

Abstract
Background and Purpose
Dioscin exhibits a range of pharmacological actions but little is known of its effects on cisplatin (CDDP)‐induced nephrotoxicity. Here, we have assessed the effects and the possible mechanisms of dioscin against CDDP‐induced nephrotoxicity.
Experimental Approach
We used an in vivo model of CDDP‐induced nephrotoxicity in rats and mice and, in vitro, cultures of NRK‐52E and HK‐2 cells. The dual luciferase reporter assay was used to demonstrate modulation, by dioscin, of the targeting of sirtuin 1 (Sirt1) by microRNA (miR)‐34a. Molecular docking assays were used to analyse the effects of dioscin with Sirt1, Keap1 and NF‐κB.
Key Results
Dioscin attenuated cell damage in vitro and decreased renal injury in rats and mice, treated with CDDP. In terms of mechanisms, dioscin reversed CDDP‐induced up‐regulation of miR‐34a and thus up‐regulated Sirt1 levels. In addition, dioscin altered levels of haem oxygenase 1, glutathione‐cysteine ligase subunits (GCLC, GCLM) and Keap1, along with increased nuclear translocation of Nrf2, thus decreasing oxidative stress. Also, dioscin affected levels of AP‐1, COX‐2, HMGB1, IκB‐α, IL‐1β, IL‐6 and TNF‐α and decreased the ratio of acetylated NF‐κB and normal NF‐κB, to suppress inflammation. From molecular docking assays, dioscin directly bound to Sirt1, Keap1 and NF‐κBp65 by hydrogen bonding and/or hydrophobic interactions.
Conclusions and Implications
Our results have linked CDDP‐induced nephrotoxicity and the miR‐34a/Sirt1 signalling pathway, which was modulated by dioscin. This natural product could be developed as a new candidate to alleviate CDDP‐induced renal injury.
Abbreviations
- AP‐1
activator protein‐1
- BUN
blood urea nitrogen
- CDDP
cisplatin
- CMC‐Na
carboxymethyl cellulose sodium
- Cr
creatinine
- GCLC
GSH cysteine ligase catalytic subunit
- GCLM
GSH cysteine ligase modifier subunit
- HMGB1
high‐mobility group box 1
- HO‐1
haem oxygenase‐1
- Nrf2
NF erythroid 2‐related factor 2
- Sirt1
Sirtuin 1
Tables of Links
| TARGETS | |
|---|---|
| Enzymes a | Other protein targets b |
| COX‐2 | Keap1 |
| HO‐1, haem oxygenase 1 | |
| Sirt1, sirtuin 1 |
These Tables list key protein targets and ligands in this article that are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in The Concise Guide to PHARMACOLOGY 2015/16 (a,bAlexander et al., 2015a,b).
Introduction
Cisplatin (CDDP), an inorganic platinum compound, has broad‐spectrum activities against different types of human tumours including breast, testicular, ovarian, lung, bladder, head and neck cancers (Arany and Safirstein, 2003). However, the severe side effects of CDDP, particularly nephrotoxicity, limit its clinical applications (Karasawa and Steyger, 2015). Therefore, it is necessary to seek effective therapeutic methods or agents to reduce CDDP‐induced nephrotoxicity.
There is evidence that oxidative stress and inflammation play critical roles in the pathogenesis of CDDP‐induced nephrotoxicity (Pérez‐Rojas et al., 2009; Hagar et al., 2015). Oxidative stress is caused by a range of oxygen‐derived free radicals (ROS), including superoxide anion, hydrogen peroxide and hydroxyl radical. Excessive production of these free radicals and lipid peroxidation, as have been found in CDDP‐induced renal dysfunction (Chirino et al., 2008), can activate many pro‐inflammatory cytokine responses and cause irreversible renal damage (Ma et al., 2015).
A variety of biological molecules and signal pathways affect oxidative stress and inflammation. MicroRNAs (miRs) inhibit mRNA transcription or promote mRNA degradation, in the initiation and development of various diseases (Martello et al., 2010). A member of miR‐34 family, miR‐34a,plays important roles in oxidative stress, inflammation and apoptosis (Do et al., 2014; Guennewig et al., 2014; Tilyek et al., 2016) and is known to interact directly with sirtuin 1 (Sirt1), an NAD+‐dependent nuclear class III histone deacetylase (Yamakuchi et al., 2008). Sirt1 is involved in diverse biological processes including metabolism, oxidative stress and inflammation by deacetylation of some genes including those for NF‐κB, FOXO3, p53 and histones (Luo et al., 2001; Vaziri et al., 2001; Motta et al., 2004; Yeung et al., 2004). Sirt1 can lead to the rapid activation of NF‐κB associated with CDDP‐induced renal inflammation through increasing the levels of pro‐inflammatory cytokines and inflammatory mediators including IL‐1β, IL‐6 and TNF‐α (Ma et al., 2015). Thus, miR‐34a/Sirt1‐mediated NF‐κB signalling is an important pathway of controlling inflammation.
Furthermore, Sirt1 can also regulate the transcription factor Nrf2 to modulate the transcription of pro‐ and antioxidant enzymes and thus affect the cellular redox state (Do et al., 2014). Under normal conditions, Nrf2 is anchored in the cytoplasm where it binds with Keap1 (Kelch‐like ECH‐associated protein 1). Upon activation, Nrf2 translocates into the nucleus and transactivates its target genes through the antioxidant response element (ARE) (Ke et al., 2013) to reduce the formation of ROS and suppress oxidative stress (Dhanda et al., 2013). Haem oxygenase‐1 (HO‐1), GSH cysteine ligase catalytic subunit (GCLC) and GSH cysteine ligase modifier subunit (GCLM) are examples of Nrf2 target genes (Zhao et al., 2009; Shah et al., 2013). HO‐1 can catalyse haem metabolism and yield the antioxidant compounds, carbon monoxide and bilirubin, to scavenge free radicals (Bataille and Manautou, 2012). GCLC and GCLM are critical enzyme components for GSH biosynthesis ansd thus control cellular redox status and play important roles in the removal of ROS (Harvey et al., 2009). In this way, miR‐34a/Sirt1‐mediated Nrf2 signalling is important in controlling oxidative stress. Therefore, increasing the Sirt1 signal by down‐regulating miR‐34a and thus suppressing both inflammation and oxidative stress, may be one potential strategy against CDDP‐induced nephrotoxicity.
Complementary and alternative medicine may have a unique role in treating CDDP‐induced nephrotoxicity and traditional Chinese medicines have been used in China to prevent diseases for thousands of years. Some active natural products including epigallocatechin‐3‐gallate, saikosaponin‐D and resveratrol (Sahin et al., 2010a; Ma et al., 2015; Reddy et al., 2016) have been used to treat CDDP‐induced nephrotoxicity. Thus, it is reasonable to develop effective natural products from medicinal plants for the treatment of CDDP‐induced nephrotoxicity.
Dioscin (see Supporting Information Figure S1), a natural steroid saponin, is found in various herbs (Yin et al., 2010b). Pharmacological investigations have shown that dioscin has anti‐tumour, anti‐hyperlipidaemic and anti‐fungal activities (Li et al., 2010; Cho et al., 2013; Hsieh et al., 2013). Our earlier studies indicated that dioscin had potent effects against carbon tetrachloride‐ and paracetamol‐induced acute liver damage (Lu et al., 2012; Zhao et al., 2012), non‐alcoholic fatty liver disease (Xu et al., 2014), hepatic ischaemia/reperfusion injury (Tao et al., 2014), cerebral ischaemia/reperfusion injury (Tao et al., 2015) and alcoholic liver fibrosis (Xu et al., 2014). Importantly, we also found that dioscin had protective effects against renal ischaemia/reperfusion injury and lipopolysaccharide‐induced inflammatory kidney injury (Qi et al., 2015; Qi et al., 2016). However, no studies have reported the effects and molecular mechanisms of dioscin in preventing CDDP‐induced nephrotoxicity.
Therefore, the aim of the present work was to investigate the protective effects, molecular mechanisms and possible targets of dioscin against CDDP‐induced nephrotoxicity.
Methods
Cell culture
The NRK‐52E normal rat kidney epithelial cell line and HK‐2 human kidney tubular epithelial cell line were purchased from the Institute of Biochemistry Cell Biology (Shanghai, China), which were maintained in DMEM supplemented with 10% FBS and antibiotics (100 IU·mL−1 penicillin and 100 mg·mL−1 streptomycin) in a humidified atmosphere of 5%CO2 and 95%O2 at 37°C.
Dioscin toxicity assay
The NRK‐52E and HK‐2 cells were plated in 96‐well plates at a density of 5 × 104 cells·mL−1 for 24 h and treated with various concentrations of dioscin (0, 50, 100, 200, 400, 800, 1600 and 3200 ng·mL−1) for 6, 12 and 24 h at 37°C, and then the cyto‐toxicity of the compound was assayed using the MTT method, as previously described (Si et al., 2017). In brief, MTT solution was added to the plates for 4 h incubation at 37°C, and then the formazan crystals were dissolved by 100 μL of DMSO. Finally, the absorbance was measured at 490 nm with a microplate reader (Thermo, Waltham, MA, USA).
