

Abstract
Objective:
To estimate the genetic risk conferred by known amyotrophic lateral sclerosis (ALS)–associated genes to the pathogenesis of sporadic ALS (SALS) using variant allele frequencies combined with predicted variant pathogenicity.
Methods:
Whole exome sequencing and repeat expansion PCR of C9orf72 and ATXN2 were performed on 87 patients of European ancestry with SALS seen at the University of Utah. DNA variants that change the protein coding sequence of 31 ALS-associated genes were annotated to determine which were rare and deleterious as predicted by MetaSVM. The percentage of patients with SALS with a rare and deleterious variant or repeat expansion in an ALS-associated gene was calculated. An odds ratio analysis was performed comparing the burden of ALS-associated genes in patients with SALS vs 324 normal controls.
Results:
Nineteen rare nonsynonymous variants in an ALS-associated gene, 2 of which were found in 2 different individuals, were identified in 21 patients with SALS. Further, 5 deleterious C9orf72 and 2 ATXN2 repeat expansions were identified. A total of 17.2% of patients with SALS had a rare and deleterious variant or repeat expansion in an ALS-associated gene. The genetic burden of ALS-associated genes in patients with SALS as predicted by MetaSVM was significantly higher than in normal controls.
Conclusions:
Previous analyses have identified SALS-predisposing variants only in terms of their rarity in normal control populations. By incorporating variant pathogenicity as well as variant frequency, we demonstrated that the genetic risk contributed by these genes for SALS is substantially lower than previous estimates.
Amyotrophic lateral sclerosis (ALS) is a progressive neurodegenerative disease of the upper and lower motor neurons, which eventually leads to death within an average of 3–5 years1 after symptom onset. ALS is classified as familial (FALS) when a clear family history of ALS exists and sporadic (SALS) when it does not. No clinical features reliably distinguish FALS from SALS. Genetic research on ALS has largely been focused on FALS, which represents 10% of ALS cases.1 Most FALS is inherited in autosomal dominant fashion. However, this transmission pattern can be complicated by the early death of unrecognized affected family members due to non-ALS causes, misdiagnoses in older affected individuals, small family sizes, incomplete penetrance of genetic risk factors, and the development of disorders associated with ALS, such as frontotemporal dementia (FTD). Thus, sporadic and familial forms of ALS can be difficult to distinguish, and much remains unknown about the roles of genetic factors in FALS and especially in SALS. The discovery of the pathogenic (G4C2)n hexanucleotide repeat expansion of C9orf72 in a large percentage of FALS and SALS patients,2–4 as well as the identification of other ALS genes in patients with SALS,5,6 has highlighted the importance of genetic risk factors in SALS pathogenesis. The significance of genetics in SALS is further supported by ALS genome-wide association studies, which estimate the heritability of ALS to be at least 21.0%.7
With the growing affordability and availability of next-generation sequencing technologies, along with the advent of specific treatments for certain genetic forms of ALS,8 it is increasingly important to understand the genetic factors in causing SALS. Currently, there is considerable variation in estimates of the percentage of SALS cases caused by genetic variants, ranging from 11%5 to 28%9 in populations of European ancestry. This variation is due largely to differences in estimation methods. In one large study,9 the percentage was derived by calculating the portion of SALS cases with a rare (minor allele frequency [MAF] <0.01), protein-altering variant in a set of known ALS genes. Using variant rarity as the main criterion for pathogenicity may have inflated the risk estimate as the majority of rare nonsynonymous variants are not thought to be pathogenic.10
In this study, we sought to better estimate the percentage of SALS cases that have an identifiable genetic factor likely responsible for disease pathogenesis. To address this, a joint approach utilizing both allele frequency and variant pathogenicity prediction was used to determine the percentage of SALS cases that possess a potentially deleterious genetic variant in an ALS-associated gene.
METHODS
Standard protocol approvals, registrations, and patient consents.
The sample collection and study design we performed was approved by the University of Utah institutional review board. Written informed consent for disease-specific genetic studies was obtained from each patient who participated in this study.
Participants.
