

Abstract
Background and Purpose
CB1 receptor signalling is canonically mediated through inhibitory Gαi proteins, but occurs through other G proteins under some circumstances, Gαs being the most characterized secondary pathway. Determinants of this signalling switch identified to date include Gαi blockade, CB1/D2 receptor co‐stimulation, CB1 agonist class and cell background. Hence, we examined the effects of receptor number and different ligands on CB1 receptor signalling.
Experimental Approach
CB1 receptors were expressed in HEK cells at different levels, and signalling characterized for cAMP by real‐time BRET biosensor –CAMYEL – and for phospho‐ERK by AlphaScreen. Homogenate and whole cell radioligand binding assays were performed to characterize AM6544, a novel irreversible CB1 receptor antagonist.
Key Results
In HEK cells expressing high levels of CB1 receptors, agonist treatment stimulated cAMP, a response not known to be mediated by receptor number. Δ9‐THC and BAY59‐3074 increased cAMP only in high‐expressing cells pretreated with pertussis toxin, and agonists demonstrated more diverse signalling profiles in the stimulatory pathway than the canonical inhibitory pathway. Pharmacological CB1 receptor knockdown and Gαi1 supplementation restored canonical Gαi signalling to high‐expressing cells. Constitutive signalling in both low‐ and high‐expressing cells was Gαi‐mediated.
Conclusion and Implications
CB1 receptor coupling to opposing G proteins is determined by both receptor and G protein expression levels, which underpins a mechanism for non‐canonical signalling in a fashion consistent with Gαs signalling. CB1 receptors mediate opposite consequences in endpoints such as tumour viability depending on expression levels; our results may help to explain such effects at the level of G protein coupling.
Abbreviations
- 2‐AG
2‐arachidonoyl glycerol
- AC
adenylate cyclase
- AEA
anandamide/N‐arachidonoylethanolamine
- AM6544
1‐(2,4‐dichlorophenyl)‐5‐(4‐(4‐isothiocyanatobut‐1‐yn‐1‐yl)phenyl)‐4‐methyl‐N‐(piperidin‐1‐yl)‐1H‐pyrazole‐3‐carboxamide
- BAY
BAY59,3074
- CAMYEL
cAMP sensor YFP‐Epac‐Rluc
- HA
haemagglutinin
- PEI
polyethylenimine
- PTX
pertussis toxin
- THC
Δ9‐tetrahydrocannabinol
Introduction
Despite longstanding consensus as to its canonical Gαi‐mediated signalling pathway, the type one cannabinoid receptor (CB1) is among several GPCRs now understood to transduce signals through more than one G protein effector – a capacity sometimes referred to as ‘promiscuous G protein coupling’ (reviewed in Maudsley et al., 2005). Environmental and pharmacological conditions that have been found to affect CB1 receptor signalling in this way include blockade of the canonical Gαi pathway with pertussis toxin (PTX) (Glass and Felder, 1997; Bonhaus et al., 1998; Scotter et al., 2010), the class of CB1 agonist (Bonhaus et al., 1998; Lauckner et al., 2005), receptor oligomers (Kearn et al., 2005; Bagher et al., 2016) and cell background (McIntosh et al., 2007).
The CB1 receptor was first seen to demonstrate Gαs‐like signalling behaviour under conditions of co‐stimulation with the type two dopamine receptor (D2) in primary rat striatal neurons: on stimulation with quinpirole (a specific D2 receptor agonist) or HU210 (a CB1 receptor agonist) alone, these neurons demonstrated the expected inhibition of forskolin‐induced cAMP, but when HU210 was applied in addition to quinpirole, this inhibitory signal was reversed in a concentration‐dependent manner (Glass and Felder, 1997). In both primary cells and transfected cell lines stably expressing CB1 receptors, PTX pretreatment has also been found to unmask this Gαs‐like signalling phenotype, in the absence of D2 receptor co‐expression/co‐stimulation (Glass and Felder, 1997; Bonhaus et al., 1998; Scotter et al., 2010). These observations gave rise to the hypothesis that Gαs‐like CB1 receptor signalling arose in circumstances when the availability of Gαi was limited (‘Gαi exhaustion’), either because Gαi was directly inactivated by PTX or because it was sequestered by other Gαi‐coupled receptors (of which the D2 receptor is an example). However, Jarrahian et al. (2004) have reported that the mere co‐expression of CB1 and D2 receptors is sufficient for a CB1 receptor‐mediated Gαs‐like cAMP phenotype to be unmasked, a finding which appears to require either G protein pre‐coupling (Ferre, 2015) or high levels of D2 receptor‐mediated constitutive signalling for the Gαi exhaustion hypothesis to hold. Subsequently, co‐immunoprecipitation data led Kearn et al. (2005) to propose that the CB1 receptor signalling switch is derived pleiotropically, from altered protein conformations caused by a physical interaction between D2 and CB1 receptors – a receptor heterodimer. Characterization of the CB1/D2 receptor heterodimer has continued using other approaches (Marcellino et al., 2008; Przybyla and Watts, 2010; Bagher et al., 2016). It remains noteworthy that the two hypotheses of the signalling switch mechanism need not be mutually exclusive, and both promiscuous (low‐efficacy activation of a non‐preferred G protein species due to lack of availability of the preferred species; i.e. Gαi exhaustion) and pleiotropic (stoichiometric/conformational) mechanisms have precedent in the literature (Zhu et al., 1994; Laugwitz et al., 1996; Cordeaux et al., 2000; Jin et al., 2001).