CDDP‐induced cell injury
CDDP was dissolved to make a series of working solutions in serum‐free DMEM. The NRK‐52E and HK‐2 cells were plated in 96‐well plates at a density of 5 × 104 cells·mL−1 for 24 h before challenge with various concentrations of CDDP (1–12 μg·mL−1). Cell viability was assessed using the MTT method as described above, and the most suitable concentration of CDDP was determined.
Cell viability assay
The NRK‐52E and HK‐2 cells were seeded in 96‐well plates at a density of 5 × 104 cells·mL−1 for 24 h and pretreated with various concentrations of dioscin (50, 100 or 200 ng·mL−1) for 6, 12 and 24 h before challenge with CDDP (9 μg·mL−1) for 24 h. The cells in model group were cultured without dioscin, and the cells in control group were cultured in serum‐free DMEM under normal conditions. Cell viability was assayed using the MTT method as described above.
Measurement of intracellular ROS level
The NRK‐52E and HK‐2 cells were plated in six‐well culture plates at a density of 5 × 104 cells·mL−1 and treated with dioscin (50, 100 and 200 ng·mL−1) for 12 h before challenge with CDDP. The cells were harvested and then resuspended in 500 μL of DCFH‐DA (10.0 μM) for the detection of ROS, which was analysed by flow cytometry (Becton‐Dickinson, Franklin Lake, NJ, USA) as described earlier (Sun et al., 2016).
Animals and ethical approval
All animal care and experimental procedures were in strict accordance with PR China Legislation Regarding the Use and Care of Laboratory Animals, and all experiments involving animals were approved by the Animal Care and Use Committee of Dalian Medical University (approval number: 20150658). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). Forty‐eight male Wistar rats weighing 200–250 g (8 weeks old) and male C57BL/6J mice weighing 18–22 g (4 weeks old) were obtained from the Experimental Animal Center at Dalian Medical University (Dalian, China) (SCXK: 2013‐0003). The animals were group‐housed with two to three rats per cage on a 12 h light/dark cycle in a temperature‐controlled (25 ± 2°C) room with free access to water and food and were allowed 1 week to acclimatize before experiment. Randomization was used to assign samples to the experimental groups and to collect and process data. The experiments were performed by investigators blinded to the treatment groups.
CDDP‐induced acute kidney injury in vivo
The rats were randomly divided into six groups (n = 8): control group (0.5% CMC‐Na), dioscin only (40 mg·kg−1) group, CDDP only group and CDDP with dioscin (10, 20 and 40 mg·kg−1) treatment groups. The rats were given dioscin orally, at doses of 10, 20 and 40 mg·kg−1 once daily for 12 consecutive days,(groups CDDP + Dio 40, CDDP + Dio 20 and CDDP + Dio 10). The mice were randomly divided into six groups (n = 8): control group (0.5% CMC‐Na), Dio (60 mg·kg−1) group, CDDP only group and Dio‐only groups (the mice were given dioscin orally at doses of 15, 30 and 60 mg·kg−1 once daily for 10 consecutive days (groups CDDP + Dio 60, CDDP + Dio 30 and CDDP + Dio 15). One hour after dioscin administration on the last day, the animals in the CDDP only and dioscin‐treated groups were injected with CDDP (7 mg·kg−1 for rats and 25 mg·kg−1 for mice; i.p.) in 0.9% saline (10 mL·kg−1). Four hours after the last dosing, blood samples were obtained from the abdominal aorta, and the kidneys were rapidly removed for histological staining and Western blot assays. Serum samples were obtained from blood samples by centrifugation (1200× g, 4°C) for 10 min and stored at −20°C until assay.
Assessment of biochemical parameters
The renal index (RI, kidney weight to body weight) was measured, and the serum blood urea nitrogen (BUN) and creatinine (Cr) levels were detected using the specific kits (Product ID C013‐2 and C011‐2) from Nanjing Jiancheng Institute of Biotechnology (Nanjing, China) according to the manufacturer's instructions. The levels of malonaldehyde (MDA), SOD, GSH and GSH‐peroxidase (GSH‐Px) in kidney tissues were determined using the commercial kits (Product IDs A003‐1, A001‐3, A006‐2 and A005) from Nanjing Jiancheng Institute of Biotechnology (Nanjing, China), following the manufacturer's instructions.
Histological and immunofluorescence assays
The kidney tissues were fixed in 10% formalin, embedded in paraffin and sectioned into 5 μm slices. The slices were stained with haematoxylin and eosin (H&E) and photographed using a light microscope (Nikon Eclipse TE2000‐U; Nikon, Chiyoda pill, Tokyo, Japan). For the immunofluorescence staining of Sirt1 and NF‐κB, the tissue slices or formalin‐fixed cells were incubated with anti‐Sirt1 and anti‐NF‐κB antibodies (Santa Cruz, Dallas, TX, USA), respectively, in a humidified chamber at 4°C overnight, followed by incubation with an Alexa fluorescein‐labelled secondary antibody at 37°C for 1 h. Cell nuclei were stained with DAPI (5 μg·mL−1). Immunostained samples were analysed by fluorescence microscopy (Olympus, Tokyo, Japan) with 200× magnification.
Quantification of miR‐34a
The total miR samples were isolated using the SanPrep Column Micro‐RNA Mini‐Prep Kit. Reverse transcription was performed using a MicroRNA First Strand cDNA Synthesis Kit, and the levels of mature miR were quantified using real‐time PCR with a MicroRNAs Quantitation PCR Kit and an ABI 7500 real‐time PCR system (Applied Biosystems, Waltham, MA, USA). The primers for miR‐34a are listed in Supporting Information Table S1, and the U6 small nucleolar RNA was used for normalization.
Dual luciferase reporter assays
Plasmid DNA (wt‐Luc‐Sirt1, mut‐Luc‐Sirt1) and miR‐34a mimics or negative control were co‐transfected into NRK‐52E and HK‐2 cells. When appropriate, the cells were incubated with or without (control) 200 ng·mL−1 of dioscin for 24 h after transfection. Luciferase activity was measured with a Double‐Luciferase Reporter Assay Kit using the Dual‐Light Chemiluminescent Reporter Gene Assay System (Berthold, Wildbad, Stuttgart, Germany), and the Renilla luciferase was used to normalize for the transfection efficacy.
Quantitative real‐time PCR assay
Total RNA samples were obtained from NRK‐52E cells, HK‐2 cells and kidney tissues using RNAiso Plus reagent following the manufacturer's protocol. Each RNA sample was reverse‐transcribed into cDNA using the PrimeScript® RT reagent Kit. The forward (F) and reverse (R) primers used in the present study are given in Supporting Information Table S2. Among the data from each sample, the Ct value of the target genes was normalized to that of GAPDH. The unknown template in our study was calculated using the standard curve for quantitative analysis.
Western blotting assay
The total, nuclear and cytoplasmic proteins from NRK‐52E cells, HK‐2 cells and kidney tissues were extracted using cold lysis buffer containing 1 mM phenylmethyl sulfonyl fluoride according to the manufacturer's protocol, and the protein content was determined using the bicinchoninic acid protein assay kit. Proteins were subjected to SDS‐PAGE (10 to15%) and then were transferred to PVDF membranes (Millipore, Danvers, MA, USA). Finally, the protein expression images were obtained by a Bio‐Spectrum Gel Imaging System (UVP, Upland, CA, USA). Intensity values expressed as the relative protein expression were normalized to GAPDH, Lamin B1 and tubulin. The primary antibodies are listed in Supporting Information Table S3.
Overexpression of miR‐34a
The NRK‐52E and HK‐2 cells were seeded in six‐well plates (5 × 104 cells·mL−1) in a serum‐free medium and transfected with the miR‐34a mimics (50 nM) or negative controls mixed with Lipofectamine 2000 according to the manufacturer's instructions. The negative controls consisting of random sequences had no detectable effects on the cell lines. The protein levels of Sirt1 in NRK‐52E and HK‐2 cells were detected to confirm whether Sirt1 is the target protein of miR‐34a after 24 h transfection. In addition, 24 h after transfection, the cells were subjected to serum deprivation for 24 h in the presence or absence of dioscin (200 ng·mL−1) before they were challenged with CDDP (9 μg·mL−1) for an additional 24 h. Then, the protein levels of Sirt1, Nrf2 and Keap1; acetylated NF‐κB; and the mRNA levels of IL‐1β, IL‐6 and TNF‐α were measured after 24 h of transfection.
Sirt1 siRNA treatment
Transfection was performed when the NRK‐52E and HK‐2 cells were cultured to 70–80% confluence in six‐well or 96‐well plates. The Sirt1‐targeted siRNA and control siRNA were dissolved in Opti‐MEM and then equilibrated for 5 min at room temperature. The cells were transfected with Sirt1 siRNA or non‐binding control siRNA using Lipofectamine2000 reagent according to the manufacturer's protocol. Then, cell viability; the protein levels of Sirt1, Nrf2 and Keap1; acetylated NF‐κB; and the mRNA levels of IL‐1β, IL‐6 and TNF‐α were measured after 24 h of transfection.
