

Abstract
Objective:
To assess changes in 3 clinical measures, the Revised ALS Functional Rating Scale (ALSFRS-R), letter fluency, and Frontal Behavioral Inventory (FBI), over time in C9orf72 mutation carriers (C9+) with varied clinical phenotypes.
Methods:
Thirty-four unrelated participants with mutations in C9orf72 were enrolled in a prospective natural history study. Participants were classified as asymptomatic, amyotrophic lateral sclerosis (ALS), ALS–familial frontotemporal dementia (FTD), or behavioral-variant FTD by clinical diagnostic criteria. Diagnostic cognitive and motor tests were repeated at 6 and 18 months. The ALSFRS-R, letter fluency, and FBI were administered at baseline and follow-up visits at 6, 12, and 18 months.
Results:
The clinical diagnosis of most patients did not change over the follow-up. ALSFRS-R scores correlated with measures of motor function. Letter fluency correlated with FBI and cognitive tests. ALSFRS-R, letter fluency, and FBI differed among the C9+ diagnostic subgroups at enrollment and worsened over follow-up in symptomatic patients, with different slopes among the subgroups. Most patients survived to the 6-month time point after enrollment. Survival of C9+ patients with ALS and C9+ patients with ALS-FTD declined over the 12- and 18-month follow-up.
Conclusions:
The pattern of scores of the ALSFRS-R, letter fluency, and FBI distinguished between ALS, ALS-FTD, and FTD presentations of C9orf72 mutation carriers and asymptomatic carriers. Longitudinal changes in these measures occurred with disease progression in a manner consistent with presenting phenotype.
Expansion mutations in the gene C9orf72 account for 25% to 40% of familial amyotrophic lateral sclerosis (ALS) and familial frontotemporal dementia (FTD), as well as 5% to 10% of sporadic cases of these disorders in the United States and Europe.1–3 Several promising approaches to gene-specific therapy are being pursued.4,5 Biochemical assays may be able to assess the effectiveness of gene targeting by these therapies,6 but assessing clinical effectiveness requires consideration of the varied phenotypes caused by the C9orf72 mutation. Patients with the C9orf72 mutation can present with ALS, FTD, mixed features of ALS and FTD, or other neurodegenerative disorders, with variable presentations within the same pedigree.7–9 Clinical measures of cognitive-behavioral and motor symptoms will be needed to determine treatment effectiveness in future clinical trials. To date, different outcome measures have been used to assess disease severity in ALS and behavioral-variant FTD (bvFTD), the 2 most common clinical phenotypes caused by C9orf72 mutations. In this study, we evaluated 3 clinical outcome measures in C9orf72 mutation carriers who were not selected for clinical phenotype to determine whether these measures provided a snapshot of impairment in motor, cognitive, and behavioral domains at enrollment and changed over time. The 3 primary clinical outcome measures, the Revised ALS Functional Rating Scale (ALSFRS-R),10 verbal fluency, and the Frontal Behavioral Inventory (FBI),11 are brief, amenable to administration in person or by phone,12 and known to decline with disease progression in ALS or FTD.13–17
METHODS
Recruitment.
We recruited participants throughout the United States through NIH Internet postings, links posted on disease advocacy group websites, the Centers for Disease Control and Prevention’s National ALS Registry (www.cdc.gov/als/, accessed July 3, 2017), Researchmatch.org (accessed July 3, 2017), and mailings to physicians associated with ALS or memory disorder clinics. Between September 2013 and July 2016, 47 respondents were eligible and 34 agreed to participate.
Standard protocol approvals, registrations, and patient consents.
Thirty-four unrelated carriers of an expansion mutation in the C9orf72 gene (20 male, 14 female; hereafter referred to as C9+ participants) gave written informed consent for participation in the research study approved by the NIH Combined Neuroscience Institutional Review Board (NCT01925196). Consent included agreement to sharing of deidentified clinical data and biospecimens. Eligibility required all participants to have Clinical Laboratory Improvement Amendments–certified clinical testing showing an expanded hexanucleotide repeat in the C9orf72 gene. All asymptomatic carriers had family members with ALS or FTD. Each participant underwent a multiday evaluation visit at enrollment. Multiday follow-up visits were held at 6 and 18 months. A phone assessment was carried out at 12 months. Table e-1 at Neurology.org shows the study schedule.
Diagnostic evaluation.
Each participant’s clinical diagnosis at enrollment was determined by neurologic examination and diagnostic testing. Neurologic examination and electromyography were used to determine whether participants met the revised El Escorial criteria for possible, probable, laboratory-supported, or definite ALS.18 A battery of cognitive testing and caregiver interviews were used to determine whether participants met the Rascovsky criteria for the diagnosis of possible, probable, or definite bvFTD.19 The testing battery included the Mattis Dementia Rating Scale (DRS-2),20,21 which assesses attention, initiation-perseveration, conceptualization, construction and memory; the Trails, Sorting, Verbal Fluency, and 20-question subtests of the Delis-Kaplan Executive Function System22 (D-KEFS); language tests from the National Alzheimer Coordinating Committee FTLD module (https://www.alz.washington.edu/NONMEMBER/FTLD/FTLD-IVP-C1F.pdf; accessed February 3, 2017); the Mini-Mental State Examination (MMSE); and the Frontal Assessment Battery5 (FAB). The testing battery was repeated to reassess clinical diagnosis at in-person follow-up visits, with the exception that electromyography was not repeated in patients who already met criteria for definite ALS. Measures of motor function, including the 9-Hole Peg Test and rate of syllable repetition (pa-ta-ka), were made at each in-person visit. Some participants met criteria for both ALS and bvFTD; one patient had an Alzheimer-type dementia with memory loss and was excluded from further analysis here. The other participants were classified into 4 diagnosis subgroups at enrollment: C9+ asymptomatic, C9+ ALSC9+ bvFTD, and C9+ ALS-FTD.
