

Abstract
Background and Purpose
The PDE enzymes (PDE1–11) hydrolyse and thus inactivate cyclic nucleotides and are important in the regulation of the cardiovascular system. Here,we have investigated the effects on the cardiovascular system, of two novel selective PDE1 inhibitors, Lu AF41228 and Lu AF58027.
Experimental Approach
We used rat mesenteric small arteries (internal diameters of 200–300 μm), RT‐PCR and measured isometric wall tension. Effects of Lu AF41228 and Lu AF58027 on heart rate and BP were assessed in both anaesthetized and conscious male rats.
Key Results
Nanomolar concentrations of Lu AF41228 and Lu AF58027 inhibited PDE1A, PDE1B and PDE1C enzyme activity, while micromolar concentrations were required to observe inhibitory effects at other PDEs. RT‐PCR revealed expression of PDE1A, PDE1B and PDE1C in rat brain, heart and aorta, but only PDE1A and PDE1B in mesenteric arteries. In rat isolated mesenteric arteries contracted with phenylephrine or U46619, Lu AF41228 and Lu AF58027 induced concentration‐dependent relaxations which were markedly reduced by inhibitors of guanylate cyclase, ODQ, and adenylate cyclase, SQ22536, and in preparations without endothelium. In anaesthetized rats, Lu AF41228 and Lu AF58027 dose‐dependently lowered mean BP and increased heart rate. In conscious rats with telemetric pressure transducers, repeated dosing with Lu AF41228 lowered mean arterial BP 10–15 mmHg and increased heart rate.
Conclusions and Implications
These novel PDE1 inhibitors induce vasodilation and lower BP, suggesting a potential use of these vasodilators in the treatment of hypertension and vasospasm.
Abbreviations
- L‐NAME
N ω‐nitro‐l‐arginine methyl ester
- ODQ
1H‐[1,2,4]oxadiazolo[4,3‐a]quinoxalin‐1‐one
- SQ22536
(9‐(tetrahydro‐2‐furanyl)‐9H–purin‐6‐amine, 9‐THF‐Ade)
Introduction
The PDE family consists of 11 enzymes (PDE1–11) which hydrolyse cyclic nucleotides (Bender and Beavo, 2006; Stangherlin and Zaccolo, 2012; Alexander et al., 2015b). PDE1 is the only PDE regulated by Ca2+–calmodulin, and the substrate specificity significantly differs among the different isoforms with PDE1A and PDE1B showing a preference for cGMP, whereas PDE1C hydrolyzes cGMP and cAMP equally well (Bender and Beavo, 2006). PDE1 in the presence of Ca2+ and calmodulin and PDE5 are the main contributors to the hydrolysis of cGMP in the human aorta (Miyahara et al., 1995), and PDE1 activity was found in rat mesenteric small arteries (Komas et al., 1991) and human ventricular cardiomyocytes (Johnson et al., 2012). Moreover, all three PDE1 isoenzymes are expressed in human and rat pulmonary, aorta and mesenteric small arteries (Murray et al., 2007; Schermuly et al., 2007). PDE1A was found to primarily regulate cGMP in rat cardiomyocytes and vascular smooth muscle cells (Bender and Beavo, 2006), and PDE1C regulates cAMP in aortic (Rybalkin et al., 2002) and pulmonary smooth muscle cells (Murray et al., 2007; Schermuly et al., 2007). Furthermore, there was up‐regulation of PDE1C in the vasculature of rats with pulmonary hypertension (Schermuly et al., 2007) and of PDE1A in rats with angiotensin II‐induced systemic hypertension (Giachini et al., 2011), and all three PDE1 enzymes are up‐regulated in senescent and human vascular smooth muscle cells (Bautista Nino et al., 2015; Cai et al., 2015). PDE1A is involved in the development of hypertrophy and fibrosis in the hypertrophic rat heart (Miller et al., 2009; Miller et al., 2011). Therefore, inhibition of PDE1 appears to be attractive for reversing pathological remodelling in the cardiovascular system (Chan and Yan, 2011).
In the past only, non‐selective PDE1‐inhibitors, for example, vinpocetine and 8‐methoxymethyl IBMX (Loughney et al., 1996) and SCH51866 and zaprinast (Vemulapalli et al., 1996; Bender and Beavo, 2006), have been used to assess cardiovascular function of PDE1 inhibitors, although IC86340 has an IC50 of 0.4 μmol·L−1 for PDE1 and 500 μmol·L−1 for PDE5 (Nagel et al., 2006). Treatment with 8‐methoxymethyl IBMX lowered pressure in the pulmonary circulation of mice and rats, while the pressure in the systemic circulation was unaltered (Schermuly et al., 2007). Infusion of SCH51866 also lowered the systemic BP in rats, whereas PDE5 inhibition had no effect (Vemulapalli et al., 1996). However, due to the previous lack of selective PDE1 inhibitors, the cardiovascular effects of selective inhibition of PDE1 are unclear.
Recently, two novel and selective inhibitors of PDE1, Lu AF41228 and Lu AF58027, have been developed (Figure 1). Lu AF41228 was discovered during a medicinal chemistry programme as an analogue of a published PDE1 ligand PF‐04471141 (Humphrey et al., 2014). Lu AF58027 was identified from a patent application claiming selective PDE1 inhibitors as radiotracers and a proposed PET ligand (Li et al., 2011). The purpose of the present study was to investigate the activity of these novel PDE1 inhibitors on isolated resistance arteries as well as the mechanisms underlying relaxations of mesenteric small arteries, and on heart rate (HR) and BP in vivo.
Figure 1.
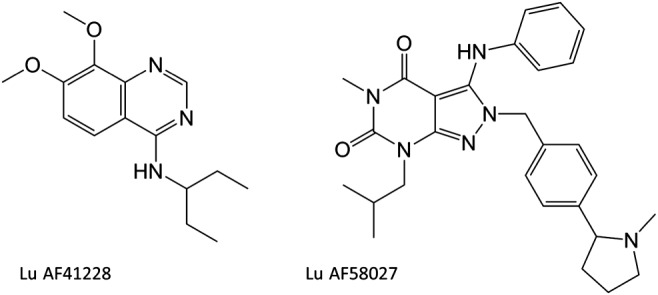
Structure of Lu AF41228 and Lu AF58027.
Methods
Animals and preparation of samples
All animal care and experimental protocols in this study were conducted under the supervision of a veterinarian and in accordance with the Danish legislation of animal use for scientific procedures as described in the ‘Animal Testing Act’ (Consolidation Act No. 726 of 9 September 1993 as amended by Act No. 1081 of 20 December 1995) and approved by the Danish Animal Experiments Inspectorate (permission 2011/561‐2011). The Danish Animal Testing Act fully and extensively covers the requirements included in the Guide for the Care and Use of Laboratory Animals as adopted and promulgated by the U.S. National Health Institute and the animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Animals were housed in the animal facility in cages (Universal Euro III type long) with standard wood bedding and space for two rats. There was a 12 h shift between light and darkness, and the animals had free access to food and drinking water.
The animals were selected randomly, and wherever possible, observations were made without knowledge of the treatments administered. For RT‐PCR studies and functional studies in isolated arteries, adult male Wistar rats (12–14 weeks) weighing 250–300 g (Taconic Aps, Ry, Denmark) were killed by cervical dislocation by trained animal staff at Aarhus University, and the mesenteric bed was removed and immediately immersed in 4°C cold physiological saline solution (pH = 7.4) of the following composition (mM): 119 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 1.5 CaCl2, 24.9 NaHCO3, 0.026 EDTA and 5.5 glucose. Mesenteric small arteries (internal diameters of 200–300 μm) were dissected.
