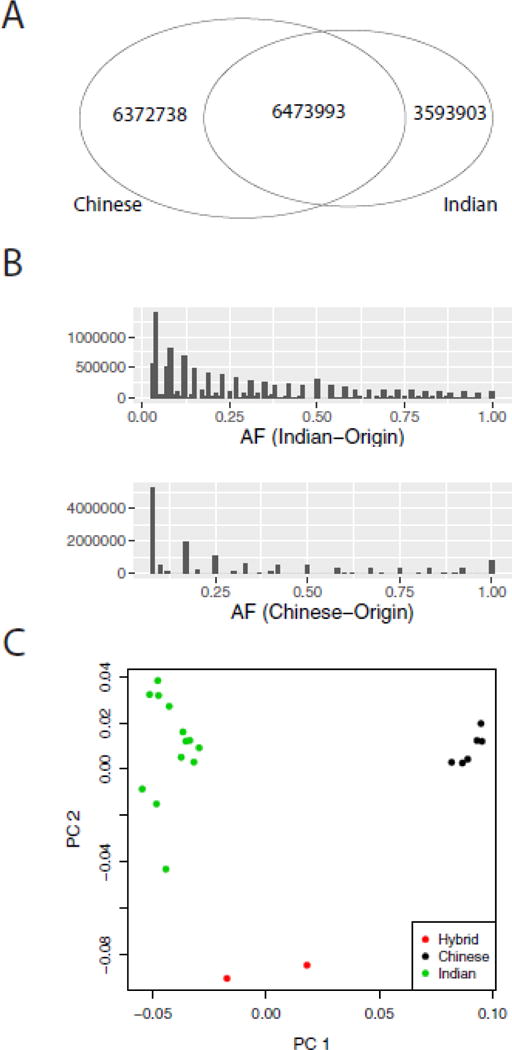
High confidence SNVs detected by population. A) Venn diagram showing overlap of high-confidence SNVs detected in Indian- and Chinese-origin animals. B) Histograms showing the distribution of allele frequency of variants detected in Indian-origin (top) and Chinese-origin (bottom) animals. C) Principal component analysis of IBS clustering using all high-confidence SNVs detected in our cohort. Indian-origin animals are in green, Chinese-origin animals are in black, and hybrid animals are in red.
Predicted Functional Consequence
To evaluate the functional consequence of the final SNV set, we employed SnpEff to calculate predicted effects [33]. As expected, the majority of SNVs were intergenic (99.1%). Of the remaining 149,693 SNVs, 36.2% are predicted to be missense, 63.6% to be silent, and 0.2% to be nonsense. A total of 613 SNVs are predicted to have high functional impact, which includes a loss of a start or stop codon, gain of a stop codon, and SNVs predicted to alter splicing (Supplemental Table II). It should be noted that predicted impacts are highly sensitive to the accuracy of the gene model, and we omitted 14 predicted high-impact SNVs because they had an allele frequency >0.9, which suggests a systematic error in gene annotations or predicted effects. Of the remaining SNVs, only 9 had an allele frequency >0.6, and a significant number, 42% (263), were detected in only a single individual within the cohort. Per animal, we observed an average of 130.8 predicted high impact SNVs (range 94–169). On average, Chinese origin animals had more SNVs predicted to have high impact with 147.8 per animal (range 137–155); however, the animal with the highest number of high impact SNVs (179) was a hybrid macaque.
The SNVs predicted to have high impact are located within 517 distinct genes (Supplemental Table III). We used the Reactome database to evaluate the pathways in which these genes are involved [34]. While no single pathway was overrepresented, the affected genes include those involved in a wide range of processes, including metabolism, the immune system, signal transduction, and DNA repair. Among the genes affected, a notable example was a mutation in the gene BTRC (chr10 97046465 C−>G), predicted to introduce a stop codon and result in a truncated protein. BTRC encodes a member of the F-box protein family, characterized by an approximately 40 amino acid motif that functions in phosphorylation-dependent ubiquitination. This stop-gain allele has a frequency of 0.024 in our cohort. The protein encoded by BTRC is involved in CD4 degradation through interaction with the HIV protein Vpu [35], and has also been shown to activate nuclear factor kappa-B (NFKB) through ubiquitination and degradation of NFKBIA [36]. A separate nonsense variant is predicted to truncate FAM120B, which has been associated with type I diabetes [37]. This variant had an allele frequency of 0.083 and was detected in a single Chinese-origin macaque.