Longitudinal motor, cognitive, and behavioral outcome measures.
The ALSFRS-R is a 12-item survey of bulbar, dexterity, gross, and respiratory motor function with a full scale score of 48.10 The ALSFRS-R was administered by neurologists at the 3 in-person visits and the 12-month phone assessment. A research nurse administered the FBI during in-person and phone visits. The FBI is a 24-item caregiver interview that measures behavioral disinhibition and negative behaviors such as apathy. Because some items of the FBI can be affected by motor function, only items unaffected by each patient’s motor limitations were scored, and the scores are reported as the percentage of the total possible score for each patient. Higher FBI scores indicate greater behavioral impairment. The letter fluency score used as a primary clinical outcome measure was the total number of correct, nonrepeated words generated in 1 minute each for 3 letters, with rules about proper nouns and numbers. During the in-person visit, letter fluency was obtained by neuropsychologists as part of the D-KEFS fluency subtest22 using alternate versions on serial visits. Two patients with ALS with bulbar dysfunction and normal dexterity provided written responses. Verbal fluency was not tested in 3 participants who were unable to speak or write. The phone assessment of letter fluency was obtained by a trained research assistant using different letters with the same rules.
Statistics.
Descriptive data are reported as means ± SDs. Differences in baseline variables between diagnosis subgroups were assessed by analysis of variance with equal or unequal variance models based on the Levene test for homogeneity of variances. The Tukey method was applied to post hoc pairwise multiple comparisons. The Pearson R was calculated in SPSS (IBM SPSS version 23, Armonk, NY).
Changes in the 3 outcome measures over time in the diagnosis subgroups were assessed by random coefficient analysis with a mixed model that included diagnosis (4 levels: asymptomatic, ALS, ALS-FTD, and bvFTD), time (continuous, in months), and the interaction between diagnosis and time as fixed effects, with intercept and slope as random effects. The F test for diagnosis (df = 3) and the interaction (df = 3) were used to test common intercept and slope, respectively. Simulation with 10,000 permutations was used to test the probability that the slope and intercept in each C9+ symptomatic diagnosis subgroup differed from those in the C9+ asymptomatic group.
The above statistical analyses were performed with SAS version 9.4 (SAS Institute Inc, Cary, NC). The threshold for significance was p < 0.05, corrected for multiple comparison for each dependent variable.
RESULTS
Clinical features: Baseline.
Demographics.
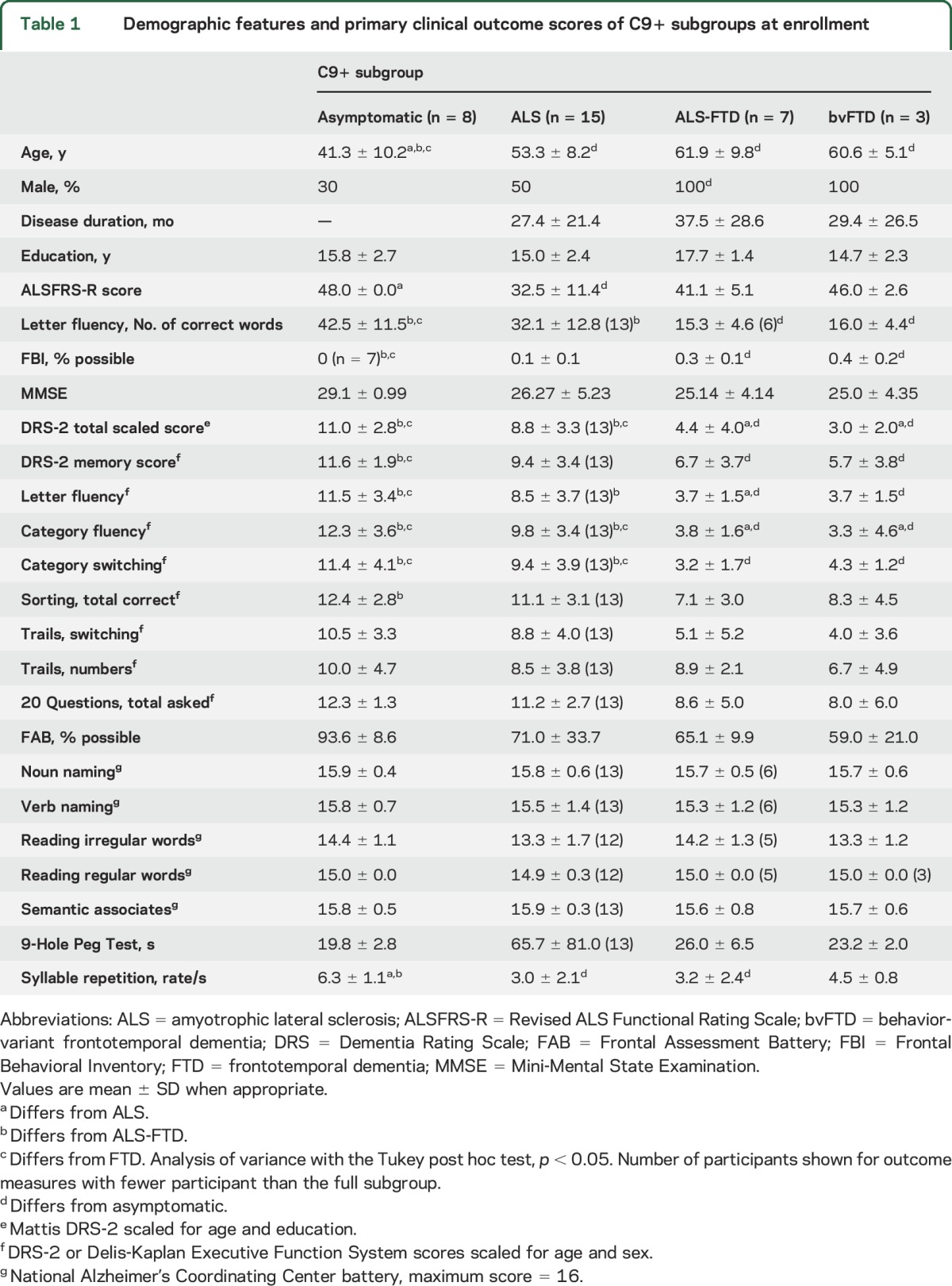
Table 1 shows the demographics, 3 primary outcome measures, and clinical and cognitive measures of the C9+ diagnosis subgroups at enrollment. More men than women were enrolled; the bvFTD and ALS-FTD subgroups consisted of only men. Eight C9+ participants were asymptomatic carriers. Asymptomatic carriers were younger than the other diagnosis subgroups, who did not differ in age. Years of education did not differ among subgroups. Among the symptomatic patients, symptoms began with cognitive dysfunction in 8 and motor symptoms in 18 (10 limb onset, 8 bulbar onset). The median symptom duration at enrollment was 24.4 months (range 1–89 months). All patients were enrolled for ≥6 months, and 23 were enrolled for ≥18 months. Data reported here were compiled from 74 NIH visits, 2 home visits, and 20 phone assessments.
