

Abstract
The Protein Data Bank (PDB) has been an integral resource for shaping our fundamental understanding of protein structure and for the advancement of such applications as protein design and structure prediction. Over the years, information from the PDB has been used to generate models ranging from specific structural mechanisms to general statistical potentials. With accumulating structural data, it has become possible to mine for more complete and complex structural observations, deducing more accurate generalizations. Motif libraries, which capture recurring structural features along with their sequence preferences, have exposed modularity in the structural universe and found successful application in various problems of structural biology. Here we summarize recent achievements in this arena, focusing on sub-domain level structural patterns and their applications to protein design and structure prediction, and suggest promising future directions as the structural database continues to grow.
Introduction
The observation that proteins exhibit recurring structural motifs, ranging from secondary structure elements (SSE) to domains, has in many ways shaped the development of structural biology, providing insights into sequence determinants of structure and function, and enabling the classification of protein structure space [1-6]. This review focuses on local structural patterns that recur at the sub-domain level, involving one or several SSE fragments. Such patterns provide a potentially potent combination of high degree of detail, allowing for the possibility to discern quantitative sequence-structure relationships, with generally high degree of recurrence in the structural database, strengthening associated statistics. Whereas some such patters may be associated with specific functions (e.g., phosphorylation [4], small molecule binding [5], or catalysis [6]), others recur in a wide range of functional and evolutionary contexts, indicating that the structural universe is fundamentally modular. This modularity has been recognized for some time [7], but more recent analyses, armed with considerably more data, have described it in higher detail, leading to new insights. For example, Kolodny and co-workers modeled domain space as a network, where domains were connected by sub-regions of similar structure and sequence, showing that this space is modular with regions of both low and high connectivity [8]. In another recent analysis of sub-domain modularity, Lupas and colleagues discovered a set of 40 super-secondary motifs representing possible remnants of a primordial RNA-peptide world by looking for similar sequence-structure patterns across unrelated proteins [9].
Fragment libraries—collections of (typically short) structural segments that are highly recurrent across proteins—have been an important means of describing protein structural modularity. In addition to being of fundamental interest, such libraries have enabled advancements in modeling, prediction, and design applications (see Figure 1). For example, Fernandez-Fuentes et al. generated the Smotif library of super-secondary motifs, which they defined as two sequence-adjacent SSEs connected by a loop [10,11]. The authors used four geometric parameters that describe the relative orientation of adjacent SSEs to group all Smotifs instances into just 324 types [12]. Importantly, they found that even novel folds can be described as combinations of previously seen Smotifs (i.e., those found in pre-existing folds), indicating that while new folds are globally different from previous ones, local structural patterns within them are saturated [12]. On the other hand, novel folds did show different patterns of Smotif utilization, including higher usage of rare motifs. The group has shown the utility of Smotifs in loop modeling [10,11], NMR structure determination [13], and structure prediction [14]. Other attempts to discern motifs that make up the structural universe have focused to classifying contigious backbone segments by root-mean-square-deviation (RMSD) upon optimal superposition. The FragBag library clustered segments of 4-7 residues by Ca RMSD [15]. The pattern of motif usage within proteins was sufficient to identify structural neighbors at a level comparable to advanced structural alignment methods like CE and STRUCTAL [16]. The BRIX project took advantage of increased data and computational power to create a thorough database of fragments grouped by length (4-14 residues) and clustered by backbone RMSD at a variety of thresholds (0.5 to 1.0 Å) [17]. While the BRIX database is a highly compressed representation of local backbone structure space (e.g. ∼2,500 clusters at 0.7 Å out of ∼260,000 7-residue fragments), it was nevertheless sufficient to reconstruct backbones of previously unseen native proteins to an average accuracy of ∼0.5 Å [17]. BRIX fragments have been extended to the modeling of loops [18], interface geometries [19], and protein-peptide docking [20].
Figure 1.
