

Abstract
Background and Purpose
Aberrant activation of the cyclin D1‐cyclin‐dependent kinase 4/6 (CDK4/6)‐Rb signalling pathway is common in oesophageal squamous cell carcinoma (ESCC). PD‐0332991, a highly specific inhibitor of CDK4/6, has potent antitumour activity against many types of cancer. The purpose of this study was to examine the in vitro and in vivo antineoplastic effect of PD‐0332991 against the growth and metastasis of ESCC cells.
Experimental Approach
Cell viability and any synergy between PD‐0332991 and 5‐fluorouracil or cisplatin were measured by MTS assay and CalcuSyn software respectively. Cell migration and invasion were detected by wound healing and transwell assays. Apoptosis was evaluated by flow cytometry after staining annexin V‐FITC/PI. Cellular senescence was assessed by measuring SA‐β‐gal activity. Nude mouse xenograft models of ESCC were employed to determine the in vivo activity of PD‐0332991 against tumour growth and lung metastasis.
Key Results
PD‐0332991 inhibited cellular growth and induced mitochondrial‐dependent apoptosis in ESCC cells. PD‐0332991 also suppressed migration, invasion and the expression of MMP‐2 in ESCC cells. Furthermore, PD‐0332991 treatment caused cell senescence in a FOXM1‐dependent manner. In addition, there was synergy between PD‐0332991 and cisplatin or 5‐fluorouracil. Importantly, the xenografted tumour experiments demonstrated that PD‐0332991 potently inhibits ESCC cell growth and lung metastasis.
Conclusions and Implications
PD‐0332991 can elicit a strong antitumour activity against ESCC growth and metastasis and may be a promising candidate drug for the treatment of patients with ESCC. Our results warrant a clinical trial to further evaluate the efficacy of PD‐0332991 in ESCC patients, even those with metastasis.
Abbreviations
- CDK4
cyclin‐dependent kinase 4
- CDK6
cyclin‐dependent kinase 6
- ESCC
oesophageal squamous cell carcinoma
- FOXM1
forkhead box M1
- MMPs
matrix metalloproteinases
- PI
propidium iodide
- SA‐β‐gal
senescence‐associated β‐galactosidase
- XIAP
X‐linked inhibitor of apoptosis
Introduction
Oesophageal squamous cell carcinoma (ESCC), a tumour of the digestive tract, is a particularly aggressive cancer and has been ranked as the fifth leading cause of cancer‐related deaths worldwide (Torre et al., 2015). Approximately 70% of global ESCC cases occur in China (Pennathur et al., 2013). Despite the various regimens implemented, such as chemotherapy, radiotherapy or their combinations, the 5 year overall survival rate of this disease is only 10–30% (Sakaeda et al., 2009; Nakajima and Kato, 2013). Moreover, 20–30% of patients newly diagnosed with ESCC had distant metastasis (Quint et al., 1995). Therefore, there is an urgent need to develop novel targeted therapeutic drugs for ESCC patients.
An immunohistochemical analysis of 90 patients with ESCC showed that cyclin D1 and cyclin‐dependent kinase 4 (CDK4) were overexpressed in 28 (34.4%) and 39 (43.3%) cases, respectively (Matsumoto et al., 1999). A comprehensive genomic analysis of 158 ESCC cases also showed that cyclin D1 and CDK4/6 were amplified in 46.4% and 23.6% of cases, respectively (Song et al., 2014). In addition, simultaneous overexpression of cyclin D1 and CDK4 in ESCC are frequently associated with venous invasion and poor prognosis (Song et al., 2014). Therefore, the cyclin D1‐CDK4/6 complex may be an attractive therapeutic target in ESCC.
PD‐0332991 (palbociclib) is an orally available, highly specific and reversible inhibitor of both CDK4 and CDK6 kinase activities (Toogood et al., 2005). PD‐0332991 exerts an anti‐proliferative effect in breast and ovarian cancer cells, myeloma cells and glioblastoma cells (Finn et al., 2009; Michaud et al., 2010; Konecny et al., 2011) and in human tumour xenografts (Fry et al., 2004; Michaud et al., 2010) or in FBXO4 (+/−) mice (Lian et al., 2015). A phase I study of PD‐0332991 in patients with Rb‐positive solid tumours or non‐Hodgkin's lymphoma also showed that PD‐0332991 is generally well tolerated (Schwartz et al., 2011). Of note, PD‐0332991 has recently been approved by the FDA for use, in combination with letrozole, as a first‐line treatment of oestrogen receptor‐positive, HER2‐negative, advanced breast cancer (Dhillon, 2015; Finn et al., 2015; 2016) or in combination with fulvestrant for the treatment of women with hormone receptor‐positive, HER2‐negative advanced or metastatic breast cancer (Walker et al., 2016). Whether PD‐0332991 is active against ESCC remains unknown.
In the present study, our purpose was to examine the antineoplastic effect of PD‐0332991 against the ESCC cells. We discovered that PD‐0332991 potently induced G1‐phase arrest in ESCC cells by inhibiting CDK4/6 activities. Moreover, PD‐0332991 suppressed MMP‐2 expression as well as the migration and invasion of EC109 and EC9706 cells. In addition, PD‐0332991 induced cellular senescence via an effect on FOXM1. Furthermore, PD‐0332991 potentiated the lethality of 5‐fluorouracil (5‐FU) and cisplatin on ESCC cells. More importantly, the in vivo studies also demonstrated that PD‐0332991 can suppress the growth and metastasis of ESCC cells in nude mice. These findings suggest that PD‐0332991 is a promising candidate drug for the treatment of ESCC.
Methods
Cell culture
Human ESCC cell lines EC109, EC9706, KYSE30, KYSE140, KYSE150, KYSE410 and CE‐81T were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China) and cultured in Dulbecco's modified Eagle medium (DMEM, Invitrogen) containing 100 U·mL−1 penicillin/streptomycin, supplemented with 10% (v.v−1) heat‐inactivated FBS at 37°C in a humidified incubator containing 5% CO2.
Blinding and data normalization
All the data were analysed by two observers who were blinded to the group assignment of animals. Cell viability measurements and quantitative analysis of colony formation were normalized relative to control (Curtis et al., 2015). The graph Y‐axis was labelled as ‘% of control’.
Cell viability assay
Cell viability was determined by MTS assay (CellTiter 96 Aqueous One Solution Cell Proliferation assay; Promega, Madison, WI) as previously described (Jin et al., 2016b). Briefly, cells were seeded in 96‐well flat‐bottom plates (Corning Inc., Corning, NY, USA) and incubated with various concentrations of PD‐0332991 for 72 h. Twenty microlitres of MTS solution was added to each well during the last 4 h of culture. Absorbance was read on a 96‐well plate reader at a wavelength of 490 nm. Control cells received DMSO (<0.1%) containing medium. The drug concentration resulting in 50% inhibition of cell growth (IC50) was determined.
The combinations were assessed in serial fixed‐ratio dilutions of the two drugs. The effects of combinations were estimated using the CalcuSyn software (Biosoft, Cambridge, UK). The combination index (CI) was the ratio of the combination dose to the sum of the single‐agent doses at an isoeffective level. Therefore, CI < 1 indicates synergy; CI > 1, antagonism; and CI = 1, additive (Pan et al., 2010).
Anchorage‐independent growth assay
For anchorage‐independence, a two‐layer soft agar‐containing media was plated in 24‐well flat‐bottom plates (Corning Inc.) as previously described (Ismail et al., 2011). The first layer (300 μL) was DMEM growth medium containing 0.8% agar and 10% FBS. The second layer contained 2000 cells in DMEM medium containing 0.4% agar and 10% FBS. After incubation for 10 to 14 days at 37°C in a humidified incubator, the colonies (containing ≥50 cells) were counted using an inverted phase‐contrast microscope.
Apoptosis assay
Apoptosis was measured using an annexin V‐FITC Apoptosis Detection Kit (Sigma‐Aldrich, Shanghai, China) by flow cytometry as previously described (Jin et al., 2016a). After PD‐0332991 treatment, cells were collected and stained with annexin V‐FITC at room temperature for 15 min in the dark. The cells were washed and resupended in 1 × binding buffer. Immediately after this the propidium iodide (PI) solution was added to the cell suspension, apoptosis was assessed using the FACS LSRFortessa flow cytometer and its software.
Cell‐cycle analysis
For measurement of effects on the cell cycle, cells were pretreated with or without PD‐0332991 for 8 h, then 20 ng·mL−1 nocodazole (Sigma‐Aldrich) was added as previously described, with modifications (Tang et al., 2012). Cells were harvested by trypsinization 16 h after addition of nocodazole, washed with PBS and fixed in 70% ethanol. PI solution (Sigma‐Aldrich) was then added; the cells were incubated at room temperature for 1 h in the dark. Cell‐cycle distribution was analysed using a FACS LSRFortessa flow cytometer with its software as previously described (Dai et al., 2016).
Western blotting analysis
Western blotting analysis was performed using the whole cell lysates prepared in RIPA buffer (Jin et al., 2014). For the detection of cytochrome c, the cytosolic fraction was prepared in digitonin extraction buffer [10 mM PIPES, 0.015% digitonin, 300 mM sucrose, 100 mM NaCl, 3 mM MgCl and 2.5 mM EDTA. 1 × protease inhibitor cocktail (Roche, Indianapolis, IN, USA)], to which 10 mM β‐glycerophosphate, 1 mM sodium orthovanadate, 10 mM NaF and 1 mM PMSF were added. Equal amounts of total proteins were separated using 10–15% SDS‐PAGE and then transferred onto nitrocellulose membranes. After being blocked with 5% dried skimmed milk, the membranes were then incubated with the primary antibodies overnight. After the probing with appropriate secondary antibodies, the membranes were scanned with the Odyssey infrared imaging system (LI‐COR, Lincoln, NE, USA) (Jin et al., 2016b).