Predicting functional consequence of the macaque SNVs is limited by incomplete genomic annotations. To overcome this, we mapped the rhesus macaque SNVs to the human genome (GRCh38) using liftover tools, with a total of 66.1% of SNVs successfully mapping. Of these, 870,174 (7.8%) mapped to sites that are polymorphic in humans (dbSNP build 144), which is slightly higher than the 3.2% overlap previously reported using dbSNP build 132 [12]. We next compared these lifted variants to ClinVar, a database of the clinical impact of genomic variants, to determine if any SNVs observed in macaques corresponded to SNVs implicated in human disease. A total of 676 macaque variants matched alleles published in ClinVar, and 45 of these were annotated as pathogenic. The latter were implicated in a range of disorders, including cardiomyopathy, immune disorders, muscular dystrophy, and neurological disorders such as early infantile epileptic encephalopathy, cancers (Supplemental Table IV).
Many functionally important mutations lie outside of coding regions, such as transcriptional regulatory elements. We therefore compared our variants to the ENCODE database of transcription factor binding sites (TFBS), and found that 1,685,536 macaque SNVs lie within regions annotated as TFBS [38]. While the presence of a variant within a TFBS does not inherently indicate an impact on transcription, and it should be noted that TFBS are frequently not conserved across species, it is likely at least some of these mutants will modulate transcriptional activity.
Discussion
The rapid decrease in the cost of sequencing and increase in computational capacity has dramatically changed the study of genetics and genomic medicine. To translate insights gained through these technologies into treatments, the genomic characterization of pre-clinical animal models will be essential. As a key pre-clinical model with a close evolutionary relationship to humans, better characterization of macaque genomics will provide direct benefits to the study of human disease. Macaques should provide a platform both to experimentally test mutations identified in humans and to detect novel disease-associated variants. From a relatively small cohort, we detected 47 variants that are identical to variants previously implicated as pathogenic in humans. As more genomic data becomes available in macaques, and as human databases of pathogenic variants grow, is it likely this will increase further. In addition to variants already implicated in human disease, we detected 613 SNVs predicted to have high impact on protein function. These data and other work [12–14] suggest existing breeding colonies may contain previously undetected models for many human disorders, which may be especially important for rare genetic disorders. Such animals may have been undetected either because individuals homozygous for a deleterious mutant do not survive, or because the symptoms of the disorder were not noticed.
The ability to capture variation across the full genome presents opportunities for established non-human primate disease models as well. Per animal, we detected an average of 131 variants predicted to significantly impact protein coding. This number is very likely an underestimate, as the impact of some mutations will be more subtle and difficult to predict from sequence alone. Previously undetected genetic differences between individuals could underlie variations in phenotype or disease outcome. For example, we identified predicted loss-of-function mutants in BTRC, a gene involved in nuclear factor kappa-B signaling, and a similar loss-of-function mutation in FAM120B, a gene implicated in type I diabetes. The increased availability and usage of genome-wide data in macaque studies will almost certainly reveal other clinically relevant variants.
Despite the potential of genomic study in macaques, the model has been slow to fully realize the benefits of genome sequencing. Most modern genomic analyses rely heavily on centralized resources developed for that species, including the reference genome, annotations, and characterization of population-level variation. Despite many efforts, macaques lag considerably behind many other organisms. While the first macaque genome was published in 2007, the next update was not published until seven years later. While the subsequent release of Mmul_8.0.1 in 2015 may indicate an increase in the pace of macaque genome improvements, the genome remains less complete than human and has far fewer annotations available. While the continued improvement in the macaque genome is clearly beneficial, new genome releases have also created difficulties and confusion for researchers in the field. Each subsequent release of the macaque genome continues to represent a much larger change than updates to more mature genomes like that of human. As such, publications generated against different macaque genome builds can be difficult to compare, and differing sets of annotations are available for each genome build. As new builds of the macaque genome become available, it is essential for these genomes to facilitate migration, such as including resources to lift variants and annotations from previous builds to that genome. The field needs greater collective effort to improve genome annotation, not only of genes, but also including regulatory regions and other features. It should also be noted that the fact that similar mapping rates are obtained from Chinese-origin and Indian-origin animals when aligned to an Indian-origin reference genome suggests that even though there are clear genetic differences between these animals, improving that quality of the existing Indian-origin genome is likely of higher priority than expanding into Rhesus macaques of distinct geographic origins.