Table 1.
Demographic features and primary clinical outcome scores of C9+ subgroups at enrollment
Family history and ethnicity.
Eleven participants reported a family history of only ALS. Seven reported a family history of only dementia. Fifteen patients reported a family history of both ALS and dementia. The pattern of inheritance appeared to be dominant in all cases, although one parent with a presumed C9orf72 expansion lived to age 90 without symptoms. Only one patient had no family history of ALS or dementia. All participants were white, and one was of Hispanic ethnicity.
Clinical features: Longitudinal changes.
Survival.
Nine patients died within 18 months of enrollment. The time from symptom onset to death ranged from 18 to 46 months (mean 31.2 ± 11.9 months). Seven who died had the clinical diagnosis at enrollment of ALS and 2 had ALS-FTD. Weakness was the first symptom in all, with bulbar onset in 4 and limb onset in 5. In 2 of the limb-onset patients, cognitive symptoms developed within a year. None of the C9+ patients with bvFTD or asymptomatic participants died during the 18 months of follow-up. Of the 17 C9+ patients with ALS and ALS-FTD still alive 18 months after enrollment, the mean symptom duration was 50.2 ± 26.1 months. Only 1 patient died before the 6-month follow-up visit. However, 8 patients were too impaired to return for a 6-month follow-up. Figure 1 shows the proportion of surviving patients of those enrolled at each interval by diagnosis subgroup.
Figure 1. Survival status of diagnosis subgroups of patients at 6, 12, and 18 months after enrollment.
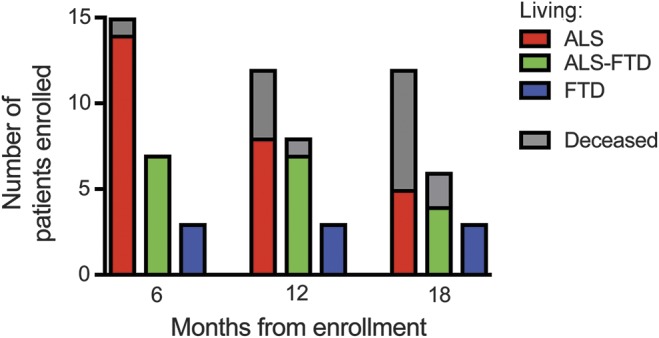
Diagnoses of C9orf72 mutation carriers are represented by colors (red, ALS; green, ALS-FTD; blue, bvFTD). The portion of surviving patients of those enrolled at each interval is shown in color; gray represents nonsurvivors. ALS = amyotrophic lateral sclerosis; bvFTD = behavior-variant frontotemporal dementia; FTD = frontotemporal dementia.
Except for 1 patient, the enrollment diagnosis remained the same on successive visits. The single patient who converted had bulbar symptoms for 26 months and met the criteria for ALS at enrollment, with normal scores on cognitive testing. Six months later, with a moderate decline in executive function, loss of insight, and disinhibited behavior, the patient met the criteria for bvFTD. Among the ALS-FTD group, 2 long-surviving patients (>7 years) had cognitive symptoms for several years before developing motor symptoms. One long-surviving patient with ALS (>7 years) had symptoms limited to one limb and normal cognition.
Primary outcome measures: Baseline.
Motor.
C9+ patients with ALS had lower ALSFRS-R scores than asymptomatic C9+ participants (p < 0.05). The patients with bvFTD lost an occasional point on the ALSFRS-R for needing assistance with dressing or hygiene. Neither the bvFTD nor ALS-FTD subgroup had ALSFRS-R scores that were significantly different from those of the asymptomatic C9+ participants.