Measurement of mitochondrial transmembrane potential
ESCC cells were exposed to 10 μM PD‐0332991 for different durations. After incubation with MitoTracker probes (CMXRos and MTGreen, Eugene, OR, USA), the cells were subjected to flow cytometry analysis for inner mitochondrial transmembrane potential (ΔΨm), as described previously (Shi et al., 2009).
Senescence‐associated β‐galactosidase staining
Cellular senescence was assayed by measuring senescence‐associated β‐galactosidase (SA‐β‐gal) activity. Cells were grown in 6‐well flat‐bottom plates (Corning Inc.) for 24 h and then treated without or with 2.5 μM PD‐0332991 for 6 days. After fixation, the cells were stained overnight according to the manufacturer's protocol (Cell Signaling #9860). The percentage of SA‐β‐gal positive cells was determined by counting the number of positive cells present in six separate microscope fields and then normalizing the results to the control (Shi et al., 2010).
Wound scratch assay
The wound scratch assay, a well‐established method to assess cell motility in two dimensions, was performed as described previously (Chen et al., 2013). Briefly, cells were inoculated in a 6‐well flat‐bottom plate (Corning Inc.) and grown to a confluent cell monolayer overnight. After the cells had been scratched with a pipette tip (200 μL), the wells were washed three times with PBS to remove the floating cells. The cells were observed and photographed under an inverted microscope at 0, 24 and 48 h post‐scratching. The percentage of wound closure was calculated as (original gap distance – gap distance at 24 or 48 h)/original gap distance × 100%.
Cell migration and invasion assays
The migration and invasion assays were performed in transwell chambers (8 μm pore size, Costar) according to the manufacturer's instructions, with modifications (Chen et al., 2013). For the migration assay, 200 μL of serum‐free medium (containing 2 × 104 cells) were placed into the upper chamber of each insert, while the lower compartment was filled with 600 μL of DMEM supplemented with 20% FBS. After incubation at 37°C for 48 h, the remaining tumour cells inside the upper chamber were removed with cotton swabs. The cells on the lower surface of the membrane were stained with 0.1% crystal violet after fixation with 4.0% paraformaldehyde, and then counted under a light microscope. The invasion assay was done using the same procedure, except that the inserts were coated with Matrigel to form a matrix barrier, and then 4 × 104 tumour cells were added to the upper chamber.
Lentiviral transduction
pLKO.1 plasmids containing shRNA sequences targeting CDK4 (shCDK4#1: TRC number: TRCN0000010520, target sequence: ACAGTTCGTGAGGTGGCTTTA; shCDK4#2: TRCN0000196698, target sequence: GAGATTACTTTGCTGCCTTAA) and CDK6 (shCDK6#1: TRC number: TRCN0000196261, target sequence: GAGAAGTTTGTAACAGATATC; shCDK6#2: TRCN0000055435, target sequence: TCTGGAGTGTTGGCTGCATAT) were obtained from Sigma‐Aldrich. A scramble shRNA (Sigma‐Aldrich, #SHC016) was used as negative control, its sequence was: GCGCGATAGCGCTAATAATTT. To generate virus, 293T cells were seeded in each 10 cm dish (Corning Inc.) and incubated at 37°C in a 5% CO2 humidified atmosphere. After 24 h, the scramble and shRNA constructs targeting CDK4 or CDK6 (10 μg each), packaging plasmid (pCMV‐dR8.91; 5 μg) and envelope plasmid (pMD2.G‐VSV‐G; 5 μg) were diluted with 1.0 mL Opti‐MEM medium (Gibco, Invitrogen, Carlsbad, CA, USA), and then 60 μL of linear polyethylenimine transfection reagent (1.0 mg·mL−1) from Polysciences (Warrington, PA, USA) was added and the samples incubated for 15 min at room temperature. Subsequently, the transfection mixture was applied to each culture dish. Twelve hours after transfection, the culture medium was changed with fresh culture medium. Fourty‐eight hours later, the virus‐containing supernatants were collected and used to infect ESCC cells in the presence of 8 μg·mL−1 polybrene (Sigma‐Aldrich). The cells were then selected in the presence of puromycin (1 μg·mL−1; Sigma‐Aldrich) to establish stable clones. Immunoblotting for CDK4 and CDK6 was performed to determine the knockdown efficiency.
Transfection with plasmid or siRNA
ESCC cells were seeded in 6‐well flat‐bottom plates (Corning Inc.) 24 h before transfection. The empty vector or pCMV6‐Entry‐FOXM1 (OriGene) plasmid (2.0 μg) and 6 μL linear polyethylenimine transfection reagent (1.0 mg·mL−1) from Polysciences (Warrington, PA) were added into 200 μL Opti‐MEM medium (Gibco). Subsequently, the transfection mixture was applied to the culture plates. Twelve hours after transfection, the culture medium was replaced with fresh culture medium, and then the ESCC cells were selected using G418 (800 μg·mL−1; Invitrogen, CA, USA). After 4‐weeks of selection, individual colonies were selected and expanded. For siRNA transfection, two different FOXM1 small interfering RNAs (siRNAs) and a non‐silencing control siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Exponentially grown untreated cells were plated in a 6‐well flat‐bottom plates (Corning Inc.) and grown for 24 h before transfection. Plated cells were transfected with FOXM1 siRNAs or a control siRNA. Fourty‐eight hours later, the cells were harvested and processed for further analysis.
Mouse models of tumour growth and metastasis
Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The experimental procedures were also conducted in compliance with the Jinan University Institutional Animal Care and Use Committee and the guidelines on the welfare and use of animals in cancer research (Workman et al., 2010). All animal care and experimental protocols were approved by the Jinan University Institutional Animal Care and Use Committee. Male nude nu/nu BALB/c mice (18–20 g, 5–6 weeks of age) were purchased from Slac Laboratory Animal Co. Ltd (Shanghai, China). All mice were bred and maintained in pathogen‐free conditions at the animal facility of Jinan University with controlled temperature (20 ± 2°C), humidity (40–50%) and a lighting cycle of 12 h light/12 h darkness. Mice were housed in isolator cages (four mice per cage). Standard pellet food and water were provided ad libitum during the experimental period.
EC109 cells (5 × 106 cells in PBS suspension) were implanted s.c. into the flank of each mouse (Wang et al., 2015). Tumours were measured every other day and calculated by the formula of a × b 2 × 0.4, where a is the smallest diameter and b is the diameter perpendicular to a. When the tumours reached 100 mm3, the mice were randomly separated into two groups: vehicle or treatment (eight mice per group). PD‐0332991 dissolved in sodium lactate buffer (50 mM, pH = 4.0) was administered daily at 150 mg·kg−1 by gavage. The control mice received the same amount of sodium lactate buffer solution. After 2 weeks of PD‐0332991 treatment, mice were anaesthetized with isoflurane before being killed by cervical dislocation. Tumours were immediately removed, weighed, fixed and kept at −80°C. The body weight, feeding behaviour and motor activity of each animal were monitored as an indicator of general health.
For the experimental metastasis assay, two groups of mice (n = 6 per group) were injected with KYSE150 cells (1 × 106 in PBS) i.v., via the lateral tail vein as previously described (Li et al., 2009). Twenty‐four hours later, the animals were treated with PD‐0332991 (150 mg·kg−1 by gavage) or vehicle for 2‐weeks. Six weeks after the injection of cells, mice were anaesthetized with isoflurane before being killed by cervical dislocation, and the lungs were removed and fixed in Bouin's solution (75% saturated picric acid, 25% formaldehyde and 5.0% acetic acid) overnight. The metastatic foci on the surface of the lung in each mouse were counted, and the presence of tumour lesions within the lungs was confirmed by haematoxylin and eosin staining.
Statistical analysis
All experiments were performed at least five times, and data are expressed as mean ± SD. Student's t‐test was used to compare the differences between two groups. We compared more than two groups with one‐way ANOVA with Tukey's post hoc test, the overall F test was significant (P < 0.05), and there was no significant variance in homogeneity. GraphPad Prism version 5.0 (GraphPad Software, San Diego, CA, USA) was employed to conduct the statistical analysis. All statistical tests were two‐sided, and P < 0.05 was considered statistically significant. The data and statistical analyses comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Materials and reagents
PD‐0332991 was purchased from Selleck Chemicals (Shanghai, China). Cisplatin and 5‐FU were purchased from Sigma‐Aldrich. Antibodies and their sources are as follows: antibodies against Rb, phospho‐Rb (Ser780), phospho‐Rb (Ser807/811) and p16 were purchased from Cell Signaling Technology (Beverly, MA, USA); antibodies against (ADP‐ribose) polymerase (PARP), Bcl‐2, X‐linked inhibitor of apoptosis (XIAP), active caspase‐3 and cytochrome c were from BD Biosciences Pharmingen (San Jose, CA, USA); antibodies against CDK4, CDK6, cyclin A, cyclin E, FOXM1, Bax, Bcl‐xL, Mcl‐1 (S‐19) and Bcl‐2 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). Anti‐rabbit immunoglobulin G and anti‐mouse immunoglobulin G fluorescent‐conjugated secondary antibodies were obtained from LI‐COR Biotechnology (Nebraska, USA).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
PD‐0332991 induces cellular Rb dephosphorylation and G1‐phase arrest in ESCC cells
The ability of PD‐0332991 to inhibit CDK4/6 was examined in ESCC cells by detecting CDK4/6‐specific phosphorylation of Rb on Ser780. As shown in Figure 1A, PD‐0332991 treatment down‐regulated Rb phosphorylation as well as E2F target genes including cyclin A and cyclin E in a concentration‐dependent fashion. However, the expression of total Rb, CDK4 and CDK6 were constant. These results suggest that PD‐0332991 markedly inhibits CDK4/6 kinase activity. If PD‐0332991 inhibited the activity of CDK4/6, then it was predicted to arrest cells at the G1‐phase of the cell cycle. Indeed, EC109 and EC9706 cells largely remained in G1‐phase upon PD‐0332991 treatment (Figure 1B). In this experiment, the growth arrest of cells treated with nocodazole alone, which blocked cell cycle in G2‐phase, was compared with cells pretreated with PD‐0332991 for 8 h, then co‐treated with PD‐0332991 and nocodazole for another 16 h. Pretreatment with PD‐0332991 resulted in a significant increase in G1‐phase arrest for both groups of ESCC cells. Collectively, these results suggest that PD‐0332991 can inhibit CDK4/6 kinase activity and halt the cell cycle at the G1‐phase in ESCC cells.