Our data also highlight a challenge that exists for all genomes. Current technologies involve the fragmentation of the subject’s genome into short segments, which are then sequenced and aligned against a single linear reference genome. Alignment errors can arise in areas of segmental duplication or where the genome of the individual differs structurally from the reference genome. This problem is especially acute for study of genes with variable copy number. Understanding the limits of variant detection from genome-wide short read sequencing is important in order to evaluate and interpret genome-wide data, and can serve to inform and prioritize regions for genome improvement. We used the short read alignment data from our cohort to identify 173 genes with evidence of significant divergence from the reference genome, including many polygenic gene families important to the immune response, such as leukocyte immunoglobulin-like receptors (LILRs), killer cell immunoglobulin-like receptors (KIRs), and the major histocompatibility class II DRB locus. Even as the remaining gaps in the macaque reference are improved, the fact remains that it is derived from a single individual, and will not represent all individuals in the population over structurally variable regions. Alignment of short reads from individuals to this reference genome will continue to produce the issues we identified. One possibility to overcome this issue would be to generate a set of representative haplotypes for complex regions and use these to augment the reference genome as alternate contigs. Longer read sequencing technologies are improving in accuracy and may reduce the cost to generate these haplotypes. Additional third-generation sequencing methods, like the “synthetic long reads” from the 10X Genomics platform could also aid these efforts. In addition, for high-value loci, such as MHC class I, targeted assays have been developed to obtain highly accurate genotype data [39–41]; however, these assays must currently be performed in parallel with genome-wide approaches and targeted assays are unlikely to become available for all complex gene regions. Similarly, computational approaches have been developed for humans to infer the complete HLA genotype from whole-genome data [42]. While it is possible similar approaches could be developed for macaques; most strategies require a comprehensive reference database of known alleles, and therefore require considerable upfront resource investment.
While whole genome sequencing has been relatively rare to date in macaques, many historic obstacles, such as cost, are being reduced or removed. The data presented here and previous work demonstrate the ability to accurately detect variation genome-wide. Expanded genomic characterization in macaques could help identify genomic variants to explain disease phenotypes in existing macaque studies, or identify novel disease models. To facilitate these studies, it is essential that the field continue to invest in resources, such as continued refinement of the reference genome, annotations and validated variation data, necessary for high quality genomic studies.
Material and Methods
Animal Selection and Welfare
Twenty one rhesus macaques, six of Chinese-origin thirteen of Indian-origin, and two hybrids, all housed at the Oregon National Primate Research Center (ONPRC), were selected for this study. The Chinese-origin animals were directly imported from laboratory animal suppliers in China (Osage Research Primates, Oriental Scientific Instruments, National Laboratory Primate Center of Kunming, Laboratory Experimental Breeding Farm, Guandong, and Guandong Scientific Instruments). Eleven of the animals were females and ten were males. Fifteen of the animals were unrelated (9 Indian and 6 Chinese), and 6 Indian-origin animals were related. The cohort included two trios (an offspring with two parents), and multiple parent/child pairs. Pedigree relationships were molecularly validated. All of the ancestries were validated using a custom 96 SNP ancestry informative marker assay [19]. The animals were also selected with consideration of their medical histories, which indicated a diversity of medical or physical traits that are common among captive bred rhesus macaques. All macaque samples used in this study were collected during routine veterinary care procedures approved by the Institutional Animal Care and Use Committee of the Oregon Health & Science University (Protocol Number: IS00002621); these samples are part of the much larger ONPRC DNA Biobank. Animal care personnel and staff veterinarians of the ONPRC provide routine and emergency health care to all animals in accordance with the Guide for the Care and Use of Laboratory Animals, and the ONPRC is certified by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Genomic DNA Isolation and Whole Genome Sequencing
DNA was extracted from either whole blood, collected by venous puncture into an EDTA vacutainer (Fischer Scientific, Waltham, MA), or from a liver sample collected and flash frozen at necropsy. All sample collection protocols were approved by the Oregon Health & Sciences University Animal Utilization and Care Committee and in accordance with the NIH and the Guide for Use and Care of Laboratory Animals. DNAs were extracted using the Gentra PureGene Blood Kit following the manufacturers protocol or the ArchivePure DNA Blood Kit (5 Prime, Inc.), following the manufacturer’s recommendations. Genomic DNA was quantified with the Qubit High Sensitivity dsDNA Assay (Life Technologies, CA). Sequence libraries with 200–300 bp inserts were prepared using Illumina’s Paired-End DNA Sample Prep Kit. Each library was sequenced to produce 100 bp paired end reads on an Illumina HiSeq2000 or HiSeq3000 to achieve an average of 25X coverage per sample (ranging from 14–48X). The genomic sequencing was carried out by the Massively Parallel Sequencing Shared Resource (MPSSR) at the Oregon Health & Science University, the Center for Genome Research and Biocomputing at Oregon State University and by Axeq Technologies, Seoul, Korea. Primary FASTQ files have been submitted to SRA, BioProject PRJNA340145.