Cognitive.
The letter fluency score was significantly worse in the ALS-FTD and bvFTD groups compared to the asymptomatic and C9+ participants with ALS (p < 0.05).
Behavioral.
The FBI score was significantly higher in the ALS-FTD and bvFTD groups compared to the asymptomatic carriers and patients with ALS (p < 0.05).
Correlation between primary outcome measures.
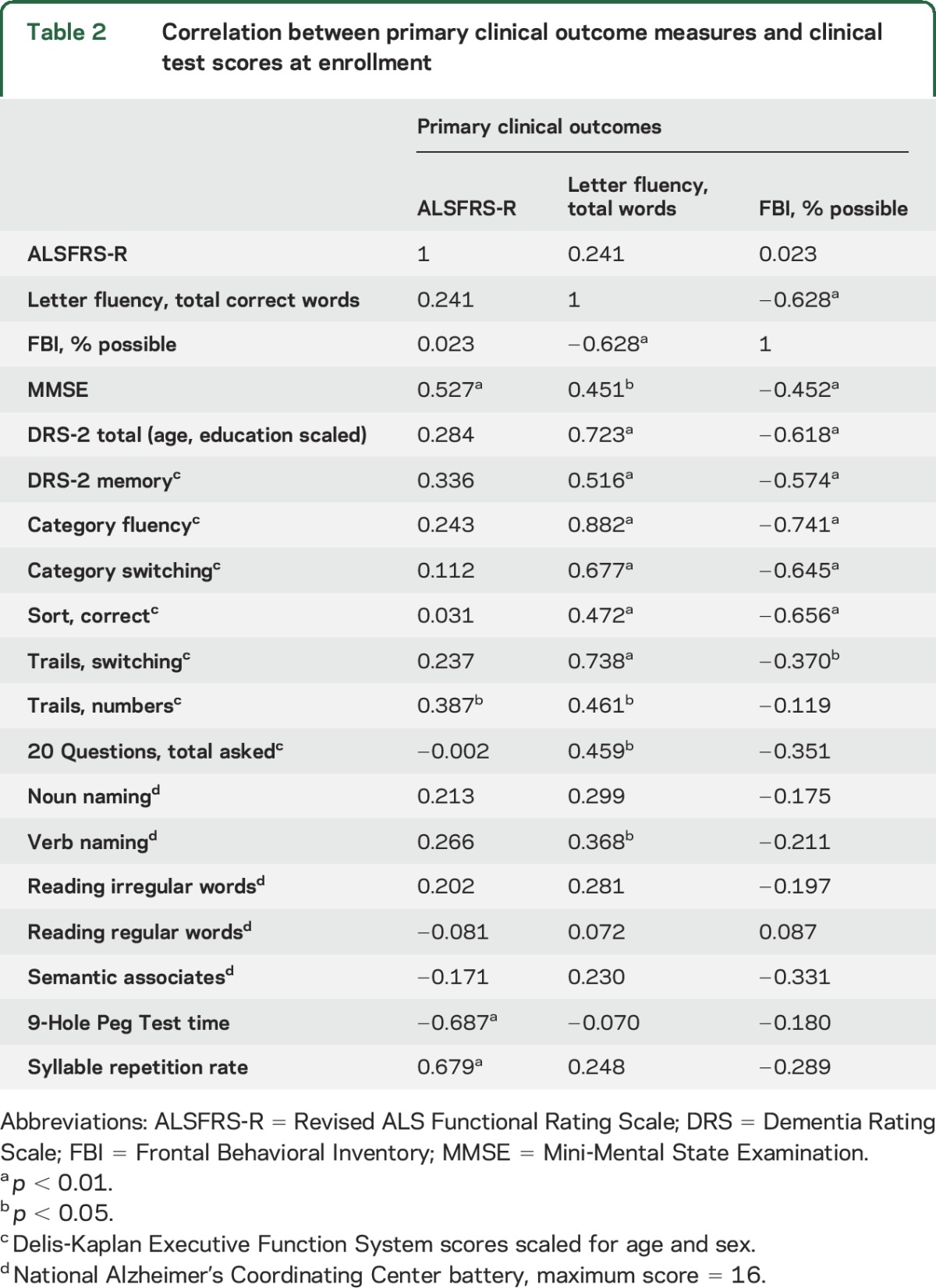
The 3 primary outcome measures were correlated with clinical measures of cognitive or motor function (table 2). High letter fluency was correlated with low FBI scores. Participants with higher letter fluency and lower FBI scores scored better on the DRS-2; D-KEFS Sort, Trails, and Fluency subtests; FAB; and MMSE. Letter fluency was not correlated with the rate of syllable repetition (r = 0.248, p = 0.187). The ALSFRS-R score was correlated with MMSE and FAB but was not correlated with letter fluency or the FBI.
Table 2.
Correlation between primary clinical outcome measures and clinical test scores at enrollment
Longitudinal outcome measures.
Changes in 3 primary outcome measures over the 18-month follow-up differed among the C9+ subgroups (figure 2). The intercept and slopes derived from the statistical mixed model were significantly different in at least one subgroup for each primary outcome measure (F test p < 0.05, table 3). The C9+ asymptomatic subgroup had little change and were used as the comparison for symptomatic subgroups by simulation testing.
Figure 2. Longitudinal changes in ALSFRS-R, letter fluency, and FBI.