Figure 1.
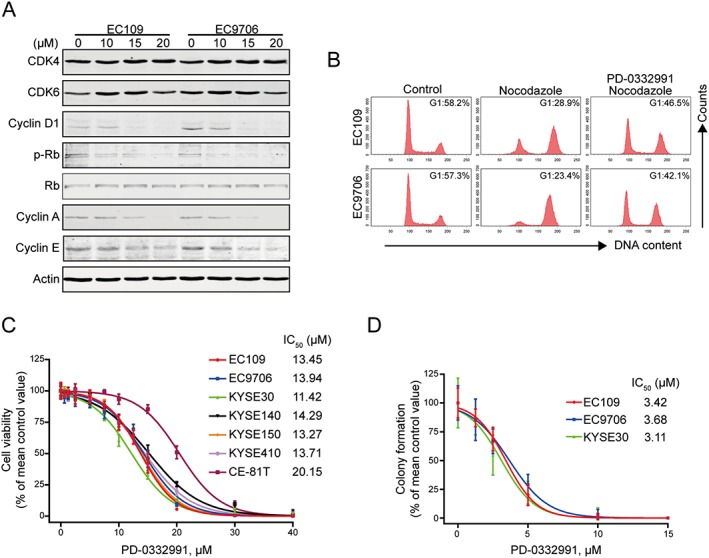
PD‐0332991 induces cellular Rb dephosphorylation and G1‐phase arrest in ESCC cells. (A) EC109 and EC9706 cells were incubated with increasing concentrations of PD‐0332991 for 48 h; immunoblotting analysis was performed with the indicated antibodies. (B) EC109 and EC9706 were treated with or without 2.5 μM PD‐0332991 for 8 h, followed by addition of 20 ng·mL−1 nocodazole for another 16 h. The cells were then fixed and stained with PI before flow cytometry analysis. (C) ESCC cells were incubated with increasing concentrations of PD‐0332991 for 72 h. Cell viability was determined with MTS assay (n = 8 per group). (D) EC109, EC9706 and KYSE30 cells were exposed to increasing concentrations of PD‐0332991 for 48 h. The cells were then washed and subjected to a drug‐free soft agar assay (n = 6 per group).
PD‐0332991 inhibits the growth of ESCC cells
To evaluate the inhibitory effect of PD‐0332991 on cell proliferation, seven lines of ESCC cells were treated with different concentrations of PD‐0332991 for 72 h, and cell viability was measured by the MTS assay. PD‐0332991 potently decreased the cell viability of ESCC cells, with IC50 values ranging from 11.42 to 20.15 μM (Figure 1C). We next determined the effect of PD‐0332991 on the clonogenicity of ESCC cells. EC109, EC9706 and KYSE30 cells were exposed to increasing concentrations of PD‐0332991 for 48 h and were then assayed for colony formation in the absence of drugs. PD‐0332991 potently inhibited the number of surviving clonogenic ESCC cells in a dose‐dependent manner (Figure 1D). Taken together, PD‐0332991 potently inhibits the growth of ESCC cells in a dose‐dependent manner.
PD‐0332991 induces apoptosis in ESCC cells
We next analysed the capacity of PD‐0332991 to induce apoptosis in ESCC cells. EC109 and EC9706 cells were treated with PD‐0332991 for 48 h, and apoptosis was assessed using flow cytometry by annexin V‐FITC/PI double staining. As shown in Figure 2A, PD‐0332991 led to significant apoptotic cell death in a concentration‐dependent fashion in both lines of ESCC cells. Moreover, PD‐0332991 could also induce apoptosis in a time‐dependent manner (Supporting Information Figure S1A). Western blotting results showed a concentration‐ and time‐dependent cleavage of PARP and caspase‐3, further indicating the occurrence of apoptosis in EC109 and EC9706 cells treated with PD‐0332991 (Figure 2B and Supporting Information Figure S1B).
Figure 2.
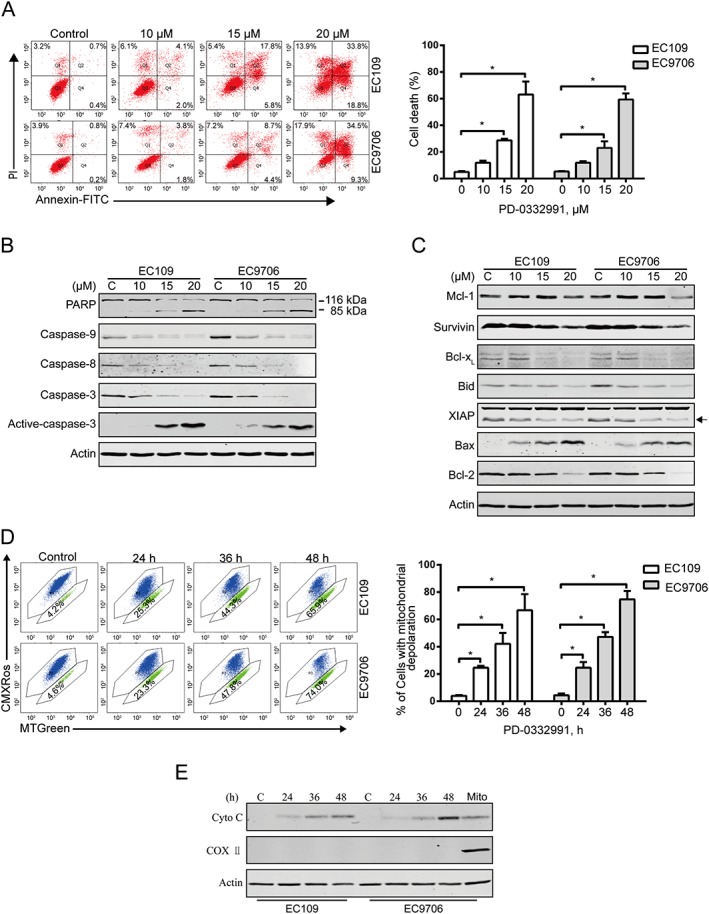
PD‐0332991 induces apoptosis in ESCC cells. (A) EC109 and EC9706 cells were treated with increasing concentrations of PD‐0332991 for 48 h and then underwent flow cytometry analysis after staining with annexin V‐FITC/PI. Right panel: the vertical axis presents the sum of the top left, top right and bottom right quadrants. Data are expressed as mean ± SD, from five independent experiments. *P < 0.05, one‐way ANOVA with post hoc intergroup comparison by the Tukey's test. (B, C) Immunoblotting analysis of apoptosis‐related proteins was performed in EC109 and EC9706 cells treated with increasing concentrations of PD‐0332991 for 48 h. Actin was used as loading control. (D) EC109 and EC9706 cells treated with or without 10 μM PD‐0332991 for 24 h, and then the mitochondrial potential was analysed by flow cytometry after staining with CMXRos and MTGreen. Right: results from five independent experiments. * P < 0.05, one‐way ANOVA with post hoc intergroup comparison by the Tukey's test. (E) PD‐0332991 led to release of cytochrome c into cytosol in ESCC cells. Levels of cytochrome c in the cytosolic extracts prepared with digitonin buffer were detected by immunoblotting. Actin was used as an internal control. The cytosolic fractionations were not contaminated as indicated by COX II.
To elucidate the mechanism of PD‐0332991‐induced apoptosis, we next evaluated the effect of PD‐0332991 on apoptosis‐related protein levels by Western blotting analysis. The results show that the levels of anti‐apoptotic proteins survivin, Bcl‐xL, Bid, XIAP and Bcl‐2 were markedly declined, whereas the level of pro‐apoptotic protein Bax was elevated (Figure 2C and Supporting Information Figure S1C). Interestingly, the expression of anti‐apoptotic protein Mcl‐1 was not significantly changed even with 20 μM PD‐0332991 treatment (Figure 2C and Supporting Information Figure S1C). The disturbance of these mitochondrial proteins prompted us to explore the effect of PD‐0332991 on mitochondrial potential. EC109 and EC9706 cells were exposed to increasing PD‐0332991 concentrations for 24 h. The proportion of ESCC cells with loss of mitochondrial potential detected by flow cytometry after staining with CMXRos and MTGreen was markedly increased (Figure 2D). The mitochondrial transmembrane potential change coincided with other apoptotic indices such as caspase‐3 activation and PARP cleavage (Figure 2B and Supporting Information Figure S1B).
Next, we examined the release of cytochrome c from mitochondria into the cytosol. EC109 and EC9706 cells were treated with PD‐0332991, and a progressive elevation of cytochrome c in cytosolic fractionations extracted using the digitonin buffer was noted (Figure 2E). Collectively, these data suggest that PD‐0332991 may trigger apoptosis through the intrinsic (mitochondrial) pathway in ESCC cells.