Assessment of PDE enzyme inhibition
PDE1A, PDE1B and PDE1C assays were performed as follows: the assays was performed in 60 μL samples containing a fixed amount of the PDE1 enzyme (sufficient to convert 20–25% of the cyclic nucleotide substrate), a buffer (50 mM HEPES, pH 7.6; 10 mM MgCl2; 0.02% Tween20), 0.1 mg·mL−1 BSA, 15 nM [3H]‐cAMP (25779 cpm per assay) and varying amounts of inhibitors. Reactions were initiated by addition of the substrate, and reactions were allowed to proceed for 1 h at room temperature before being terminated through mixing with 20 μL (0.2 mg) yttrium silicate SPA beads (PerkinElmer, Waltham, MA, USA). The beads were allowed to settle for 1 h in the dark before the plates were counted in a Wallac 1450 Microbeta counter (PerkinElmer). The measured signals were converted to activity relative to an uninhibited control (100%), and IC50 values were calculated using XlFit (model 205; IDBS, Guildford, Surrey, UK). All other PDE assays were performed in a similar fashion using either [3H]‐cAMP or [3H]‐labelled cGMP (PDE5 and PDE9).
RT‐PCR
Immediately after killing, rat tissue and dissected small arteries were placed and kept in RNAlater (Qiagen, Copenhagen, Denmark) until homogenization in 350 μL RLT with β‐mercaptoethanol and 5 μL carrier RNA and homogenized for 3 min in TissueLyser (Qiagen). The samples were centrifuged for 2 min at 13 000× g. The isolation of total RNA from the arteries was done with the RNeasy Plus Mini Kit by using QIAcube (Qiagen) according to the manufacturer's instructions, after which cDNA was synthesized from total RNA using SuperScriptIII reverse transcriptase, SuperAse In and random decamer primers according to the manufacturer's instructions. cDNA (2μL)was amplified in a thermal cycler (Eurofins MWG Operon; Peqlab) in a reaction (25 μL) containing 0.2 mm deoxynucleoside 5′‐triphosphate mix, 0.4 μm of each primer, and 0.03 U·L−1 of TaKaRa Ex Taq Hot start version DNA polymerase. A ‘hot‐start’ procedure was employed (2 min at 95°C), and thermal cycling conditions were 95°C, 12 s; 57°C, 40 s; and 70°C, 30 s, for 40 cycles with a final extension of 4 min at 72°C. PCR reaction products were resolved by agarose gel electrophoresis (2.5% w·v−1) and stained with ethidium bromide (0.5 μg·mL−1). The primers and expected product size are listed in Supporting Information Table S1. For the identity of the amplification products to be determined, the agarose gel bands were excised and the DNA was purified using a Qiaex II gel extraction kit (Qiagen). The purified PCR product was sequenced by Eurofins MWG Operon (Ebersberg, Germany).
Isometric tension recordings
Mesenteric and femoral small arteries with internal diameters of 200–300 μm and with a length of approximately 2 mm were mounted on two 40 μm wires in microvascular myographs (Danish Myotechnology, Aarhus, Denmark) for isometric tension recordings and stretched to their optimal diameter, which corresponded to an internal circumference of 90% of that achieved when the vessels were exposed to a passive tension yielding a transmural pressure of 100 mmHg (Simonsen et al., 1999). Segments were discarded if noradrenaline‐induced (5 μM) contraction was less than 13.3 kPa or if acetylcholine‐induced (10 μM) relaxation on noradrenaline‐induced (5 μM) contraction was less than 50%, and exclusion following these criteria explains that unequal group numbers are reported. The baths were heated to 37°C and equilibrated with 5% CO2 to maintain the desired pH of 7.4. Segments were allowed to equilibrate for 10 min thereafter.
After the initial test of viability, arterial segments were pre‐contracted with the thromboxane analogue, U46619 or noradrenaline corresponding to 60–70% of the maximal contraction induced by a combination of KPSS plus noradrenaline (10 μM). After reaching a level of stable pre‐contraction, concentration–response curves (10−10–10−6 M) were constructed for the PDE5 and PDE3 inhibitors sildenafil and milrinone (2‐methyl‐6‐oxo‐1,6‐dihydro‐3,4′‐bipyridine‐5‐carbonitrile), respectively, and Lu AF41228 and Lu AF58207. In parallel, a time control curve was obtained where only vehicle was added.
For investigation of the mechanisms involved in the relaxations induced by Lu AF41228 and Lu AF58027, the vessels were contracted with phenylephrine instead of noradrenaline to avoid interference from other mechanisms than the activation of α1‐adrenoceptors. For investigation of the role of the endothelium in relaxations induced by the PDE1 inhibitors, mesenteric arteries with and without endothelium were examined. The endothelial cells were removed by introducing into the lumen a human scalp hair and rubbing back and forth several times. The effectiveness of the procedure was assessed by absence of relaxation to acetylcholine in noradrenaline‐contracted arteries. Vessels with and without endothelium were contracted with phenylephrine, and concentration–response curves were constructed for Lu AF41228 and Lu AF58207. In another series of experiments, mesenteric arteries were incubated with an inhibitor of NOS, L‐NAME (100 μM); an inhibitor of guanylate cyclase, ODQ (3 μM); an inhibitor of adenylate cyclase, SQ22546 (100 μM); or the combination for 30 min, and concentration–response curves for Lu AF41228 and Lu AF58207 were constructed in arteries contracted with either phenylephrine or U46619.
Anaesthetized rat studies
Infusion of drugs in anaesthetized rats is an established model for evaluation of effects on BP (Kun et al., 2009). Male Wistar Han rats (Charles River, Sulzfeld, Germany), weighing 250–350 g, were anaesthetized using 5% isoflurane in an induction chamber followed by 3.5% maintenance on mask. Catheters were surgically implanted into the jugular vein for infusion of test compounds and the and carotid artery for recording of BP (reported as mean arterial pressure). The arterial catheter was connected to an SP844 pressure transducer (Memscap, Crolles Cedex, France) and then via a BridgeAmp and a Powerlab 4/25 (ADInstruments) to a computer where data were recorded using LabChart7 (ADInstruments). After surgery, the isoflurane concentration was reduced to 2–2.5% and arterial pressure and HR were allowed to stabilize for 20–30 min before pre‐dose baseline data acquisition was started. All animals had a mean arterial pressure BP >75 mmHg after the stabilization period. Mean arterial pressure was calculated as 10 s running average values of 1/3 systolic pressure + 2/3 diastolic pressure. Compound or vehicle was infused via the venous catheter at a constant rate aimed at reaching a maximal concentration after 60 min. For an impression of the drug exposure for each compound to be obtained, blood samples for plasma exposure measurement were obtained from two satellite animals 10, 20, 30 and 60 min after start of the drug infusion. At the end of the experiment, rats were killed with an overdose of i.v. pentobarbital.
Rat telemetry studies
To eliminate the effects of anaesthesia on haemodynamics, telemetric recordings have been used in conscious rats to evaluate the haemodynamic effects and to allow repeated dosing of drugs (Bentzen et al., 2013). The effects on haemodynamics were only investigated for Lu AF41228 to reduce the number of animals used. We instead focused on possible adaptive mechanisms of repeated dosing. Male Sprague Dawley rats (Charles River, Kent, UK) were implanted with TL11M2‐C50‐PXT telemetry transmitters [Data Sciences International (DSI), New Brighton, MN, USA] in aseptic conditions by the animal supplier. The transmitters are capable of transmitting a BP signal via a pressure catheter inserted into the abdominal aorta. Following surgery, each rat was held in a stock colony for a minimum of 2 weeks to allow sufficient recovery prior to experimental use and monitored by trained animal staff. Rats were administered carprofen (Rimadyl, 5 mg·kg−1 s.c.) at induction of anaesthesia, at end of day of surgery and for 3 days post‐surgery. An antibiotic Baytril (2 mg·kg−1 s.c.) (enrofloxacin 25 mg·mL−1 and benzylalkohol 14 mg·mL−1) was administered 24 h before surgery, at induction of anaesthesia and for 5 days post‐surgery. The measured BP signal was recorded using a DSI Dataquest OpenART data capture system, and the digital data capture system was linked with a DSI Ponemah data acquisition and analysis system. The capture system allowed recording of the BP, whilst the rat was freely moving in the recording cage. For recording, cages were placed on receivers connected to a DSI data exchange matrix. The data exchange matrices were connected to a computer interface with DSI Dataquest ART Analogue telemetry software. Pre‐dose baseline (60 min) was established and recorded, and each rat was then removed from its recording cage and was dosed by p.o. gavage with vehicle or compound, and the animals were then returned to the recording cage. Data sampling for BP and HR was carried out in intervals of approximately 60 s duration at selected time points for the duration of the experiment (24 h recordings). Blood samples for plasma exposure measurement were obtained from three satellite animals.