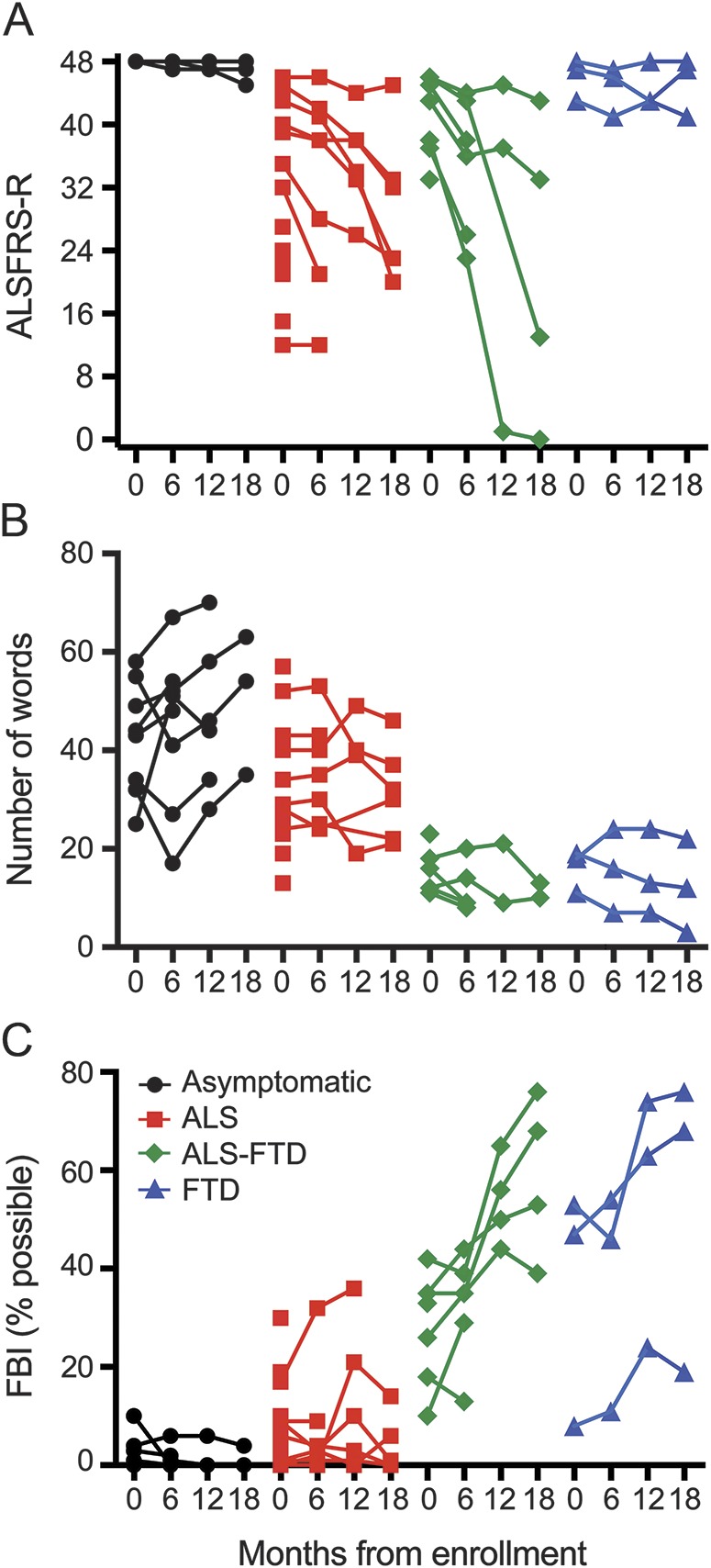
Individual C9orf72 mutation carriers are grouped by their clinical diagnosis at enrollment with values at baseline and 6-, 12-, and 18-month follow-up. Black circles represent asymptomatic carriers; red squares, ALS; green diamonds, ALS-FTD; and blue triangles, bvFTD. (A) ALS functional rating scale. (B) Letter fluency. (C) FBI, expressed as percent of possible total score because items affected by motor function in each patient were excluded. ALS = amyotrophic lateral sclerosis; ALSFRS-R = Revised ALS Functional Rating Scale; bvFTD = behavior-variant frontotemporal dementia; FBI = Frontal Behavioral Inventory; FTD = frontotemporal dementia.
Table 3.
Intercepts and slopes for longitudinal clinical measures for each diagnosis subgroup
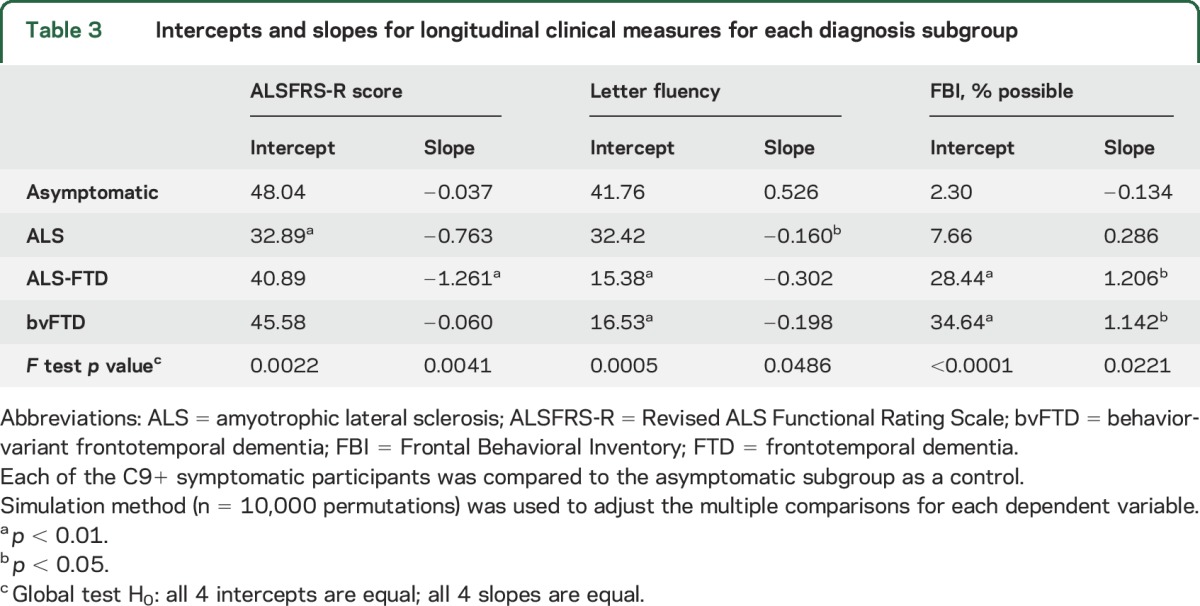
Motor.