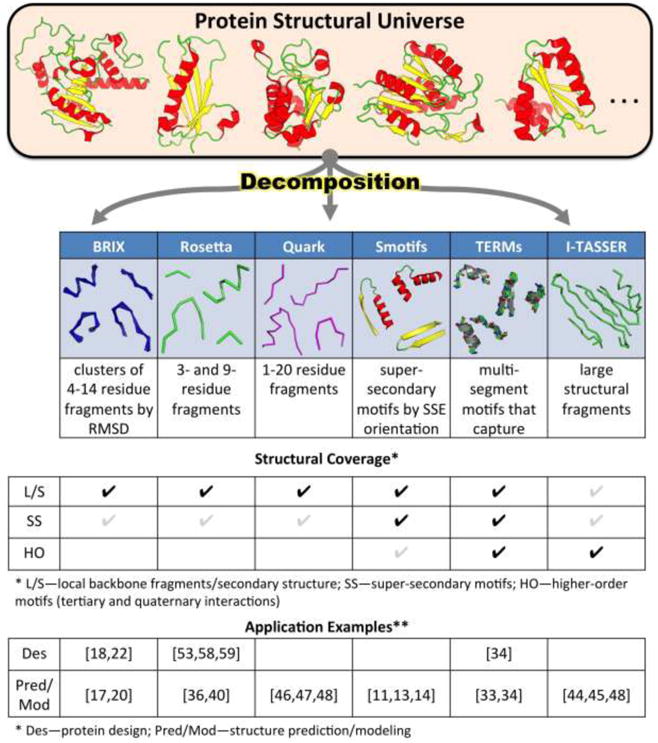
Decomposition of protein structure space into motif libraries has revealed considerable modularity across the structural hierarchy, providing useful insights for protein design and structure prediction applications. Shown are six recent examples of motif libraries, each covering local-backbone and secondary (L/S), super-secondary (SS), or higher-order (HO) structural information to different extents (black, gray, or missing checkmarks in the structural coverage table correspond to detailed, sparse, or low coverage). Shown on the bottom are examples of uses of each motif library in either design or prediction/modeling applications (examples denoted by references).
Beyond local backbone geometries, there have been efforts towards describing modularity at the tertiary and quaternary structural levels [19,21-30], which requires consideration of motifs with multiple disjoint segments. Several studies have characterized helix-helix associations, showing that they can be described with a small set of structural classes or restricted parameters [23-27]. Grigoryan and Degrado used a generalized set of parameters to describe the structural space of helical bundles [25], showing that coiled coils largely sampled near-ideal parametric structures. By breaking multipass transmembrane (TM) proteins into interacting three-segment helical motifs, Feng and Barth found that the TM regions of these proteins consist of six major structural classes, each dominated by only a few sequence motifs [26]. Another recent study revealed that interacting helical segments in TM and soluble proteins can be described with a small number of shared structural classes, even though there were important differences in sequence preferences and hydrogen bonding patterns [27]. Recent work has also found considerable modularity at the level of quaternary structure [19,21,22,28-30]. For example, pairs of interacting BRIX fragments from monomeric proteins were used to show that 65% of protein-peptide interactions were similar to structural motifs in monomeric folds [19]. Also, protein-protein interactions were characterized with ∼2,000 motifs created by clustering two-segment fragments built around quaternary contacts [30], with many of these found in a wide range of contexts (e.g., quaternary interactions between different domains).
Still, in comparison to the many of ways in which secondary and super-secondary structure has been classified, understanding of the degeneracy and modularity of tertiary and quaternary structural levels (higher-order degeneracy) has lagged behind. In part, this is due to the greater ambiguity associated with both defining and classifying tertiary structural motifs—i.e., motifs that are not necessarily contiguous in sequence. In recent years, we have been working towards addressing these issues to enable a more thorough and general decomposition of structure space across all structural levels. We have developed efficient structure search algorithms, MaDCaT and MASTER, that find all matches to a given query, composed of one or more disjoint segments, within a user-specified similarity cutoff [31,32]. We have used MASTER, which searches by backbone RMSD, to describe and take advantage of higher-order degeneracies [33,34]. Towards this, we adopted a common definition of a motif that captures the secondary, tertiary, and quaternary structural environments around a given central residue. Referred to as a TERM (tertiary motif), this motif is defined as the union of the local backbone fragment around the central residue (e.g., ± 2 residues around it) with that around all residues with which the central residue can form interactions [33]. We have shown that TERMs can be used to blindly evaluate the quality of structural models. Specifically, a TERM was defined for every residue in a given model, and MASTER was used to search for closely matching geometries in the PDB. From this, we quantified whether 1) the TERM represented a common designable geometry, and 2) sequence features emergent from the TERM's matches (from diverse proteins) agreed with the corresponding sequence region of the modeled protein [33]. The resulting score showed a strong correlation with model quality, evaluated as its distance from the native structure, and was further able to identify poorly predicted regions. We have also described a minimal set of TERMs that captures the observable structural universe (Figure 1) [34]. This has revealed considerable degeneracy beyond the secondary-structural level, such that only ∼600 TERMs are sufficient to describe over 50% of the known protein structural universe at sub-Angstrom resolution. Further, we have shown that TERMs, along with their structural matches from unrelated proteins, provide an effective mapping between sequence and structure. For example, TERM-based statistics alone recapitulate close-to-native sequences given either NMR or X-ray backbones, with corresponding predicted sequence variations in close agreement with evolutionary ones [34].