Molecular docking assay
To predict the targets of the action of dioscin against CDDP‐induced nephrotoxicity, docking studies were performed using AutoDock 4.2.6 software. The 3D structure of dioscin was produced by Maestro, and the crystal structures of Sirt1, Keap1 and NF‐κB were drawn from Protein Data Bank (PDB, http://www.rcsb.org/pdb/home/home.do). Following the requirement of docking study, water molecules, ions and non‐standard amino acid residues were removed from the proteins and the hydrogen atoms were added followed by computing Gasteiger charge. Furthermore, the grid size and AutoDock parameters were set for the docking study, and the model with the lowest energy was optimized and chosen for further assay.
Data and statistical analysis
The data and statistical analysis in this study comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). The data are expressed as the mean ± SD. GraphPad Prism 6.01 software (Paragraph Software, Inc, La Jolla, CA, USA) was used to handle these data and only when a minimum of n = 5 independent samples was acquired. Data were normalized to control for unwanted sources of variation as follows. First, the data werenormalized to bring all of the variation into proportion with one another, after removing the outliers. Then, the coefficients associated with each variable were scaled appropriately to adjust for the disparity in the variable sizes. Statistical analysis was performed with one‐way ANOVA followed by Tukey's post hoc test when comparing multiple independent groups, and when comparing two different groups, the unpaired t‐test was carried out. Post hoc tests were run only if F achieved P < 0.05 and there was no significant variance inhomogeneity. Statistical significance was considered to be P < 0.05.
Materials
Dioscin was obtained from Shanghai Tauto Biochemical Technology Co., Ltd. (Shanghai, China) and was dissolved in 0.5% carboxymethyl cellulose sodium (CMC‐Na) for in vivo experiments and in 0.1% DMSO for in vitro tests. Tissue Protein Extraction Kit was obtained from KEYGEN Biotech. Co., Ltd. (Nanjing, China). The bicinchoninic acid (BCA) Protein Assay Kit was purchased from Beyotime Institute of Biotechnology (Jiangsu, China). CCDP, Tris, SDS, CMC‐Na and DAPI were purchased from Sigma (St. Louis, MO, USA). TransZolTM, TransScript® All‐in‐One First‐Strand cDNA Synthesis SuperMix for qPCR (One‐Step gDNA Removal) and TransStart® Top Green qPCR SuperMix and Double‐Luciferase Reporter Assay Kit were supplied by Beijing TransGen Biotech Co., Ltd. (Beijing, China). The SanPrep Column MicroRNA Mini‐Prep Kit, MicroRNA First Strand cDNA Synthesis Kit and MicroRNA Quantitation PCR Kit were purchased from Sangon Biological Engineering Technology and Services Co., Ltd. (Shanghai, China). Plasmids including wild‐type miR‐34a–Sirt1 response element (wt‐Luc‐Sirt1), the corresponding mutant (mut‐Luc‐Sirt1), the miR‐34a mimics and Lipofectamine 2000 were purchased from RiboBio Co., Ltd. (Guangzhou, China).
Results
Dioscin protects NRK‐52E and HK‐2 cells from CDDP‐induced injury
As shown in Figure 1A, B, the viability of NRK‐52E and HK‐2 cells was increased or decreased slightly compared with that of control group after treatment with dioscin (50–400 ng·mL−1) for 6–24 h with no significant difference, indicating that dioscin was non‐toxic to NRK‐52E and HK‐2 cells, under the treatment conditions. As shown in Figure 1C, exposure to CDDP (1–12 μg·mL−1) for 24 h induced a concentration‐dependent decrease in the viability of NRK‐52E and HK‐2 cells. Compared with control cells not exposed to CDDP, the viability of NRK‐52E and HK‐2 cells incubated with 9.0 μg·mL−1 of CDDP were by about 50% and this concentration of CDDP was used as the standard treatment in the rest of the experiments. After pretreatment with dioscin (50‐200 ng·mL−1) for 6, 12 or 24 h, the NRK‐52E and HK‐2 cells were significantly protected against CDDP‐induced damage (Figure 1D, E). Therefore, dioscin at the concentrations of 50, 100 and 200 ng·mL−1 for 12 h were selected to protect NRK‐52E and HK‐2 cells against CDDP‐induced injury.
Figure 1.
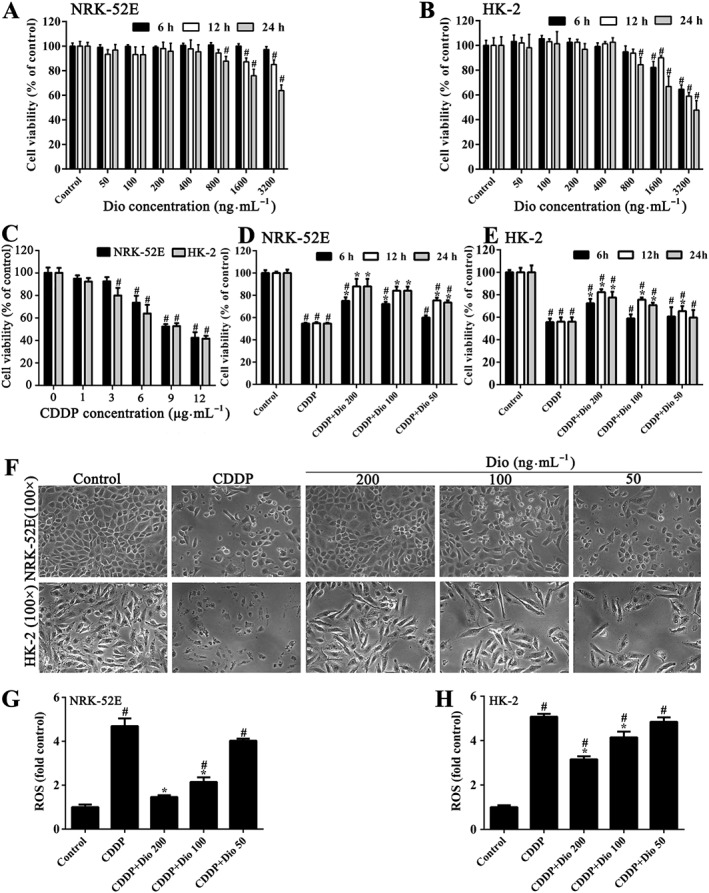
Effects of dioscin on protecting against CDDP‐induced cytotoxicity in NRK‐52E and HK‐2 cells. (A) Cytotoxicity of dioscin on NRK‐52E cells. (B) Cytotoxicity of dioscin on HK‐2 cells. (C) CDDP‐induced toxicity in NRK‐52E and HK‐2 cells. (D) Effects of dioscin on loss of cell viability in NRK‐52E cells, induced by CDDP. (E) Effects of dioscin on loss of cell viability in HK‐2 cells, induced by CDDP. (F) Effects of incubation with dioscin (50, 100 and 200 ng∙mL−1) for 12 h, on the cellular morphology and structure of NRK‐52E and HK‐2 cells by bright image (100× magnification) investigation. (G) Effects of dioscin on the levels of ROS in NRK‐52E cells after CDDP exposure. (H) Effects of dioscin on the levels of ROS in HK‐2 cells after CDDP exposure. Data are presented as the mean ± SD (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from CDDP groups.
Effects of dioscin on cellular morphology and structure
As shown in Figure 1F, pretreatment with dioscin (100 and 200 ng·mL−1 for 12 h) significantly restored the morphological changes of NRK‐52E and HK‐2 cells including cell shrinkage, nuclear pyknosis and loss of structure, caused by CDDP.
Effects of dioscin on ROS level in vitro
As shown in Figure 1G, H and Supporting Information Figure S2, compared with the CDDP only group, pre‐incubation with dioscin (100 and 200 ng·mL−1) for 12 h significantly decreased the ROS levels in NRK‐52E and HK‐2 cells.
Dioscin protects against CDDP‐induced nephrotoxicity in vivo
As shown in Figure 2A, B, compared with CDDP only group, the levels of RI, BUN and Cr in rats treated with dioscin (40 mg·kg−1) were significantly reduced, as were the levels in mice treated with 60 mg·kg−1 of dioscin. As shown in Figure 2C (200× magnification) and Supporting Information Figure S3 (400× magnification), the histopathological changes including partial renal tubular epithelial vacuole degeneration or hyaline degeneration, swelling in the renal tubular epithelial cells, a narrowed lumen, interstitial angiectasis hyperemia and the large number of infiltrated inflammatory cells, in the groups treated with CDDP only, were all markedly reversed by dioscin.
Figure 2.