The ALS-FTD subgroup had the fastest decline in the ALSFRS-R (p < 0.01), although scores were lowest at baseline in the C9+ ALS subgroup (p < 0.01). The ALSFRS-R declined on average by 0.76 points per month in the ALS group and 1.26 points per month in the patients with ALS-FTD.
Cognitive.
Letter fluency scores were lower at baseline in the C9+ ALS and ALS-FTD subgroups (p < 0.01). Letter fluency declined less in the C9+ ALS (0.16 words per month) than in the ALS-FTD subgroup (0.30 words per month), but the slope of decline was significant only for the C9+ ALS subgroup (p < 0.05). The 3 patients with bvFTD had varied changes in letter fluency; 2 lost ≈0.4 words/month, whereas the third patient, who was less impaired, improved between the first and second visit. The asymptomatic C9+ subgroup also improved slightly over time, possibly reflecting a practice effect for this task.
Behavioral.
The FBI score was significantly higher at baseline in the C9+ ALS-FTD and bvFTD subgroups (p < 0.01) and increased faster in both subgroups than for C9+ asymptomatic participants (p < 0.05). The FBI scores increased on average by >1% per month in the ALS-FTD and bvFTD subgroups.
Correlations between changes in primary outcome measures and clinical measures were examined for the 24 C9+ participants with a baseline and 6-month visit. The change in letter fluency scores was correlated with changes in the DRS-2 but not with changes in the rate of syllable repetition (table e-2). Changes in the FBI were correlated with changes in the FAB, ALSFRS-R, and time to complete the 9-Hole Peg Test. Changes in the ALSFRS-R were not correlated with changes in the DRS-2 or syllable repetition rate but were correlated with changes in the FBI, FAB, 9-Hole Peg Test, and category fluency.
DISCUSSION
The pattern of scores on the ALSFRS-R, letter fluency, and FBI differentiated among C9orf72 diagnostic subgroups that were defined by clinical criteria. Over 18 months of follow-up, all 3 measures remained relatively stable in asymptomatic C9+ participants. In symptomatic C9+ participants, these measures worsened in a manner consistent with their clinical diagnosis at enrollment. The rate of decline in the ALSFRS-R score in C9+ carriers with clinically defined ALS or ALS-FTD was similar to that reported for patients with sporadic ALS.13 The ALSFRS-R remained stable in the C9+ asymptomatic carriers and bvFTD subgroups. C9+ bvFTD and ALS-FTD were characterized by worsening behavioral function, as measured by a rising FBI score. The ALSFRS-R and FBI were uncorrelated and together provided a snapshot of disease symptom severity and worsening over time. Letter fluency performed less well than the FBI as a longitudinal measure. Although letter fluency is a known deficit in ALS cognitive impairment and ALS-FTD23,24 and differed among subgroups at baseline, the slopes of decline in C9+ ALS-FTD and C9+ bvFTD did not differ from that in C9+ asymptomatic participants. The similar flat slopes of decline may be due to a floor effect in C9+ participants with bvFTD and ALS-FTD, who generated few words at the first visit, or a practice effect in asymptomatic participants.
Most instruments used to screen for cognitive dysfunction in patients with ALS and FTD include measures of fluency, particularly letter fluency.16,17,25–27 One retrospective study of C9+ carriers found abnormal letter fluency in more than half of the C9+ participants, most of whom had FTD.7 Another retrospective study found that verbal fluency scores had a faster annualized rate of decline in C9+ patients with FTD and ALS compared to noncarriers with the same diagnosis.25 The rates of decline in letter fluency in 2 of the 3 C9+ patients with bvFTD and the patients with ALS-FTD were similar to those in the retrospective study by Irwin et al.25 Although letter fluency declined in the C9+ patients with ALS, it is limited as a longitudinal measure because of data loss in patients with worsening motor function. Our finding that letter fluency was correlated with FBI scores is consistent with previous reports showing overlap between language and behavioral symptoms in patients with pathologically confirmed FTD.28 Because letter fluency and FBI are collected from different sources, i.e., patient and caregiver, future studies may find it valuable to obtain both measures.