The exact identity of the G protein subtype that mediates the novel effects was not originally shown directly (Glass and Felder, 1997), although the stimulatory cAMP effect and absence of PTX sensitivity suggested CB1 receptor coupling to Gαs. Abadji et al. (1999) demonstrated that the CB1 receptor contains a conserved two amino acid motif that mediates Gαs coupling of the β‐adrenoceptor, but Bash et al. (2003) provided the first direct evidence of some Gαs protein involvement by demonstrating that anti‐Gαs antibodies could knock down CB1 receptor‐mediated Ca2+ flux. Very recently, Eldeeb et al. (2016) showed specific, direct Gαs protein involvement in CB1 receptor signalling using a scintillation proximity variant of a [35S]‐GTPγS assay, in a study that underscores the relevance of the CB1 receptor‐mediated Gαs signalling pathway by demonstrating its presence under experimental conditions where agonism does not induce a nett stimulatory cAMP signal.
Recently, we have observed Gαs‐like signalling of CB1 receptor in assays where the receptor is expressed transiently. This expression method frequently results in very high transgene expression primarily due to cellular uptake of multiple copies of the transfected construct. In stably expressing cell lines, high levels of transgene expression can be produced by molecular engineering of an N‐terminal signal sequence tag such as the preprolactin signal sequence (pplss). This is a short (30 amino acid) peptide, which substantially increases the efficiency of nascent protein secretion by acting as a recognition sequence for the signal recognition particle (SRP) (Kurzchalia et al., 1986), but is cleaved during maturation and therefore does not alter protein function (Belin et al., 1996). Based on our observations, we hypothesized that CB1 receptor number is a novel determinant of the Gαi‐Gαs signalling switch and have therefore systematically characterized the relationship between receptor expression and CB1 receptor signalling. A panel of six chemically distinct CB1 agonists was compiled to allow delineation of ligand‐dependence of the two signalling pathways.
Methods
Molecular biology
The pEF4A (Thermo Fisher Scientific, Waltham, MA, USA) plasmid encoding human CB1 chimerized with three N‐terminal haemagglutinin (HA) tags (3HA‐hCB1) has been described previously (Cawston et al., 2013). The coding region for bovine pplss was amplified by PCR from a plasmid kindly gifted by Professor Ken Mackie (Indiana University, Bloomington, IN, USA) with primers designed to incorporate KpnI restriction sites at either end of the amplicon. The pplss sequence was then incorporated into the 3HA‐hCB1 pEF4A plasmid by non‐directional KpnI restriction cloning, and sequence verified.
Cell culture and transfection
Cell culture media and reagents were purchased from Thermo Fisher Scientific, and plasticware was purchased from Corning (Corning, NY, USA). HEK293 cell lines were cultured in DMEM supplemented with 10% FBS and an appropriate selection of antibiotics and were cultured in 5% CO2, at 37°C in a humidified incubator.
The HEK cell line stably expressing 3HA‐hCB1 receptors has been described previously (Cawston et al., 2013). This cell line was used as the background to generate a HEK cell line that co‐expresses CB1 and D2 receptors, and is first described in Hunter et al. (2016). The HEK cell line stably expressing the human 3HA‐hCB2 receptor has also been described previously (Grimsey et al., 2011). The pplss‐3HA‐hCB1 pEF4A construct (described above) was linearized and stably transfected into the same wild‐type HEK cell background using Lipofectamine 2000. A clonal population of cells expressing receptors at qualitatively high levels was then isolated and propagated for subsequent experiments. Wild‐type HEK Flp‐In™‐293 cells (Thermo Fisher Scientific) were also stably transfected with a modified pcDNA3L‐His‐cAMP sensor Venus‐Epac‐Rluc8 (V8‐CAMYEL) plasmid (Hunter et al., 2017) (as distinct from the original CAMYEL biosensor which was used in assays described below, using the same random incorporation method of stable transfection (i.e. the Flp Recombination Target site (FRT) was left unoccupied). This cell line was used for assays involving transient receptor expression and FACS.
Assays for cAMP where CB1 receptors were expressed stably
Cellular cAMP was measured using a commercially available kinetic BRET assay (CAMYEL), as previously described (Jiang et al., 2007; Cawston et al., 2013). In brief, sufficient cells were seeded in 10 cm dishes such that they would be approximately 60% confluent the next day. The day after seeding, culture medium was replaced and 5 μg of an expression construct encoding the CAMYEL biosensor (pcDNA3L‐His‐CAMYEL) was transfected using linear polyethylenimine (PEI, MW 25 kDa; Polysciences, Warrington, PA, USA), where the transfection ratio (DNA : PEI) was 1:6. Approximately 24 h after transfection, transfected cells were lifted from their culture dishes by trypsinization and plated in poly‐D‐lysine (Sigma Aldrich) coated, white 96‐well CulturPlates™ (Perkin Elmer, Waltham, MA, USA) at a density of 8 × 104 cells per well. For experiments involving PTX pretreatment to irreversibly inactivate Gαi proteins, cells were plated in half the usual volume of culture medium, and then 6 h later, 2× concentrated PTX (Sigma Aldrich) or PTX vehicle (50% glycerol, 50 mM Tris–HCl pH 7.5, 10 mM glycine, 500 mM NaCl) was prepared in culture medium and added on top of the plating medium to give a final concentration of 100 ng·mL−1 PTX. The cells were then cultured overnight (>16 h if treated with PTX) prior to beginning detection. Cells were washed once with HBSS and then equilibrated for 30 min in HBSS supplemented with 1 mg·mL−1 BSA (ICPBio, Auckland, NZ, USA). Coelenterazine H (NanoLight Technologies, Pinetop, AZ, USA) was added to a final concentration of 5 μM for 5 min prior to dispensing drugs (2.5 μM forskolin and agonist, etc.) and initiating luminescence reading at 460 nm (Rluc) and 535 nm (YFP) in either a VICTOR™ X Light luminometer (Perkin Elmer), or a LUMIstar® Omega luminometer (BMG Labtech, Ortenberg, Germany). Each series of stimulations was detected in real‐time for approximately 20 min. All drugs (including coelenterazine H) were prepared in HBSS +1 mg·mL−1 BSA, and all incubations and stimulations were performed at 37°C.