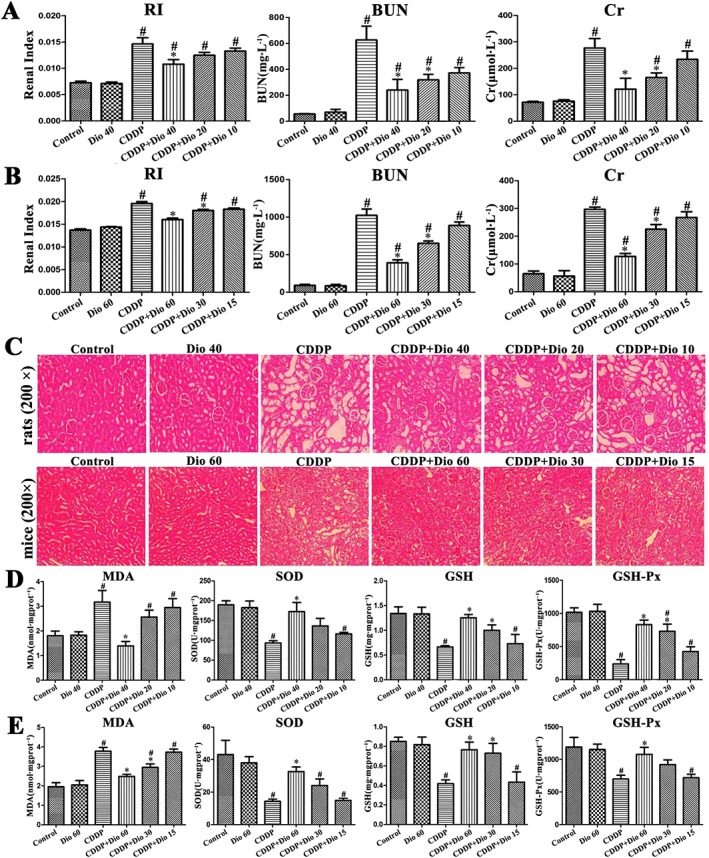
Effects of dioscin on CDDP‐induced nephrotoxicity in rats and mice. (A) Effects of dioscin on RI and serum Cr and BUN levels in rats. (B) Effects of dioscin on RI and serum Cr and BUN levels in mice. (C) H&E staining (200× original magnification) of the kidney tissue in rats and mice. (D) Effects of dioscin on MDA, SOD, GSH and GSH‐Px levels in renal tissues from rats. (E) Effects of dioscin on MDA, SOD, GSH and GSH‐Px levels in renal tissues from mice. Data are presented as the mean ± SD (n = 8). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from CDDP groups.
Dioscin suppresses oxidative stress caused by CDDP in vivo
As shown in Figure 2D, E, the level of MDA was significantly increased, and the levels of SOD, GSH and GSH‐Px were clearly decreased in rats and mice after CDDP. These changes were all markedly reversed by 40 (in rats) or 60 mg·kg−1 (in mice) of dioscin.
Dioscin down‐regulates the levels of miR‐34a in vitro and in vivo
As shown in Figure 3A, the expression levels of miR‐34a were markedly increased by CDDP in vitro and in vivo compared with the values in control conditions . These increased levels were clearly down‐regulated by dioscin (50‐200 ng·mL−1 or 10‐60 mg kg‐1).
Figure 3.
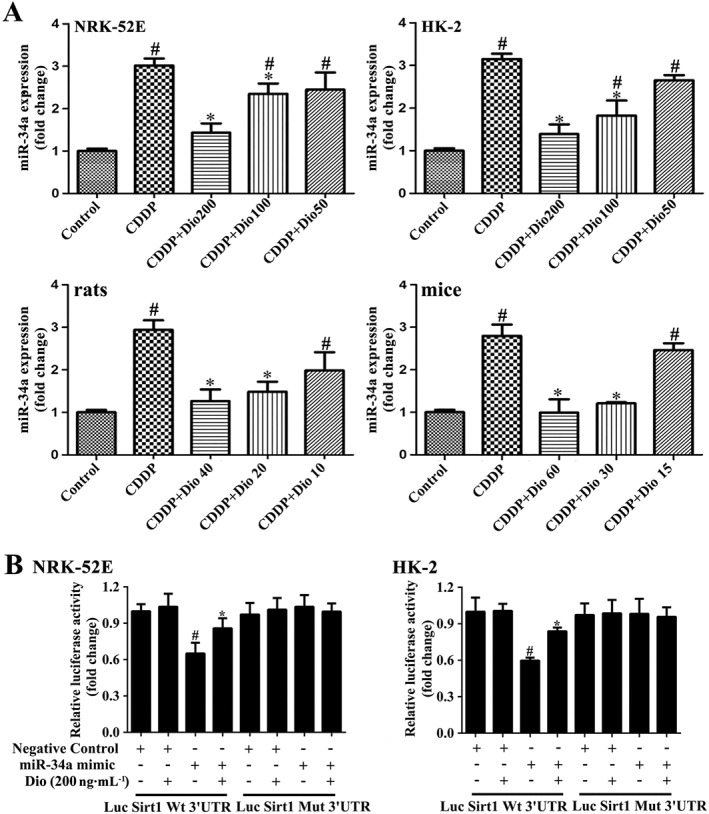
Dioscin regulates the Sirt1 signalling pathway in a miR‐34a‐dependent manner. (A) Effects of dioscin on miR‐34a levels in vitro and in vivo. Data are presented as the mean ± SD (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from CDDP groups. (B) Dioscin modulated Sirt1 via miR‐34a, based on dual luciferase reporter assay in NRK‐52E and HK‐2 cells. Data are presented as the mean ± SD (n = 5). # P < 0.05, significantly different from the negative control group; *P < 0.05, significantly different from the group transfected with miR‐34a mimic.
Dioscin inhibits miR‐34a targeting Sirt1 in NRK‐52E cells
In order to further investigate the effects of dioscin on Sirt1, via miR‐34a, we co‐transfected miR‐34a mimics and luciferase reporter plasmids containing wt‐Luc‐Sirt1 or mut‐Luc‐Sirt1 in the presence or absence of dioscin in NRK‐52E cells and HK‐2 cells. As shown in Figure 3B, the luciferase activity was significantly repressed by miR‐34a overexpression. Dioscin increased the luciferase activity compared with miR‐34a‐overexpressed cells. However, these effects were not observed with the mutated Sirt1–3′‐UTR. The above data revealed that dioscin increased Sirt1 expression at least partly by a miR‐34a‐dependent manner.
Dioscin activates the levels of Sirt1 in vitro and in vivo
Based on immunofluorescence staining shown in Figure 4A, B, compared with control groups, the expression levels of Sirt1 in cells and in kidney sections were markedly decreased after CDDP treatment in vivo and in vitro. These levels were significantly up‐regulated by pre‐treatments with dioscin, especially in rats and mice.
Figure 4.
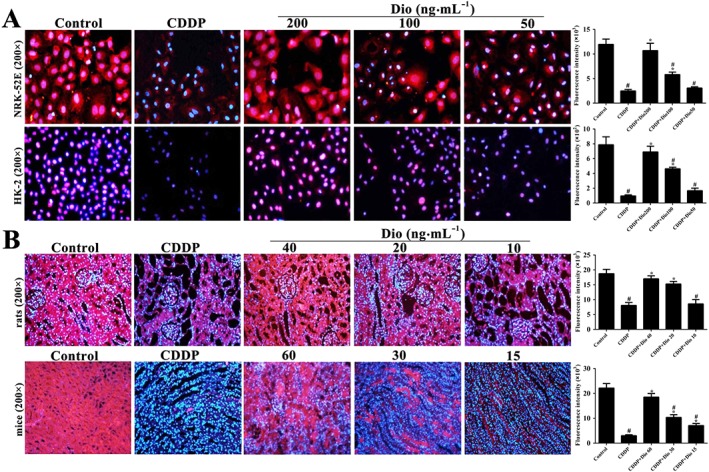
Dioscin activates Sirt1 levels in vitro and in vivo. (A) Effects of dioscin on Sirt1 levels based on immunofluorescence staining in NRK‐52E and HK‐2 cells (200× magnification). (B) Effects of dioscin on Sirt1 levels based on immunofluorescence staining in renal tissues from rats and mice (200× magnification). Data are presented as the mean ± SD (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from CDDP groups.
Dioscin regulates Sirt1/Nrf2‐mediated pathway in vivo and in vitro
As shown in Figure 5A, pretreatment of with dioscin (200 ng·mL−1) for 12 h significantly increased the protein levels of Sirt1 and total Nrf2 in cells and in renal tissue, compared with those from groups treated with CDDP only. In addition, the levels of HO‐1, GCLC and GCLM were significantly decreased, and the levels of Keap1 were significantly increased after CDDP, compared with control groups. These changes were significantly attenuated by dioscin. Moreover, dioscin clearly increased the nuclear levels and decreased cytoplasmic levels of Nrf2, suggesting that dioscin promoted the nuclear translocation of Nrf2. As shown in Figure 5B, in animal experiments, dioscin noticeably increased the expression levels of Sirt1, total Nrf2 and nuclear Nrf2 and decreased cytoplasmicNrf2 levels, compared with CDDP only groups. Furthermore, dioscin (40 mg·kg−1 in rats and 60 mg·kg−1 in mice) also markedly up‐regulated the levels of HO‐1, GCLC and GCLM and down‐regulated the levels of Keap1, compared with the CDDP only groups. These results suggested that dioscin modulated the Sirt1/Nrf2 signalling pathway and reduced ROS activation.
Figure 5.