Survival is a typical outcome measure in ALS clinical trials.29 All but 1 of the 21 symptomatic patients survived for 6 months after enrollment in the study. By 18 months, half of the 16 patients with ALS or ALS-FTD had died, whereas the 3 C9+ participants with only bvFTD survived. These data highlight the need for stratifying patients entering clinical trials if survival is to be used as an outcome. The survival of the C9+ patients with ALS over the 18-month follow-up interval was similar to reports from sporadic ALS populations13,30,31 but differs from the shorter survival of C9+ carriers than patients with sporadic ALS found in retrospective and pathologic studies.8,25,32 This difference may reflect the enrollment bias of our study toward more slowly progressing patients who were able to travel. Shorter survival has been reported to be associated with hexanucleotide repeat length in the cerebellum, which may differ from the repeat length in blood and peripheral tissues.33 Unfortunately, repeat length was not measured in blood samples or autopsy material in this study. Some studies found slower progression of C9+ patients with bvFTD compared to bvFTD noncarriers.34 Our study had too few C9+ patients with bvFTD to evaluate survival.
Clinical trials for ALS tend to be relatively short, lasting 6 to 12 months, and most set eligibility criteria on the basis of disease severity or duration.29 This natural history study included C9+ participants with early and advanced disease. Despite these differences, it is notable that the clinical diagnoses remained stable over the 18 months of follow-up in all but one patient. The single exception was a patient with ALS with a slight decline in cognition over follow-up. Patients entering the study with motor impairment became weaker, and patients entering with cognitive symptoms worsened. There were no abrupt changes in symptoms or diagnosis. This is consistent with a study that found that patients with sporadic ALS with normal cognition at presentation tended to remain cognitively intact or to decline only slowly.35 The absence of cognitive-behavioral abnormalities in the C9+ asymptomatic participants, who were on average >10 years younger than C9+ symptomatic patients, would be consistent with an earlier study comparing asymptomatic carriers and noncarriers of FTD genes from the same families.36 One caution is that cognitive-behavioral symptoms can develop gradually and may not be appreciated by families until pronounced. Caregiver histories indicated that some patients developed additional symptoms of motor or cognitive impairment only late in their course. Nevertheless, our finding that diagnoses remained stable over the course of 6 or 12 months is encouraging for future clinical trials.
The goal of this study was to assess candidate measures that would potentially be useful for following changes in different phenotypes of C9orf72 mutation carriers in clinical trials. The 3 candidate outcome measures, ALSFRS-R, verbal fluency, and FBI, were selected to represent motor, cognitive, and behavioral function on the basis of literature on ALS and FTD.13–17 Their representation of these domains is supported by the correlation with a battery of additional motor measures and cognitive tests. It was encouraging that 6-month longitudinal changes were correlated with changes in other clinical measures such as the DRS-2, FAB, and 9-Hole Peg Test, but the sample size was too small to determine the sensitivity of the 3 measures to clinical decline and longer time periods. Further studies with larger cohorts are needed to confirm our findings, but these data suggest that the 3 brief outcome measures studied here, the ALSFRS-R, letter fluency, and the FBI, may be useful to stratify the most common phenotype-associated C9orf72 mutations and to follow changes over time.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and families for participation and the neurologists who cared for and referred patients to this study. Recruitment was made possible in part by the Agency for Toxic Substances & Disease Registry’s National ALS Registry Research Notification Mechanism (http://wwwn.cdc.gov/ALS/ALSClinicalResearch.aspx).
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- ALSFRS-R
Revised ALS Functional Rating Scale
- bvFTD
behavioral-variant frontotemporal dementia
- D-KEFS
Delis-Kaplan Executive Function System
- DRS
Dementia Rating Scale
- FAB
Frontal Assessment Battery
- FBI
Frontal Behavioral Inventory
- FTD
frontotemporal dementia
- MMSE
Mini-Mental State Examination
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
Dr. Floeter: study concept and design, acquisition, analysis of data, drafted manuscript. Dr. Traynor: acquisition and interpretation of data, critical revision of manuscript. J. Farren: acquisition, organization, and analysis of data, study coordination. L.E. Braun: acquisition and analysis of data, critical review neuropsychological data. Dr. Wiggs and M. Tierney: acquisition and interpretation of neuropsychological testing. Dr. Wu: statistical analysis of data, critical revision of manuscript.
STUDY FUNDING
The study was supported by the intramural program of the National Institute of Neurological Disorders and Stroke, NIH Z01 NS003146.