PD‐0332991 suppresses the migration and invasion of ESCC cells
We next determined the effect of PD‐0332991 on wound healing ability. The migration ability of EC109 and EC9706 cells that were treated with PD‐0332991 at 5 μM for 24 h was significantly inhibited, the inhibitory effect of PD‐0332991 was further evident at 48 h (Figure 3A). Moreover, the Boyden chamber transwell assay also showed that the migration ability of both lines of ESCC cells was significantly impaired by PD‐0332991 at the concentration of 5 μM (Figure 3B). We also analysed whether PD‐0332991 could inhibit cell invasion by conducting transwell invasion assays. As shown in Figure 5E, PD‐0332991 strongly inhibited the invasion of EC109 and EC9706 cells (Figure 3C).
Figure 3.
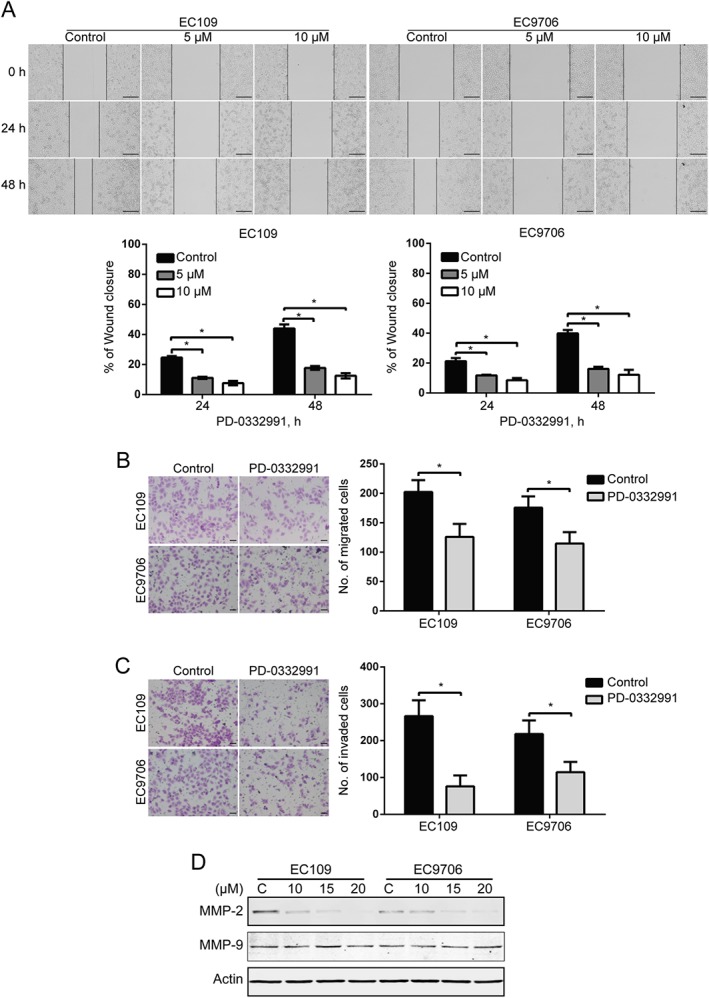
PD‐0332991 suppresses the migration and invasion of ESCC cells. (A) EC109 and EC9706 cells were treated with 0, 5 or 10 μM PD‐0332991 for 0, 24 and 48 h after scratching. Scale bar: 200 μm. The average gap width was used to evaluate migration. Bottom: quantitative analysis of the relative breadth of the wound. The wound breadth was normalized to the initial time point (0 h). Columns and error bars represent mean ± SD (n = 6 per group). * P < 0.05, one‐way ANOVA with post hoc intergroup comparison with the Tukey's test. (B, C) EC109 and EC9706 cells were treated with 5 μM PD‐0332991 for 48 h and then underwent transwell migration (B) and invasion (C) assay. Left: representative images; Right: quantitative analysis from six random fields. Scale bar: 50 μm. Mean; error bar, SD. *P < 0.05 by Student's t‐test. (D) Western blotting analysis of whole cell lysates of EC109 and EC9706 cells that were treated with PD‐0332991 for 48 h.
Matrix metalloproteinases (MMPs), a family of zinc‐dependent proteolytic enzymes capable of degrading the extracellular matrix, play a key role in tumour invasion and metastatic processes in various types of cancer including ESCC (Wang et al., 2013). We therefore tested whether PD‐0332991inhibited the expression of MMP‐2 and MMP‐9, two critical members of the MMP family, in EC109 and EC9706 cells. Western blotting analysis indicated that PD‐0332991 dramatically decreased the protein level of MMP‐2 with minimal impact on MMP‐9 (Figure 3D). Taken together, these data indicate that PD‐0332991 inhibits MMP‐2 expression, as well as the migration and invasion of ESCC cells.
CDK4/6 shRNA attenuates ESCC cell proliferation, migration and invasion
To specifically characterize the effect of CDK4 and CDK6 inhibition, EC109 and EC9706 cells were stably transduced with lentiviruses encoding shRNA against CDK4, CDK6 or a scrambled control; a decrease in CDK4 and CDK6 in the cells transduced with shRNA against CDK4, CDK6 or the combination was observed by immunoblotting (Figure 4A). In comparison with scramble, specific knockdown of CDK4 alone just had a very weak inhibitory effect on ESCC cells. Silencing CDK6 on its own significantly suppressed the anchorage‐independent growth of EC109 and EC9706 cells. In contrast, the inhibitory effect was markedly enhanced after knockdown of both CDK4 and CDK6 (Figure 4B). Similarly, simultaneously silencing CDK4 and CDK6 markedly diminished the wound healing ability of EC109 and EC9706 cells (Figure 4C). The inhibitory effect of knockdown of both CDK4 and CDK6 on migration was further confirmed by the transwell migration assay (Figure 4D). Moreover, knockdown of both CDK4 and CDK6 significantly halted the transwell invasion capacity (Figure 4E). Additionally, the level of MMP‐2 was markedly decreased in ESCC cells transduced with lenti‐shRNA against both CDK4 and CDK6 (Figure 4F). These findings suggest that, like PD‐0332991, specifically knocking down both CDK4 and CDK6 diminishes the aggressiveness of ESCC cells.
Figure 4.
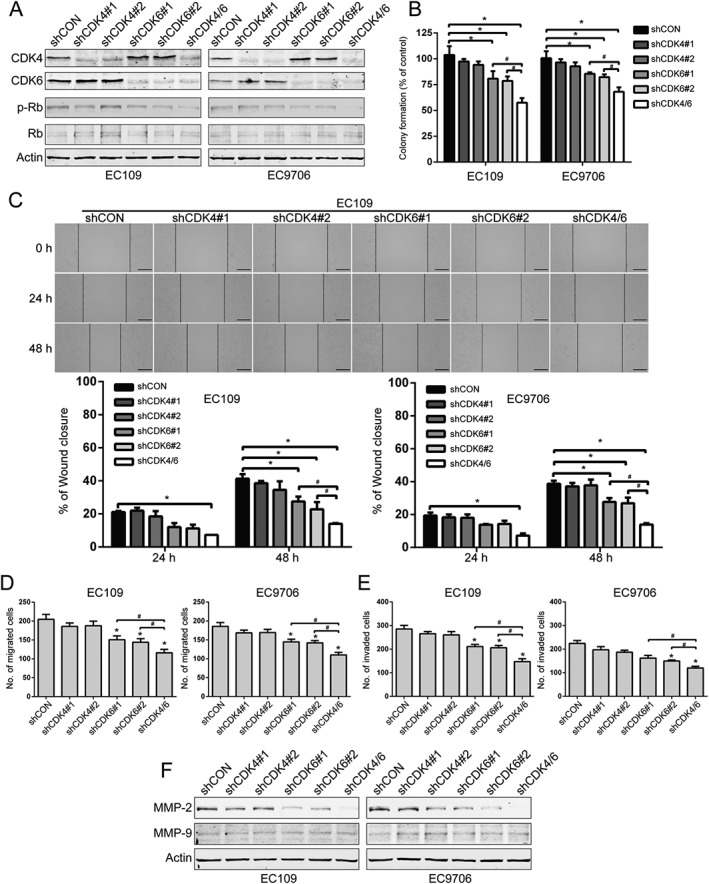
Knockdown of CDK4/6 inhibits the proliferation and migration of ESCC cells. (A) Western blotting analysis of CDK4 and CDK6, as well as the phosphorylated and total levels of Rb in EC109 and EC9706 cells stably transfected with the indicated shRNA. (B) Tumourigenicity was detected by colony formation in ESCC cells that was stably knocked down for CDK4/6 by shRNA (n = 6 per group). shCON: control shRNA; shCDK4#1 and shCDK4#2: two different shRNAs targeting CDK4; shCDK6#1 and shCDK6#2, two different shRNAs targeting CDK6; shCDK4/6, both CDK4 and CDK6 shRNA. * P < 0.05 versus shCON; # P < 0.05 versus shCDK4/6; P values were obtained by one‐way ANOVA with post hoc intergroup comparison by the Tukey's test. (C–E) Wound healing (C), transwell migration (D) and invasion (E) assays of EC109 and EC9706 cell stable clones with knockdown of CDK4, CDK6 alone or both (n = 6 per group). Scale bar for wound healing: 200 μm. *, P < 0.05 versus shCON; #, P < 0.05 versus shCDK4/6; P values were obtained by one‐way ANOVA with post hoc intergroup comparison with Tukey's test. (F) Western blotting analysis of MMP‐2 and MMP‐9 expression in CDK4/6‐knockdown ESCC cells by shRNA. Actin served as a loading control.