Patients with ALS diagnosed at the University of Utah from 2011 to 2013 were invited to participate in genetic studies. All participants were seen by neuromuscular specialists and diagnosed with probable or definite ALS according to revised El Escorial criteria.11 These patients were followed longitudinally in our motor neuron disease clinic. Patients with SALS were identified as having no self-reported family history of ALS, probable ALS, or FTD. In total, 96 patients with SALS were enrolled in this study. DNA was obtained from whole blood of each participant using the Gentra Puregene Blood Kit (Qiagen, Venlo, Netherlands).
Identification of deleterious ATXN2 and C9orf72 repeat expansions.
ATXN2 CAG repeat size was determined by fluorescent PCR amplification. Repeat lengths between 29 and 33 were considered to be of intermediate length and deleterious.12 The detection of C9orf72 GGGGCC repeat expansions was performed by using previously established repeat primed-PCR and amplicon length analysis criteria.13
Whole exome sequencing.
Patient DNA was exome enriched by the SeqCap EZ Exome Enrichment Kit v3.0 (Roche [Basel, Switzerland] NimbleGen) and sequenced by the Illumina (San Diego, CA) HiSeq platform to generate 101-bp, paired-end reads that covered target regions to an average depth ranging from 41× to 224× per sample. Reads were then aligned to the GRCh37 reference genome using BWA-MEM v0.7.12. Picard Tools v1.130 was used to perform indexing, coordinate sorting, and duplicate read marking of all aligned genomic reads. Variant calling and quality filtering were performed using Genome Analysis Toolkit's (v3.3-0) HaplotypeCaller and variant quality score recalibration (VQSR) methods.14 In order to properly power VQSR filtering, 99 CEU (Utah residents [CEPH] with northern and western European ancestry) and 92 GBR (British in England and Scotland) individuals from the 1000 Genomes Project15 with exome sequencing data were included in the genotyping steps.
Quality control.
Utah's population is outbred and genetically resembles other populations of northern European ancestry.15–17 As a result, we focused our analysis on patients with SALS who were of European ancestry in order to limit population stratification effects. Patient ancestry derived from genetic data is more reliable than self-reported ancestry, which has been used in previous ALS studies.9,18 An Admixture19 analysis (K = 3) was performed to determine the genetic ancestry of each patient with SALS. Any participants with less than 90% European ancestry were removed from further analysis. Next, principal components analysis (PCA) was performed using smartpca20 to remove poor-quality samples. Any sample with an eigenvector value more than 6 SDs from the mean for the first 10 principal components was discarded. Finally, the sex of each patient with SALS was inferred by PLINK 1.921 and compared to the reported sex to identify sample identification errors.
Variant annotation.
SnpEff22 (v4.1) was used to identify protein-coding and splice-site altering genetic variants. These variants were then annotated with information from the database for nonsynonymous single nucleotide polymorphism functional predictions (dbNSFP; sites.google.com/site/jpopgen/dbNSFP) v2.9.23 dbNSFP contains 11 different in silico functional prediction methods that determine which single nucleotide variants (SNVs) are likely to alter protein function. MetaSVM was chosen as the primary method to determine variant pathogenicity as it has been shown to have a better predictive ability than other methods.24 Insertion, deletion, and splice-site acceptor/donor variants were classified as deleterious. Variants were also annotated with European-specific MAF estimates from the Exome Aggregation Consortium (ExAC)25 by dbNSFP. A manual search of the Amyotrophic Lateral Sclerosis Online Database (ALSoD),26 the Single Nucleotide Polymorphism database (dbSNP),27 and the Human Gene Mutation Database (HGMD)28 was performed to identify known ALS pathogenic variants.
Genetic risk analysis.