Analysis of Sequence Data
Whole genome data were processed using a pipeline following the best practice recommendations from the Broad Institute’s Genome Analysis Toolkit (GATK; [21, 43]), adapted for rhesus macaque. Briefly, paired-end reads were trimmed using Trimmomatic adaptive quality trimming [44], and aligned to the MacaM reference genome [9], using BWA-MEM [45]. The MacaM genome can be downloaded from: http://www.unmc.edu/rhesusgenechip/index.htm. BAM post-processing included local re-alignment around indels using GATK [43], and marking of duplicate reads using Picard tools [46]. GATK’s HaplotypeCaller was used to produce gVCF files, followed by genotype calling using GenotypeGVCFs. For the latter, a score of 20 was used as the threshold for calling and emitting variants. The resulting VCF was filtered at the site level using the following criteria: quality by depth (QD < 5.0), strand bias (FS > 15.0), mapping quality (MQ < 50.0), proximity to the read end (ReadPosRankSum < −8.0), the difference in mapping quality between reference and alternate reads (MQRankSum < −12.5), and single nucleotide variant (SNV) clusters of three SNVs within a 10 bp span were also filtered. In addition, and SNVs located within repetitive regions (identified using RepeatMasker [47] were filtered for removal. In addition, individual genotypes were filtered if the genotype was supported by fewer than 10X read depth, sites with greater than 100X coverage, or the genotype quality score was less than 20. To identify regions of the genome where our empirical data systematically differs from the reference sequence, we wrote a custom GATK Walker designed to iterate across a series of BAM files and report any positions where three or more alleles are detected in a single subject (available at: https://cpas:cpas@hedgehog.fhcrc.org/tor/stedi/trunk/externalModules/labModules/SequenceAnalysis/pipeline_code/gatk/MultipleAllelesAtLoci.java). These analyses also employed Picard tools [46] and FASTQC [48] for quality control of the raw data, and JBrowse [49] to visualize the resulting data. Sequence data were managed and analyzed using DISCVR-Seq [50], a LabKey Server-based system [51]. All pipelines and tools used in this manuscript have been incorporated into DISCVR-Seq. SnpEff was utilized to calculate predicted effects for variants [33]. Variant data were submitted to dbSNP under BioProject PRJNA340145.
Identity by state genotype clustering and principal component analysis were performed using PLINK [52, 53]. Macaque SNVs were mapped to human coordinates using LiftOver tools [54]. Lifted variants were annotated against the ClinVar database [55], ENCODE transcription factor binding sites [38] and dbSNP [56] using Annovar [57].
SNP Validation
Selected SNVs were PCR amplified for sequence validation using flanking primers. Amplification products were sequenced on an ABI 3130XL Genetic Analyzer (Applied Biosystems, Inc., Foster City, CA). The sequences were analyzed using CodonCode Aligner software to detect variant alleles (CodonCode Corporation, Centerville, MA). A list of primers is available in Supplemental Table I.
Supplementary Material
Supplemental Figure 1: Scree plot of eigenvalues associated with principal component analysis shown in Figure 3C.
{kind=link}
Supplemental Table I: Sequence verification of WGS data. The genomic position, and the primers used for amplification are listed. In addition, the total number of animals sequence verified, and the number of these animals with a non-WT genotype are shown.