PD‐0332991 induces ESCC cell senescence by inhibiting FOXM1
Previous studies have shown that prolonged incubation with PD‐0332991 can induce senescence‐like morphological changes in glioblastoma, osteosarcoma (Anders et al., 2011), melanoma (Leontieva and Blagosklonny, 2013; Leontieva et al., 2013), fibrosarcoma (Leontieva and Blagosklonny, 2013) and breast cancer cells (Dean et al., 2010). We therefore investigated whether PD‐0332991 induced senescence in ESCC cells or not. As shown in Figure 5A, treatment with PD‐0332991 drastically increased the activity of SA‐β‐gal, a marker for senescent cells in EC109 and EC9706 cells. Because forkhead box M1 (FOXM1), a critical substrate of CDK4/6 signalling, plays a key role in protecting tumour cells from senescence (Anders et al., 2011; Wang et al., 2011), we examined whether it is involved in PD‐0332991‐mediated senescence in ESCC cells. Western blotting analysis revealed that the protein level of FOXM1 was decreased by PD‐0332991 in a concentration‐dependent manner (Figure 5B). To further substantiate the involvement of FOXM1, we reconstituted ESCC cells with FOXM1 plasmid (Figure 5C). Enforced expression of FOXM1 attenuated PD‐0332991‐induced cell senescence in EC109 and EC9706 cells (Figure 5D, E). By contrast, there was no significant difference between cells transfected with empty vector and FOXM1‐encoding plasmid (Figure 5D, E). However, silencing FOXM1 with two different FOXM1siRNA duplexes resulted in a substantial increase in basal senescence (Figure 5F–H). Importantly, compared with si‐control transfection, knockdown of FOXM1 obviously potentiated the PD‐0332991‐induced cellular senescence, as indicated by a significant increase in the percentage of SA‐β‐gal positive cells (Figure 5G, H). These results suggest that PD‐0332991 may trigger ESCC cell senescence by inhibiting FOXM1.
Figure 5.
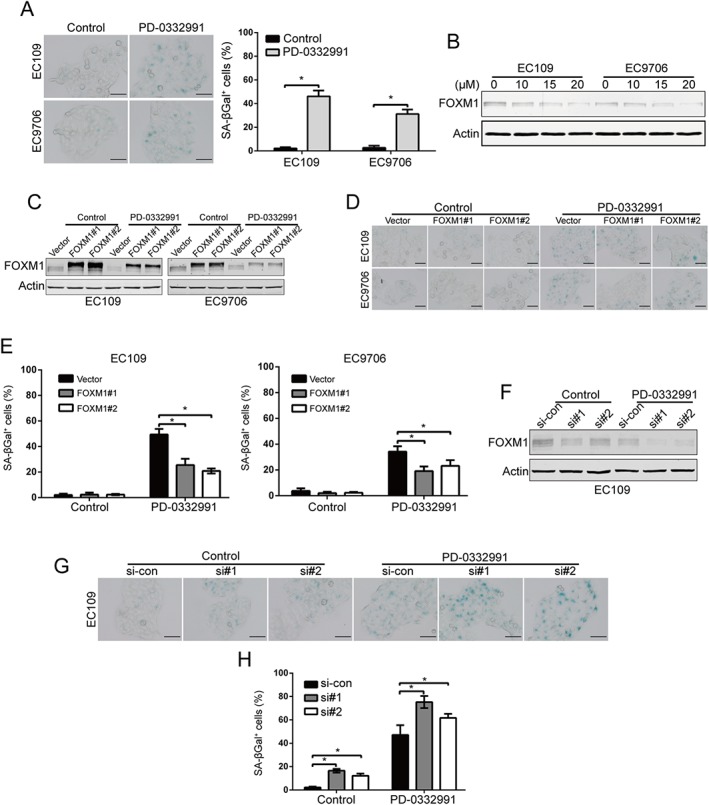
PD‐0332991 induces FOXM1‐dependent senescence in ESCC cells. (A) Senescence phenotype analysis was conducted in ESCC cells after treatment with 2.5 μM PD‐0332991 for 6 days. Left: representative images of SA‐β‐galactosidase assay. Right: quantitative analysis of SA‐β‐galactosidase positive cells. The percentages of cells with senescence morphology are from six random fields. Scale bar: 100 μm. Mean; error bar, SD. * P < 0.05 by Student's t‐test. (B) Immunoblotting of FOXM1 in EC109 and EC9706 cells was analysed after treatment with increasing concentrations of PD‐0332991 for 48 h. (C–E) ESCC cells ectopically overexpressing FOXM1 were exposed to 2.5 μM PD‐0332991 for 6 days, Western blotting of FOXM1 (C) and SA‐β‐galactosidase positive cells (D, E) were analysed. (D) Representative images of SA‐β‐galactosidase assay. Scale bar: 100 μm. (E) Quantitative analysis of SA‐β‐galactosidase positive (n = 6 per group). * P < 0.05, one‐way ANOVA with post hoc intergroup comparison with Tukey's test. (F–H) EC109 cells was transiently transfected with the siRNA duplexes against Control or FOXM1 for 48 h and then treated with 2.5 μM PD‐0332991 for 6 days. si‐CON, control siRNA; si#1 and si#2, two different siRNA dulplexes against FOXM1. Western blotting of FOXM1 (F) and SA‐β‐galactosidase positive cells (G‐H) were analysed. (G) Representative images of SA‐β‐galactosidase assay. Scale bar: 100 μm. (H) Quantitative analysis of SA‐β‐galactosidase positive cells (n = 6 per group). *P < 0.05, one‐way ANOVA with post hoc intergroup comparison with Tukey's test.
PD‐0332991 is synergistic with 5‐FU and cisplatin
Given that the combination of molecule‐targeted therapeutic agents and conventional therapeutic agents is an effective strategy to improve the quality of life of ESCC patients (Ekman et al., 2008), we next explored whether there was a synergy between PD‐0332991 and the front‐line chemotherapeutic agents for ESCC, 5‐FU and cisplatin, at inhibiting the growth of the cells. EC109 and EC9706 cells were treated with a serially diluted mixture (at a fixed ratio) of PD‐0332991 and 5‐FU or cisplatin for 72 h; cell viability was detected by MTS assay, and the synergistic effect was evaluated using the median‐effect method of Chou and Talalay (Chou, 2010). The results suggest that PD‐0332991 acts synergistically with both drugs in causing growth inhibition of ESCC cells (Figure 6A). Using another approach, EC109 and EC9706 cells were exposed to PD‐0332991 (10 μM) in combination with 5‐FU (200 μM) or cisplatin (12.5 μM) for 48 h, and the number of live cells was counted with the trypan blue exclusion assay. As shown in Figure 6B, D, 5‐FU exhibited no toxic effects on ESCC cells, and either PD‐0332991 or cisplatin induced minimal lethality. However, the combination treatments of PD‐0332991 and 5‐FU or cisplatin led a significantly enhanced ratio of dead cells (Figure 6B, D). Consistent with these results, Western blotting results also showed that the combinations of PD‐0332991 and 5‐FU or cisplatin led to enhanced apoptosis, as indicated by specific cleavage of PARP (Figure 6C, E). These findings encourage a combination regimen of PD‐0332991 and 5‐FU/cisplatin for the treatment of ESCC patients.
Figure 6.
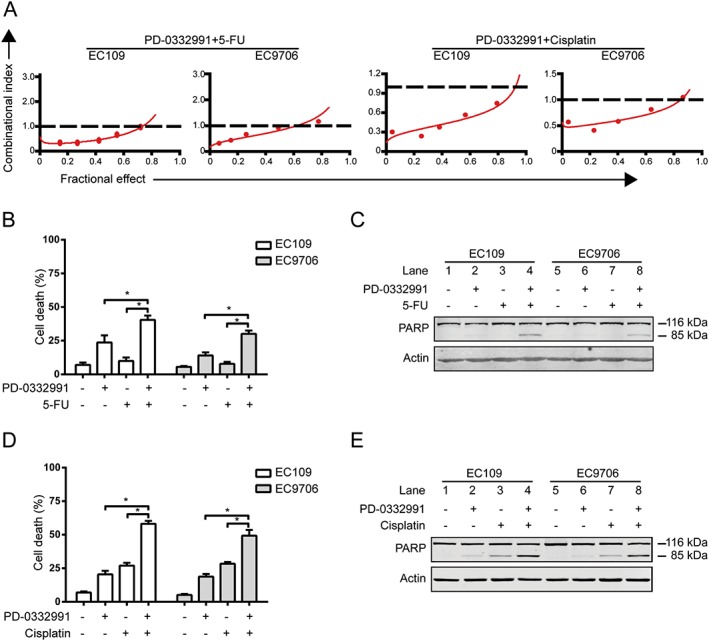
PD‐0332991 is synergistic with cisplatin and 5‐FU. (A) EC109 and EC9706 cells were incubated in a serially diluted mixture (a fixed ratio) of PD‐0332991 and cisplatin or 5‐FU for 72 h; the cell viability was determined by MTS assay. The synergistic effect was estimated using the median‐effect method of Chou and Talalay. The CI was the ratio of the combined dose to the sum of the single‐agent doses at an isoeffective level. CI < 1 indicates synergy; CI > 1, antagonism; and CI = 1, additive. (B, D) EC109 and EC9706 cells were exposed to PD‐0332991 (10 μM) combined with 5‐FU (200 μM) or cisplatin (12.5 μM) for 48 h and then examined with a haemocytometer by trypan blue exclusion assay (n = 6 per group). Column, mean; error bar, SD. * P < 0.05, one‐way ANOVA with post hoc intergroup comparison with Tukey's test. (C, E) The cleavage of PARP was detected by Western blotting analysis. + indicates the presence, and – indicates the absence of the drugs.