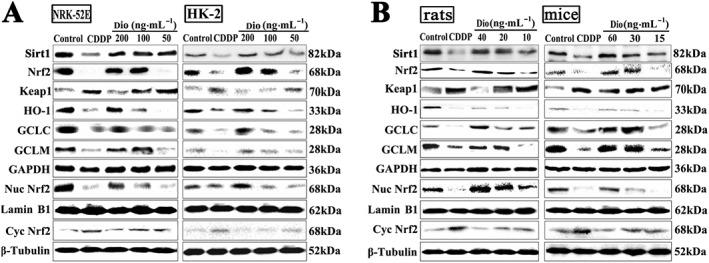
Dioscin regulates the Sirt1/Nrf2‐mediated pathway in vivo and in vitro. (A) Effects of dioscin on the protein levels of Keap1, total Nrf2, nuclear Nrf2, cytoplasmic Nrf2, HO‐1, GCLC, GCLM in NRK‐52E and HK‐2 cells (n = 5). (B) Effects of dioscin on the protein levels of Keap1, total Nrf2, nuclear Nrf2, cytoplasmic Nrf2, HO‐1, GCLC and GCLM in renal tissues from rats and mice (n = 5).
Dioscin suppresses inflammation caused by CDDP in vitro and in vivo
As shown in Figure 6A, high doses of dioscin significantly down‐regulated the expression levels of the transcription factor AP‐1, COX‐2 and high‐mobility group box 1 (HMGB1), up‐regulated the protein levels of IκB‐α and clearly decreased the ratios of acetylated to normal NF‐κB, compared with CDDP groups in vivo and in vitro. As shown in Figure 6B, the mRNA levels of TNF‐α, IL‐1β and IL‐6 in vitro and in vivo were significantly increased after CDDP alone and these levels were all clearly restored by dioscin.
Figure 6.
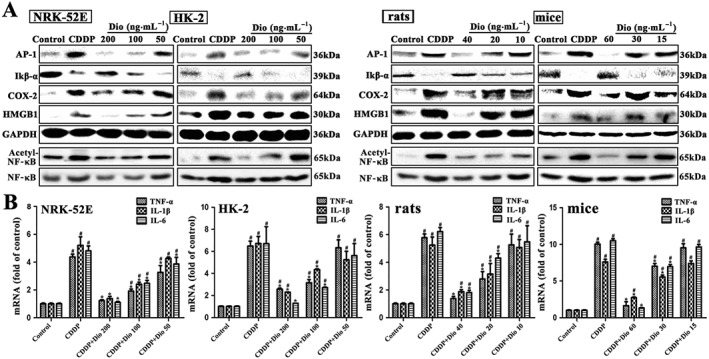
Dioscin suppresses inflammation caused by CDDP in vitro and in vivo. (A) Effects of dioscin on the protein levels of AP‐1, COX‐2, IκB‐α degradation, HMGB1 and acetylated NF‐κB in vitro and in vivo. (B) Effects of dioscin on the mRNA levels of IL‐1β, IL‐6 and TNF‐α in vitro and in vivo. Data are presented as the mean ± SD (n = 5). # P < 0.05, significantly different from control group; *P < 0.05, significantly different from CDDP groups.
Dioscin reverses the effects of miR‐34a mimics
For further investigation of the effect of dioscin on the activation of Sirt1‐mediated signalling, we used miR‐34a. Compared with the control group, miR‐34a mimics markedly decreased the protein levels of Sirt1 in CDDP‐treated NRK‐52E and HK‐2 cells (Figure 7A). Furthermore, as shown in Figure 7B, transfecting miR‐34a mimics exacerbated CDDP‐induced nephrotoxicity, compared with control siRNA groups. Compared with the cells treated with miR‐34a mimics, the viability of NRK‐52E and HK‐2 cells was still significantly increased by dioscin (200 ng·mL−1). The protein levels of Sirt1 and Nrf2 were markedly increased and the protein level of Keap1 was markedly decreased in dioscin‐treated groups after transfection (Figure 7C). In addition, dioscin also notably decreased the ratio of acetylated NF‐κB and normal NF‐κB in NRK‐52E and HK‐2 cells, indicating that dioscin inhibited CDDP‐induced NF‐κB acetylation. As the same, dioscin significantly decreased the mRNA levels of IL‐1β, IL‐6 and TNF‐α (Figure 7D). These results showed that the effect of dioscin on the Sirt1 signalling pathway might be mediated by decreasing miR‐34a. Based on the immunofluorescence staining assay shown in Figure 7E, the expression levels of Sirt1 in NRK‐52E and HK‐2 cells were markedly increased by dioscin with or without miR‐34a mimics. These results suggested that dioscin reversed the down‐regulating effect of miR‐34a mimics on Sirt1 signal pathway.
Figure 7.
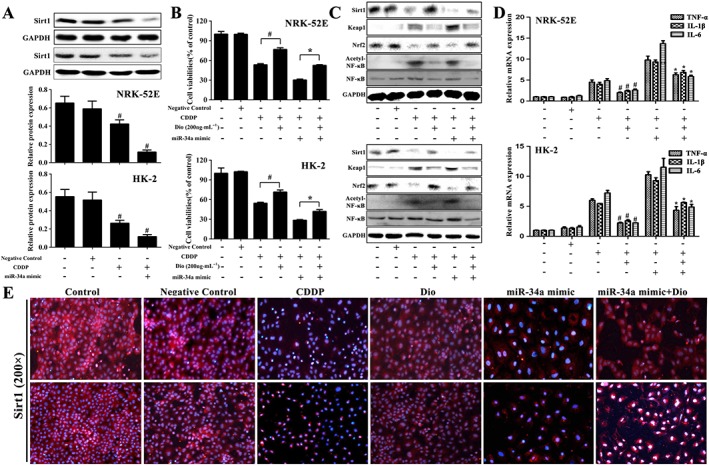
Dioscin reverses the effects of miR‐34a mimics. (A) Effects of miR‐34a on the protein levels of Sirt1 in CDDP‐treated NRK‐52E and HK‐2 cells. (B) Effects of dioscin on the viability of NRK‐52E and HK‐2 cells with or without transfection of miR‐34a mimic in vitro. (C) Effects of dioscin on the protein levels of Sirt1, Keap1, Nrf2 and acetylated NF‐κB after the transfection of miR‐34a mimic in NRK‐52E and HK‐2 cells. (D) Effects of dioscin on the mRNA levels of IL‐1β, IL‐6 and TNF‐α after transfection of miR‐34a mimic in NRK‐52E and HK‐2 cells. (E) Effects of dioscin on the expression levels of Sirt1 in NRK‐52E and HK‐2 cells with or without transfection of miR‐34a mimic, based on immunofluorescence staining in vitro (200× magnification). Data are presented as the mean ± SD (n = 5). # P < 0.05, significantly different from CDDP groups; *P < 0.05, significantly different from CDDP group transfected with miR‐34a mimic.
Dioscin reverses the effects of Sirt1 siRNA
To investigate the mechanisms of attenuating oxidation and inflammation caused by dioscin, we hypothesized that the anti‐inflammatory capability of the compound might primarily result from up‐regulating Sirt1 signal pathway. For the hypothesis to be tested, the NRK‐52E and HK‐2 cells were transfected with Sirt1 siRNA and pretreated with dioscin (200 ng·mL−1) for 12 h before CDDP test. As shown in Figure 8A, transfecting Sirt1 siRNA exacerbated CDDP‐induced injury compared with control siRNA groups. Compared with Sirt1 siRNA groups, the viability of NRK‐52E and HK‐2 cells was still significantly attenuated by dioscin (200 ng·mL−1) after transfection with Sirt1 siRNA. As shown in Figure 8B, the protein levels of Sirt1 and Nrf‐2 were markedly increased and Keap1 levels were markedly decreased in dioscin‐treated groups after transfection. In addition, dioscin significantly down‐regulated the ratios of acetylated NF‐κB and normal NF‐κB in NRK‐52E and HK‐2 cells, suggesting that dioscin had the ability to inhibit CDDP‐induced NF‐κB acetylation. Dioscin also significantly decreased the mRNA levels of IL‐1β, IL‐6 and TNF‐α (Figure 8C). Based on the immunofluorescence staining shown in Figure 8D, the expression levels of Sirt1 in NRK‐52E and HK‐2 cells were markedly increased by dioscin with or without Sirt1 siRNA. These results further confirmed that dioscin up‐regulated Sirt1 expression.
Figure 8.