Pharmacokinetic analysis
Rat plasma concentrations of Lu AF41228 and Lu AF58027 were determined using Ultra PLC coupled to MS/MS. See the Supporting Information for detailed description of methods and results.
Data evaluation and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). All data are presented as mean ± SEM with a significance level of P < 0.05 and n represents the number of individual animals. A lower number of animals were included in the vehicle groups due to low variability, while still providing adequate data for statistical analysis. Two‐way ANOVA followed by a post hoc Bonferroni t‐test was used to test for differences in concentration–response curves in isolated vessel segments using GraphPad Prism 7.0a (GraphPad Software Inc., La Jolla, CA, USA). The EC50 for relaxation was calculated in GraphPad Prism 7.0a using a nonlinear curve fit model:
Langendorff isolated heart data were evaluated using repeated measures, one‐way ANOVA with a Dunnett's multiple comparisons post test. Anaesthetized rat data were evaluated using two‐way ANOVA. Single‐dose rat telemetry data were evaluated using ANCOVA for each post‐dose time point separately, with treatment, period and animal as the factors in the model and baseline (0 h time point) included as a covariate, and using either a Williams or Dunnett's post test. Repeat‐dose rat telemetry data were evaluated using ANCOVA for each post‐dose time point separately, with treatment and animal as the factors in the model and baseline included as a covariate. The vehicle on days 1, 3 and 7 was compared with the compound treatments on days 8, 10 and 14, respectively, using t‐tests based on the error mean square from the ANCOVA.
Materials
The following drugs ‐ acetylcholine; L‐NAME; noradrenaline hydrochloride, milrinone (2‐methyl‐6‐oxo‐1,6‐dihydro‐3,4′‐bipyridine‐5‐carbonitrile), ODQ; phenylephrine hydrochloride; sildenafil (5‐[2‐ethoxy‐5‐(4‐methylpiperazin‐1‐ylsulfonyl)phenyl]‐1‐methyl‐3‐propyl‐1,6‐dihydro‐7H‐pyrazolo[4,3‐d]pyrimidin‐7‐one); SQ22546, and U46619 (9α‐epoxymethanoprostaglandin F2α) ‐ were supplied by Sigma (St Louis, MO, USA). [3H] cAMP ((NET275001MC, 31.3 Ci mmol‐1) was from PerkinElmer Danmark A/S (Skovlunde, Denmark). Lu AF58027 and Lu 41228 were synthesized by Lundbeck A/S (Copenhagen, Denmark). ODQ, Lu AF58027 and Lu 41228 were dissolved in DMSO and further diluted in distilled water. Unless otherwise stated, the substances were dissolved in distilled water. The DMSO concentration in the bath was low, and parallel control curves were run to examine whether vehicle affected vascular contractility.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a, 2015b).
Results
Effect of Lu AF41228 and Lu AF58027 on PDE activity
PDE1 enzymes were inhibited with half maximal inhibitory concentrations (IC50) of Lu AF41228 (39–170 nM) and Lu AF58027 (2–45 nM), while other PDEs were only affected at μM concentrations (Table 1). For examination for off‐target effects, a screening of binding of Lu AF41228 (10 μM) against 100 different receptors and transporters was performed (CEREP, Le Bois l'Evêque, France), and it showed that at this concentration it inhibited binding of marked ligands to adenosine A1, A2A, A2B and A3 receptors by 22.6 ± 0.4, 21.8 ± 2.4, 17.6 ± 6.1 and 34.7 ± 5.5% respectively.
Table 1.
Inhibition of PDE enzymic activity by Lu AF41228 or Lu AF58027
| Lu AF41228 | Lu AF58027 | ||
|---|---|---|---|
| PDE1A (IC50, nM) | 170 | PDE1A (IC50, nM) | 13 |
| PDE1B (IC50, nM) | 39 | PDE1B (IC50, nM) | 45 |
| PDE1C (IC50, nM) | 78 | PDE1C (IC50, nM) | 1.4 |
| PDE2 (% inh @ 2 μM) | 13 | PDE2 (% inh @ 2 μM) | 0 |
| PDE3 (% inh @ 2 μM) | 11.5 | PDE3 (% inh @ 2 μM) | 48 |
| PDE4D (IC50, nM) | 2600 | PDE4D (% inh @ 10 μM) | 52 |
| PDE5A (% inh @ 10 μM) | 23 | PDE5A (IC50, nM) | 910 |
| PDE7A (% inh @ 10 μM) | 9 | PDE7A (% inh @ 10 μM) | 42 |
| PDE7B (% inh @ 2 μM) | 13 | PDE7B (% inh @ 2 μM) | 38 |
| PDE8A (% inh @ 2 μM) | 17 | PDE8A (% inh @ 2 μM) | 19 |
| PDE9 (% inh @ 10 μM) | 9 | PDE9 (% inh @ 10 μM) | 0 |
| PDE10 (% inh @ 2 μM) | 36.5 | PDE10 (% inh @ 10 μM) | 63 |
| PDE11 (% inh @ 10 μM) | 15.5 | PDE11 (% inh @ 10 μM) | 25 |
Inhibition of enzymic activity was determined in vitro either as dose–response (IC50 determination) or as single‐point. Note that for PDE4 and PDE10, results are not reported similarly between Lu AF41228 and Lu AF58027.
Expression of PDE1
The primer sequences and expected product size are listed in Supporting Information Table S1. The PCR product was sequenced, and only bands corresponding to the respective PDE1 subtypes are reported. RT‐PCR revealed the expression of PDE1A, PDE1B and PDE1C in rat aorta, lung, heart and brain, while only PDE1A and PDE1B were expressed in mesenteric small arteries (Figure 2).
Figure 2.

RT‐PCR of PDE1A–C expression. Expression in rat (1) mesenteric arteries, (2) aorta, (3) lung, (4) heart and (5) brain. PDE1A–C were expressed in all investigated tissues with the exception of PDE1C, which was not expressed in mesenteric arteries. The bands were sequenced and confirmed as the correct product. The gels are representative of tissue from three animals, run in three different experiments. Primers can be seen in Supporting Information Table S1.
Studies in isolated vascular segments
In noradrenaline‐ and U46619‐contracted mesenteric and femoral small arteries, Lu AF41228 and Lu AF58207 caused concentration‐dependent relaxations (Figure 3). In the U46619‐contracted mesenteric arteries, the PDE inhibitors induced relaxations with the following potency order (−logEC50): sildenafil (9.02 ± 0.11) > Lu AF58207 (7.22 ± 0.20) = milrinone (7.17 ± 0.31) ≥ Lu AF41228 (7.01 ± 0.19) (Supporting Information Table S2). In the U46619‐contracted small femoral arteries, the PDE inhibitors induced relaxations with the following potency order (−logEC50): sildenafil (7.88 ± 0.23) > milrinone (7.15 ± 0.31) ≥ Lu AF58207 (6.11 ± 0.21) ≥ Lu AF41228 (5.86 ± 0.32). In the noradrenaline‐contracted mesenteric and femoral arteries, the curves were shifted to the right, but potency order was similar (Figure 3).
Figure 3.