Protein Structure Prediction
Fragment libraries have a long history of use in protein structure prediction. Typically, short fragments are selected based on the predicted secondary structure of the target and are used to bias structural sampling towards models more likely to be compatible with the target sequence [35,36]. Combined with other forms of structural sampling and knowledge-based potentials, fragment-based prediction methods are among the best performers in the free modeling (FM) section of the semi-annual CASP competition (Critical Assessment of Structure Prediction) [37,38]. The highly successful structure prediction program Rosetta uses contiguous fragments of 3-9 residues to enhancing structural sampling [39,40] and has been a top performer at CASP meetings [37,38]. Rosetta's fragment sampling method, which uses torsion angles from fragments to replace those in the modeled structure guided by a Monte Carlo simulation, has also been applied to NMR structure determination [41], molecular replacement [42], and cryo-EM structure refinement [43].
Another top CASP performer, I-TASSER, threads the protein onto a set of representative structures to identify larger fragments (from super-secondary motifs to folds) that are compatible with regions of the target sequence and predicted secondary structure [44,45]. These fragments are assembled into structural models in a Monte Carlo simulation while ab-initio modeling is used for unaligned regions. The preliminary structural models are clustered and structurally matching fragments from the PDB are identified for another round of fragment assembly. The Quark structure prediction method, which was ranked number one in free modeling in CASP 9-11, uses short fragments to construct models in a Monte Carlo simulation with a knowledge based potential [46,47]. Unlike I-TASSER, it does not require templates from homologs to generate fragments. Instead, it finds fragments from a database of known (but non-homologous) structures based on sequence and predicted secondary structure similarity to short regions of the target sequence. In addition, an inter-residue distance-based energy term is derived from pairs of fragments in the PDB. A pipeline has been developed in which Quark is used to produce initial models for I-TASSER to refine, improving the accuracy for models in both the FM and Template-Based Modeling categories of CASP [48]. Smotifs have also been applied to structure prediction, showing that they can identify the correct global fold from sequence alone about half of the time, in close competition with other state-of-the-art methods [14]. In this approach, locations of putative Smotifs within the target are predicted from sequence alone, and structural models are generated by replacing these predictions with combinations of actual Smotifs, with matching SSE types, generated from structures of remote homologues.
By taking advantage of higher-order structural degeneracies, it may be possible to identify contacting structural segments that are not necessarily close in sequence—a highly challenging aspect of structure prediction. Although methods based on evolutionary co-variation have shown considerable promise towards predicting contacts in recent years [49-51], such prediction applies only to native proteins and works best in cases with available deep evolutionary sequence alignments. On the other hand, universal structural degeneracies could apply equally well to evolutionarily ancient, relatively novel, or even engineered proteins. In our own work, we have shown that the presence and location of multi-segment TERMs can be identified within proteins based purely on sequence information [34]. We built sequence models for each multi-segment TERM, from corresponding structural matches, and scored all alignments in previously unseen sequences to identify likely TERM positions. Despite the number of possible alignments growing exponentially with the number of disjoint TERM segments, structurally correct alignments were highly enriched in the resulting predictions, whether the method was applied to native or de novo designed proteins [34].
Protein Design
Motifs can also be used to limit structure space in design. Reusing naturally occurring local geometries effectively focuses on parts of structural space more likely to be designable—i.e., realizable with natural amino acids. Several recent studies have reported using structural statistics of constituent fragments to filter sets of de novo generated templates for high designability [52-54]. For example, Brunette et al. estimated the designability of de novo repeat protein templates by calculating the frequency with which backbones from pairs of positions within these structures matched residue pairs with interacting side-chains in the PDB [53]. Kuhlman and co-workers have further pushed the idea of using fragments from existing proteins to generate designable templates [55]. In their SEWING approach, novel proteins were computationally assembled from single or multi-segment fragments extracted from the PDB [55]. Models were generated by combining fragments that superimposed well and did not clash with the growing structure. For models assembled from multi-segment motifs, loop design was required to connect segments. With this method, the authors successfully designed two novel proteins in good agreement with their corresponding X-ray or NMR structures. While this study focused on helical motifs and only combined a small number of fragments, this method is general and could be extended to larger assemblies with different fragments. This study is especially exciting as an important step towards using multi-segment motifs for generating novel templates and assuring their designability. A study by Azatoie and colleagues showcased another use of discontinuous motifs in design, whereby the authors transplanted a disjoint fragment consisting of two loops from HIV gp120 to an unrelated scaffold [56]. The PDB was scanned for scaffolds that could structurally accommodate this fragment and a library was computationally designed and screened in vitro, resulting in a novel protein that bound a gp120-targeting antibody with affinity and specificity near that of gp120 itself.