CAMYEL assays involving pharmacological CB1 receptor knockdown with AM6544 were performed as noted above, except that an AM6544 serial dilution series was prepared in serum‐free medium supplemented with 1 mg·mL−1 BSA and applied to cells for 1 h. The AM6544 dilutions were then removed and wells washed twice with HBSS, before the commencement of the 30 min equilibration in HBSS +1 mg·mL−1 BSA as described above.
For CAMYEL assays involving transient transfection of GNAI1 pcDNA3.1+ (or empty pcDNA3.1+) DNA, the cell lines of interest were seeded into poly‐D‐lysine coated, white CulturPlates™. Approximately 24 h later, an in‐well Lipofectamine 2000 protocol was used to transiently transfect either GNAI1 in pcDNA3.1+ (cDNA Resource Center, Bloomsburg, PA, USA) or empty pcDNA3.1+ (Thermo Fisher Scientific), followed by a second Lipofectamine 2000 in‐well transfection of CAMYEL biosensor DNA approximately 6–8 h later. Lipofectamine reagent was selected for these assays because it results in higher transfection efficiency than linear PEI. Cells were cultured thus for 48 h post‐transfection, prior to stimulation and detection in accordance with the protocol described above.
Cell staining and FACS for cAMP assays where receptor was expressed transiently
The aforementioned HEK Flp‐In™ cell line stably expressing the V8‐CAMYEL biosensor was plated in a 10 cm dish at a density such that the cells would be approximately 60% confluent the next day. The day after plating, transient transfection of the pplss‐3HA‐hCB1 pEF4A construct was performed using Lipofectamine 2000, in order to produce a population of V8‐CAMYEL biosensor‐expressing cells, which also expressed CB1 receptors over a wide range of expression levels. Following transfection, cells were cultured for 2 days, then detached with Versene and resuspended in culture medium. The cells were sedimented and washed in culture medium, then labelled with mouse anti‐HA clone 16B12 monoclonal antibody (1:500; Covance, Princeton, NJ, USA) and secondary Alexa Fluor® 647 goat anti‐mouse antibody (1:300; Thermo Fisher Scientific). Following the secondary antibody incubation, the cells were washed and resuspended to a concentration of approximately 2.5 × 106 mL−1 in HBSS supplemented with 1 mg·mL−1 BSA. FACS was performed using a FACSVantage Cell Sorter (Becton Dickinson, Franklin Lakes, NJ, USA). A ‘no primary antibody’ control was used to define the 647 background (low signal detection limit), and gating was used such that ‘above background’ 647 staining intensity was assessed during sorting. The transiently expressed population of cells was sorted into six sub‐populations on the basis of receptor expression (647 staining intensity). The sorted cells were then sedimented and resuspended in HBSS +1 mg·mL−1 BSA, and counted. A white 96‐well CulturPlate™ was prepared with diluted coelenterazine H (see CAMYEL assay details above) and drugs (forskolin and CP55,940) such that 1 × 104 cells could be subsequently dispensed into each well to give the desired drug concentrations, and detection in a VICTOR™ X Light luminometer was initiated immediately, as described above for other CAMYEL assays. The FACS and subsequent cAMP assay was repeated in three independent experiments.
Cell membrane preparation and quantification
HEK cells were harvested when semi‐confluent in 175cm2 flasks, with ice‐cold 5 mM EDTA in PBS. Cells were then sedimented, and cell pellets were snap frozen at −80°C. Pellets were thawed on ice and resuspended in sucrose buffer (200 mM sucrose, 50 mM Tris–HCl pH 7.4, 5 mM MgCl2, 2.5 mM EDTA), and then thoroughly homogenized by plunging (40 strokes) with a tight‐fitting pestle in a glass dounce homogenizer. The homogenate suspension was sedimented by centrifugation at 1000 × g for 10 min (‘P1 pellet’), and the supernatant subsequently sedimented by centrifugation at 26 916 × g for 30 min (‘P2 pellet’). Membrane pellets were resuspended in sucrose buffer, aliquoted and stored at −80°C. Multiple membrane preparations were produced for each cell line, in order that biological (independent) replicates of binding assays reflected cell line variability relevant to functional assays, rather than just experimental error.
Membrane protein concentrations were quantified by Amido Black 10B protein assay. In brief, 1 μL of membrane preparation was spiked into 1 M Tris–HCl pH 7.5 (contained 2% w.v‐1 SDS), and protein was precipitated by addition of concentrated trichloroacetic acid. The precipitated protein samples, alongside standards of 0–20 μg BSA, were then spotted onto 0.45 μm HA filters of a 96‐well MultiScreen‐HA Filter Plate (Merck Millipore, Billerica, MA, USA) and stained with 0.1% w.v‐1 amido black 10B dye (Sigma Aldrich) in a 45:10:4 solution of methanol : glacial acetic acid : milliQ water. The stain was washed off with milliQ water, then destained twice with a 90:2:8 solution of methanol : glacial acetic acid : milliQ water and washed again with milliQ. The stain was eluted with a solution of 25 mM NaOH, 0.05 mM EDTA and 50% v.v‐1 absolute ethanol, and absorbance measurements taken for BSA standards and unknowns at 630 nm on an EnSpire® plate reader (Perkin Elmer). To increase confidence in interpolated sample protein concentrations, the protein assay was performed at least twice on each membrane preparation.
