

Graphical abstract
Graphical abstract key: Wt: weight, Ht: height, OFC: open fontanelle, DD: developmental delay, ID: intellectual disability, EEG: electroencephalogram, OCD: obsessive compulsive disorder, ADHD: attention deficit hyperactivity disorder, ASD: atrial septal defect, VSD: ventricular septal defect, PDA: patent ductus arteriosis, PFO: patent foramen ovale.
Keywords: de novo, intellectual disability, PACS1, seizures, whole exome sequencing
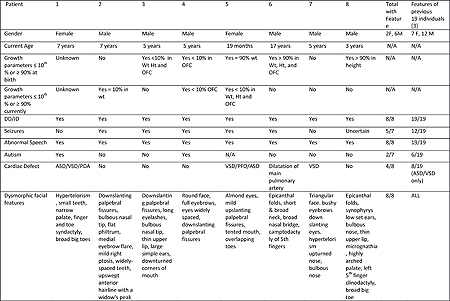
A recurrent de novo missense variant, p.Arg203Trp was identified within the binding domain of PACS1 in nineteen individuals [1-3] who all share an overlapping phenotype including intellectual disability (ID), delayed language development, dysmorphic craniofacial features, and seizures [1-3]. Using whole exome sequencing (WES), we identified eight additional individuals with the same p.Arg203Trp missense variant in PACS1 that we demonstrated were de novo in seven individuals with available parental DNA. Consistent with the previous studies, all eight individuals of our series have ID, impaired language development, and similar dysmorphic facial features (Table 1)[3]. The dysmorphic features include hypertelorism, downslanting palpebral fissures, epicanthal folds, flat and long philtrum, and bulbous nasal tip. Speech and language in our cohort was universally delayed but developed in most, and three demonstrated dysarthria. Four individuals were unable to speak more than a few words within the first 3 years of life. Autism was diagnosed in two individuals, and four individuals have behavioral challenges including obsessive compulsive disorder, attention deficit hyperactivity disorder, self-injurious behaviors, outbursts, aggression, impulsivity, tactile aversions, and hand flapping. Seizures were observed in five individuals, which included Rolandic epilepsy, complex partial seizures, absence seizures, generalized and focal tonic-clonic seizures, and drop seizures. Cardiac defects were noted in four individuals, including two individuals with atrial septal defects, three with ventricular septal defects, and one individual with a dilated pulmonary artery. The majority of individuals in our series had gastroesophageal reflux, and constipation was common. Additionally, one individual required a gastrostomy-tube, and oral aversion was reported in two individuals. At their current ages, several individuals had lower than average height, weight, and/or head circumference.
The p.Arg203Trp allele is predicted to be damaging by prediction algorithms including PolyPhen2, SIFT, Mutation Taster, and CADD. PACS1 is intolerant to variation and has a Residual Variance Intolerance Score (RVIS) score of 4.1% and subRVIS suggests that it falls within an intolerant region of PACS1. Additionally, ExAC gene-level constraint scores of misZ = 4.32 (4-sigma conservation against missense variations) and pLI = 1.0 (100% likelihood the gene is loss-of-function intolerant), based on ExACv0.3.1 non-TCGA data, indicating that PACS1 is strongly constrained against both missense and loss-of-function variants.
PACS1 is responsible for trafficking proteins in the trans-Golgi-network [4]. PACS1 localizes proteins such as furin, and cation independent mannose-6-phosphate receptor (CI-MPR) by recognizing and binding to their acidic clusters detected by PACS1's furin-binding region (FBR) from amino acids 117 to 266 [2,4]. Within the FBR, there is a CK-2-binding motif from amino acids 196 to 200 [2]. The p.Arg203Trp missense variant is located within the FBR and three amino acid residues downstream from the CK2 binding motif [2]. The CK-2 binding motif is a target of CK-2 phosphorylation and PP2A dephosphorylation, which regulates cargo binding and PACS1 activation [5]. The close proximity of this important regulatory region and the p.Arg203Trp missense variant suggests that the variant could disrupt regulation leading to mislocalization of proteins [2, 3].
Due to the recurrence of the same specific p.Arg203Trp missense variant, similar phenotypes across the twenty seven patients, the intolerance of PACS1 to genetic variation, and consistent in silico predictions of pathogenicity, Shuurs-Hoeijmakers' proposal that this variant is acting via a dominant-negative or a gain–of-function is likely correct [2, 3].
Acknowledgments
We thank the patients and their families for their generous participation, Jessica Hoffman and Tara Funari for compiling clinical information, and support from NIH2T35HL007616-36 and the Simons Foundation.
Footnotes
Conflicts of Interest: Megan Cho, Rashmi Chikarmane, Rebecca Willaert, Kyle Retterer, Berivan Baskin, Maria Guillen Sacoto, Ingrid Wentzensen, Heather McLaughlin, and Dianalee McKnight are employees of GeneDx. Wendy Chung is a former employee of GeneDx and a member of the Scientific Advisory Board Regeneron Genetics Center. Dr. Schrier Vergano is a member of the Scientific Advisory Board for Ambry Genetics.
Ethical Statement: This study was approved by the Institutional Review Board of Columbia University.
References
- 1.Gadzicki D, Docker D, Schubach M, et al. Expanding the phenotype of a recurrent de novo variant in PACS1 causing intellectual disability. Clin Genet. 2015;88(3):300–2. doi: 10.1111/cge.12544. [DOI] [PubMed] [Google Scholar]
- 2.Schuurs-Hoeijmakers JH, Landsverk ML, Foulds N, et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectual-disability syndrome. Am J Hum Genet. 2012;91(6):1122–7. doi: 10.1016/j.ajhg.2012.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schuurs-Hoeijmakers JH, Oh EC, Vissers LE, et al. Clinical delineation of the PACS1-related syndrome--Report on 19 patients. Am J Med Genet A. 2016;170(3):670–5. doi: 10.1002/ajmg.a.37476. [DOI] [PubMed] [Google Scholar]
- 4.Wan L, Molloy SS, Thomas L, et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998;94(2):205–16. doi: 10.1016/s0092-8674(00)81420-8. [DOI] [PubMed] [Google Scholar]
- 5.Scott GK, Gu F, Crump CM, et al. The phosphorylation state of an autoregulatory domain controls PACS-1-directed protein traffic. EMBO J. 2003;22(23):6234–44. doi: 10.1093/emboj/cdg596. [DOI] [PMC free article] [PubMed] [Google Scholar]