DISCLOSURE
M. Floeter reports no disclosures relevant to the manuscript. B. Traynor and the NIH have applied for a patent on the C9orf72 gene. J. Farren, L. Braun, E. Wiggs, M. Tierney, and T. Wu report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Riboldi G, Zanetta C, Ranieri M, et al. Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD diseases. Mol Neurobiol 2014;50:721–732. [DOI] [PubMed] [Google Scholar]
- 5.Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 2014;37:433–442. [DOI] [PubMed] [Google Scholar]
- 6.Gendron TF, Cosio DM, Petrucelli L. c9RAN translation: a potential therapeutic target for the treatment of amyotrophic lateral sclerosis and frontotemporal dementia. Expert Opin Ther Targets 2013;17:991–995. [DOI] [PubMed] [Google Scholar]
- 7.Boeve BF, Boylan KB, Graff-Radford NR, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Byrne S, Elamin M, Bede P, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9orf72 repeat expansion: a population-based cohort study. Lancet Neurol 2012;11:232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chio A, Calvo A, Moglia C, Mazzini L, Mora G. Phenotypic heterogeneity of amyotrophic lateral sclerosis: a population based study. J Neurol Neurosurg Psychiatry 2011;82:740–746. [DOI] [PubMed] [Google Scholar]
- 10.Cedarbaum JM, Stambler N, Malta E, et al. The ALSFRS-R: a revised ALS functional rating scale that incorporates assessments of respiratory function: BDNF ALS Study Group (phase III). J Neurol Sci 1999;169:13–21. [DOI] [PubMed] [Google Scholar]
- 11.Milan G, Lamenza F, Iavarone A, et al. Frontal Behavioural Inventory in the differential diagnosis of dementia. Acta Neurol Scand 2008;117:260–265. [DOI] [PubMed] [Google Scholar]
- 12.Christodoulou G, Gennings C, Hupf J, et al. Telephone based cognitive-behavioral screening for frontotemporal changes in patients with amyotrophic lateral sclerosis (ALS). Amyotroph Lateral Scler Frontotemporal Degener 2016;17:482–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Atassi N, Berry J, Shui A, et al. The PRO-ACT database: design, initial analyses, and predictive features. Neurology 2014;83:1719–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Proudfoot M, Jones A, Talbot K, Al-Chalabi A, Turner MR. The ALSFRS as an outcome measure in therapeutic trials and its relationship to symptom onset. Amyotroph Lateral Scler Frontotemporal Degener 2016;17:414–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Possin KL, Feigenbaum D, Rankin KP, et al. Dissociable executive functions in behavioral variant frontotemporal and Alzheimer dementias. Neurology 2013;80:2180–2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranasinghe KG, Rankin KP, Lobach IV, et al. Cognition and neuropsychiatry in behavioral variant frontotemporal dementia by disease stage. Neurology 2016;86:600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Knopman DS, Kramer JH, Boeve BF, et al. Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain 2008;131:2957–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 19.Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jurica SJ, Leitten CL, Mattis S. Dementia Rating Scale: Professional Manual. Odessa: Psychological Assessment Resources; 2001. [Google Scholar]
- 21.Rascovsky K, Salmon DP, Hansen LA, Galasko D. Distinct cognitive profiles and rates of decline on the Mattis Dementia Rating Scale in autopsy-confirmed frontotemporal dementia and Alzheimer’s disease. J Int Neuropsychol Soc 2008;14:373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delis DC, Kramer JH, Kaplan E, Holdnack J. Reliability and validity of the Delis-Kaplan Executive Function System: an update. J Int Neuropsychol Soc 2004;10:301–303. [DOI] [PubMed] [Google Scholar]
- 23.Abrahams S, Newton J, Niven E, Foley J, Bak TH. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener 2014;15:9–14. [DOI] [PubMed] [Google Scholar]
- 24.Heidler-Gary J, Hillis AE. Distinctions between the dementia in amyotrophic lateral sclerosis with frontotemporal dementia and the dementia of Alzheimer’s disease. Amyotroph Lateral Scler 2007;8:276–282. [DOI] [PubMed] [Google Scholar]
- 25.Irwin DJ, McMillan CT, Brettschneider J, et al. Cognitive decline and reduced survival in C9orf72 expansion frontotemporal degeneration and amyotrophic lateral sclerosis. J Neurol Neurosurg Psychiatry 2013;84:163–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bak TH, Hodges JR. Cognition, language and behaviour in motor neurone disease: evidence of frontotemporal dysfunction. Dement Geriatr Cogn Disord 1999;10(suppl 1):29–32. [DOI] [PubMed] [Google Scholar]
- 27.Woolley SC, York MK, Moore DH, et al. Detecting frontotemporal dysfunction in ALS: utility of the ALS Cognitive Behavioral Screen (ALS-CBS). Amyotroph Lateral Scler 2010;11:303–311. [DOI] [PubMed] [Google Scholar]
- 28.Harris JM, Jones M, Gall C, et al. Co-occurrence of language and behavioural change in frontotemporal lobar degeneration. Dement Geriatr Cogn Dis Extra 2016;6:205–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Miller RG, Munsat TL, Swash M, Brooks BR. Consensus guidelines for the design and implementation of clinical trials in ALS: World Federation of Neurology Committee on Research. J Neurol Sci 1999;169:2–12. [DOI] [PubMed] [Google Scholar]
- 30.Chio A, Logroscino G, Hardiman O, et al. Prognostic factors in ALS: a critical review. Amyotroph Lateral Scler 2008;10:310–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Czaplinski A, Yen AA, Simpson EP, Appel SH. Slower disease progression and prolonged survival in contemporary patients with amyotrophic lateral sclerosis: is the natural history of amyotrophic lateral sclerosis changing? Arch Neurol 2006;63:1139–1143. [DOI] [PubMed] [Google Scholar]
- 32.Umoh ME, Fournier C, Li Y, et al. Comparative analysis of C9orf72 and sporadic disease in an ALS clinic population. Neurology 2016;87:1024–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.van Blitterswijk M, DeJesus-Hernandez M, Niemantsverdriet E, et al. Association between repeat sizes and clinical and pathological characteristics in carriers of C9ORF72 repeat expansions (Xpansize-72): a cross-sectional cohort study. Lancet Neurol 2013;12:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Devenney E, Hornberger M, Irish M, et al. Frontotemporal dementia associated with the C9ORF72 mutation: a unique clinical profile. JAMA Neurol 2014;71:331–339. [DOI] [PubMed] [Google Scholar]
- 35.Elamin M, Bede P, Byrne S, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology 2013;80:1590–1597. [DOI] [PubMed] [Google Scholar]
- 36.Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal Dementia Initiative (GENFI) study: a cross-sectional analysis. Lancet Neurol 2015;14:253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.