Radioligand binding assays
Competition binding assays
Homogenate competition/displacement binding assays were performed on ‘P2’ HEK cell membrane preparations (see above). In brief, concentration series of non‐tritiated drugs, dilutions of [3H]‐CP55,940 and [3H]‐ SR141716A (rimonabant) (Perkin Elmer) and P2 membrane dilutions were prepared separately in binding buffer (50 mM HEPES pH 7.4, 1 mM MgCl2, 1 mM CaCl2, 2 mg·mL−1 BSA) and dispensed into 96‐well, polypropylene V‐shaped plates (Hangzhou Gene Era Biotech Co Ltd, Zhejiang, China), with the membrane dilution dispensed last. Final reaction volumes were 200 μL per well. Plates were sealed and incubated for 1 h at 30°C. During the incubation, the 1.2 μm pore fibreglass filters of a 96‐well Harvest Plate (Perkin Elmer) were blocked with a solution of 0.1% w.v‐1 branched PEI (Sigma Aldrich) in water. At the conclusion of the incubation, the PEI solution was washed through the filters using a filter plate vacuum manifold (Pall Corporation, Port Washington, NY, USA), and all filters were washed with 200 μL ice cold wash buffer (50 mM HEPES pH 7.4, 500 mM NaCl, 1 mg·mL−1 BSA). The 200 μL binding assay mixture was then transferred onto the filter plate (under vacuum); V‐well plate wells were immediately washed once with wash buffer, and then unbound 3H‐ligand was washed through the filter by washing each well three more times with wash buffer. Harvest plate filters were then dried overnight, and the underside of the plate was sealed. Irgasafe Plus scintillation fluid (Perkin Elmer) was then dispensed to each well, and after a 30 min delay, scintillation was read for 2 min per well in a Wallac MicroBeta® TriLux Liquid Scintillation Counter (Perkin Elmer), with counts corrected for detector efficiency.
Saturation binding assays to characterize AM6544
Saturation binding assays were performed in the Makriyannis laboratory to characterize the novel irreversible antagonist of CB1 receptors, AM6544 (Figure 4A) (Patent US8084451, 2011). Irreversible labelling assays were performed as described previously (Janero et al., 2015; Hua et al., 2016). HEK293F cell P2 membranes containing the hCB1 receptor were purified as described previously (Xu et al., 2005) and were resuspended at a concentration of 1 mg·mL−1 in TME buffer (25 mM Tris Base, 5 mM MgCl2, 1 mM EDTA, pH 7.45) containing 1 mg·mL−1 w.v‐1 BSA and incubated with 10× Ki (~100 nM) of AM6544 or SR141716A (or DMSO vehicle) at 30°C for 1 h with gentle agitation. The reaction was terminated by centrifugation at 27 000 × g for 10 min, followed by removal of excess unbound ligand by washing. Pellets were resuspended in TME buffer containing 1 mg·mL−1 BSA and incubated at 30°C for 30 min with gentle agitation. This centrifugation and resuspension was repeated. After a final centrifugation, membrane pellets were resuspended in TME containing 1 mg·mL−1 BSA, and 25 μg of protein were added to each assay well of a 96‐well plate. [3H]‐CP55,940 was diluted in TME/BSA to yield ligand concentrations ranging from 1.58 to 50 nM, and [3H]‐SR141716A was diluted in TME/BSA to yield ligand concentrations ranging from 3.77 to 119 nM. Nonspecific binding was assayed in the presence of 5 μM unlabelled CP55,940 or SR141716A. Membranes were incubated at 30°C for 1 h with gentle agitation. Samples were then transferred to Unifilter GF/B filter plates and filtered using a Packard Filtermate‐96 Cell Harvester (Perkin Elmer, Shelton, CT, USA). Filter plates were washed four times with ice‐cold wash buffer (50 mM Tris base, 5 mM MgCl2 containing 5 mg·mL−1 BSA, pH 7.4). Bound radioactivity was quantified in a Packard Top Count Scintillation Counter. Nonspecific binding was subtracted from the total bound radioactivity to calculate specific binding of [3H]‐CP55,940 and [3H]‐SR141716A.
Figure 4.
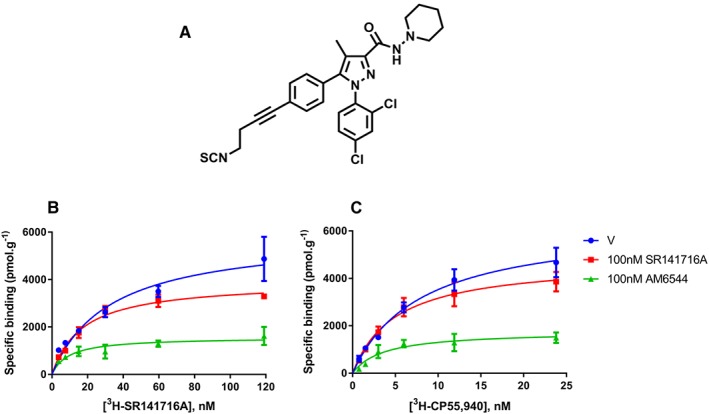
Chemical structure of AM6544 (A), and non‐specific binding‐corrected [3H]‐SR141716A (B) and [3H]‐CP55,940 (C) saturation binding curves on hCB1 HEK293F membranes pretreated for 1 h with vehicle (‘V’, DMSO), SR141716A (100 nM) or AM6544 (100 nM) and then washed three times before the incubation with the radioligand. Representative data are presented, demonstrating mean ± SEM of technical duplicates.