To determine the proportion of patients with SALS who have a potentially disease-causing variant in a SALS gene, all annotated rare (European MAF <0.001), protein-coding, and splice-site altering variants in 31 ALS-associated genes (ANG, CHCHD10, CHMP2B, DAO, DCTN1, ELP3, ERBB4, EWSR1, FIG4, FUS, GLE1, GRN, HNRNPA1, HNRNPA2B1, MATR3, NEFH, NEK1, OPTN, PFN1, SETX, SOD1, SPAST, SQSTM1, SS18L1, TAF15, TARDBP, TBK1, TUBA4A, UBQLN2, VAPB, VCP) were assessed. A MAF of 0.001 corresponds roughly to the European allele frequency of SOD1:p.Asp91Ala,29 which is the most common known pathogenic variant we could identify in ALSoD. The proportion of patients with SALS who possessed a rare and deleterious variant, as determined by MetaSVM, in at least 1 of the 31 ALS-associated genes or had a deleterious repeat expansion in C9orf72 or ATXN2 was then calculated. The proportion of patients with SALS who possessed a rare variant, deleterious or not, or a repeat expansion in an ALS-associated gene was also calculated as a reference. All variants were assumed to act in a dominant fashion, like most ALS-causing variants.30
Odds ratio analysis.
Genetic burden analysis determines if there is a difference in the amount of pathogenic variation, or burden, in a set of genes between cases and controls. To determine whether the combination of variant frequency and MetaSVM predictions identified variant pathogenicity better than variant frequency alone, we estimated the excess burden of ALS-associated genes in SALS cases vs healthy controls. To do so, whole exome sequence data from 714 individuals from 181 families of the Simons Simplex Collection (SSC)31 were analyzed. The SSC dataset contains exome sequence data from children with autism, an unaffected sibling, and their unaffected parents. These samples underwent joint variant calling with the SALS exomes using the same pipeline as described above. Admixture and PCA were performed as previously described on 362 unaffected parents to select for high-quality controls of European ancestry. Variant calls were limited to exome capture regions with at least 5× coverage on average in both the SALS and SSC cohorts. The proportion of SSC controls with a rare and deleterious variant in at least 1 of the same 31 ALS-associated genes was calculated. An odds ratio (OR) analysis was then performed to determine whether the burden of ALS-associated genes is higher in patients with SALS than in normal controls. An OR analysis comparing the genetic burden when only variant frequency was utilized was also performed. The significance of this OR was determined by a one-tailed Fisher exact test.
RESULTS
Patient cohort characteristics.
The Admixture (figure 1A) and PCA (figure 1B) results showed that 9 of the 96 patients with SALS possessed significant non-European ancestry or were genetic outliers. No sex mismatches were detected in the data. The 87 patients with SALS of European ancestry were selected for analysis, and characteristics of these patients are detailed in table 1. We selected 324 SSC parents as high-quality European controls because Admixture showed that 38 of the parents were likely non-European.
Figure 1. Admixture and principal components analysis (PCA) plots show the ancestry and sample quality of the sporadic amyotrophic lateral sclerosis (SALS) cohort.

(A) An Admixture plot where each bar represents a patient with SALS (in total 96 patients). The height of each colored bar represents the amount of ancestry each individual derives from. Blue = European (CEU), green = East Asian (CHB + JPT), and red = African (YRI). Individuals with less than 90% European ancestry (yellow bar) were removed from further analysis. The 9 patients with SALS with less than 90% European ancestry are indicated with a red asterisk. (B) PCA plot of all 96 individuals with 1,000 genomes data (CEU = Utah residents [CEPH] with northern and western European ancestry; CHB = Han Chinese in Beijing, China; JPT = Japanese in Tokyo, Japan; YRI = Yoruba in Ibadan, Nigeria). Shaded areas represent the area over which the kernel density of each respective 1000 genomes population spans. SALS samples are listed as purple circles. Arrows indicate non-European individuals who were removed from further analysis.
Table 1.
Detailed summary of the sporadic amyotrophic lateral sclerosis cohort before and after selecting for European patients

Known ALS-associated genetic variants.