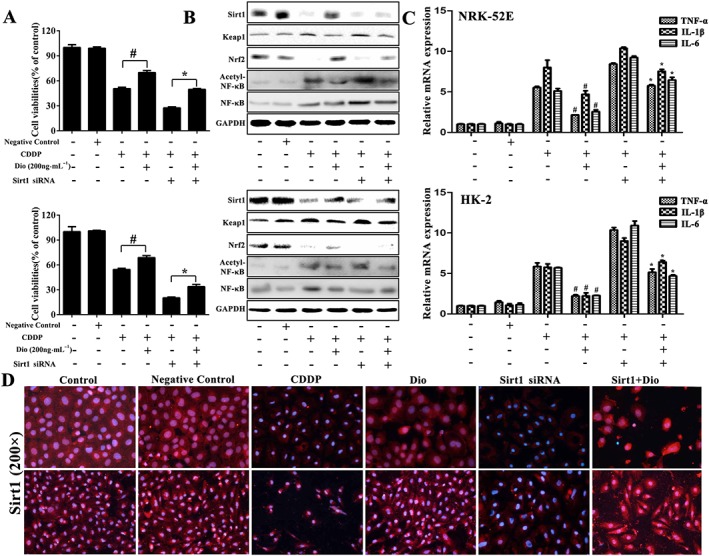
Dioscin reverses the effects of Sirt1 siRNA. (A) Effects of dioscin on the viability of NRK‐52E and HK‐2 cells with or without transfection of Sirt1 siRNA in vitro. (B) Effects of dioscin on the protein levels of Sirt1, Keap1, Nrf2 and acetylated NF‐κB after the transfection of Sirt1 siRNA in NRK‐52E and HK‐2 cells. (C) Effects of dioscin on the mRNA levels of IL‐1β, IL‐6 and TNF‐α after transfection of Sirt1 siRNA in NRK‐52E and HK‐2 cells. (D) Effects of dioscin on the expression of Sirt1 in NRK‐52E and HK‐2 cells with or without transfection of Sirt1 siRNA based on immunofluorescence staining in vitro (200× magnification). Data are presented as the mean ± SD (n = 5). # P < 0.05, significantly different from CDDP groups; *P < 0.05, significantly different from CDDP group transfected with Sirt1 siRNA.
Prediction of drug targets of dioscin against CDDP‐induced nephrotoxicity
As shown in Figure 9A, B and Supporting Information Figure S4, the 3D structure of dioscin was employed and the 3D diagram of Sirt1 was given in Supporting Information Figure S5A. The agonist structure and the binding pocket of Sirt1 were confirmed from PDB (ID: 5BTR), which was utilized to evaluate the binding affinity of dioscin towards Sirt1 compared with three molecules of resveratrol. As shown in Figure 9C, D and Supporting Information Figure S5B, the inhibitor structure of Keap1 and the binding modes of Kelch's structural domain in PPDB (ID: 5FNU and 5CGJ) were employed, which was used to evaluate the binding affinity of dioscin towards Keap1, compared with RA839. As shown in Figure 9E, F and Supporting Information Figure S5C, the binding modes with DNA of NF‐κBp65 homologous dimers and NF‐κBp65/NF‐κB50 heterogeneous dimers in PDB (ID: 2RAM and 1VKX) were employed. The binding pocket of NF‐κB p65 was predicted and the molecular docking sites were confirmed. The information of molecular docking sites is listed in Supporting Information Table S4. We considered Site1 as the best binding site to evaluate the binding affinity of dioscin towards NF‐κBp65. By molecular docking, the binding energy of dioscin towards Sirt1 was −10.99 kcal·mol−1, which was lower than the binding energy produced by three molecules of resveratrol (−9.33 kcal·mol−1) (Supporting Information Table S5). The binding energy of dioscin towards Keap1 was −9.73 kcal·mol−1, which was −9.70 kcal·mol−1 for RA839 (Supporting Information Table S6), and the binding energy of dioscin towards NF‐κBp65 Site4 was −7.0 kcal·mol−1, which was −7.8 kcal·mol−1 for Site1 (Supporting Information Table S7). Moreover, hydrogen bonding, hydrophobic effect and electrostatic interaction analysis were carried out to clarify the dynamics behind the strong binding affinity. From the docking results, due to the strong hydrogen bonding and/or hydrophobic effects, dioscin with lower binding energy showed powerful affinity towards to Sirt1, Keap1 and NF‐κBp65, suggesting that the compound may directly bind to these proteins to exert its biological activities.
Figure 9.
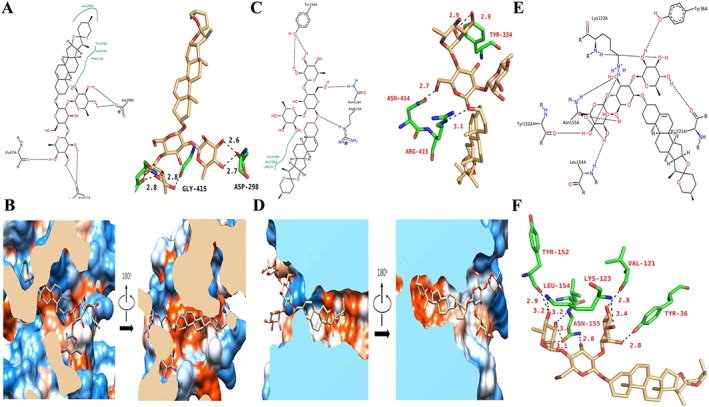
Targets of dioscin against CDDP‐induced nephrotoxicity. (A) The schematic and the three‐dimensional diagram of the hydrogen bond interaction between dioscin and Sirt1. (B) Hydrophobic effect between dioscin and Sirt1. The strong hydrophobic regions are orange, and the strong hydrophilic regions are blue. The stick model of dioscin is coloured in grey. (C) The schematic diagram and the three‐dimensional diagram of the hydrogen bond interaction between dioscin and Keap1. The three‐dimensional diagram of the hydrogen bond interaction between dioscin and Keap1. (D) Hydrophobic interactions between dioscin and Keap1. The strong hydrophobic regions are orange, and the strong hydrophilic regions are blue. The stick model of dioscin is coloured in light yellow. (E, F) The schematic diagram and the three‐dimensional diagram of the hydrogen bond interaction between dioscin and NF‐κBp65.
Discussion
CDDP‐induced nephrotoxicity results in decreased renal function including severe reductions in glomerular filtration and Cr clearance (Brahmi et al., 2012). About 25–35% of patients suffer from a significant decline in renal function after a single dose of CDDP (Sahin et al., 2010b). Several studies have demonstrated the protective effect of antioxidant agents against CDDP‐induced nephrotoxicity, through effects on inflammation and oxidative stress (Saad et al., 2007).
Dioscin, a natural product, has already been shown to exert nephroprotective effects against renal ischaemia/reperfusion injury and lipopolysaccharide‐induced inflammatory kidney injury (Qi et al., 2015; Qi et al., 2016). In the present work, dioscin protected against CDDP‐induced injury to NRK‐52E and HK‐2 cells in vitro and, in vivo, against CDDP‐induced renal damage in rats and mice, as shown by decreasing serum levels of BUN and Cr and alleviating histopathological changes. These data showed that dioscin could inhibit CDDP‐induced nephrotoxicity.
Recently, miRNAs have emerged as regulators of many biological processes through mRNA degradation or translational suppression (Martello et al., 2010). One member of the miR‐34 family, miR‐34a, directly targeted the histone deacetylase Sirt1 (Yamakuchi et al., 2008). In the present study, the levels of miR‐34a were markedly decreased by dioscin in vitro and in vivo, accompanied by increased levels of Sirt1. Then, we investigated the relationship between dioscin‐induced down‐regulation of miR‐34a and Sirt1 activation. Dual luciferase assays revealed that dioscin significantly decreased miR‐34a expression and subsequently increased Sirt1 expression in wild‐type plasmids. However, no change was observed in the cells transfected with the mutant plasmid. The results suggested that miR‐34a was markedly decreased by dioscin, which directly targeted Sirt1 to regulate its expression. Furthermore, dioscin reversed the lowering of Sirt1 levels, induced by miR‐34a overexpression. Taken together, these data suggested that Sirt1 is a target of miR‐34a, and the effect of dioscin on inhibition of miR‐34a against CDDP‐induced nephrotoxicity was related to the activation of the Sirt1 signalling pathway.
The processes of oxidative stress and inflammation have been found to be relevant to CDDP‐induced nephrotoxicity (Khan et al., 2012; Basu et al., 2016). Thus, new ways of suppressing oxidative stress should be potential methods of treating CDDP‐induced nephrotoxicity. The level of MDA, an end‐product of lipid hydroperoxide, has been frequently used as an indicator of oxidative stress. SOD is an enzyme that catalytically reduces O2 − to hydrogen peroxide and GSH can catalyse the reduction of hydrogen peroxide as well as other peroxides (Cao et al., 2015). Thus, high levels of SOD and GSH can protect against hepatic ischaemia/reperfusion injury. In the present work, CDDP promoted oxidative stress as shown by the high levels of GSH, GSH‐Px and SOD and low level of MDA in rats and mice, which were reversed by dioscin . In addition, dioscin markedly reduced ROS levels in NRK‐52E and HK‐2 cells caused by CDDP. Thus, suppression of oxidative stress may be one mechanism by which dioscin protects against CDDP‐induced nephrotoxicity.
Many studies have shown that Sirt1 exerts potent antioxidant effects (Feng et al., 2016) through enhancing the transcriptional activity of Nrf2. This transcription factor inhibits cellular oxidative stress by regulating intracellular redox homeostasis, and can also activate phase II antioxidants including HO‐1 (Chi et al., 2015). Nrf2 translocates from the cytosol to the nucleus and then binds to the ARE. Thus, up‐regulation of antioxidant enzymes can decrease the sensitivity of cells to oxidative stress‐induced damage (Oliveira et al., 2008). In this study, dioscin significantly increased the expression levels of Sirt1, total Nrf2, nuclear Nrf2, HO‐1, GCLC and GCLM in vitro and in vivo. Taken together, these results suggested that the nephroprotective effect of dioscin might be attributable to its antioxidant properties through increasing the levels of Sirt1 and activating the Nrf2/HO‐1 pathway.