Whole cell binding assays
For whole cell binding assays, pplss‐3HA‐hCB1 HEK cells were plated in poly‐D‐lysine coated 24‐well plates and were cultured overnight. The next day, cell confluency was approximately 80%. For live cell assays to determine AM6544 washout (‘live cell affinity/irreversibility’), a concentration series of AM6544 was prepared in serum‐free medium supplemented with 1 mg·mL−1 BSA and incubated with the cells under conditions described above for the CAMYEL assays (volume adjusted for same well surface area : volume ratio, and 37°C for 1 h). At the end of the AM6544 pretreatment, wells were washed twice with warm HBSS, and the binding assay was initiated. [3H]‐SR141716A was diluted in serum‐free medium supplemented with 1 mg·mL−1 BSA; dilution series of non‐tritiated drugs were prepared in this mixture where applicable and dispensed into the 24‐well plate. Cells were incubated with radioligand for 1 h at 30°C. At the end of the incubation, the plate was moved to ice, well contents were removed with a vacuum and wells were gently washed twice with ice‐cold PBS. Cells were lysed with 250 μL per well 0.1 M NaOH in milliQ water. Lysates (200 μL per well) were moved to 4 mL scintillation tubes and diluted in excess volumes of scintillation fluid. Scintillation was read in the TriLux Counter (as above) following a 6 h delay, to allow time for the aqueous lysate to fully disperse in the scintillation fluid.
Phospho‐ERK assays
Characterization of the activation by phosphorylation of ERK (pERK) was performed using the commercially available AlphaScreen SureFire kit (Perkin Elmer). In brief, cells were plated in 96‐well culture plates and then after 24 h of culture were serum starved for >16 h (in serum free medium supplemented with 1 mg·mL−1 BSA) prior to stimulation, in the presence or absence of PTX (100 ng·mL−1). Drugs were prepared in serum‐free medium supplemented with 1 mg·mL−1 BSA, and then pERK time courses were performed with plates resting on a barely submerged stage in a 37°C waterbath. At the conclusion of the time course, plates were placed on ice, well contents were removed by aspiration, and cells were immediately lysed in 30 μL of lysis buffer. Subsequently, AlphaScreen detection was performed according to manufacturer instructions, and plates were read in a CLARIOstar® plate reader (BMG Labtech, Ortenberg, Germany).
Data and statistical analysis
BRET cAMP time course data were analysed using AUC analysis, as described in Finlay et al. (2016). Data were normalized to basal (0%) and forskolin alone (100%) in order to enable a combination of independent experiments (accounting for small differences in forskolin‐induced cAMP levels between replicates). Concentration–response parameters were obtained by fitting three‐parameter (Hill coefficient constrained to 1) nonlinear regression curves. Where concentration–response curves could not be fitted, connecting lines were drawn between data points to help visualize trends, including for U‐shaped responses. Plots show representative data (mean ± SEM) of technical replicates, unless otherwise specified. All statistical analyses (see below) were performed using data from biological (independent) replicates. Operational analysis for ligand bias (van der Westhuizen et al., 2014) and all plots and curve‐fits were obtained using GraphPad Prism v6 (GraphPad Software Inc., La Jolla, CA, USA).
Homologous binding data were modelled utilizing the predefined equations in GraphPad Prism, producing log KD and BMAX values. The resulting BMAX values (in disintegrations per min) were converted to milli‐Curies per sample point. The specific activity of [3H]‐CP55,940 (175 Ci·mmol−1) was then used to convert bound radioligand into mmol radioligand bound, and this was normalized for protein amount.
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Power analysis for cAMP assays was performed using data from pilot experiments, for both EC50 (minimum effect difference of 0.7 log M) and EMAX (minimum effect difference of 20% of the matched forskolin response). The values for α and power were 0.05 and 0.80 respectively. All final datasets were tested for compliance with parametric test assumptions of normality (Shapiro–Wilk) and equality of variance (Levene Median). If the tested population failed to meet either assumption, data were transformed and re‐tested. Where statistical tests were utilized (one‐ or two‐way ANOVA, as appropriate, unless otherwise specified), data were obtained from at least five biological replicates of each experiment. Where statistically significant differences (P < 0.05) were detected across the entire group in the test, the Holm–Šídák post hoc test was used. All statistical analysis was performed using SigmaPlot™ v12.5 (Systat Software Inc., San Jose, CA, USA). Data presented in figures are either ‘representative’ (illustrative data from a single experiment, consistent with data from independent replicates) or ‘combined’ (summary data, where all replicates' results were averaged) of five independent experiments unless otherwise indicated. This approach was necessary in order to preserve the statistical meaningfulness of parameters from concentration–response curves, as explained in detail by Hall and Langmead (2010).