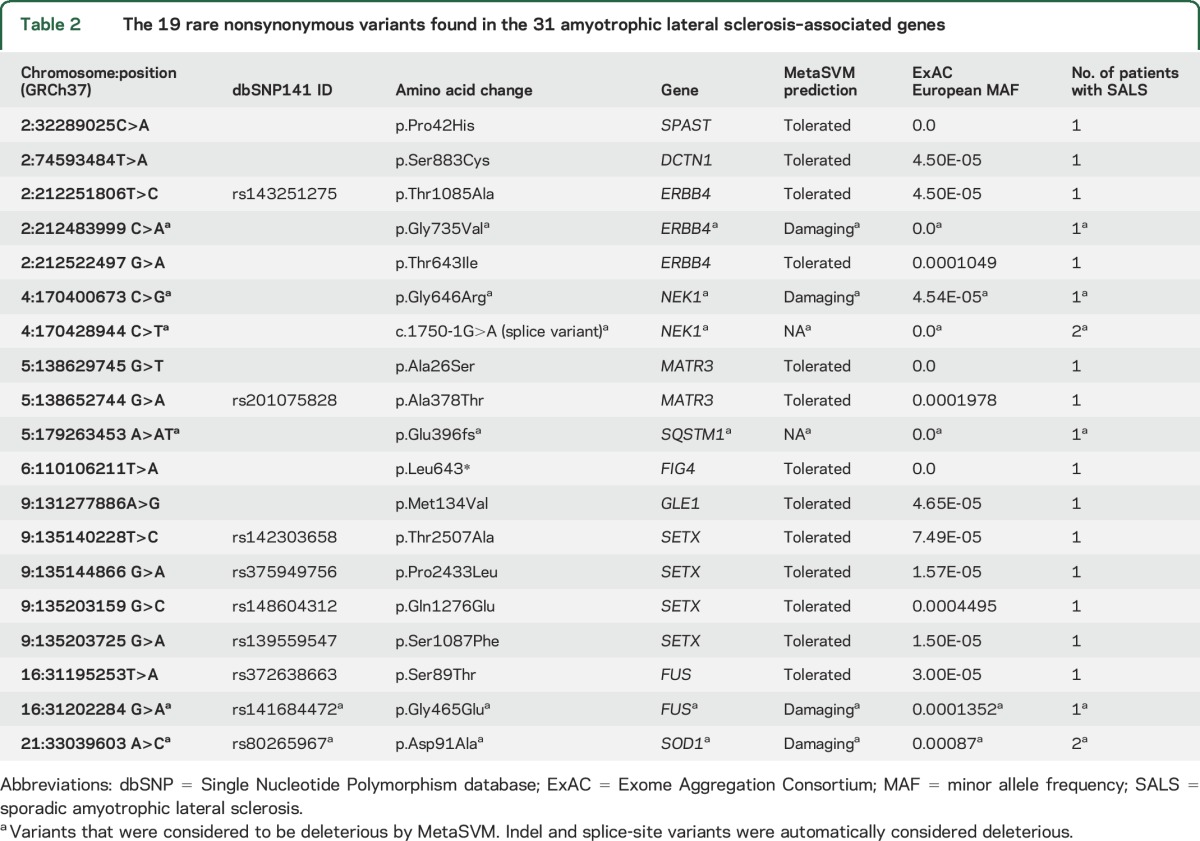
We identified pathogenic C9orf72 hexanucleotide repeat expansions in 5 of 87 patients with SALS (5.7%). Two patients with SALS (2.3%) possessed ALS-associated trinucleotide repeat expansions in ATXN2 (31 and 32 repeats in length, respectively). We compared the rare (European MAF <0.001) coding variants in 31 ALS-associated genes uncovered in the SALS cohort to known ALS risk variants contained in ALSoD, dbSNP, and HGMD. This comparison revealed only one known ALS-associated rare SNV (SOD1:p.Asp91Ala29), which was found in 2 heterozygous patients (table 2).
Table 2.
The 19 rare nonsynonymous variants found in the 31 amyotrophic lateral sclerosis–associated genes
Potentially novel ALS variants.