Supplemental Table II: Predicted high-impact macaque SNVs. List of all SNVs that are predicted to have a damaging protein impact, including genomic position, gene, allele frequency, and predicted effect.
Supplemental Table III: SNVs matching pathogenic variants from the ClinVar database. Macaque SNVs were lifted to the human genome and compared to the ClinVar database. All variants annotated as pathogenic are listed below, including the genomic position (build GRCh38), accession, and disorder listed in ClinVar.
Supplemental Table IV: Genes with evidence of copy number variation relative to the reference genome. All alignments against the MacaM reference genome were analyzed, and genomic positions for which more than two alleles were detected, suggesting mismapping, were identified. Any genes with exons intersecting these positions are shown, including the number of distinct animals and number of distinct genomic positions with that gene where conflicting reads were detected.
Highlights.
Whole genome sequencing was performed on 21 rhesus macaques, including Indian- and Chinese-origin animals.
Identified >16 million high-confidence SNVs using complementary approaches to generate accurate genome-wide genotypes.
Per animal, we identified an average of 131 SNVs predicted to have high-impact on protein coding, along with 45 SNVs that coincide with known human pathogenic variants.
Suggests that expanded screening of existing breeding colonies will identify novel models of human disease, and increased genomic characterization can help inform research studies in macaques
Acknowledgments
The authors thank Benjamin Popescu and Elizabeth Swanson for technical support in this project, and Dr. Lina Gao for statistical advice. This work was funded by NIH grants P51OD011092, R24RR017444, and R24OD021324.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Francis PJ, Appukuttan B, Simmons E, Landauer N, Stoddard J, Hamon S, et al. Rhesus monkeys and humans share common susceptibility genes for age-related macular disease. Hum Mol Genet. 2008;17(17):2673–80. doi: 10.1093/hmg/ddn167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rogers J, Shelton SE, Shelledy W, Garcia R, Kalin NH. Genetic influences on behavioral inhibition and anxiety in juvenile rhesus macaques. Genes Brain Behav. 2008;7(4):463–9. doi: 10.1111/j.1601-183X.2007.00381.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goulder PJ, Watkins DI. Impact of MHC class I diversity on immune control of immunodeficiency virus replication. Nat Rev Immunol. 2008;8(8):619–30. doi: 10.1038/nri2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Genomes Project C. Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International HapMap C. The International HapMap Project. Nature. 2003;426(6968):789–96. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 6.Rhesus Macaque Genome S. Analysis C, Gibbs RA, Rogers J, Katze MG, Bumgarner R, et al. Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316(5822):222–34. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- 7.Zhang X, Goodsell J, Norgren RB., Jr Limitations of the rhesus macaque draft genome assembly and annotation. BMC Genomics. 2012;13:206. doi: 10.1186/1471-2164-13-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Norgren RB., Jr Improving genome assemblies and annotations for nonhuman primates. ILAR J. 2013;54(2):144–53. doi: 10.1093/ilar/ilt037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zimin AV, Cornish AS, Maudhoo MD, Gibbs RM, Zhang X, Pandey S, et al. A new rhesus macaque assembly and annotation for next-generation sequencing analyses. Biol Direct. 2014;9(1):20. doi: 10.1186/1745-6150-9-20.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yan G, Zhang G, Fang X, Zhang Y, Li C, Ling F, et al. Genome sequencing and comparison of two nonhuman primate animal models, the cynomolgus and Chinese rhesus macaques. Nat Biotechnol. 2011;29(11):1019–23. doi: 10.1038/nbt.1992. [DOI] [PubMed] [Google Scholar]
- 11.Groves C. Primate Taxonomy. Washington, DC: Smithsonian Institution Press; 2001. p. 350. [Google Scholar]