Drugs
Forskolin, CP55,940, WIN55,212‐2 and BAY59‐3074 (BAY) were purchased from Tocris Bioscience (Bristol, UK); anandamide (AEA) and 2‐arachidonoyl glycerol (2‐AG) were purchased from Cayman Chemical Company (Ann Arbour, MI, USA); and (‐)‐trans‐Δ9‐tetrahydrocannabinol (THC) was purchased from THC Pharm GmbH (Frankfurt, Germany). U0126 was purchased from Cell Signalling Technology (Danvers, MA, USA), and PMA and quinpirole hydrochloride were purchased from Sigma Aldrich (St Louis, MO, USA). SR141716A (rimonabant) was a gift from Roche (Basel, Switzerland). Drug stocks were prepared in absolute ethanol (CP55,940, WIN55,212‐2, AEA, 2‐AG, THC, BAY) or DMSO (forskolin, AM6544, SR141716A) and were stored in aliquots at −80°C prior to use. Drug aliquots used for experiments involving serial dilutions were always single‐use, to minimize assay drift due to solvent evaporation or chemical decay from freeze–thaw cycles. Vehicle controls for serial dilutions were maintained constant within experiments, and cell exposure to solvents was minimized (0.1–0.3%). In order to assuage concerns about endogenous cannabinoids in serum, all drug stimulations were performed in the absence of FBS and following a period of serum starvation.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Gαs‐mediated signalling unmasks agonist bias and is specific for CB1 receptors
Using the CAMYEL assay, cAMP concentration–response curves were obtained for six CB1 agonists in (not pplss‐tagged) 3HA‐hCB1 HEK cell line, following pretreatment both in the absence and presence of PTX. These data revealed that all six agonists act with substantial efficacy in the Gαi pathway (Table 1; Figure 1A), with THC and BAY, reported partial agonists (De Vry et al., 2004; Pertwee, 2008), inhibiting cAMP with significantly lesser efficacies than CP55,940 (P < 0.05). Following PTX pretreatment, however, inhibition of cAMP synthesis was no longer evident. Instead, the majority of agonists stimulated cAMP synthesis above that produced by forskolin alone. Agonist potencies were significantly reduced in comparison with their potencies at inhibiting cAMP production (interaction effects, P < 0.05; Table 1). Wider variations in EMAX values were also observed (Figure 1B), with significant differences in efficacies proceeding 2AG (most efficacious) >WIN55,212‐2 > AEA ≈ CP55,940 in the stimulatory pathway (P < 0.05; Table 1). Interestingly, THC and BAY did not measurably stimulate Gαs‐like cAMP signalling (reliable concentration–response curves could not be fitted to enable statistical comparisons of EMAX values). Operational analysis (Table 2) was performed to compare agonists' signalling in the Gαi pathway (Figure 1A) with signalling in the Gαs pathway (Figure 1B), but bias was not detected for any ligand (Table 2). To determine whether the signalling switch was specific for the CB1 receptor, concentration‐responses with traditional full agonists were performed for hCB2 receptors (Figure 1C) and hD2 receptors (Figure 1D) following pretreatment in the absence and presence of PTX. No evidence was seen for non‐Gαi signalling, as PTX pretreatment resulted in a loss of all receptor‐mediated cAMP signalling.
Table 1.
CB1 receptor agonist potencies/efficacies under various stimulation conditions (summary data)
| 3HA‐hCB1 HEK | ||||||
|---|---|---|---|---|---|---|
| ‐PTX | +PTX | |||||
| pEC50 (M) (± SEM) | EMAX (% of FSK) (± SEM) | n | pEC50 (M) (± SEM) | EMAX (% of FSK) (± SEM) | n | |
| CP55,940 | 9.19 (0.18) | 43.79 (1.28) | 5 | 8.04 (0.03) | 134.86 (5.82) | 5 |
| AEA | 7.47 (0.14) | 48.18 (3.61) | 5 | 5.36 (0.19) | 142.06 (4.69) | 5 |
| WIN55,212‐2 | 7.82 (0.11) | 51.29 (2.83) | 5 | 5.93 (0.11) | 173.03 (3.28) | 5 |
| 2‐AG | 6.99 (0.05) | 55.39 (2.48) | 5 | 5.20 (0.16) | 192.31 (6.14) | 5 |
| THC | 7.74 (0.17) | 59.97 (3.50) | 5 | Not Measurable | Not Measurable | 5 |
| BAY | 7.25 (0.03) | 62.93 (3.09) | 5 | Not Measureable | Not Measureable | 5 |
| pplss‐3HA‐hCB1 HEK | ||||||
|---|---|---|---|---|---|---|
| ‐PTX | +PTX | |||||
| pEC50 (M) (± SEM) | EMAX (% of FSK) (± SEM) | n | pEC50 (M) (± SEM) | EMAX (% of FSK) (± SEM) | n | |
| CP55,940 | 7.42 (0.07) | 230.00 (12.23) | 5 | 7.93 (0.11) | 288.66 (8.29) | 5 |
| AEA | 5.79 (0.14) | 183.33 (7.03) | 5 | 6.24 (0.10) | 287.96 (11.25) | 5 |
| WIN55,212‐2 | 6.169 (0.06) | 341.98 (12.69) | 5 | 6.82 (0.07) | 331.22 (7.22) | 5 |
| 2‐AG | 5.41 (0.08) | 326.82 (13.46) | 5 | 5.99 (0.06) | 327.64 (17.94) | 5 |
| THC | Not Measurable | Not Measurable | 5 | 6.78 (0.06) | 184.59 (5.66) | 5 |
| BAY | Not Measureable | Not Measureable | 5 | 6.20 (0.17) | 186.17 (12.08) | 5 |
Figure 1.