Of course, motifs can focus the sampling of not only structure but also sequence. This is because sequence statistics emergent from motif instances can report on important determinants of the targeted structure. Whereas contiguous fragments have been widely used for deriving such empirical sequence constraints in design [22,53,57], applications of multi-segment motifs towards this end sparser [52,56] [34]. Still, this represents a promising direction, especially as more structural data accumulate, with the prospect of providing quantitative sequence determinants of tertiary and quaternary structure, just as sequence statistics of local backbone fragments enabled the quantification of secondary-structural propensities. Such higher-order sequence statistics would be greatly beneficial to protein design and structure prediction applications.
Fragment libraries have also been increasingly used to assess the conformational landscape of designed proteins by sampling over large number of structural possibilities to verify that the target state is preferred for the designed sequence [53,58,59]. In what is often referred to as forward folding simulations, the designed sequence is subjected to broad conformational sampling via Rosetta using fragments biased by the secondary structure of the targeted template. The presence of an energy funnel leading to the target state is then treated as a necessary condition for design success [39]. In an approach targeted towards the redesign of antibody binding, Fleishman and co-workers have exploited PDB fragments for both of the abovementioned benefits—to focus structural sampling on designable space and to discern relevant sequence-structure relationships. Relying on the relatively large number of antibody entries in the PDB, their computational method, AbDesign, uses backbone fragments from aligned regions of antibody structures to combinatorially generate new putative templates. The sequence for each combination is optimized with a modified Rosetta design procedure, in which amino acids are constrained to those naturally found in the fragments [57,60].
Koga et al. reported a set of rules relating SSE patterns in super-secondary motifs and used these to successfully design a number of novel proteins [58]. These rules, which were derived from observations in the PDB and Rosetta folding experiments, explained super-secondary motif geometries in terms of constituent SSEs, their lengths and registers, and the length of the loop connecting them. This information was used to select SSE and loop lengths to favor each of five design blueprints prior to sequence optimization. NMR structures of designs corresponding to each of these five folds showed close agreement with computational models. Building on this work, packing geometries between successive SSEs were described using more detailed rules that accounted for the co-dependence between the length of SSEs and the loop connecting them, and the backbone torsion angles of the loop [59]. Rules were developed based on the frequencies of each pattern in naturally occurring proteins and extensive Rosetta folding simulations. These insights were used to computationally design seven folds based on SSE blueprints, six of which led to NMR structures that agreed with design models to within 1.1-2.4 Å of backbone RMSD. Similar design rules were recently used to aid in the design of hyperstable disulfide-crosslinked peptides, some of which were heterochiral and/or backbone-cyclized [61]. Using fragments biased towards the detailed backbone geometry of the desired structures, hyperstable peptides were computationally designed and experimentally validated with 12 X-ray and NMR structures highly similar to corresponding design models.
Conclusions
In recent years, structural patterns from the PDB, across a variety of scales, have enhanced our understanding of the structural universe and led to considerable advances in protein structure prediction and design. Fragment libraries have served as an important tool for expressing and taking advantage of the modularity in the structural universe. Such libraries have been used both to limit structural space in sampling, focusing on designable templates in design or native-like models in structure prediction, and to develop sequence-structure relationships at the sub-domain level. Incorporation of tertiary information into fragment libraries appears to be the key next step in building more complete fragment libraries for use in both applications. There has been some work in this direction recently, but many new developments will likely emerge in the near future. In general, as the structural database continues to grow, we anticipate that the reductionist approach of studying protein structure—i.e., decomposing the protein structural universe into its recurrent building blocks to synthesize emergent principles—will provide increasingly accurate and complete insights.
Highlights.