After examining the 31 ALS-associated genes in our patient cohort, we identified 18 rare coding variants (European MAF <0.001) not previously described in ALS (table 2). Of these variants, 10 were not found in dbSNP (v141). Furthermore, 6 were novel, as they were not found in ExAC, the 1000 Genomes Project dataset,15 or the National Heart, Lung, and Blood Institute Exome Sequencing Project dataset. One novel single nucleotide frameshift insertion was found in SQSTM1 (chr5:179263453A>AT). A novel splice-site acceptor variant in NEK1 (chr4:170428944C>T) was found in 2 patients.
Genetic risk analysis.
Among the 31 ALS-associated genes, 19 rare variants were found in 21 patients. The FIG4:p.Leu643* variant was the only variant not Sanger-validated due to a lack of high-quality DNA. When combined with the 5 C9orf72 and 2 ATXN2 deleterious repeat expansions, 28 rare variants or repeat expansions were found in 25 patients across all ALS-associated genes. Three patients had 2 rare variants in an ALS gene. One patient had a GLE1:p.Met134Val (chr9:131277886A>G) variant in addition to a pathologic C9orf72 repeat expansion. Another patient had a C9orf72 repeat expansion in addition to a rare SETX:p.Thr2507Ala (chr9:135140228T>C) missense variant. Finally, one patient possessed SPAST:p.Pro42His (chr2:32289025C>A) and ERBB4:p.Thr643Ile (chr2:212522497 G>A) missense variants.
These 28 rare variants were used to calculate the proportion of patients with SALS who have a rare mutation in at least one ALS-associated gene. A total of 25 patients with SALS (28.7%) had at least one rare variant or pathogenic repeat expansion in an ALS gene when variant deleteriousness was not considered. However, only 4 of the 17 SNVs annotated with MetaSVM were considered deleterious (table 2). As a result, the proportion of patients with SALS with a rare and deleterious SNV or repeat expansion in an ALS-associated gene was 17.2% (15/87 patients; figure 2). Variant predictions from 10 other methods were also used (table e-1 at Neurology.org), which yielded proportions ranging between 14.9% and 21.8%.
Figure 2. Percentage of sporadic amyotrophic lateral sclerosis (SALS) cases with an identifiable genetic variant likely responsible for disease.

The percentage next to each gene indicates what percentage of SALS cases have a rare and pathogenic variant in that gene. A majority (82.8%) of SALS cases have no identifiable genetic variants potentially responsible for their disease.
OR analysis.
The genetic burden of ALS-associated genes in patients with SALS was compared with the burden among 324 SSC controls. Using only rare variant frequency as a criterion for assessing burden, patients with SALS had a modest increase in burden compared to controls (OR 1.90, p < 0.025; table 3). However, when variant pathogenicity was added by incorporating MetaSVM results and variant frequency, SALS cases showed a much higher burden in ALS-associated genes compared to controls (OR 4.98, p < 9 × 10−5; table 3). Other variant prediction methods in dbNSFP yielded similar findings, but the OR analysis using MetaSVM predictions resulted in the highest p value (table e-2).
Table 3.
Odds ratio (OR) analyses comparing the genetic burden of amyotrophic lateral sclerosis (ALS)–associated genes of patients with sporadic ALS (SALS) vs controls

DISCUSSION
We report findings from a genomic analysis of 87 patients with SALS of European origin. In total, 28 rare variants were found in 33 ALS genes in our patient cohort. Only one non-repeat variant that has been previously described in ALS pathogenesis was observed (SOD1:p.Asp91Ala). This variant is known to cause autosomal recessive ALS and was predicted to be deleterious by MetaSVM. SOD1:p.Asp91Ala has also been suggested to act in a dominant fashion; however, few instances of this have been reported.32 In addition, we identified 18 rare variants in ALS-associated genes that have not been described previously in patients with ALS. Of these 18 variants, 5 either caused a protein loss of function or were predicted to be deleterious by MetaSVM. One is a frameshift variant in the ubiquitin-associated domain of SQSTM1. This change likely ablates SQSTM1's ability to bind ubiquitinated substrates, which is often seen in SQSTM1 variants that cause ALS.33 NEK1:c.1750-1G>A is a novel loss of function SNV that ablates the splice acceptor site of intron 19, which is located approximately in the middle of the gene. NEK1:p.Gly646Arg is another damaging variant that was discovered in NEK1; however, it does not occur in a defined protein domain. FUS:p.Gly465Glu is predicted to be damaging by MetaSVM and affects an amino acid one position upstream from previously reported SALS variant (FUS:p.Met464Ile).34 ERBB4:p.Gly735Val is an SNV predicted to be deleterious and occurs in the tyrosine kinase domain of erbB-4. The tyrosine kinase function of erbB-4 is required for protein autophosphorylation and triggering downstream signaling cascades upon activation. A variant in the tyrosine kinase domain of erbB-4, which was identified from a FALS family, has been shown to reduce protein autophosphorylation and likely causes ALS.35 Additional studies will determine the functional importance of these variants on cellular and molecular mechanisms.