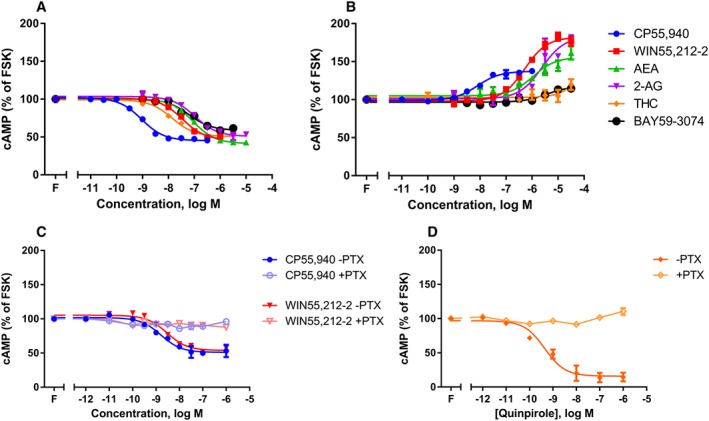
Concentration–response curves for cAMP formation showing 3HA‐hCB1 HEK signalling on stimulation with 2.5 μM forskolin (‘F’; FSK) and a panel of six agonists, following >16 h pretreatment in the absence (A) or presence (B) of PTX. Panel (C) shows the signalling of 3HA‐hCB2 HEK cells stimulated with CP or WIN, following pretreatment with or without PTX; panel (D) shows the signalling of 3HA‐hCB1/FLAG‐D2 HEK cells stimulated with quinpirole, following pretreatment with or without PTX. Representative data are presented, demonstrating mean ± SEM of technical duplicates. Curves were generated by AUC analysis of kinetic CAMYEL biosensor data, which were normalized to basal (0%) and forskolin (100%).
Table 2.
Operational analysis with bias factors (ΔΔLogR ± error) comparing Gαi (no PTX) with Gαs (with PTX) signalling of moderate expression 3HA‐hCB1 HEK cells
| CP55,940 | WIN55,212‐2 | AEA | 2‐AGa | THC | BAY | |
|---|---|---|---|---|---|---|
| ΔΔLogR | 0.29 ± 0.37 | 0.19 ± 0.29 | 0.53 ± 0.27 | 0.00 ± 0.29 | NDb | NDb |
2‐AG was used as the reference ligand.
As no Gαs signal was observed for THC or BAY, a bias factor could not be determined.
High CB1 receptor expression is sufficient to induce nett Gαs signalling
To determine whether receptor number was sufficient to result in a switch in cAMP signalling from mostly Gαi‐mediated (inhibitory) to mostly Gαs‐mediated (stimulatory), cAMP concentration–response stimulations were performed on the HEK cell line stably expressing pplss‐3HA‐hCB1 (see below for quantification of receptor expression differences). In these cells (Figure 2A), the effect of CB1 receptor activation on cAMP signalling was stimulatory, with most agonists demonstrating greater Gαs pathway efficacies than those seen following PTX‐treatment of the non‐pplss CB1‐expressing HEK cells described above (all P < 0.05). THC and BAY again produced negligible stimulation of cAMP (Figure 2A, Table 1). PTX pretreatment of the pplss‐3HA‐hCB1 HEK cells did not increase maximum efficacy in the Gαs pathway for WIN55,212‐2 or 2AG (P > 0.05; Figure 2B, Table 1), despite potencies appearing to increase for all agonists for which curves could be fitted in both pathways (possibly indicating some degree of signalling pathway competition at sub‐maximal signalling activities). However, PTX pretreatment did result in increased efficacy for some agonists: most notably THC and BAY (which shifted from no nett change in cAMP, to low‐efficacy stimulation of cAMP following PTX treatment), and also (to a lesser extent) AEA and CP55,940 (all P < 0.05; Figure 2A, B; Table 1).
Figure 2.
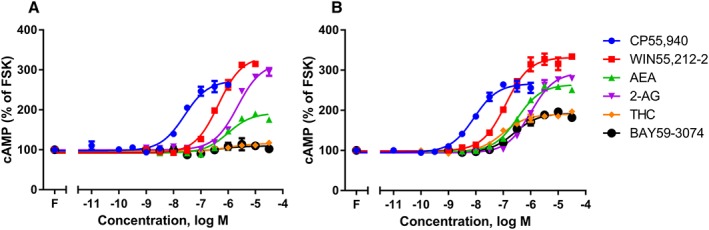
Concentration–response curves for cAMP formation showing pplss‐3HA‐hCB1 HEK signalling, on stimulation with 2.5 μM FSK (‘F’; FSK) and a panel of six agonists, following >16 h pretreatment in the absence (A) or presence (B) of PTX. Representative data are presented, demonstrating mean ± SEM of technical duplicates. Curves were created by AUC analysis of kinetic CAMYEL biosensor data, which were normalized to basal (0%) and FSK (100%).
To quantify the difference in receptor expression between the 3HA‐hCB1 and pplss‐3HA‐hCB1 HEK cell lines, and to determine whether changes in receptor number influenced ligand affinity, homologous competition binding assays were performed with [3H]‐CP55,940 on membrane homogenates from each of the two cell lines of interest, and pKd and BMAX were calculated. The pKd of CP55,940 was not significantly different between the two cell lines, at 8.82 (SEM ± 0.11) and 8.76 (SEM ± 0.12) respectively (n = 6, t‐test P > 0.05). BMAX was therefore calculated using a shared average pKd of 8.79 (SEM ± 0.08). Membranes from the clonal 3HA‐hCB1 HEK cell line contained 1005.96 fmol·mg−1 (SEM ± 85.87, n = 6) of CB1, while membranes from the clonal pplss‐3HA‐hCB1 cell line were found to contain 2713.57 fmol·mg−1 (SEM ± 319.20, n = 6) of CB1 receptors; a 2.7‐fold difference in receptor number. Homologous competition assays for [3H]‐CP55,940 were also performed in membranes from the 3HA‐hCB2 cell line (Figure 1C) to determine whether the lack of stimulatory signalling following PTX treatment could be explained by a substantially lower receptor number. However, membranes from this cell line were found to contain substantially greater CB2 receptor protein than the 3HA‐hCB1 HEK cells, at 2541.08 fmol·mg−1 (SEM ± 91.77, n = 6; pKd 9.33, SEM ± 0.12).