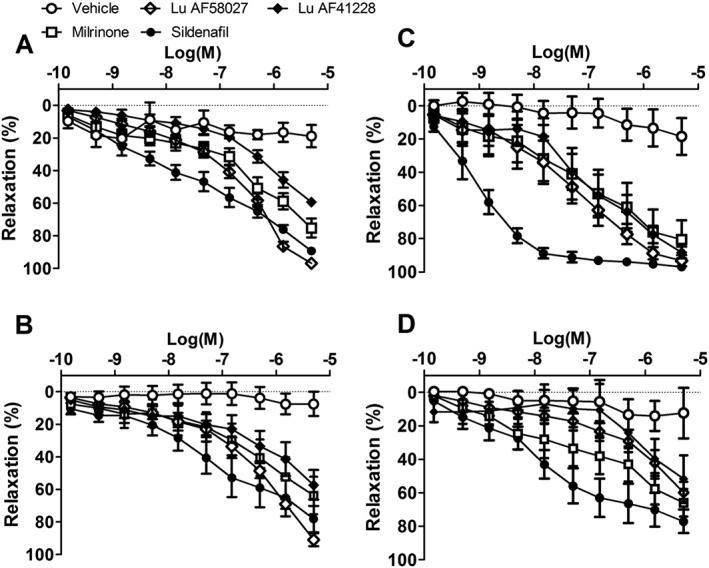
Vasodilatation induced by PDE1 inhibition in rat small arteries. Concentration–response curves for sildenafil (n = 6), a PDE5 inhibitor; milrinone (n = 5), a PDE3 inhibitor; and two PDE1 inhibitors, Lu AF41228 (n = 5) and Lu AF58027 (n = 5), and vehicle (n = 7) in (A, B) noradrenaline‐ and (C, D) U46619‐contracted, (A, C) mesenteric and (B, D) femoral arteries. Data are means ± SEM, where n indicates the number of animals.
Role of the endothelium and nucleotide pathways in relaxations induced by Lu AF58207 and Lu AF41228 in mesenteric arteries
In phenylephrine‐contracted preparations, relaxations induced by Lu AF58207 and Lu AF41228 were markedly reduced in mesenteric arteries without endothelium compared with arteries with endothelium (Figure 4). In arteries with endothelium contracted with either phenylephrine or U46619, incubation with an inhibitor of NOS, L‐NAME, reduced relaxations induced by Lu AF58207 or Lu AF41228 to the same degree as endothelial cell removal, suggesting that endothelium‐derived NO plays a role in these relaxations (Figure 4).
Figure 4.
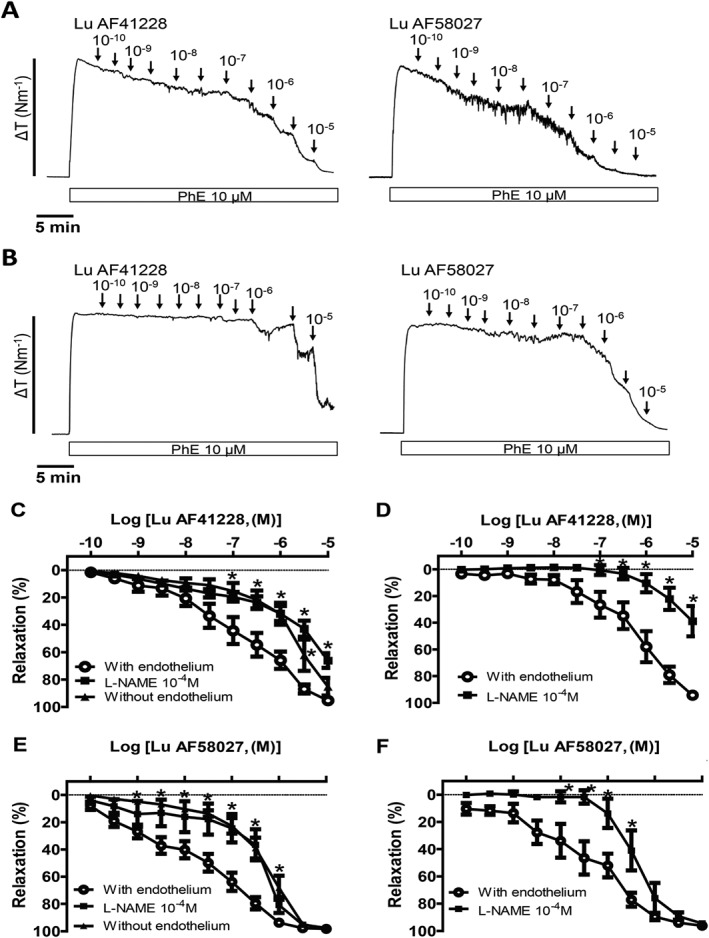
Effect of endothelial cell removal and NOS inhibition on vasodilatation induced by PDE1 inhibition in rat mesenteric arteries. Original recordings showing concentration–response curves for Lu AF41228 and Lu AF58027 in phenylephrine (PhE)‐contracted mesenteric arteries with endothelium (A) and without endothelium (B). Average concentration–response curves for Lu AF41228 in (C) PhE‐contracted arteries with endothelium (n = 12), in the presence of the NOS inhibitor, L‐NAME (n = 6), and in arteries without endothelium (n = 6), (D) U46619‐contracted arteries with endothelium (n = 6) and in the presence of L‐NAME (n = 6). Concentration–response curves for Lu AF58027 in (E) PhE‐contracted arteries with endothelium (n = 10), in the presence of L‐NAME (n = 6) and in arteries without endothelium (n = 7), (F) U46619‐contracted arteries with endothelium (n = 6) and in the presence of L‐NAME (n = 6). Data are means ± SEM, where n indicates the number of animals. *P < 0.05, significantly different from arteries with endothelium.
ODQ and SQ22536 are inhibitors of, respectively, guanylate and adenylate cyclase and markedly inhibited relaxations induced by Lu AF58207 or Lu AF41228 (Figure 5), while the combination of ODQ and SQ22536 caused further inhibition of relaxations induced by Lu AF58207 and Lu AF41228 (Figure 5). Incubation with the inhibitors (ODQ and SQ22536) did not change the contractions induced by adding phenylephrine or U46619 (Supporting Information Table S3).
Figure 5.
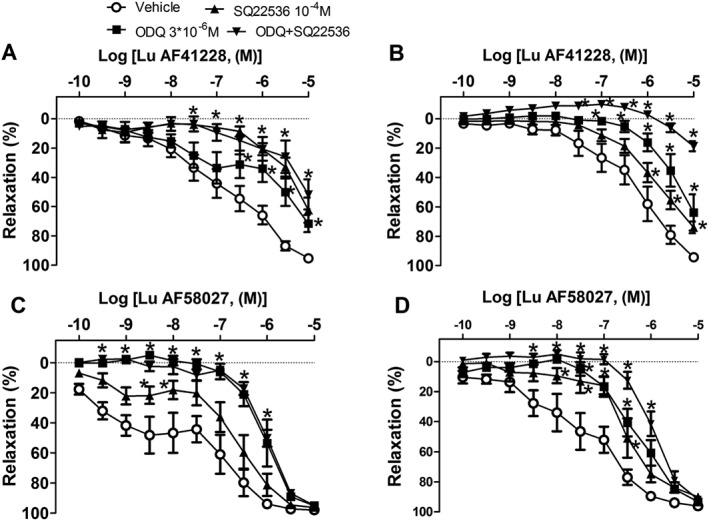
Effect of guanylate cyclase and adenylate cyclase inhibition on vasodilatation induced by PDE1 inhibition in rat mesenteric arteries. Concentration–response curves for Lu AF41228 in (A) phenylephrine‐contracted arteries incubated with vehicle (n = 6); an inhibitor of guanylate cyclase, ODQ (n = 6); an inhibitor of adenylate cyclase, SQ22536 (n = 6); or the combination of ODQ and SQ22536 (n = 5), and (B) U46619‐contracted arteries incubated with vehicle (n = 6), ODQ (n = 6), SQ22536 (n = 6) or the combination of ODQ and SQ22536 (n = 5). Concentration–response curves for Lu 58027 in (C) phenylephrine‐contracted arteries incubated with vehicle (n = 6); an inhibitor of guanylate cyclase, ODQ (n = 7); an inhibitor of adenylate cyclase, SQ22536 (n = 5); or the combination of ODQ and SQ22536 (n = 6), and (D) U46619‐contracted arteries incubated with vehicle (n = 6), ODQ (n = 7), SQ22536 (n = 5) or the combination of ODQ and SQ22536 (n = 6). Data are means ± SEM. *P < 0.05, significantly different from vehicle control.