We have demonstrated that using variant pathogenicity predictions is more reliable than variant frequency alone to determine the proportion of patients with SALS whose disease is likely caused by a variant in an ALS-associated gene. The relative effect of ALS-associated genes is stronger when variant pathogenicity is considered instead of only variant rarity. This follows from the fact that an appreciable proportion of rare nonsynonymous variants are not predicted to be functionally damaging.10 Thus, only a subset of rare variants in ALS-associated genes are pathogenic.
Our approach to estimating the genetic contribution of a large panel of known ALS-associated genes by directly predicting variant pathogenicity differs from earlier approaches. The first attempts at determining the proportion of genetically caused SALS cases did so by calculating the proportion of patients who had a protein-coding variant in a panel of 5–7 ALS-associated genes.18,36–38 These analyses yielded estimates ranging from 2.8% to 11%, which are lower than our estimate of 17.2%. A more recent study, in which variant rarity (MAF <1%) was used as the sole criterion for pathogenicity in a panel of 17 ALS-associated genes, concluded that genetic factors may cause 27.8% of SALS cases,9 a figure similar to our estimate when only variant rarity is considered (table e-1). However, these variants (MAF <1%) are not significantly more common in our patients with SALS than in unaffected controls (OR 1.25, p > 0.25), suggesting that many of them are not pathogenic. The same 17 ALS-associated genes are significantly more burdened in patients with SALS than controls when variant rarity (MAF <1%) and pathogenicity (estimated by MetaSVM) are combined (OR 2.61, p < 0.02). These OR differences support our conclusion that variant frequency alone is not a sufficient predictor of SALS risk.
Another analysis of 33 ALS-associated genes defined only novel and extremely rare variants (MAF ≈ 0.0002) as pathogenic and found that 14.5% of SALS cases could be attributed to genetic causes.39 In our sample, the genetic burden of ALS-associated genes in patients with SALS is less when pathogenicity is defined in the same way than when MetaSVM is integrated (OR 2.24, p < 0.01 vs OR 5.52, p < 2 × 10−4). These results demonstrate that direct predictions of variant pathogenicity are important for defining genetic risk in SALS and other genetic diseases.
Our results also highlight that genetic factors play an important role in the disease, the clinical relevance of which will become even more important as genetic specific treatments become available. Further, exome or targeted sequencing of patients with SALS and their family members is likely warranted to provide adequate genetic counseling. In addition, our results suggest the distinction between SALS and FALS may be problematic as heritable risk factors are found in a significant proportion of patients with SALS. Future genetic investigations of patients with SALS are needed to broaden the scope of SALS-associated loci. Studies with larger patient cohorts that incorporate measures of variant pathogenicity will also be needed to further pinpoint the proportion of SALS cases with an identifiable probable genetic cause of disease, especially as more ALS-associated genetic loci are discovered.
Our study has several limitations. First, the size of the SALS cohort was limited, especially given the genetically heterogeneous nature of ALS. Second, because we focused on individuals of European ancestry, our findings may not be completely applicable to ALS found in other populations. Third, 13 of the 324 (4.0%) healthy control samples used in this study had at least one rare and deleterious variant in ALS-associated genes as predicted by MetaSVM (table 3; table e-3). The mean age of these individuals was 41.76 (SD 5.92) years, which is much lower than the average age at onset of SALS at 56 years of age.40 It is possible that some of the control individuals with these variants could develop ALS later in life.