- 12.Fawcett GL, Raveendran M, Deiros DR, Chen D, Yu F, Harris RA, et al. Characterization of single-nucleotide variation in Indian-origin rhesus macaques (Macaca mulatta) BMC Genomics. 2011;12:311. doi: 10.1186/1471-2164-12-311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan Q, Zhou Z, Lindell SG, Higley JD, Ferguson B, Thompson RC, et al. The rhesus macaque is three times as diverse but more closely equivalent in damaging coding variation as compared to the human. BMC Genet. 2012;13:52. doi: 10.1186/1471-2156-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cornish AS, Gibbs RM, Norgren RB., Jr Exome screening to identify loss-of-function mutations in the rhesus macaque for development of preclinical models of human disease. BMC Genomics. 2016;17:170. doi: 10.1186/s12864-016-2509-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vallender EJ. Expanding whole exome resequencing into non-human primates. Genome Biol. 2011;12(9):R87. doi: 10.1186/gb-2011-12-9-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xue C, Raveendran M, Harris RA, Fawcett GL, Liu X, White S, et al. The population genomics of rhesus macaques (Macaca mulatta) based on whole-genome sequences. Genome Res. 2016;26(12):1651–62. doi: 10.1101/gr.204255.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross CT, Roodgar M, Smith DG. Evolutionary distance of amino acid sequence orthologs across macaque subspecies: identifying candidate genes for SIV resistance in Chinese rhesus macaques. PLoS One. 2015;10(4):e0123624. doi: 10.1371/journal.pone.0123624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou Y, Bao R, Haigwood NL, Persidsky Y, Ho WZ. SIV infection of rhesus macaques of Chinese origin: a suitable model for HIV infection in humans. Retrovirology. 2013;10:89. doi: 10.1186/1742-4690-10-89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kanthaswamy S, Johnson Z, Trask JS, Smith DG, Ramakrishnan R, Bahk J, et al. Development and validation of a SNP-based assay for inferring the genetic ancestry of rhesus macaques (Macaca mulatta) Am J Primatol. 2014;76(11):1105–13. doi: 10.1002/ajp.22290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ng J, Trask JS, Smith DG, Kanthaswamy S. Heterospecific SNP diversity in humans and rhesus macaque (Macaca mulatta) J Med Primatol. 2015;44(4):194–201. doi: 10.1111/jmp.12174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11 0 1–33. doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jansen A, Gemayel R, Verstrepen KJ. Unstable microsatellite repeats facilitate rapid evolution of coding and regulatory sequences. Genome Dyn. 2012;7:108–25. doi: 10.1159/000337121. [DOI] [PubMed] [Google Scholar]
- 23.Rebollo R, Horard B, Hubert B, Vieira C. Jumping genes and epigenetics: Towards new species. Gene. 2010;454(1–2):1–7. doi: 10.1016/j.gene.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 24.Dennis MY, Eichler EE. Human adaptation and evolution by segmental duplication. Curr Opin Genet Dev. 2016;41:44–52. doi: 10.1016/j.gde.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol. 2005;5(3):201–14. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 26.Hudson LE, Allen RL. Leukocyte Ig-Like Receptors – A Model for MHC Class I Disease Associations. Front Immunol. 2016;7:281. doi: 10.3389/fimmu.2016.00281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gardiner CM. NK cell function and receptor diversity in the context of HCV infection. Front Microbiol. 2015;6:1061. doi: 10.3389/fmicb.2015.01061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martin MP, Carrington M. Immunogenetics of HIV disease. Immunol Rev. 2013;254(1):245–64. doi: 10.1111/imr.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moore T, Dveksler GS. Pregnancy-specific glycoproteins: complex gene families regulating maternal-fetal interactions. Int J Dev Biol. 2014;58(2–4):273–80. doi: 10.1387/ijdb.130329gd. [DOI] [PubMed] [Google Scholar]
- 30.Liu WB, Han F, Jiang X, Yang LJ, Li YH, Liu Y, et al. ANKRD18A as a novel epigenetic regulation gene in lung cancer. Biochem Biophys Res Commun. 2012;429(3–4):180–5. doi: 10.1016/j.bbrc.2012.10.116. [DOI] [PubMed] [Google Scholar]
- 31.Street SL, Kyes RC, Grant R, Ferguson B. Single nucleotide polymorphisms (SNPs) are highly conserved in rhesus (Macaca mulatta) and cynomolgus (Macaca fascicularis) macaques. BMC Genomics. 2007;8:480. doi: 10.1186/1471-2164-8-480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cingolani P, Platts A, Wang le L, Coon M, Nguyen T, Wang L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly (Austin) 2012;6(2):80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, et al. The Reactome pathway knowledgebase. Nucleic Acids Res. 2014;42:D472–7. doi: 10.1093/nar/gkt1102. (Database issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bour S, Perrin C, Akari H, Strebel K. The human immunodeficiency virus type 1 Vpu protein inhibits NF-kappa B activation by interfering with beta TrCP-mediated degradation of Ikappa B. J Biol Chem. 2001;276(19):15920–8. doi: 10.1074/jbc.M010533200. [DOI] [PubMed] [Google Scholar]