Anaesthetized rat studies
In anaesthetized rats, Lu AF41228 induced a dose‐dependent effect on mean BP already reported in methods and HR. At 10 mg·kg−1·h−1 (n = 6), Lu AF41228 induced an immediate, minor decrease in BP and an increase in HR (Supporting Information Figure S1). BP returned to baseline after 3–4 min, while HR remained elevated (36 bpm increase vs. vehicle after 5 min; Figure 6A, B). From 5 min onwards, there was a slower decline in BP to a ~20 mmHg decrease compared to vehicle at the end of the experiment. The increase in HR persisted throughout the experiment but gradually declined in magnitude. Unbound plasma exposure at 10, 20, 30 and 60 min was 3.3 ± 0.1, 3.6 ± 0.3, 5.7 ± 0.1 and 7.7 ± 0.1 μM (Supporting Information Figure S2A).
Figure 6.
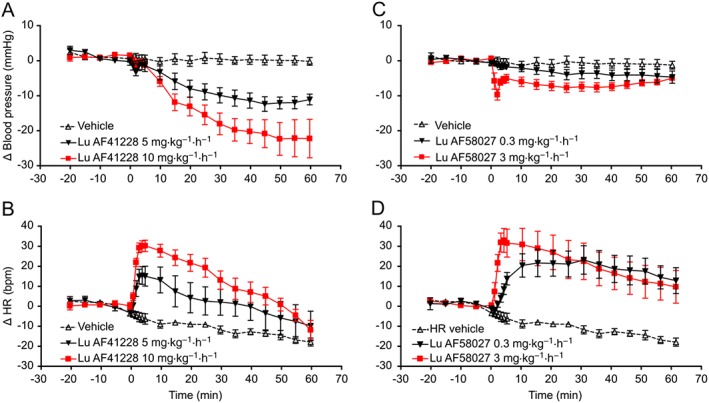
BP decrease and HR increase in response to PDE1 inhibition in anaesthetized rats. After equilibrium was reached, baseline was recorded for 20 min and animals were then given i.v. infusions of either compound or vehicle (continuous infusion over 60 min). Plasma exposure was measured in separate time‐matched animals. (A) Decrease in BP in response to Lu AF41228 (5 and 10 mg·kg−1·h−1). A minor response occurred immediately after start of the drug infusion at both doses followed by a return towards baseline after 5 min. BP then dose‐dependently decreased compared with vehicle throughout the experiment (5 mg·kg−1·h−1: P < 0.05, n = 4, 10 mg·kg−1·h−1: P < 0.05, n = 6, vehicle: n = 4). (B) Dose‐dependent increase in HR in response to Lu AF41228 (5 and 10 mg·kg−1·h−1). Response occurred immediately after start of the drug infusion (5 mg·kg−1·h−1: P < 0.05, n = 4, 10 mg·kg−1·h−1: P < 0.05, n = 6, vehicle: n = 4). (C) Decrease in BP in response to Lu AF58027 (0.3 and 3 mg·kg−1·h−1). Only a small but significant decrease was observed at 0.3 mg·kg−1·h−1. At 3 mg·kg−1·h−1, a minor response occurred immediately after start of the drug infusion followed by a return towards baseline after 5 min. BP then remained stably decreased compared with vehicle throughout the experiment (0.3 mg·kg−1·h−1: P < 0.05, n = 4, 3 mg·kg−1·h−1: P < 0.05, n = 6, vehicle: n = 4). (D) Dose‐dependent increase in HR in response to Lu AF58027 (0.3 and 3 mg·kg−1·h−1). Response occurred immediately after start of the drug infusion (0.3 mg·kg−1·h−1: P < 0.05, n = 4, 3 mg·kg−1·h−1: P < 0.05, n = 6, vehicle: n = 4).
At 5 mg·kg−1·h−1, Lu AF41228 induced an immediate, minor decrease in BP after 2 min with an incomplete recovery at 5 min (Figure 6A). At this dose, the effect on BP was also accompanied by an immediate increase in HR (21 bpm increase vs. vehicle at 5 min; Figure 6B). This was followed by a slower decline in BP to max 12 mmHg decrease compared to vehicle at 45 min. Using computer simulation, unbound plasma exposure of Lu AF41228 at 5 mg·kg−1 was predicted to be in the range between 1.2 and 3.8 μM during the 60 min infusion based on the pharmacokinetic data obtained at 10 mg·kg−1 (Supporting Information Figure S2A).
In anaesthetized rats, Lu AF58027 also induced a dose‐dependent effect on mean BP and HR. At 3 mg·kg−1·h−1 (n = 6), Lu AF58027 induced an immediate, minor decrease in BP and a large increase in HR. BP partly returned to baseline after 3–4 min, while HR remained elevated (37 bpm increase vs. vehicle after 5 min; Figure 6C, D). From 5 min onwards, BP remained stable at −4 to −7 mmHg compared with vehicle. The increase in HR persisted throughout the experiment. Unbound plasma exposure at 10, 20, 30 and 60 min was 66 ± 2, 67 ± 10, 86 ± 2 and 75 ± 1 nM (Supporting Information Figure S2B). At 0.3 mg·kg−1·h−1, Lu AF58027 only induced a small but significant decrease in BP (<4 mmHg vs. vehicle) throughout the experiment. However, an immediate, sustained increase in HR was observed. The increase in HR had a slower onset than the 3 mg·kg−1·h−1 dose and reached steady state after 10 min (35 bpm increase vs. vehicle at 30 min). Unbound plasma exposure at 10, 20, 30 and 60 min was 5.4 ± 0.9, 5.5 ± 0.5, 8.1 ± 1.1 and 6.6 ± 0.9 nM (Supporting Information Figure S2B).
Rat telemetry studies
In freely moving telemetered rats, the cardiovascular effects were only tested for one of the selective compounds. Lu AF41228, administered once p.o. at 10, 30 and 60 mg·kg−1 (n = 6 per test session), had no statistically significant effects on mean BP, compared with vehicle‐treated animals, at any of the measured time points (Figure 7A). However, Lu AF41228, administered once p.o. at 10, 30 and 60 mg·kg−1, significantly increased HR compared with vehicle‐treated animals at 0.25 and 0.5 h (10 mg·kg−1), at 0.25, 0.5, 1 and 2 h (30 mg·kg−1) and at 0.25, 0.5, 1, 2 and 4 h (60 mg·kg−1), P < 0.05 (Figure 7B). These effects were dose dependent in terms of effect and duration, but the maximum increase in HR was equal at the two high doses. Maximum unbound plasma exposure at 10, 30 and 60 mg·kg−1 was 374, 1010 and 1938 nM, respectively (Supporting Information Figure S3A).
Figure 7.
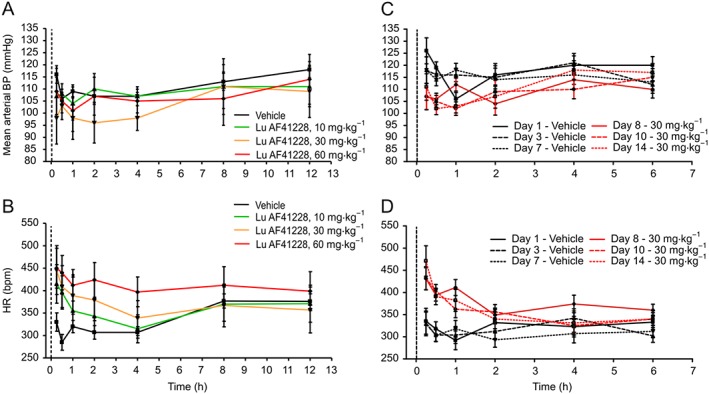
BP and HR changes in response to PDE1 inhibition in telemetered rats. Rats were dosed once with three different doses of Lu AF41228 (A, B, C) or twice daily with a single dose of Lu AF41228 for 7 days (D, E, F). Dotted vertical line represents time of dosing. All experiments are n = 6 except for exposure evaluations. (A) Lu AF41228 did not lead to statistically significant changes in mean arterial BP at any dose tested (maximum 60 mg·kg−1). (B) Lu AF41228 resulted in dose‐dependent increases in HR immediately after dosing. (C) After repeated dosing, Lu AF41228 resulted in a minor decrease in mean arterial BP (P < 0.05 after 0.25 and 0.5 h at day 8 and after 0.5 and 1 h at days 10 and 14). (D) After repeated dosing, Lu AF41228 resulted in an increase in HR immediately after dosing. The effect was similar at days 8, 10 and 14 (P < 0.05 after 0.25 and 0.5 and 1 h at day 8 and after 0.25 and 0.5 h at days 10 and 14).