statistical analyses of the PDB have shaped our understanding of protein structure
recurrent motifs have been used to great effect in structure prediction and design
increasing amounts of data give new insights into structure-sequence relationships
tertiary structural motifs are poised to provide new knowledge and capabilities
decompositions of tertiary structure space have revealed strong degeneracies
Acknowledgments
This work was supported in part by National Science Foundation award DMR-1534246 (GG) and an award from the Neukom Institute at Dartmouth College (GG).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bystroff C, Baker D. Prediction of local structure in proteins using a library of sequence-structure motifs. Journal of Molecular Biology. 1998;281:565–577. doi: 10.1006/jmbi.1998.1943. [DOI] [PubMed] [Google Scholar]
- 2.Cuff A, Redfern OC, Greene L, Sillitoe I, Lewis T, Dibley M, Reid A, Pearl F, Dallman T, Todd A, et al. The CATH Hierarchy Revisited—Structural Divergence in Domain Superfamilies and the Continuity of Fold Space. Structure. 2009;17:1051–1062. doi: 10.1016/j.str.2009.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fox NK, Brenner SE, Chandonia JM. SCOPe: Structural Classification of Proteins—extended, integrating SCOP and ASTRAL data and classification of new structures. Nucleic Acids Research. 2014;42:D304–D309. doi: 10.1093/nar/gkt1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amanchy R, Periaswamy B, Mathivanan S, Reddy R, Tattikota SG, Pandey A. A curated compendium of phosphorylation motifs. Nature Biotechnology. 2007;25:285–286. doi: 10.1038/nbt0307-285. [DOI] [PubMed] [Google Scholar]
- 5.Kufareva I, Ilatovskiy AV, Abagyan R. Pocketome: an encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Research. 2012;40:D535–D540. doi: 10.1093/nar/gkr825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nilmeier JP, Kirshner DA, Wong SE, Lightstone FC. Rapid Catalytic Template Searching as an Enzyme Function Prediction Procedure. PLoS ONE. 2013;8:e62535. doi: 10.1371/journal.pone.0062535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Orengo C, Michie A, Jones S, Jones D, Swindells M, Thornton J. CATH – a hierarchic classification of protein domain structures. Structure. 1997;5:1093–1109. doi: 10.1016/s0969-2126(97)00260-8. [DOI] [PubMed] [Google Scholar]
- 8*.Nepomnyachiy S, Ben-Tal N, Kolodny R. Global view of the protein universe. Proceedings of the National Academy of Sciences. 2014;111:11691–11696. doi: 10.1073/pnas.1403395111. This study considers the idea of evolutionary paths between protein domains. It uses the structures available today to build a network of either domains or motifs, in which connections indicate shared regions of similar sequence and structure. The authors vary the threshold used to define edges to study the connectivity of the networks, finding (for example) that patterns of alternating alpha and beta elements are highly shared among proteins, inducing large connected components in the networks. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9*.Alva V, Söding J, Lupas AN. A vocabulary of ancient peptides at the origin of folded proteins. eLife. 2015:4. doi: 10.7554/eLife.09410. By searching for structural and sequence patterns across unrelated domains, the authors identified 40 motifs that may represent the remnants of ancient peptides which gave rise to present-day domains. This study illustrates how sub-domain fragments can be used to uncover modularity and putative evolutionary relationships. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fernandez-Fuentes N, Oliva B, Fiser A. A supersecondary structure library and search algorithm for modeling loops in protein structures. Nucleic Acids Research. 2006;34:2085–2097. doi: 10.1093/nar/gkl156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fernandez-Fuentes N, Fiser A. A Modular Perspective of Protein Structures: Application to Fragment Based Loop Modeling. In: Kister AE, editor. Protein Supersecondary Structures. Vol. 932. Humana Press; 2012. pp. 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fernandez-Fuentes N, Dybas JM, Fiser A. Structural Characteristics of Novel Protein Folds. PLoS Computational Biology. 2010;6:e1000750. doi: 10.1371/journal.pcbi.1000750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Menon V, Vallat Brinda K, Dybas Joseph M, Fiser A. Modeling Proteins Using a Super-Secondary Structure Library and NMR Chemical Shift Information. Structure. 2013;21:891–899. doi: 10.1016/j.str.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14*.Vallat B, Madrid-Aliste C, Fiser A. Modularity of Protein Folds as a Tool for Template-Free Modeling of Structures. PLOS Computational Biology. 2015;11:e1004419. doi: 10.1371/journal.pcbi.1004419. This study uses an existing super-secondary fragment library and a simple scoring function topredict protein structure. In preliminary tests on targets with remote homologs it preformsequally well as I-TASSER and better than Rosetta. It suggests that using complex yet generalfragments may benefit structure prediction. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolodny R, Koehl P, Guibas L, Levitt M. Small Libraries of Protein Fragments Model Native Protein Structures Accurately. Journal of Molecular Biology. 2002;323:297–307. doi: 10.1016/s0022-2836(02)00942-7. [DOI] [PubMed] [Google Scholar]