Supplementary Material
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- ALSoD
Amyotrophic Lateral Sclerosis Online Database
- dbNSFP
database for nonsynonymous single nucleotide polymorphism functional predictions
- dbSNP
Single Nucleotide Polymorphism database
- ExAC
Exome Aggregation Consortium
- FALS
familial amyotrophic lateral sclerosis
- FTD
frontotemporal dementia
- HGMD
Human Gene Mutation Database
- MAF
minor allele frequency
- OR
odds ratio
- PCA
principal components analysis
- SALS
sporadic amyotrophic lateral sclerosis
- SNV
single nucleotide variant
- SSC
Simons Simplex Collection
- VQSR
variant quality score recalibration
Footnotes
Supplemental data at Neurology.org
Editorial, page 220
AUTHOR CONTRIBUTIONS
Dr. Gibson made a substantive contribution to the design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising of the manuscript. J.M. Downie made a substantive contribution to the design and conceptualization of the study, analysis and interpretation of the data, and drafting and revising of the manuscript. Dr. Tsetsou made a substantive contribution to the drafting and revising of the manuscript. J.E. Feusier made a substantive contribution to the analysis and interpretation of the data and revising of the manuscript. K.P. Figueroa made a substantive contribution to the analysis and interpretation of the data as well as revising of the manuscript. Dr. Bromberg made a substantive contribution to the revising of the manuscript. Dr. Pulst made a substantive contribution to the design and conceptualization of the study, interpretation of the data, and revising of the manuscript. Dr. Jorde made a substantive contribution to the design and conceptualization of the study, interpretation of the data, and revising the manuscript.
STUDY FUNDING
Supported by NIH (TR001066, GM59290, GM104390, GM118335, and NS033123) and Target ALS. Research reported in this publication was supported by the National Center for Advancing Translational Sciences of the NIH under award TR001066. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Partial funding support for S.M.P. was provided by Target ALS and by the NIH under award NS033123. L.B.J. was supported by NIH grants GM59290, GM104390, and GM118335. The control data used in this analysis can be found in the National Database for Autism Research (ndar.nih.gov/) under study DOI:10.15154/1149697.
DISCLOSURE
S. Gibson is a shareholder of Recursion Pharmaceuticals. J. Downie, S. Tsetsou, J. Feusier, K. Figueroa, M. Bromberg, and L. Jorde report no disclosures relevant to the manuscript. S. Pulst reports the following disclosures: Progenitor Life Sciences, shareholder; Cedars-Sinai, royalties; University of Utah, royalties; and Ataxion Therapeutics, consultant. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Rowland LP, Shneider NA. Amyotrophic lateral sclerosis. N Engl J Med 2001;344:1688–1700. [DOI] [PubMed] [Google Scholar]
- 2.DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Majounie E, Renton AE, Mok K, et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Renton AE, Chio A, Traynor BJ. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 2014;17:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science 2015;347:1436–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Keller MF, Ferrucci L, Singleton AB, et al. Genome-wide analysis of the heritability of amyotrophic lateral sclerosis. JAMA Neurol 2014;71:1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.van Zundert B, Brown RH Jr. Silencing strategies for therapy of SOD1-mediated ALS. Neurosci Lett 2017;636:32–39. [DOI] [PubMed] [Google Scholar]
- 9.Cady J, Allred P, Bali T, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol 2015;77:100–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li MX, Kwan JS, Bao SY, et al. Predicting mendelian disease-causing non-synonymous single nucleotide variants in exome sequencing studies. PLoS Genet 2013;9:e1003143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brooks BR, Miller RG, Swash M, Munsat TL; World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 12.Neuenschwander AG, Thai KK, Figueroa KP, Pulst SM. Amyotrophic lateral sclerosis risk for spinocerebellar ataxia type 2 ATXN2 CAG repeat alleles: a meta-analysis. JAMA Neurol 2014;71:1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Akimoto C, Volk AE, van Blitterswijk M, et al. A blinded international study on the reliability of genetic testing for GGGGCC-repeat expansions in C9orf72 reveals marked differences in results among 14 laboratories. J Med Genet 2014;51:419–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DePristo MA, Banks E, Poplin R, et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 2011;43:491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, et al. A global reference for human genetic variation. Nature 2015;526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McLellan T, Jorde LB, Skolnick MH. Genetic distances between the Utah Mormons and related populations. Am J Hum Genet 1984;36:836–857. [PMC free article] [PubMed] [Google Scholar]