For evaluation of the effects on the cardiovascular system after repeated administration, Lu AF41228 was then administered twice daily (b.i.d) at 30 mg·kg−1 for 7 days. Specifically, vehicle was administered to rats (5 mL·kg−1, nominally 12 h apart) on days 1–7. Lu AF41228 was dosed 30 mg·kg−1 (5 mL·kg−1, nominally 12 h apart) on days 8–14 of the study. Lu AF41228 (30 mg·kg−1 p.o., b.i.d.) evoked statistically significant decreases in mean BP compared with vehicle‐treated animals on their respective comparative days (day 8 vs. day 1, day 10 vs. day 3 and day 14 vs. day 7; mean BP in Figure 7C). At the same time, Lu AF41228 (30 mg·kg−1 p.o., b.i.d.) evoked statistically significant increases in HR in telemetered rats compared with vehicle‐treated animals on their respective comparative days (day 8 vs. day 1, day 10 vs. day 3 and day 14 vs. day 7; Figure 7D). Changes in mean BP and HR evoked by Lu AF41228 compared with vehicle‐treated animals were similar in pattern and magnitude on days 8, 10 and 14 after p.o. dosing of Lu AF41228 (30 mg·kg−1 p.o., b.i.d). Maximum unbound plasma exposure at days 8, 10 and 14 was 722, 516 and 734 nM respectively (Supporting Information Figure S3B).
For the direct effects on the isolated heart to further be explored, Lu AF41228 and Lu AF58027 were infused in spontaneously beating Langendorff guinea pig hearts. Both compounds markedly increased the perfusion rate, but they did not change HR, ECG and cardiac contraction (Supporting Information Figure S4).
Discusssion
The main findings of the present study were that Lu AF41228 and Lu AF58207 inhibited the activity of PDE1 enzymes in the nM range, while there was only a small effect on other PDEs, in the μM range. In mesenteric as well as femoral arteries from rats, both compounds induced relaxations in the nM range comparable in magnitude to relaxations induced by the PDE3 inhibitor, milrinone. Moreover, endothelial cell removal and inhibition of NOS or of guanylate cyclase reduced relaxations induced by Lu AF58027 and Lu AF41228, indirectly suggesting that inhibition of PDE1 increases the cGMP levels in the vascular smooth muscle and thereby potentiates the vasodilatation of endothelium‐derived NO in small arteries. These findings agree with our observations of PDE1A and PDE1B expression in rat mesenteric arteries. The vasodilator effect of Lu AF41228 and Lu AF58027 was substantiated by the observations that infusion of the compounds in anaesthetized animals lowered mean BP and increased HR as the unbound plasma concentrations increased.
In the past, non‐selective and not very potent PDE1 inhibitors, for example, vinpocetine and 8‐methoxymethyl IBMX (Loughney et al., 1996) and SCH51866 and zaprinast (Vemulapalli et al., 1996; Bender and Beavo, 2006), have been used to assess the cardiovascular function of PDE1 inhibitors (Bender and Beavo, 2006; Alexander et al., 2015b). The present study reveals that in isolated enzyme assays, Lu AF41228 and Lu AF58207 inhibit all three PDE1 isoforms in the nM range, while μM concentrations are required to observe an effect on other PDE enzymes including PDE3 and PDE5. These results suggest that these compounds have a unique profile allowing specific investigation of the functional effects of inhibiting the PDE1 enzymes.
Previous studies have found PDE1A, PDE1B and PDE1C expression in pulmonary vasculature, aorta and mesenteric arteries from rats (Murray et al., 2007; Schermuly et al., 2007; Giachini et al., 2011). Further, up‐regulation of PDE1A in hypertension (Giachini et al., 2011) and PDE1C in pulmonary hypertension has also been established (Schermuly et al., 2007). We found expression of PDE1A, PDE1B and PDE1C in the brain, lungs and heart of healthy rats. All three PDE1 subtypes were also expressed in rat aorta, while only PDE1A and PDE1B mRNA were expressed in the rat mesenteric arteries. Taken together, these results suggest that at least PDE1A and PDE1B are expressed in arteries from healthy animals, but that all three PDE1 subtypes may be of importance in hypertensive animals.
In man, the PDE3 and PDE5 inhibitors, milrinone and sildenafil, have previously been shown to decrease systemic arterial BP by 5–10 mmHg (Ludmer et al., 1986; Vardi et al., 2002) and to induce vasodilatation in pulmonary and systemic arteries from rat and man (Andersen et al., 2005; Grzesk et al., 2014; Rieg et al., 2014). In agreement with these findings, we found that sildenafil and milrinone relaxed rat mesenteric and femoral arteries. Lu AF41228 and Lu AF58027 were less potent than sildenafil, but they induced relaxations of the same potency and magnitude as milrinone. That the PDE1 inhibitors have a vasodilator effect is also supported by the observations in anaesthetized animals that infusion of Lu AF41228 and Lu AF58027 dose‐dependently reduced mean arterial BP. These findings are in agreement with similar observations showing vasodilatory and antihypertensive effects of a sulfonamide compound with effect on PDE1 (Pontes et al., 2012), but lower concentrations and doses of Lu AF41228 and Lu AF58027 were required to obtain the same effects, suggesting that these selective PDE1 inhibitors are potent vasodilators.
PDE1A and PDE1B preferentially hydrolyze cGMP (Bender and Beavo, 2006). In the present study, there are several observations supporting an involvement of the NO/cGMP pathway in relaxations induced by Lu AF41228 and Lu AF58027. Relaxations induced by Lu AF41228 and Lu AF58027 were reduced in mesenteric arteries without endothelium, and in the presence of L‐NAME, supporting the inhibition of Lu AF41228 and Lu AF58027 relaxations can be ascribed to reduction of NO availability. Moreover, Lu AF41228 relaxations were reduced in noradrenaline‐contracted, compared with phenylephrine‐contracted, mesenteric arteries (Figure 3 vs. Figure 4A) and can be ascribed to decreased NO availability due to direct chemical interactions at μM concentrations of noradrenaline as we have previously reported for nitrite‐induced NO‐mediated relaxations (Dalsgaard et al., 2007). Although other more direct measurements will be required, our findings indirectly suggest that inhibition of PDE1A and PDE1B by Lu AF41228 in the smooth muscle cells increases cGMP levels in the vascular smooth muscle and thereby potentiates the vasorelaxant effect of endothelium‐derived NO.
In mesenteric arteries, the removal of endothelium and inhibition of NOS caused a rightward shift of the concentration‐response curves for Lu AF58027, while the maximal relaxations were unchanged. These results suggest both a dependence on endothelium and a direct smooth muscle effect with Lu AF58027.
The adenylate cyclase inhibitor, SQ22536, also reduced Lu AF58027 and Lu AF41228 relaxations, suggesting that the adenylate cyclase/cAMP pathway is also involved in the vasodilator effect. The effect of SQ22536 was less in phenylephrine‐contracted than in U46619‐contracted mesenteric arteries. U46619 activation of thromboxane receptors in rat mesenteric arteries has previously been observed to lead to the formation of PGE2 (Bolla et al., 2004). Although further studies would be required, PGE2 may activate the adenylate cyclase/cAMP pathway in the vascular smooth muscle and hence explain the larger effect of SQ22536 on Lu AF58027 and Lu AF41228 relaxations in U46619 versus phenylephrine‐contracted small arteries. Another possibility is that the increase in smooth muscle cell calcium induced by activation of thromboxane receptors by U46619 is less than with α1‐adrenoceptor activation (Plane and Garland, 1996). The PDE1 enzymes are activated by intracellular calcium concentrations, and therefore, we cannot exclude that this may explain the lower effect of Lu AF41228 in phenylephrine‐ versus U46619‐contracted arteries.