Activation of the Sirt1/NF‐κB pathway releases pro‐inflammatory cytokines including TNF‐α, IL‐1β and IL‐6 in CDDP‐induced nephrotoxicity (Yeung et al., 2004). Meanwhile, these pro‐inflammatory cytokines can further stimulate the activation of NF‐κB signalling (Wang et al., 2015). In the present work, dioscin significantly down‐regulated the expression levels of HMGB1, COX‐2, AP‐1, TNF‐α, IL‐1β and IL‐6, dramatically up‐regulated the expression levels of IκB‐α and decreased the ratios of acetylated NF‐κB and normal NF‐κB. Thus, suppression of inflammation via the Sirt1/NF‐κB signalling pathway may be another relevant mechanism underlying of the actions of dioscin against CDDP‐induced nephrotoxicity.
In addition, from the molecular docking assay results, due to the strong hydrogen bonding and/or hydrophobic interactions, dioscin showed powerful affinities towards to Sirt1, Keap1 and NF‐κBp65, indicating that the compound may directly bind to these proteins, to exert its biological activities.
In summary, we have investigated a simplified pathway to describe the possible involvement of miR‐34a/Sirt1 signalling in the actions of dioscin against CDDP‐induced renal injury. Dioscin significantly alleviated CDDP‐induced nephrotoxicity, and this natural product should be developed as a new potential candidate for clinical use. Of course, further analysis of the mechanisms involved and assessments of the clinical applications of this natural product against CDDP‐induced renal injury are required.
Author contributions
Y.M.Z., X.F.T. and J.Y.P. designed the experiments and wrote the manuscript. Y.M.Z., X.F.T., L.H.Y. and L.N.X. performed the animal and cell experiments. Y.M.Z., Y.W.X. and Y.Q. performed the quantitative real‐time PCR and Western blotting assays. X.H. and S.S.S. performed the gene transfection experiments. Y.Y.Z. and Y.L. performed the molecular docking assay. K.X.L. and J.Y.P. edited the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 The information of the sequences used in the present work.
Table S2 The primer sequences used for real‐time PCR assay in the present work.
Table S3 The information of the antibodies used in the present work.
Table S4 The binding energy of dioscin towards to Sirt1 by molecular docking assay.
Table S5 The binding energy of dioscin towards to Keap1 by molecular docking assay.
Table S6 The information of binding sites.
Table S7 The binding energy of dioscin towards to the binding sites of NF‐κB p65 by molecular docking assay.
Figure S1 The chemical structure of dioscin.
Figure S2 Effects of dioscin on the levels of ROS based on flow cytometry assay (n = 5).
Figure S3 H&E staining (400× original magnification) of the kidney tissue in rats and mice.
Figure S4 The 3D structure of dioscin.
Figure S5 Drug‐target prediction of dioscin against CDDP‐induced nephrotoxicity. (A) The three‐dimensional diagram of Sirt1. (B) The inhibitor structure of Keap1 protein and binding modes of Kelch's structural domain. The binding pocket of Keap1 protein. The comparison of binding modes of dioscin and RA839 in the active pocket of Keap1. The white cartoon model is Keap1 (the active pocket is green and dioscin is yellow), and RA839 is shown in magenta stick model. (C) The binding modes with DNA of NF‐κB p65 homologous dimmers and NF‐κB p65/NF‐κB50 heterogeneous dimers. The prediction of binding pocket and molecular docking site of NF‐κB p65. The binding mode of dioscin in the active pocket of NF‐κB p65. The white cartoon model is NF‐κB p65 (the active pocket is green and dioscin is yellow), and the DNA molecule is shown in orange cartoon model.
Acknowledgements
This work was financially supported by National Natural Science Foundation of China (no. 81603178) and the Special Grant for Translational Medicine, Dalian Medical University (2015004).
Zhang, Y. , Tao, X. , Yin, L. , Xu, L. , Xu, Y. , Qi, Y. , Han, X. , Song, S. , Zhao, Y. , Lin, Y. , Liu, K. , and Peng, J. (2017) Protective effects of dioscin against cisplatin‐induced nephrotoxicity via the microRNA‐34a/sirtuin 1 signalling pathway. British Journal of Pharmacology, 174: 2512–2527. doi: 10.1111/bph.13862.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Overview. Br J Pharmacol 172: 5734–5143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arany I, Safirstein RL (2003). Cisplatin nephrotoxicity. Semin Nephrol 23: 460–464. [DOI] [PubMed] [Google Scholar]
- Basu A, Singha Roy S, Bhattacharjee A, Bhuniya A, Baral R, Biswas J et al. (2016). Vanadium(III)‐l‐cysteine protects cisplatin‐induced nephropathy through activation of Nrf2/HO‐1 pathway. Free Radic Res 50: 39–55. [DOI] [PubMed] [Google Scholar]
- Bataille AM, Manautou JE (2012). Nrf2: a potential target for new therapeutics in liver disease. Clin Pharmacol Ther 92: 340–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brahmi D, Ayed Y, Hfaiedh M, Bouaziz C, Mansour HB, Zourgui L et al. (2012). Protective effect of cactus cladode extract against cisplatin induced oxidative stress, genotoxicity and apoptosis in balb/c mice: combination with phytochemical composition. BMC Complement Altern Med 12: 111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YW, Jiang Y, Zhang DY, Wang M, Chen WS, Su H et al. (2015). Protective effects of Penthorum chinense Pursh against chronic ethanol‐induced liver injury in mice. J Ethnopharmacol 161: 92–98. [DOI] [PubMed] [Google Scholar]
- Chi X, Zhang R, Shen N, Jin Y, Alina A, Yang S et al. (2015). Sulforaphane reduces apoptosis and oncosis along with protecting liver injury‐induced ischemic reperfusion by activating the Nrf2/ARE pathway. Hepatol Int 9: 321–329. [DOI] [PubMed] [Google Scholar]
- Chirino YI, Sanchez‐Gonzalez DJ, Martinez‐Martinez CM, Cruz C, Pedraza‐Chaverri J (2008). Protective effects of apocynin against cisplatin‐induced oxidative stress and nephrotoxicity. Toxicology 245: 18–23. [DOI] [PubMed] [Google Scholar]
- Cho J, Choi H, Lee J, Kim MS, Sohn HY, Lee DG (2013). The antifungal activity and membrane‐disruptive action of dioscin extracted from Dioscorea nipponica . Biochim Biophys Acta 1828: 1153–1158. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanda S, Kaur S, Sandhir R (2013). Preventive effect of N‐acetyl‐l‐cysteine on oxidative stress and cognitive impairment in hepatic encephalopathy following bile duct ligation. Free Radic Biol Med 56: 204–215. [DOI] [PubMed] [Google Scholar]
- Do MT, Kim HG, Choi JH, Jeong HG (2014). Metformin induces microRNA‐34a to downregulate the Sirt1/Pgc‐1alpha/Nrf2 pathway, leading to increased susceptibility of wild‐type p53 cancer cells to oxidative stress and therapeutic agents. Free Radic Biol Med 74: 21–34. [DOI] [PubMed] [Google Scholar]
- Feng J, Li SS, Chen HW (2016). Tanshinone IIA inhibits myocardial remodeling induced by pressure overload via suppressing oxidative stress and inflammation: possible role of silent information regulator 1. Eur J Pharmacol 791: 632–639. [DOI] [PubMed] [Google Scholar]
- Guennewig B, Roos M, Dogar AM, Gebert LF, Zagalak JA, Vongrad V et al. (2014). Synthetic pre‐microRNAs reveal dual‐strand activity of miR‐34a on TNF‐ alpha. RNA 20: 61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagar H, Medany AE, Salam R, Medany GE, Nayal OA (2015). Betaine supplementation mitigates cisplatin‐induced nephrotoxicity by abrogation of oxidative/nitrosative stress and suppression of inflammation and apoptosis in rats. Exp Toxicol Pathol 67: 133–141. [DOI] [PubMed] [Google Scholar]
- Harvey CJ, Thimmulappa RK, Singh A, Blake DJ, Ling G, Wakabayashi N et al. (2009). Nrf2‐regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic Biol Med 46: 443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh MJ, Tsai TL, Hsieh YS, Wang CJ, Chiou HL (2013). Dioscin‐induced autophagy mitigates cell apoptosis through modulation of PI3K/Akt and ERK and JNK signaling pathways in human lung cancer cell lines. Arch Toxicol 87: 1927–1937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karasawa T, Steyger PS (2015). An integrated view of cisplatin‐induced nephrotoxicity and ototoxicity. Toxicol Lett 237: 219–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke B, Shen XD, Zhang Y, Ji H, Gao F, Yue S et al. (2013). KEAP1–NRF2 complex in ischemia‐induced hepatocellular damage of mouse liver transplants. J Hepatol 59: 1200–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan R, Khan AQ, Qamar W, Lateef A, Ali F, Rehman MU et al. (2012). Chrysin abrogates cisplatin‐induced oxidative stress, p53 expression, goblet cell disintegration and apoptotic responses in the jejunum of Wistar rats. Br J Nutr 108: 1574–1585. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Huang W, Wen YQ, Gong GH, Zhao QB, Yu G (2010). Anti‐thrombotic activity and chemical characterization of steroidal saponins from Dioscorea zingibere ‐nsis C.H. Wright. Fitoterapia 81: 1147–1156. [DOI] [PubMed] [Google Scholar]