- 16.Budowski-Tal I, Nov Y, Kolodny R. FragBag, an accurate representation of protein structure, retrieves structural neighbors from the entire PDB quickly and accurately. Proceedings of the National Academy of Sciences. 2010;107:3481–3486. doi: 10.1073/pnas.0914097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Baeten L, Reumers J, Tur V, Stricher F, Lenaerts T, Serrano L, Rousseau F, Schymkowitz J. Reconstruction of Protein Backbones from the BriX Collection of Canonical Protein Fragments. PLoS Computational Biology. 2008;4:e1000083. doi: 10.1371/journal.pcbi.1000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanhee P, Verschueren E, Baeten L, Stricher F, Serrano L, Rousseau F, Schymkowitz J. BriX: a database of protein building blocks for structural analysis, modeling and design. Nucleic Acids Research. 2011;39:D435–D442. doi: 10.1093/nar/gkq972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vanhee P, Stricher F, Baeten L, Verschueren E, Lenaerts T, Serrano L, Rousseau F, Schymkowitz J. Protein-Peptide Interactions Adopt the Same Structural Motifs as Monomeric Protein Folds. Structure. 2009;17:1128–1136. doi: 10.1016/j.str.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 20.Verschueren E, Vanhee P, Rousseau F, Schymkowitz J, Serrano L. Protein-Peptide Complex Prediction through Fragment Interaction Patterns. Structure. 2013;21:789–797. doi: 10.1016/j.str.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 21.Vanhee P, Reumers J, Stricher F, Baeten L, Serrano L, Schymkowitz J, Rousseau F. PepX: a structural database of non-redundant protein-peptide complexes. Nucleic Acids Research. 2010;38:D545–D551. doi: 10.1093/nar/gkp893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verschueren E, Vanhee P, van der Sloot AM, Serrano L, Rousseau F, Schymkowitz J. Protein design with fragment databases. Current Opinion in Structural Biology. 2011;21:452–459. doi: 10.1016/j.sbi.2011.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Walters RFS, DeGrado WF. Helix-packing motifs in membrane proteins. Proceedings of the National Academy of Sciences. 2006;103:13658–13663. doi: 10.1073/pnas.0605878103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grigoryan G, Keating A. Structural specificity in coiled-coil interactions. Current Opinion in Structural Biology. 2008;18:477–483. doi: 10.1016/j.sbi.2008.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grigoryan G, DeGrado WF. Probing Designability via a Generalized Model of Helical Bundle Geometry. Journal of Molecular Biology. 2011;405:1079–1100. doi: 10.1016/j.jmb.2010.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26*.Feng X, Barth P. A topological and conformational stability alphabet for multipass membrane proteins. Nature Chemical Biology. 2016;12:167–173. doi: 10.1038/nchembio.2001. This work shows that a small number of structural classes are sufficient to describe three-helix interactions in membrane proteins. Each structural class contained a small number of sequence motifs, information that may be valuable for the prediction and design of such proteins. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27*.Zhang SQ, Kulp DW, Schramm CA, Mravic M, Samish I, DeGrado WF. The membrane- and soluble-protein helix-helix interactome: similar geometry via different interactions. Structure (London, England: 1993) 2015;23:527–541. doi: 10.1016/j.str.2015.01.009. In this study, interacting helix pairs in TM and soluble proteins were each described with a small number of structural classes. While there were shared geometries between the two groups, important differences in sequence preferences and hydrogen bonding patterns within those classes exist. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tuncbag N, Gursoy A, Guney E, Nussinov R, Keskin O. Architectures and Functional Coverage of Protein–Protein Interfaces. Journal of Molecular Biology. 2008;381:785–802. doi: 10.1016/j.jmb.2008.04.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao M, Skolnick J. Structural space of protein-protein interfaces is degenerate, close to complete, and highly connected. Proceedings of the National Academy of Sciences. 2010;107:22517–22522. doi: 10.1073/pnas.1012820107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie ZR, Chen J, Zhao Y, Wu Y. Decomposing the space of protein quaternary structures with the interface fragment pair library. BMC Bioinformatics. 2015;16 doi: 10.1186/s12859-014-0437-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Grigoryan G. Mining Tertiary Structural Motifs for Assessment of Designability. In: Elsevier, editor. Methods in Enzymology. Vol. 523. 2013. pp. 21–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhou J, Grigoryan G. Rapid search for tertiary fragments reveals protein sequence-structure relationships: Tertiary Motif Search gives Structure Rules. Protein Science. 2015;24:508–524. doi: 10.1002/pro.2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33*.Zheng F, Zhang J, Grigoryan G. Tertiary Structural Propensities Reveal Fundamental Sequence/Structure Relationships. Structure. 2015;23:961–971. doi: 10.1016/j.str.2015.03.015. This study shows that statistics of tertiary motifs (TERMs) can be harnessed to describe sequence-structure relationship in a general manner. Relying solely on these relationships, the authors were able to successfully evaluate the quality of structure prediction models from CASP. [DOI] [PubMed] [Google Scholar]