The incubation with the combination of ODQ and SQ22536 is considered to reduce cGMP and cAMP to insignificant levels, but at high concentrations, Lu AF41228 and Lu AF58027 still induced relaxations in the rat mesenteric arteries. At high concentrations (10 μM), which are far above the nM concentrations of Lu AF41228 and Lu AF58027 inhibiting the PDE1 enzymes, we would suggest that these relaxations to Lu AF41228 and Lu AF58027 which were insensitive to ODQ and SQ22536 may be mediated by off‐target effects. Thus, biphenyl pyridazinone derivatives which inhibit PDE4 were described to have affinity for adenosine receptors (Gracia et al., 2016) and, in our work, screening also revealed that Lu AF41228 at a high concentration (10 μM) binds to several adenosine receptor subtypes.
In the present study, infusion of the PDE1 inhibitors Lu AF41228 and Lu AF58027 dose‐dependently lowered BP, by as much as ~25 mmHg in anaesthetized rats. The BP‐lowering effect of Lu AF41228 was also present in conscious animals, but with a smaller magnitude, than in anaesthetized animals. However, it has also to be taken into account that Lu AF41228 was administered i.v. in anaesthetized rats but given orally in the conscious animals. Overall, our findings suggest a BP‐lowering effect of both Lu AF41228 and Lu AF58027.
Importantly, Lu AF41228 and Lu AF58027 induced increases in HR with a rapid onset in anaesthetized rats and, in the case of Lu AF41228, also in freely moving rats. Although the possible species difference should be considered, the lack of effect in isolated guinea pig hearts combined with our findings in intact animals suggest that the effect on HR may result from such a rapid drop in BP, resulting in baroreceptor‐mediated increase in HR. However, the baroreceptor reflex may be involved in only the first few min after infusion of the compounds, and other mechanisms may contribute to the increases in HR seen throughout the 60 min recordings (see Figure 6). A more direct effect on HR is also supported by the different PDE1 subtype inhibition and the more pronounced increase in HR observed with Lu AF58027 compared with Lu AF41228, despite similar decreases in mean arterial pressure. Therefore, we cannot exclude central or direct effects of the compounds, leading to increased HR in intact rats.
There is substantial evidence for the involvement of increased PDE1A and PDE1B activity in vascular remodelling (Nagel et al., 2006; Schermuly et al., 2007), PDE1A and PDE1C are up‐regulated in systemic and pulmonary hypertension (Schermuly et al., 2007; Giachini et al., 2011) and PDE1B and PDE1C are up‐regulated in senescent human vascular smooth muscle cells (Bautista Nino et al., 2015). Thus, specific inhibition of PDE1 offers an interesting approach to explore whether there is a therapeutic potential for PDE1 inhibitors in the treatment of vascular diseases. The increase in HR may constitute a limitation for the use of Lu AF41228 and Lu AF58027 for hypertension, but given that it is an indirect effect, it can be blocked by use of a selective β1‐adrenoceptor antagonist, thus allowing full advantage to be taken of the pronounced vasodilator and BP‐lowering effect of these PDE1 inhibitors.
In conclusion, our findings suggest that two novel inhibitors of PDE1, Lu AF41228 and Lu AF58027, induce vasodilatation in resistance arteries followed by BP lowering, which is compensated for by an increase in HR in male rats. From the presumed physiological function of PDE1, these selective inhibitors could offer interesting therapeutic possibilities, in hypertensive disease and in patients with vasospasms.
Author contributions
S.M. performed the RT‐PCR studies of PDE1 enzyme expression. E.R.H. performed immunoblotting for the PDE1A enzyme expression. L.B., E.P. and J.S.K. performed the functional studies of small arteries in vitro. M.L., M.G. and T.M. were responsible for studies in the isolated heart and the in vivo studies and wrote the corresponding parts. J.K. was responsible for chemistry and wrote the corresponding parts. C.T.C. was responsible for the PDE enzymatic evaluation and wrote the corresponding parts. C.B. was responsible for the plasma kinetic assessment of the compounds and wrote the corresponding parts. All authors contributed in the design of the study and in the analysis of the data. A first draft of the manuscript was written by M.L., L.B. and U.S. and further elaborated on by the other authors.
Conflict of interest
This work was supported in part by Lundbeck A/S, Copenhagen (in conjunction with the Danish Research Council), which holds the patents pertaining to the structure and clinical use of Lu AF41228 and Lu AF58027.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Original recordings showing the effect on systolic/diastolic blood pressure (BP), mean arterial BP, and heart rate after infusion of respectively, (A) Lu AF41228 and (B) Lu AF58027 in anesthetized rats.
Figure S2 Plasma exposure to Lu AF41228 and Lu AF58027 after intravenous administration in anesthetized rats.
Figure S3 Plasma exposure to Lu AF41228 after oral administration in telemetered rats.
Figure S4 Coronary flow increases in response to PDE1 inhibition in guinea pig isolated perfused heart. After equilibrium was reached, baseline was recorded for 15 minutes and hearts were then perfused with vehicle solution for 15 minutes followed by consecutive infusions of compound at 15 minute intervals.
Table S1 Primers used for detection of PDE1 subtypes.
Table S2 Potency and maximal relaxation of PDE inhibitors in mesenteric and femoral small arteries.
Table S3 Contraction level induced by (A) phenylephrine and (B) U46619 and potency and maximal relaxation of PDE1inhibitors in mesenteric small arteries in the absence and the presence of an inhibitor of guanylate cyclase, ODQ, an inhibitor of adenylate cyclase, SQ22536 or the combination.
Acknowledgements
U.S. was supported by the Danish Heart Foundation, the Danish Research Council and the NovoNordisk Foundation.