PD‐0332991 inhibits ESCC cell growth and metastasis in nude mice
To evaluate the in vivo effect of PD‐0332991, we inoculated nu/nu BALB/c nude mice with EC109 cells s.c. Six days later, when the tumour xenografts were palpable (about 100 mm3), the mice were randomly divided into two groups: vehicle or PD‐0332991 (eight mice per group; 150 mg·kg−1, oral gavage, daily) for about 2 weeks. The growth curve (tumour volume versus time curve) of EC109 tumours was significantly inhibited by the PD‐0332991 treatment (Figure 7A). In addition, the tumour weight in the PD‐0332991‐treated mice was significantly lower than the vehicle‐treated mice (Figure 7B). Furthermore, assessment of the Ki67 proliferation marker by immunohistochemistry showed that the proliferation of EC109 cells was significantly impaired by PD‐0332991 (Figure 7C). Moreover, tumours in the PD‐0332991‐treated animals demonstrated an increase in active caspase‐3 (an indicator of apoptotic cell death) compared with those from the vehicle‐treated mice (Figure 7C). The level of phospho‐Rb was substantially diminished in the PD‐0332991‐treated group, which indicates that PD‐0332991 inhibited the activity of CDK4/6 kinase (Figure 7C). Consistent with these results, Western blotting analysis of cell lysates from xenografted tumours showed that PD‐0332991 dramatically inhibited the phospho‐Rb levels, while the total amount of Rb protein was not altered (Figure 7D). Notably, analysis of tumour tissues also demonstrated that PD‐0332991 markedly decreased FOXM1 expression but increased p16 protein which is a senescence marker (Figure 7D).
Figure 7.
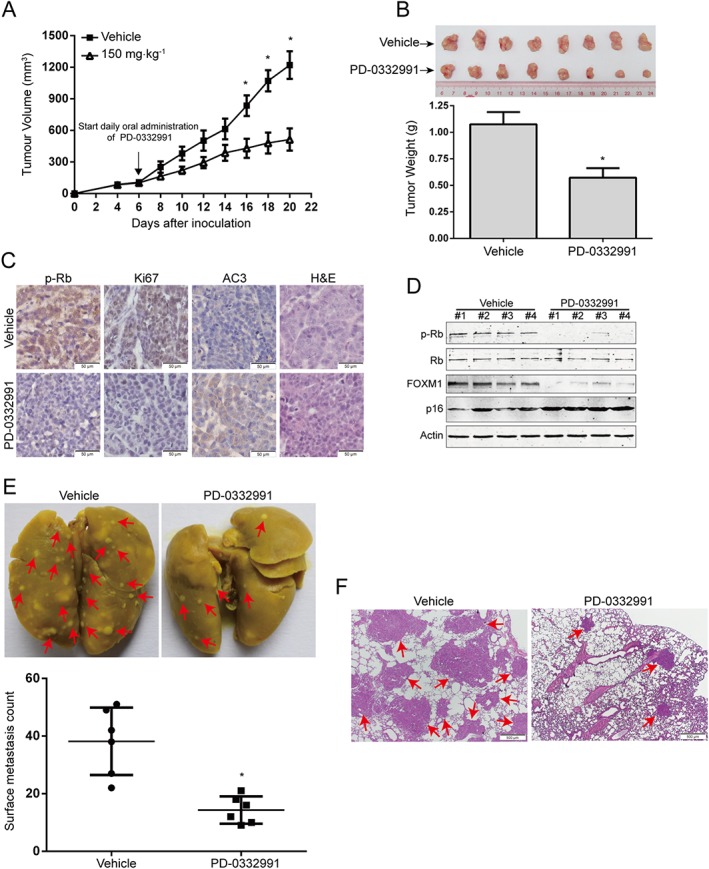
PD‐0332991 inhibits ESCC cell growth and metastasis in nude mice. (A) The growth curves of s.c. xenografts of EC109 cells are shown. Nude mice bearing EC109 xenograft tumours were treated with vehicle or PD‐0332991 (150 mg·kg−1, administered by oral gavage daily) from day 6 to 20 after inoculation of EC109 cells. Point, mean; error bar, SD. n = 8 per group. * P < 0.05 by Student's t‐test. (B) After 14 days of PD‐0332991 treatment, the mice were killed, and tumours were dissected, weighed and photographed. Top: representative tumours from the control and experiment group are shown; bottom: comparison of tumour weights in control and experimental group (n = 8 per group). * P < 0.05 by Student's t‐test. (C) Immunohistochemical analysis of phospho‐Rb, Ki67 and active caspase‐3 (AC3) in xenograft tissues from mice. Haematoxylin and eosin (H&E)‐stained serial sections of the same xenografts are presented. Scale bar: 50 μm. (D) Immunoblotting of the phospho‐Rb, total Rb, FOXM1 and p16 proteins in xenograft tissues is shown. (E) KYSE150 cells were injected i.v. into nude mice via the tail vein. Top: representative images of lungs harvested 6 weeks post‐injection (arrows indicate the metastatic nodules). Bottom: surface metastatic nodules in the lungs were counted. Error bars represent mean ± SD (n = 6 per group). * P < 0.05 by Student's t‐test. (F) Representative images of lung sections stained with H&E (arrows indicate the metastatic colonization of tumour cells in the lung tissues). Scale bar, 500 μm.
Tumour metastasis is one of the important reasons that patients with ESCC often have a poor survival rate. Because CDK4/6 inhibition decreased the migration and invasive potential of ESCC cells in vitro, we next evaluated the effect of PD‐0332991 on metastasis of ESCC cells in vivo. Compared with vehicle treatment, PD‐0332991 significantly attenuated the activity of lung metastasis in nude mice, as evidenced by the decreased number of metastatic tumour nodules in the lungs (Figure 7E). Moreover, histological analysis also revealed an appreciable decrease in size and number of metastatic foci in the lungs of PD‐0332991‐treated mice, compared with that of the vehicle‐treated mice (Figure 7F). These results therefore support the possibility that PD‐0332991 can inhibit the growth and metastasis of ESCC cells in vivo.
Discussion
In this study, we analysed the inhibitory effect of PD‐0332991 on ESCC cells in vitro and in vivo. We found that PD‐0332991 significantly suppressed the growth, migration and invasion of ESCC cells. The expression of MMP‐2 was simultaneously down‐regulated. Moreover, knockdown of CDK4/6 using shRNA mimicked the inhibitory effects of PD‐0332991 on cell growth, migration and invasion. PD‐0332991 treatment also triggered ESCC cell senescence by decreasing FOXM1 expression. In addition, PD‐0332991 displayed a synergistic effect on cell growth inhibition with cisplatin or 5‐FU. More importantly, the in vivo studies also demonstrated that PD‐0332991 can inhibit the growth and metastasis of ESCC cells in nude mice, which was consistent with the findings from our in vitro study. To our knowledge, this is the first report to elucidate the role of PD‐0332991 in ESCC cells, using both in vitro and in vivo models.
PD‐0332991 is an orally active, water soluble cell‐permeable pyridopyrimidine, that potently inhibits the activity of recombinant CDK4 and CDK6 (IC50 = 11–15 nM) by competing with ATP‐binding sites (Fry et al., 2004). The anti‐proliferative effects of PD‐0332991 have been demonstrated in a number of preclinical studies (Fry et al., 2004; Finn et al., 2009; Konecny et al., 2011). PD‐0332991 was reported to induce the cell cycle arrest at G1‐phase by suppressing phosphorylation of Rb tumour suppressor protein (Malumbres and Barbacid, 2009). Rb phosphorylation facilitates the release of Rb from E2F transcription factor, enabling E2F to transcribe the genes that are required for cell cycle progression from the G1 to the S phase and ultimately for cellular proliferation (Musgrove et al., 2011; Reynolds et al., 2014). In this study, our data also showed that Rb phosphorylation was inhibited and the cell cycle was blocked at the G1‐phase by PD‐0332991. The MTS and clonogenicity assays also confirmed that PD‐0332991 prevented the proliferation of ESCC cells at low concentrations. Furthermore, our in vivo data revealed that PD‐0332991, at an oral dose of 150 mg·kg−1·day−1, potently inhibited the growth of xenografted EC109 cells, and markedly suppressed Rb phosphorylation, suggesting that PD‐0332991 may be an attractive therapeutic candidate for ESCC.
In addition, PD‐0332991 induced apoptosis in ESCC cells, as demonstrated by annexin V binding, specific cleavage of PARP and activation of caspase‐3. The apoptosis was triggered by the mitochondrial‐dependent pathway because PD‐0332991 evoked the release of cytochrome c into the cytosol. The levels of survivin, XIAP, Bcl‐2 and Bcl‐xL were decreased in PD‐0332991‐mediated apoptotic ESCC cells. Consistent with our results, it is reported that disruption of the cyclinD1/CDK4 complex by chemical inhibitors can reduce the expression of the ant‐apoptotic survivin (Baughn et al., 2006; Retzer‐Lidl et al., 2007). Future work needs to further define the precise mechanism of this effect of PD‐0332991.
ESCC has a poor prognosis because invasion and metastasis are prevalent. In this study, we found that PD‐0332991 inhibited ESCC cell migration and invasion and decreased the expression of MMP‐2. Consistent with our results, it was shown that PD‐0332991 inhibits breast cancer cell migration and invasion in both ER‐positive (T47D) and ‐negative (MDA‐MB‐231) cells (Qin et al., 2015). However, a previous report has shown that CDK4/6 inhibition by PD‐0332991 can induce epithelial–mesenchymal transition and enhance invasiveness in pancreatic cancer cells by activating Smad‐dependent TGF‐β signalling (Liu and Korc, 2012). Taken together, these findings suggest that whether PD‐0332991 inhibits or induces migration and invasion may depend on the tumour context.