- Lu BN, Xu YW, Xu LN, Cong XN, Yin LH, Li H et al. (2012). Mechanism investigation of dioscin against CCl4‐induced acute liver damage in mice. Environ Toxicol Pharmacol 34: 127–135. [DOI] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A et al. (2001). Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107: 137–148. [DOI] [PubMed] [Google Scholar]
- Ma X, Dang C, Kang H, Dai Z, Lin S, Guan H et al. (2015). Saikosaponin‐D reduces cisplatin‐induced nephrotoxicity by repressing ROS‐mediated activation of MAPK and NF‐kappaB signalling pathways. Int Immunopharmacol 28: 399–408. [DOI] [PubMed] [Google Scholar]
- Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S et al. (2010). A MicroRNA targeting dicer for metastasis control. Cell 141: 1195–1207. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W et al. (2004). Mammalian SIRT1 represses forkhead transcription factors. Cell 116: 551–563. [DOI] [PubMed] [Google Scholar]
- Oliveira CS, Rigon AP, Leal RB, Rossi FM (2008). The activation of ERK1/2 and p38 mitogen‐activated protein kinases is dynamically regulated in the developing rat visual system. Int J Dev Neurosci 26: 355–362. [DOI] [PubMed] [Google Scholar]
- Pérez‐Rojas JM, Cruz C, García‐López P, Sánchez‐González DJ, Martínez‐Martínez CM, Ceballos G et al. (2009). Renoprotection by alpha‐Mangostin is related to the attenuation in renal oxidative/nitrosative stress induced by cisplatin nephrotoxicity. Free Radic Res 43: 1122–1132. [DOI] [PubMed] [Google Scholar]
- Qi M, Yin L, Xu L, Tao X, Qi Y, Han X et al. (2016). Dioscin alleviates lipopolysaccharide‐induced inflammatory kidney injury via the microRNA let‐7i/TLR4/MyD88 signaling pathway. Pharmacol Res 111: 509–522. [DOI] [PubMed] [Google Scholar]
- Qi M, Zheng L, Qi Y, Han X, Xu Y, Xu L et al. (2015). Dioscin attenuates renal ischemia/reperfusion injury by inhibiting the TLR4/MyD88 signaling pathway via up‐regulation of HSP70. Pharmacol Res 100: 341–352. [DOI] [PubMed] [Google Scholar]
- Reddy KP, Madhu P, Reddy PS (2016). Protective effects of resveratrol against cisplatin‐induced testicular and epididymal toxicity in rats. Food Chem Toxicol 91: 65–72. [DOI] [PubMed] [Google Scholar]
- Saad SY, Arafah MM, Najjar TA (2007). Effects of mycophenolate mofetil on cisplatin‐induced renal dysfunction in rats. Cancer Chemother Pharmacol 59: 455–460. [DOI] [PubMed] [Google Scholar]
- Sahin K, Tuzcu M, Gencoglu H, Dogukan A, Timurkan M, Sahin N et al. (2010a). Epigallocatechin‐3‐gallate activates Nrf2/HO‐1 signaling pathway in cisplatin‐induced nephrotoxicity in rats. Life Sci 87: 240–245. [DOI] [PubMed] [Google Scholar]
- Sahin K, Tuzcu M, Sahin N, Ali S, Kucuk O (2010b). Nrf2/HO‐1 signaling pathway may be the prime target for chemoprevention of cisplatin‐induced nephrotoxicity by lycopene. Food Chem Toxicol 48: 2670–2674. [DOI] [PubMed] [Google Scholar]
- Shah NM, Rushworth SA, Murray MY, Bowles KM, MacEwan DJ (2013). Understanding the role of NRF2‐regulated miRNAs in human malignancies. Oncotarget 4: 1130–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si L, Xu L, Yin L, Qi Y, Han X, Xu Y et al. (2017). Potent effects of dioscin against pancreatic cancer via miR‐149‐3P‐mediated inhibition of the Akt1 signalling pathway. Br J Pharmacol 174: 553–1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun B, Gao L, Ahsan A, Chu P, Song Y, Li H et al. (2016). Anticancer effect of SZC015 on lung cancer cells through ROS‐dependent apoptosis and autophagy induction mechanisms in vitro . Int Immunopharmacol 40: 400–409. [DOI] [PubMed] [Google Scholar]
- Tao X, Sun X, Yin L, Han X, Xu L, Qi Y et al. (2015). Dioscin ameliorates cerebral ischemia/reperfusion injury through the downregulation of TLR4 signaling via HMGB‐1 inhibition. Free Radic Biol Med 84: 103–115. [DOI] [PubMed] [Google Scholar]
- Tao X, Wan X, Xu Y, Xu L, Qi Y, Yin L et al. (2014). Dioscin attenuates hepatic ischemia–reperfusion injury in rats through inhibition of oxidative–nitrative stress, inflammation and apoptosis. Transplantation 98: 604–611. [DOI] [PubMed] [Google Scholar]
- Tilyek A, Chai C, Hou X, Zhou B, Zhang C, Cao Z et al. (2016). The protective effects of Ribes diacanthum Pall on cisplatin‐induced nephrotoxicity in mice. J Ethnopharmacol 178: 297–306. [DOI] [PubMed] [Google Scholar]
- Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK et al. (2001). hSIR2 (SIRT1) functions as an NAD‐dependent p53 deacetylase. Cell 107: 149–159. [DOI] [PubMed] [Google Scholar]
- Wang W, Zhou PH, Xu CG, Zhou XJ, Hu W, Zhang J (2015). Baicalein attenuates renal fibrosis by inhibiting inflammation via down‐regulating NF‐kappaB and MAPK signal pathways. J Mol Histol 46: 283–290. [DOI] [PubMed] [Google Scholar]
- Xu LN, Wei YL, Dong DS, Yin LH, Qi Y, Han X et al. (2014). iTRAQ‐based proteomics for studying the effects of dioscin against nonalcoholic fatty liver disease in rats. RSC Adv 4: 30704–30711. [Google Scholar]
- Yamakuchi M, Ferlito M, Lowenstein CJ (2008). miR‐34a repression of SIRT1 regulates apoptosis. Proc Natl Acad Sci U S A 105: 13421–13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA et al. (2004). Modulation of NF‐kappaB‐dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23: 2369–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin LH, Xu YS, Xu LN, Qi Y, Han X, Peng JY (2010b). A green and efficient protocol for industrial‐scale preparation of dioscin from Dioscorea nipponica Makino by two‐step macroporous resin column chromatograph. Chem Eng J 165: 28–289. [Google Scholar]
- Zhao F, Wu T, Lau A, Jiang T, Huang Z, Wang XJ et al. (2009). Nrf2 promotes neuronal cell differentiation. Free Radic Biol Med 47: 867–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao X, Cong X, Zheng L, Xu L, Yin L, Peng J (2012). Dioscin, a natural steroid saponin, shows remarkable protective effect against acetaminophen‐induced liver damage in vitro and in vivo. Toxicol Lett 214: 69–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 The information of the sequences used in the present work.
Table S2 The primer sequences used for real‐time PCR assay in the present work.
Table S3 The information of the antibodies used in the present work.
Table S4 The binding energy of dioscin towards to Sirt1 by molecular docking assay.
Table S5 The binding energy of dioscin towards to Keap1 by molecular docking assay.
Table S6 The information of binding sites.
Table S7 The binding energy of dioscin towards to the binding sites of NF‐κB p65 by molecular docking assay.
Figure S1 The chemical structure of dioscin.
Figure S2 Effects of dioscin on the levels of ROS based on flow cytometry assay (n = 5).
Figure S3 H&E staining (400× original magnification) of the kidney tissue in rats and mice.
Figure S4 The 3D structure of dioscin.
Figure S5 Drug‐target prediction of dioscin against CDDP‐induced nephrotoxicity. (A) The three‐dimensional diagram of Sirt1. (B) The inhibitor structure of Keap1 protein and binding modes of Kelch's structural domain. The binding pocket of Keap1 protein. The comparison of binding modes of dioscin and RA839 in the active pocket of Keap1. The white cartoon model is Keap1 (the active pocket is green and dioscin is yellow), and RA839 is shown in magenta stick model. (C) The binding modes with DNA of NF‐κB p65 homologous dimmers and NF‐κB p65/NF‐κB50 heterogeneous dimers. The prediction of binding pocket and molecular docking site of NF‐κB p65. The binding mode of dioscin in the active pocket of NF‐κB p65. The white cartoon model is NF‐κB p65 (the active pocket is green and dioscin is yellow), and the DNA molecule is shown in orange cartoon model.