Laursen, M. , Beck, L. , Kehler, J. , Christoffersen, C. T. , Bundgaard, C. , Mogensen, S. , Mow, T. J. , Pinilla, E. , Knudsen, J. S. , Hedegaard, E. R. , Grunnet, M. , and Simonsen, U. (2017) Novel selective PDE type 1 inhibitors cause vasodilatation and lower blood pressure in rats. British Journal of Pharmacology, 174: 2563–2575. doi: 10.1111/bph.13868.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen CU, Mulvany MJ, Simonsen U (2005). Lack of synergistic effect of molsidomine and sildenafil on development of pulmonary hypertension in chronic hypoxic rats. Eur J Pharmacol 510: 87–96. [DOI] [PubMed] [Google Scholar]
- Bautista Nino PK, Durik M, Danser AH, de Vries R, Musterd‐Bhaggoe UM, Meima ME et al. (2015). Phosphodiesterase 1 regulation is a key mechanism in vascular aging. Clin Sci (Lond) 129: 1061–1075. [DOI] [PubMed] [Google Scholar]
- Bender AT, Beavo JA (2006). Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev 58: 488–520. [DOI] [PubMed] [Google Scholar]
- Bentzen BH, Grunnet M, Hyveled‐Nielsen L, Sundgreen C, Lassen JB, Hansen HH (2013). Anti‐hypertensive treatment preserves appetite suppression while preventing cardiovascular adverse effects of tesofensine in rats. Obesity (Silver Spring) 21: 985–992. [DOI] [PubMed] [Google Scholar]
- Bolla M, You D, Loufrani L, Levy BI, Levy‐Toledano S, Habib A et al. (2004). Cyclooxygenase involvement in thromboxane‐dependent contraction in rat mesenteric resistance arteries. Hypertension 43: 1264–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Nagel DJ, Zhou Q, Cygnar KD, Zhao H, Li F et al. (2015). Role of cAMP‐phosphodiesterase 1C signaling in regulating growth factor receptor stability, vascular smooth muscle cell growth, migration, and neointimal hyperplasia. Circ Res 116: 1120–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan S, Yan C (2011). PDE1 isozymes, key regulators of pathological vascular remodeling. Curr Opin Pharmacol 11: 720–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard T, Simonsen U, Fago A (2007). Nitrite‐dependent vasodilation is facilitated by hypoxia and is independent of known NO‐generating nitrite reductase activities. Am J Physiol Heart Circ Physiol 292: H3072–H3078. [DOI] [PubMed] [Google Scholar]
- Giachini FR, Lima VV, Carneiro FS, Tostes RC, Webb RC (2011). Decreased cGMP level contributes to increased contraction in arteries from hypertensive rats: role of phosphodiesterase 1. Hypertension 57: 655–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracia J, Buil MA, Castro J, Eichhorn P, Ferrer M, Gavalda A et al. (2016). Biphenyl pyridazinone derivatives as inhaled PDE4 inhibitors: structural biology and structure–activity relationships. J Med Chem 59: 10479–10497. [DOI] [PubMed] [Google Scholar]
- Grzesk G, Szadujkis‐Szadurska K, Matusiak G, Malinowski B, Gajdus M, Wicinski M et al. (2014). Influence of celecoxib on the vasodilating properties of human mesenteric arteries constricted with endothelin‐1. Biomed Rep 2: 412–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey J, Yang E, am Ende CW, Arnold E (2014). Small‐molecule phosphodiesterase probes: discovery of potent and selective CNS‐penetrable quinazoline inhibitors of PDE1. Med Chem Commun 5: 1290–1296. [Google Scholar]
- Johnson WB, Katugampola S, Able S, Napier C, Harding SE (2012). Profiling of cAMP and cGMP phosphodiesterases in isolated ventricular cardiomyocytes from human hearts: comparison with rat and guinea pig. Life Sci 90: 328–336. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komas N, Lugnier C, Andriantsitohaina R, Stoclet JC (1991). Characterisation of cyclic nucleotide phosphodiesterases from rat mesenteric artery. Eur J Pharmacol 208: 85–87. [DOI] [PubMed] [Google Scholar]
- Kun A, Matchkov VV, Stankevicius E, Nardi A, Hughes AD, Kirkeby HJ et al. (2009). NS11021, a novel opener of large‐conductance Ca(2+)‐activated K(+) channels, enhances erectile responses in rats. Br J Pharmacol 158: 1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Wennogle LP, Zhao J, Zheng, H (2011). Phosphodiesterase 1‐targeting tracers and methods. Google Patents, WO2011043816, A1.
- Loughney K, Martins TJ, Harris EA, Sadhu K, Hicks JB, Sonnenburg WK et al. (1996). Isolation and characterization of cDNAs corresponding to two human calcium, calmodulin‐regulated, 3′,5′‐cyclic nucleotide phosphodiesterases. J Biol Chem 271: 796–806. [DOI] [PubMed] [Google Scholar]
- Ludmer PL, Wright RF, Arnold JM, Ganz P, Braunwald E, Colucci WS (1986). Separation of the direct myocardial and vasodilator actions of milrinone administered by an intracoronary infusion technique. Circulation 73: 130–137. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Cai Y, Oikawa M, Thomas T, Dostmann WR, Zaccolo M et al. (2011). Cyclic nucleotide phosphodiesterase 1A: a key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res Cardiol 106: 1023–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller CL, Oikawa M, Cai Y, Wojtovich AP, Nagel DJ, Xu X et al. (2009). Role of Ca2+/calmodulin‐stimulated cyclic nucleotide phosphodiesterase 1 in mediating cardiomyocyte hypertrophy. Circ Res 105: 956–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyahara M, Ito M, Itoh H, Shiraishi T, Isaka N, Konishi T et al. (1995). Isoenzymes of cyclic nucleotide phosphodiesterase in the human aorta: characterization and the effects of E4021. Eur J Pharmacol 284: 25–33. [DOI] [PubMed] [Google Scholar]
- Murray F, Patel HH, Suda RY, Zhang S, Thistlethwaite PA, Yuan JX et al. (2007). Expression and activity of cAMP phosphodiesterase isoforms in pulmonary artery smooth muscle cells from patients with pulmonary hypertension: role for PDE1. Am J Physiol Lung Cell Mol Physiol 292: L294–L303. [DOI] [PubMed] [Google Scholar]
- Nagel DJ, Aizawa T, Jeon KI, Liu W, Mohan A, Wei H et al. (2006). Role of nuclear Ca2+/calmodulin‐stimulated phosphodiesterase 1A in vascular smooth muscle cell growth and survival. Circ Res 98: 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plane F, Garland CJ (1996). Influence of contractile agonists on the mechanism of endothelium‐dependent relaxation in rat isolated mesenteric artery. Br J Pharmacol 119: 191–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pontes LB, Antunes F, Sudo RT, Raimundo JM, Lima LM, Barreiro EJ et al. (2012). Vasodilatory activity and antihypertensive profile mediated by inhibition of phosphodiesterase type 1 induced by a novel sulfonamide compound. Fundam Clin Pharmacol 26: 690–700. [DOI] [PubMed] [Google Scholar]
- Rieg AD, Suleiman S, Perez‐Bouza A, Braunschweig T, Spillner JW, Schroder T et al. (2014). Milrinone relaxes pulmonary veins in guinea pigs and humans. PLoS One 9: e87685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybalkin SD, Rybalkina I, Beavo JA, Bornfeldt KE (2002). Cyclic nucleotide phosphodiesterase 1C promotes human arterial smooth muscle cell proliferation. Circ Res 90: 151–157. [DOI] [PubMed] [Google Scholar]
- Schermuly RT, Pullamsetti SS, Kwapiszewska G, Dumitrascu R, Tian X, Weissmann N et al. (2007). Phosphodiesterase 1 upregulation in pulmonary arterial hypertension: target for reverse‐remodeling therapy. Circulation 115: 2331–2339. [DOI] [PubMed] [Google Scholar]
- Simonsen U, Wadsworth RM, Buus NH, Mulvany MJ (1999). In vitro simultaneous measurements of relaxation and nitric oxide concentration in rat superior mesenteric artery. J Physiol 516 (Pt 1): 271–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stangherlin A, Zaccolo M (2012). Phosphodiesterases and subcellular compartmentalized cAMP signaling in the cardiovascular system. Am J Physiol Heart Circ Physiol 302: H379–H390. [DOI] [PubMed] [Google Scholar]
- Vardi Y, Klein L, Nassar S, Sprecher E, Gruenwald I (2002). Effects of sildenafil citrate (viagra) on blood pressure in normotensive and hypertensive men. Urology 59: 747–752. [DOI] [PubMed] [Google Scholar]
- Vemulapalli S, Watkins RW, Chintala M, Davis H, Ahn HS, Fawzi A et al. (1996). Antiplatelet and antiproliferative effects of SCH 51866, a novel type 1 and type 5 phosphodiesterase inhibitor. J Cardiovasc Pharmacol 28: 862–869. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Original recordings showing the effect on systolic/diastolic blood pressure (BP), mean arterial BP, and heart rate after infusion of respectively, (A) Lu AF41228 and (B) Lu AF58027 in anesthetized rats.
Figure S2 Plasma exposure to Lu AF41228 and Lu AF58027 after intravenous administration in anesthetized rats.
Figure S3 Plasma exposure to Lu AF41228 after oral administration in telemetered rats.
Figure S4 Coronary flow increases in response to PDE1 inhibition in guinea pig isolated perfused heart. After equilibrium was reached, baseline was recorded for 15 minutes and hearts were then perfused with vehicle solution for 15 minutes followed by consecutive infusions of compound at 15 minute intervals.
Table S1 Primers used for detection of PDE1 subtypes.
Table S2 Potency and maximal relaxation of PDE inhibitors in mesenteric and femoral small arteries.
Table S3 Contraction level induced by (A) phenylephrine and (B) U46619 and potency and maximal relaxation of PDE1inhibitors in mesenteric small arteries in the absence and the presence of an inhibitor of guanylate cyclase, ODQ, an inhibitor of adenylate cyclase, SQ22536 or the combination.