- 17.Jorde LB. Inbreeding in the Utah Mormons: an evaluation of estimates based on pedigrees, isonomy, and migration matrices. Ann Hum Genet 1989;53:339–355. [DOI] [PubMed] [Google Scholar]
- 18.Lattante S, Conte A, Zollino M, et al. Contribution of major amyotrophic lateral sclerosis genes to the etiology of sporadic disease. Neurology 2012;79:66–72. [DOI] [PubMed] [Google Scholar]
- 19.Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 2009;19:1655–1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet 2006;2:e190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cingolani P, Platts A, Wang le L, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 2012;6:80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat 2013;34:E2393–E2402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong C, Wei P, Jian X, et al. Comparison and integration of deleteriousness prediction methods for nonsynonymous SNVs in whole exome sequencing studies. Hum Mol Genet 2015;24:2125–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016;536:285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Abel O, Shatunov A, Jones AR, Andersen PM, Powell JF, Al-Chalabi A. Development of a smartphone app for a genetics website: the amyotrophic lateral sclerosis online genetics database (ALSoD). JMIR Mhealth Uhealth 2013;1:e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sherry ST, Ward MH, Kholodov M, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 2001;29:308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stenson PD, Mort M, Ball EV, Shaw K, Phillips A, Cooper DN. The human gene mutation database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Hum Genet 2014;133:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Andersen PM, Nilsson P, Ala-Hurula V, et al. Amyotrophic lateral sclerosis associated with homozygosity for an Asp90Ala mutation in CuZn-superoxide dismutase. Nat Genet 1995;10:61–66. [DOI] [PubMed] [Google Scholar]
- 30.Robberecht W, Philips T. The changing scene of amyotrophic lateral sclerosis. Nat Rev Neurosci 2013;14:248–264. [DOI] [PubMed] [Google Scholar]
- 31.Iossifov I, O'Roak BJ, Sanders SJ, et al. The contribution of de novo coding mutations to autism spectrum disorder. Nature 2014;515:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Al-Chalabi A, Andersen PM, Chioza B, et al. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor. Hum Mol Genet 1998;7:2045–2050. [DOI] [PubMed] [Google Scholar]
- 33.Rea SL, Majcher V, Searle MS, Layfield R. SQSTM1 mutations: bridging Paget disease of bone and ALS/FTLD. Exp Cell Res 2014;325:27–37. [DOI] [PubMed] [Google Scholar]
- 34.Nagayama S, Minato-Hashiba N, Nakata M, et al. Novel FUS mutation in patients with sporadic amyotrophic lateral sclerosis and corticobasal degeneration. J Clin Neurosci 2012;19:1738–1739. [DOI] [PubMed] [Google Scholar]
- 35.Takahashi Y, Fukuda Y, Yoshimura J, et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet 2013;93:900–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kwon MJ, Baek W, Ki CS, et al. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiol Aging 2012;33:e1017–e1023. [DOI] [PubMed] [Google Scholar]
- 37.van Blitterswijk M, van Es MA, Hennekam EA, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet 2012;21:3776–3784. [DOI] [PubMed] [Google Scholar]
- 38.Chio A, Calvo A, Mazzini L, et al. Extensive genetics of ALS: a population-based study in Italy. Neurology 2012;79:1983–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kenna KP, McLaughlin RL, Byrne S, et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet 2013;50:776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kinsley L, Siddique T. Amyotrophic lateral sclerosis overview. In: Pagon RA, Adam MP, Ardinger HH, et al., editors. GeneReviews [Internet]. Seattle: University of Washington; 1993. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.