- 34**.Mackenzie C, Zhou J, Grigoryan G. Tertiary alphabet for the observable protein structural universe. Proceedings of the National Academy of Sciences. 2016;113:E7438–E7447. doi: 10.1073/pnas.1607178113. This work derives a universal library of multi-segment motifs (TERMs) that describes secondary, tertiary, and quaternary structural information in the PDB. This shows, in a very general manner, the extent to which tertiary and quaternary structure space in native proteins is degenerate, demonstrating that it can be described to sub-Angstrom accuracy with relatively small motif sets. Preliminary computational studies show that TERMs have promise in design and structure prediction applications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bowie JU, Eisenberg D. An evolutionary approach to folding small alpha-helical proteins that uses sequence information and an empirical guiding fitness function. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:4436–4440. doi: 10.1073/pnas.91.10.4436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simons KT, Kooperberg C, Huang E, Baker D. Assembly of protein tertiary structures from fragments with similar local sequences using simulated annealing and bayesian scoring functions. Journal of Molecular Biology. 1997;268:209–225. doi: 10.1006/jmbi.1997.0959. [DOI] [PubMed] [Google Scholar]
- 37.Kinch LN, Li W, Monastyrskyy B, Kryshtafovych A, Grishin NV. Evaluation of free modeling targets in CASP11 and ROLL: Targets in CASP11 and ROLL. Proteins: Structure, Function, and Bioinformatics. 2016;84:51–66. doi: 10.1002/prot.24973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tai CH, Bai H, Taylor TJ, Lee B. Assessment of template-free modeling in CASP10 and ROLL: CASP10 FM and ROLL Assessment. Proteins: Structure, Function, and Bioinformatics. 2014;82:57–83. doi: 10.1002/prot.24470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rohl CA, Strauss CEM, Misura KMS, Baker D. Protein structure prediction using Rosetta. Methods in Enzymology. 2004;383:66–93. doi: 10.1016/S0076-6879(04)83004-0. [DOI] [PubMed] [Google Scholar]
- 40.Gront D, Kulp DW, Vernon RM, Strauss CEM, Baker D. Generalized Fragment Picking in Rosetta: Design, Protocols and Applications. PLoS ONE. 2011;6:e23294. doi: 10.1371/journal.pone.0023294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lange OF, Baker D. Resolution-adapted recombination of structural features significantly improves sampling in restraint-guided structure calculation. Proteins: Structure, Function, and Bioinformatics. 2012;80:884–895. doi: 10.1002/prot.23245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.DiMaio F, Terwilliger TC, Read RJ, Wlodawer A, Oberdorfer G, Wagner U, Valkov E, Alon A, Fass D, Axelrod HL, et al. Improved molecular replacement by density- and energy-guided protein structure optimization. Nature. 2011;473:540–543. doi: 10.1038/nature09964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DiMaio F, Song Y, Li X, Brunner MJ, Xu C, Conticello V, Egelman E, Marlovits TC, Cheng Y, Baker D. Atomic-accuracy models from 4.5-Å cryo-electron microscopy data with density-guided iterative local refinement. Nature Methods. 2015;12:361–365. doi: 10.1038/nmeth.3286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang Y, Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proceedings of the National Academy of Sciences. 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu S, Zhang Y. LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Research. 2007;35:3375–3382. doi: 10.1093/nar/gkm251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu D, Zhang Y. Ab initio protein structure assembly using continuous structure fragments and optimized knowledge-based force field. Proteins: Structure, Function, and Bioinformatics. 2012;81:1715–1735. doi: 10.1002/prot.24065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xu D, Zhang Y. Toward optimal fragment generations for ab initio protein structure assembly. Proteins: Structure, Function, and Bioinformatics. 2013;81:229–239. doi: 10.1002/prot.24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y. Interplay of I-TASSER and QUARK for template-based and ab initio protein structure prediction in CASP10: Composite Protein Structure Prediction in CASP10. Proteins: Structure, Function, and Bioinformatics. 2014;82:175–187. doi: 10.1002/prot.24341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kamisetty H, Ovchinnikov S, Baker D. Assessing the utility of coevolution-based residue-residue contact predictions in a sequence- and structure-rich era. Proceedings of the National Academy of Sciences. 2013;110:15674–15679. doi: 10.1073/pnas.1314045110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morcos F, Hwa T, Onuchic JN, Weigt M. Direct Coupling Analysis for Protein Contact Prediction. In: Kihara D, editor. Protein Structure Prediction. Vol. 1137. Springer; New York: 2014. pp. 55–70. [DOI] [PubMed] [Google Scholar]