CDK4 and CDK6, being highly homogeneous, can be bound and activated by the D‐type cyclins (cyclin D1, D2 and D3). In this respect, their functions may be redundant. Indeed, previous reports have shown that mice deficient in CDK4 are viable and only display proliferative defects in specific endocrine cell types, an observation attributed to a putative compensatory role by CDK6 (Martin et al., 2003). Similarly, CDK6‐knockout mice are viable and develop normally despite a slightly impaired haematopoiesis (Malumbres et al., 2004), whereas CDK4/CDK6 double‐knockout embryos die during the late stages of embryonic development due to severe anaemia (Malumbres et al., 2004). Moreover, in oesophageal adenocarcinoma (EAC), a combined model of both CDK6 and CDK4 expression is a superior predictor of survival over either alone. Specific knockdown of CDK4 and/or CDK6 in EAC cells by siRNAs demonstrated that their function is additive (Ismail et al., 2011). Consistently, our results revealed that the combined knockdown of both CDK4 and CDK6 led to a stronger suppression of ESCC cell growth, migration and invasion than knockdown of either CDK4 or CDK6 alone. These experiments are consistent with previous studies showing that CDK4 and CDK6 have redundant roles in controlling the G1 to S transition and proliferation (Malumbres et al., 2004). Together, our data further suggest the functional redundancy of CDK4 and CDK6 and highlight the requirement of simultaneous inhibition of CDK4 and CDK6 for suppression of ESCC.
Cellular senescence is a tumour‐suppressive mechanism, which must be overcome during cell immortalization and transformation (Anders et al., 2011). Prolonged incubation with PD‐0332991 has been shown to induce a senescence‐like morphological change in melanoma, glioblastoma and breast cancer cells, as indicated by enhanced activity of SA‐β‐Gal (Dean et al., 2010; Michaud et al., 2010; Anders et al., 2011). In this study, we found that treatment with PD‐0332991 induced cellular senescence in EC109 and EC9706 cells. FOXM1 is a forkhead box family transcription factor that regulates the transcription of cell cycle genes critical for progression into S‐phase and mitosis. A previous study has indicated that early‐passage FOXM1−/− mouse embryonic fibroblasts display premature senescence (Wang et al., 2005). Moreover, a systematic screen for CDK4/6 substrates has demonstrated that FOXM1 is a potential target of CDK4/6 signalling, and CDK4/6 can stabilize and activate FOXM1 to protect cancer cells from senescence (Anders et al., 2011). Consistent with these reports, our results showed that the decrease in FOXM1 by PD‐0332991 in ESCC cells might facilitate cellular senescence, because forced expression of FOXM1 protected ESCC cells against senescence in response to PD‐0332991, which is in line with the previous finding showing that FOXM1 exhibits senescence‐suppressing activity (Wang et al., 2005). However, silencing FOXM1 with siRNAs resulted in a substantial increase in basal senescence. Of importance, FOXM1 knockdown further enhanced PD0332991‐induced SA‐β‐Gal activities in EC109 cells. These findings may strengthen the rationale for the treatment of ESCC with small molecule inhibitors of CDK4/6. In contrast, Lian et al. did not observe senescence following PD‐0332991 treatment in ESCC cells (Lian et al., 2015). The possible reasons might include shorter exposure of ESCC cells to PD‐0332991 and the different lines (TE7, TE8 and TE10) used in their system.
It is widely recognized that combination approaches to therapy are helpful for making targeted agents successful in the treatment of ESCC (Ekman et al., 2008). There are numerous preclinical data regarding the synergy between PD‐0332991 and anti‐oestrogen agents including tamoxifen, trastuzumab in breast cancer (Perou et al., 2000; Finn et al., 2009). Moreover, PD‐033299, when used in combination with cytotoxic drugs, such as bortezomib and dexamethasone, markedly enhances the killing of myeloma cells by the cytotoxic agents (Baughn et al., 2006; Menu et al., 2008). In agreement with these findings, we found a notable synergistic anti‐proliferative effect between PD‐0332991 and 5‐FU or cisplatin in ESCC cells. During the revision of this manuscript, Zhou et al. reported that an increase in cell cycle‐related genes including CDK6 and cyclin D1 accompanies the amplification of the EGFR gene in primary ESCC tumours, and that blocking CDK4/6 activity with PD‐0332991 can prevent the resistance of these cells to the EGFR inhibitor erlotinib in vitro and improve the response of both OE21 and KYSE140 xenografts to erlotinib (Zhou et al., 2017). Taken together, dual inhibition of CDK4/6 by PD‐0332991 in combination with either cytotoxic chemotherapy or targeted therapy might be a promising strategy for ESCC patients.
Conclusion
In conclusion, PD‐0332991, a potent inhibitor of CDK4 and CDK6, inhibits cell growth, induces apoptosis and senescence and suppresses migration, invasion and metastasis in ESCC cells. Our results warrant a clinical trial to further evaluate its efficacy in ESCC patients, even in those with metastasis.
Author contributions
L.C. performed the research, analysed the data and wrote the paper. J.P. designed and performed the research, analysed the data and wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Time‐course observation of apoptosis in ESCC cells after treatment with PD‐0332991. (A) EC109 and EC9706 cells were treated with 20 μM PD‐0332991 for indicated durations, flow cytometry analysis was used to detect the cell death after staining with annexin V‐FITC/PI. Data were expressed as mean ± SD (n = 5 per group). *, P < 0.05, one‐way ANOVA with post hoc intergroup comparison with the Tukey's test. (B) and (C) Immunoblotting of apoptosis‐related proteins was shown in EC109 and EC9706 cells treated with 20 μM PD‐0332991.
Acknowledgements
This study was supported by grants from National Natural Science Funds (nos U1301226 and 81373434 to J. P.), the Research Foundation of Education Bureau of Guangdong Province, China (Grant cxzd1103 to J. P.), the Research Foundation of Guangzhou Bureau of Science and Technology and the Natural Science Foundation of Guangdong province (Grant 2015A030312014 to J. P.).
Chen, L. , and Pan, J. (2017) Dual cyclin‐dependent kinase 4/6 inhibition by PD‐0332991 induces apoptosis and senescence in oesophageal squamous cell carcinoma cells. British Journal of Pharmacology, 174: 2427–2443. doi: 10.1111/bph.13836.
References
- Alexander SP, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SP, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5729–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders L, Ke N, Hydbring P, Choi YJ, Widlund HR, Chick JM et al. (2011). A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20: 620–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughn LB, Di Liberto M, Wu K, Toogood PL, Louie T, Gottschalk R et al. (2006). A novel orally active small molecule potently induces G1 arrest in primary myeloma cells and prevents tumor growth by specific inhibition of cyclin‐dependent kinase 4/6. Cancer Res 66: 7661–7667. [DOI] [PubMed] [Google Scholar]
- Chen L, Li M, Li Q, Wang CJ, Xie SQ (2013). DKK1 promotes hepatocellular carcinoma cell migration and invasion through β‐catenin/MMP7 signaling pathway. Mol Cancer 12: 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TC (2010). Drug combination studies and their synergy quantification using the Chou–Talalay method. Cancer Res 70: 440–446. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SPA, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai W, Zhou J, Jin B, Pan J (2016). Class III‐specific HDAC inhibitor Tenovin‐6 induces apoptosis, suppresses migration and eliminates cancer stem cells in uveal melanoma. Sci Rep 6: 22622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean JL, Thangavel C, McClendon AK, Reed CA, Knudsen ES (2010). Therapeutic CDK4/6 inhibition in breast cancer: key mechanisms of response and failure. Oncogene 29: 4018–4032. [DOI] [PubMed] [Google Scholar]
- Dhillon S (2015). Palbociclib: first global approval. Drugs 75: 543–551. [DOI] [PubMed] [Google Scholar]
- Ekman S, Dreilich M, Lennartsson J, Wallner B, Brattstrom D, Sundbom M et al. (2008). Esophageal cancer: current and emerging therapy modalities. Expert Rev Anticancer Ther 8: 1433–1448. [DOI] [PubMed] [Google Scholar]
- Finn RS, Crown JP, Ettl J, Schmidt M, Bondarenko IM, Lang I et al. (2016). Efficacy and safety of palbociclib in combination with letrozole as first‐line treatment of ER‐positive, HER2‐negative, advanced breast cancer: expanded analyses of subgroups from the randomized pivotal trial PALOMA‐1/TRIO‐18. Breast Cancer Res 18: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RS, Crown JP, Lang I, Boer K, Bondarenko IM, Kulyk SO et al. (2015). The cyclin‐dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first‐line treatment of oestrogen receptor‐positive, HER2‐negative, advanced breast cancer (PALOMA‐1/TRIO‐18): a randomised phase 2 study. Lancet Oncol 16: 25–35. [DOI] [PubMed] [Google Scholar]
- Finn RS, Dering J, Conklin D, Kalous O, Cohen DJ, Desai AJ et al. (2009). PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor‐positive human breast cancer cell lines in vitro. Breast Cancer Res 11: R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E et al. (2004). Specific inhibition of cyclin‐dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3: 1427–1438. [PubMed] [Google Scholar]