- 36.Latres E, Chiaur DS, Pagano M. The human F box protein beta-Trcp associates with the Cul1/Skp1 complex and regulates the stability of beta-catenin. Oncogene. 1999;18(4):849–54. doi: 10.1038/sj.onc.1202653. [DOI] [PubMed] [Google Scholar]
- 37.Bradfield JP, Qu HQ, Wang K, Zhang H, Sleiman PM, Kim CE, et al. A genome-wide meta-analysis of six type 1 diabetes cohorts identifies multiple associated loci. PLoS Genet. 2011;7(9):e1002293. doi: 10.1371/journal.pgen.1002293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Consortium EP. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489(7414):57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heimbruch KE, Karl JA, Wiseman RW, Dudley DM, Johnson Z, Kaur A, et al. Novel MHC class I full-length allele and haplotype characterization in sooty mangabeys. Immunogenetics. 2015;67(8):437–45. doi: 10.1007/s00251-015-0847-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Westbrook CJ, Karl JA, Wiseman RW, Mate S, Koroleva G, Garcia K, et al. No assembly required: Full-length MHC class I allele discovery by PacBio circular consensus sequencing. Hum Immunol. 2015;76(12):891–6. doi: 10.1016/j.humimm.2015.03.022. [DOI] [PubMed] [Google Scholar]
- 41.Wiseman RW, Karl JA, Bimber BN, O’Leary CE, Lank SM, Tuscher JJ, et al. Major histocompatibility complex genotyping with massively parallel pyrosequencing. Nat Med. 2009;15(11):1322–6. doi: 10.1038/nm.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 2014;30(23):3310–6. doi: 10.1093/bioinformatics/btu548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20(9):1297–303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–20. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(5):589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tools P. Picard Tools: A set of command line tools (in Java) for manipulating high-throughput sequencing (HTS) data. Available from: http://broadinstitute.github.io/picard/
- 47.Smit A, Hubley R, Green P. RepeatMasker Open-4.0, 2013–2015. Available from: http://www.repeatmasker.org.
- 48.Andrews S. FastQC: a quality control tool for high throughput sequence data 2010. Available from: http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- 49.Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Res. 2009;19(9):1630–8. doi: 10.1101/gr.094607.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bimber B. DISCVR-Seq: LabKey Server Extensions for Management and Analysis of Sequencing Data 2015. Available from: https://github.com/bbimber/discvr-seq/wiki.
- 51.Nelson EK, Piehler B, Eckels J, Rauch A, Bellew M, Hussey P, et al. LabKey Server: an open source platform for scientific data integration, analysis and collaboration. BMC Bioinformatics. 2011;12:71. doi: 10.1186/1471-2105-12-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. doi: 10.1186/s13742-015-0047-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81(3):559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kent WJ. BLAT–the BLAST-like alignment tool. Genome Res. 2002;12(4):656–64. doi: 10.1101/gr.229202. Article published online before March 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–8. doi: 10.1093/nar/gkv1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29(1):308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Scree plot of eigenvalues associated with principal component analysis shown in Figure 3C.
Supplemental Table I: Sequence verification of WGS data. The genomic position, and the primers used for amplification are listed. In addition, the total number of animals sequence verified, and the number of these animals with a non-WT genotype are shown.
Supplemental Table II: Predicted high-impact macaque SNVs. List of all SNVs that are predicted to have a damaging protein impact, including genomic position, gene, allele frequency, and predicted effect.
Supplemental Table III: SNVs matching pathogenic variants from the ClinVar database. Macaque SNVs were lifted to the human genome and compared to the ClinVar database. All variants annotated as pathogenic are listed below, including the genomic position (build GRCh38), accession, and disorder listed in ClinVar.
Supplemental Table IV: Genes with evidence of copy number variation relative to the reference genome. All alignments against the MacaM reference genome were analyzed, and genomic positions for which more than two alleles were detected, suggesting mismapping, were identified. Any genes with exons intersecting these positions are shown, including the number of distinct animals and number of distinct genomic positions with that gene where conflicting reads were detected.