- 51**.Ovchinnikov S, Kinch L, Park H, Liao Y, Pei J, Kim DE, Kamisetty H, Grishin NV, Baker D. Large-scale determination of previously unsolved protein structures using evolutionary information. eLife. 2015;4:e09248. doi: 10.7554/eLife.09248. By incorporating contact information predicted from evolutionary co-variation into Rosetta, this study yielded some of the best performance to date on predicting the structures of large targets without templates. These results reveal the power that even imperfect knowledge of long-range contacts would have towards advancing structure prediction, motivating the development of methods to take advantage of degeneracies at tertiary and higher structural levels. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Grigoryan G, Kim YH, Acharya R, Axelrod K, Jain RM, Willis L, Drndic M, Kikkawa JM, DeGrado WF. Computational design of virus-like protein assemblies on carbon nanotube surfaces. Science. 2011;332:1071–1076. doi: 10.1126/science.1198841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53**.Brunette T, Parmeggiani F, Huang PS, Bhabha G, Ekiert DC, Tsutakawa SE, Hura GL, Tainer JA, Baker D. Exploring the repeat protein universe through computational protein design. Nature. 2015;528:580–584. doi: 10.1038/nature16162. Rosetta was used to successfully design 53 de-novo proteins, representing a wide range of geometries, many not similar to anything found in nature. Fragment libraries and observations from the PDB were used in design at various stages, illustrating their power to explore structural space beyond the PDB. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54**.Bale JB, Gonen S, Liu Y, Sheffler W, Ellis D, Thomas C, Cascio D, Yeates TO, Gonen T, King NP, et al. Accurate design of megadalton-scale two-component icosahedral protein complexes. Science. 2016;353:389–394. doi: 10.1126/science.aaf8818. Building on previous work, the authors mine the PDB for subunits that can be assembled into large protein cages through docking and interface design. Interaction motifs from the PDB were used in filtering for designable interfaces. The study also designed cargo-loading function into such protein cages, suggesting possible uses of this technology in real-life applications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55**.Jacobs TM, Williams B, Williams T, Xu X, Eletsky A, Federizon JF, Szyperski T, Kuhlman B. Design of structurally distinct proteins using strategies inspired by evolution. Science. 2016;352:687–690. doi: 10.1126/science.aad8036. The authors propose a novel method for piecing together fragments from the PDB, both local backbone fragments and two-segment motifs. Using the method, two of novel structures were computationally designed and experimentally validated. This represents a key step in the direction of exploiting general fragments, beyond sequence-contiguous motifs, towards building novel design templates. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Azoitei ML, Correia BE, Ban YEA, Carrico C, Kalyuzhniy O, Chen L, Schroeter A, Huang PS, McLellan JS, Kwong PD, et al. Computation-Guided Backbone Grafting of a Discontinuous Motif onto a Protein Scaffold. Science. 2011;334:373–376. doi: 10.1126/science.1209368. [DOI] [PubMed] [Google Scholar]
- 57.Lapidoth GD, Baran D, Pszolla GM, Norn C, Alon A, Tyka MD, Fleishman SJ. AbDesign: An algorithm for combinatorial backbone design guided by natural conformations and sequences: Combinatorial Backbone Design in Antibodies. Proteins: Structure, Function, and Bioinformatics. 2015;83:1385–1406. doi: 10.1002/prot.24779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Koga N, Tatsumi-Koga R, Liu G, Xiao R, Acton TB, Montelione GT, Baker D. Principles for designing ideal protein structures. Nature. 2012;491:222–227. doi: 10.1038/nature11600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59**.Lin YR, Koga N, Tatsumi-Koga R, Liu G, Clouser AF, Montelione GT, Baker D. Control over overall shape and size in de novo designed proteins. Proceedings of the National Academy of Sciences. 2015;112:E5478–E5485. doi: 10.1073/pnas.1509508112. Building on previous work by the same group, rules that relate SSE patterns to tertiary motifs were used to successfully design a number of novel proteins with atomistic detail. This procedure allowed for more control over the size and relative orientations of the SSEs in the protein. The methods in this study have been applied to other successful design goals. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Khersonsky O, Fleishman SJ. Why reinvent the wheel? Building new proteins based on ready-made parts: New Proteins from Old Parts. Protein Science. 2016;25:1179–1187. doi: 10.1002/pro.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bhardwaj G, Mulligan VK, Bahl CD, Gilmore JM, Harvey PJ, Cheneval O, Buchko GW, Pulavarti SVSRK, Kaas Q, Eletsky A, et al. Accurate de novo design of hyperstable constrained peptides. Nature. 2016;538:329–335. doi: 10.1038/nature19791. [DOI] [PMC free article] [PubMed] [Google Scholar]