- Ismail A, Bandla S, Reveiller M, Toia L, Zhou ZR, Gooding WE et al. (2011). Early G1 cyclin‐dependent kinases as prognostic markers and potential therapeutic targets in esophageal adenocarcinoma. Clin Cancer Res 17: 4513–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Ding K, Li H, Xue M, Shi X, Wang C et al. (2014). Ponatinib efficiently kills imatinib‐resistant chronic eosinophilic leukemia cells harboring gatekeeper mutant T674I FIP1L1‐PDGFRα: roles of Mcl‐1 and β‐catenin. Mol Cancer 13: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Yao Y, Chen L, Zhu X, Jin B, Shen Y et al. (2016a). Depletion of γ‐catenin by histone deacetylase inhibition confers elimination of CML stem cells in combination with imatinib. Theranostics 6: 1947–1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Zhou J, Xu F, Jin B, Cui L, Wang Y et al. (2016b). Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest 126: 3961–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konecny GE, Winterhoff B, Kolarova T, Qi J, Manivong K, Dering J et al. (2011). Expression of p16 and retinoblastoma determines response to CDK4/6 inhibition in ovarian cancer. Clin Cancer Res 17: 1591–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontieva OV, Blagosklonny MV (2013). CDK4/6‐inhibiting drug substitutes for p21 and p16 in senescence duration of cell cycle arrest and MTOR activity determine geroconversion. Cell Cycle 12: 3063–3069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leontieva OV, Demidenko ZN, Blagosklonny MV (2013). MEK drives cyclin D1 hyperelevation during geroconversion. Cell Death Differ 20: 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Li YY, Tsao SW, Cheung AL (2009). Targeting NF‐kappaB signaling pathway suppresses tumor growth, angiogenesis, and metastasis of human esophageal cancer. Mol Cancer Ther 8: 2635–2644. [DOI] [PubMed] [Google Scholar]
- Lian Z, Lee EK, Bass AJ, Wong KK, Klein‐Szanto AJ, Rustgi AK et al. (2015). FBXO4 loss facilitates carcinogen induced papilloma development in mice. Cancer Biol Ther 16: 750–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Korc M (2012). Cdk4/6 inhibition induces epithelial‐mesenchymal transition and enhances invasiveness in pancreatic cancer cells. Mol Cancer Ther 11: 2138–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malumbres M, Barbacid M (2009). Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9: 153–166. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Sotillo R, Santamaria D, Galan J, Cerezo A, Ortega S et al. (2004). Mammalian cells cycle without the D‐type cyclin‐dependent kinases Cdk4 and Cdk6. Cell 118: 493–504. [DOI] [PubMed] [Google Scholar]
- Martin J, Hunt SL, Dubus P, Sotillo R, Nehme‐Pelluard F, Magnuson MA et al. (2003). Genetic rescue of Cdk4 null mice restores pancreatic beta‐cell proliferation but not homeostatic cell number. Oncogene 22: 5261–5269. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Furihata M, Ishikawa T, Ohtsuki Y, Ogoshi S (1999). Comparison of deregulated expression of cyclin D1 and cyclin E with that of cyclin‐dependent kinase 4 (CDK4) and CDK2 in human oesophageal squamous cell carcinoma. Br J Cancer 80: 256–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menu E, Garcia J, Huang X, Di Liberto M, Toogood PL, Chen I et al. (2008). A novel therapeutic combination using PD 0332991 and bortezomib: study in the 5T33MM myeloma model. Cancer Res 68: 5519–5523. [DOI] [PubMed] [Google Scholar]
- Michaud K, Solomon DA, Oermann E, Kim JS, Zhong WZ, Prados MD et al. (2010). Pharmacologic inhibition of cyclin‐dependent kinases 4 and 6 arrests the growth of glioblastoma multiforme intracranial xenografts. Cancer Res 70: 3228–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musgrove EA, Caldon CE, Barraclough J, Stone A, Sutherland RL (2011). Cyclin D as a therapeutic target in cancer. Nat Rev Cancer 11: 558–572. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Kato H (2013). Treatment options for esophageal squamous cell carcinoma. Expert Opin Pharmacother 14: 1345–1354. [DOI] [PubMed] [Google Scholar]
- Pan J, Cheng C, Verstovsek S, Chen Q, Jin Y, Cao Q (2010). The BH3‐mimetic GX15‐070 induces autophagy, potentiates the cytotoxicity of carboplatin and 5‐fluorouracil in esophageal carcinoma cells. Cancer Lett 293: 167–174. [DOI] [PubMed] [Google Scholar]
- Pennathur A, Gibson MK, Jobe BA, Luketich JD (2013). Oesophageal carcinoma. Lancet 381: 400–412. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA et al. (2000). Molecular portraits of human breast tumours. Nature 406: 747–752. [DOI] [PubMed] [Google Scholar]
- Qin G, Xu F, Qin T, Zheng QF, Shi DB, Xia W et al. (2015). Palbociclib inhibits epithelial‐mesenchymal transition and metastasis in breast cancer via c‐Jun/COX‐2 signaling pathway. Oncotarget 6: 41794–41808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quint LE, Hepburn LM, Francis IR, Whyte RI, Orringer MB (1995). Incidence and distribution of distant metastases from newly diagnosed esophageal carcinoma. Cancer 76: 1120–1125. [DOI] [PubMed] [Google Scholar]
- Retzer‐Lidl M, Schmid RM, Schneider G (2007). Inhibition of CDK4 impairs proliferation of pancreatic cancer cells and sensitizes towards TRAIL‐induced apoptosis via downregulation of survivin. Int J Cancer 121: 66–75. [DOI] [PubMed] [Google Scholar]
- Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG et al. (2014). Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33: 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaeda T, Yamamori M, Kuwahara A, Nishiguchi K (2009). Pharmacokinetics and pharmacogenomics in esophageal cancer chemoradiotherapy. Adv Drug Deliv Rev 61: 388–401. [DOI] [PubMed] [Google Scholar]
- Schwartz GK, LoRusso PM, Dickson MA, Randolph SS, Shaik MN, Wilner KD et al. (2011). Phase I study of PD 0332991, a cyclin‐dependent kinase inhibitor, administered in 3‐week cycles (Schedule 2/1). Br J Cancer 104: 1862–1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Jin Y, Cheng C, Zhang H, Zou W, Zheng Q et al. (2009). Triptolide inhibits Bcr‐Abl transcription and induces apoptosis in STI571‐resistant chronic myelogenous leukemia cells harboring T315I mutation. Clin Cancer Res 15: 1686–1697. [DOI] [PubMed] [Google Scholar]
- Shi X, Wang D, Ding K, Lu Z, Jin Y, Zhang J et al. (2010). GDP366, a novel small molecule dual inhibitor of survivin and Op18, induces cell growth inhibition, cellular senescence and mitotic catastrophe in human cancer cells. Cancer Biol Ther 9: 640–650. [DOI] [PubMed] [Google Scholar]
- Song Y, Li L, Ou Y, Gao Z, Li E, Li X et al. (2014). Identification of genomic alterations in oesophageal squamous cell cancer. Nature 509: 91–95. [DOI] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang LH, Contractor T, Clausen R, Klimstra DS, Du YC, Allen PJ et al. (2012). Attenuation of the retinoblastoma pathway in pancreatic neuroendocrine tumors due to increased cdk4/cdk6. Clin Cancer Res 18: 4612–4620. [DOI] [PubMed] [Google Scholar]
- Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H et al. (2005). Discovery of a potent and selective inhibitor of cyclin‐dependent kinase 4/6. J Med Chem 48: 2388–2406. [DOI] [PubMed] [Google Scholar]
- Torre LA, Bray F, Siegel RL, Ferlay J, Lortet‐Tieulent J, Jemal A (2015). Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108. [DOI] [PubMed] [Google Scholar]
- Walker AJ, Wedam S, Amiri‐Kordestani L, Bloomquist E, Tang S, Sridhara R et al. (2016). FDA approval of palbociclib in combination with fulvestrant for the treatment of hormone receptor‐positive, HER2‐negative metastatic breast cancer. Clin Cancer Res 22: 4968–4972. [DOI] [PubMed] [Google Scholar]
- Wang C, Jiang L, Wang S, Shi H, Wang J, Wang R et al. (2015). The antitumor activity of the novel compound Jesridonin on human esophageal carcinoma cells. PLoS One 10: e0130284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ et al. (2005). Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2‐Cks1) ubiquitin ligase. Mol Cell Biol 25: 10875–10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Park HJ, Carr JR, Chen YJ, Zheng Y, Li J et al. (2011). FoxM1 in tumorigenicity of the neuroblastoma cells and renewal of the neural progenitors. Cancer Res 71: 4292–4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZP, Zhu SJ, Shen M, Liu JJ, Wang M, Li C et al. (2013). STAT3 is involved in esophageal carcinogenesis through regulation of Oct‐1. Carcinogenesis 34: 678–688. [DOI] [PubMed] [Google Scholar]
- Workman P, Aboagye EO, Balkwill F, Balmain A, Bruder G, Chaplin DJ et al. (2010). Guidelines for the welfare and use of animals in cancer research. Br J Cancer 102: 1555–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wu Z, Wong G, Pectasides E, Nagaraja A, Stachler M et al. (2017). CDK4/6 or MAPK blockade enhances efficacy of EGFR inhibition in oesophageal squamous cell carcinoma. Nat Commun 8: 13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Time‐course observation of apoptosis in ESCC cells after treatment with PD‐0332991. (A) EC109 and EC9706 cells were treated with 20 μM PD‐0332991 for indicated durations, flow cytometry analysis was used to detect the cell death after staining with annexin V‐FITC/PI. Data were expressed as mean ± SD (n = 5 per group). *, P < 0.05, one‐way ANOVA with post hoc intergroup comparison with the Tukey's test. (B) and (C) Immunoblotting of apoptosis‐related proteins was shown in EC109 and EC9706 cells treated with 20 μM PD‐0332991.