The pplss motif is not an inherent determinant of the switch to Gαs signalling
To further investigate the dependence of cAMP signalling on CB1 receptor expression level and confirm that the observed stimulatory cAMP signalling was not the result of the N‐terminal pplss tag, HEK Flp‐In™ cells stably expressing V8‐CAMYEL biosensor were transiently transfected with the pplss‐3HA‐hCB1 construct to generate a population of cells over which receptor expression ranged widely. Cells were then sorted by FACS into six subpopulations on the basis of surface CB1 receptor expression level determined by antibody binding to N‐terminal HA tags (647 staining intensity, Figure 3A). The sorted cells were stimulated (CP55,940 concentration‐response in the presence of 5 μM forskolin) and cAMP levels assayed. In cells with ‘very low’ to ‘moderate’ expression, a progressive increase in the magnitude of Gαi‐associated cAMP signalling was observed as receptor staining increased (Figure 3B). Further increases in receptor expression led to alterations in the overall signalling profile; less efficacious Gαi signalling at ‘intermediate’ receptor expression, a U‐shaped signalling response at ‘high’ receptor expression (where lower CP55,940 concentrations exerted nett Gαi cAMP effects and higher concentrations exerted nett Gαs cAMP effects) and finally a nett switch in cAMP phenotype to Gαs signalling at ‘very high’ receptor expression (Figure 3B).
Figure 3.
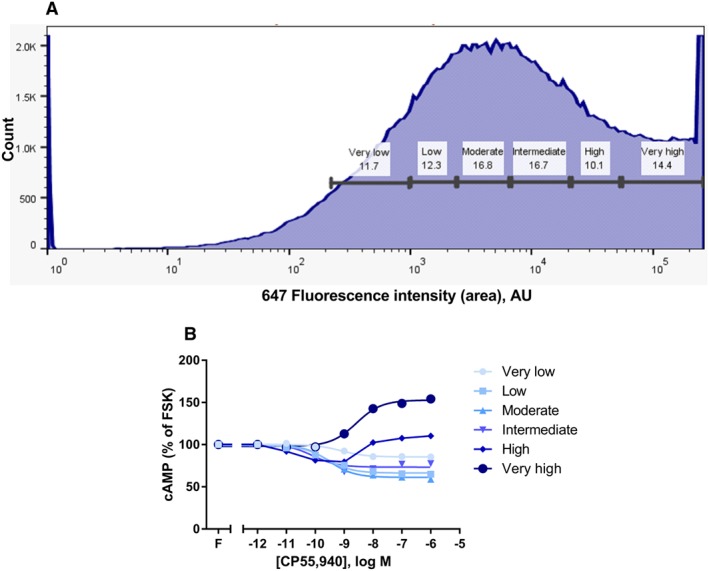
FACS frequency distribution (A), showing live‐gated HEK Flp‐In™ cells stably expressing V8‐CAMYEL, sorted into six subpopulations on the basis of transient pplss‐3HA‐hCB1 surface expression level (647 fluorophore staining intensity). Panel (B) shows cAMP concentration–response curves for CP55,940 in the presence of 5 μM forskolin (FSK; ‘F’), performed on cells from each sorted cell population. Representative data are presented, demonstrating mean ± SEM of technical duplicates.
Characterization of AM6544: an irreversible antagonist of CB1 receptors
Pharmacological knockdown involves irreversible binding of an antagonist to the orthosteric binding site, effectively eliminating receptors from the pool available to orthosterically bind ligand. As there are currently no well‐described irreversible CB1 antagonists available commercially, the binding of a novel CB1 irreversible antagonist, AM6544 (Figure 4A), was characterized under both homogenate and whole cell binding conditions, using [3H]‐SR141716A as the probe so that conditions could be kept as constant as possible between homogenate and whole cell assays. Homogenate binding assays were performed on membranes from the same cell line used for whole cell binding assays, pplss‐3HA‐hCB1 HEK, and also in membranes from hCB1 receptor‐expressing HEK293F cells (Figure 4B, C). These hCB1 receptor‐expressing HEK293F cells were not used in any functional studies reported in this study.
In homogenate binding assays, the pKd of [3H]‐SR141716A was determined empirically by homologous competition and was found to be 9.00 (SEM ± 0.24, n = 5). This value was then used for fitting one‐site, heterologous binding curves for AM6544 displacement. The resulting pKi for AM6544 was 8.93 (SEM ± 0.08, n = 5). This result was confirmed by replication in the Makriyannis laboratory on HEK293F cells expressing hCB1 receptors (pKi 8.38 ± 0.22). Irreversibility of AM6544 in membrane homogenates was then determined by saturation binding assay, with a comparison of SR141716A versus AM6544 washout at ~100× Ki (100 nM). After extensive washing to remove unbound and reversibly bound ligand, AM6544 caused a 79% reduction in [3H]‐SR141716A BMAX, relative to BMAX in vehicle pretreated hCB1 HEK293F membranes (Figure 4B). Similarly, AM6544 caused an 83% reduction in [3H]‐CP55,940 BMAX, relative to that of vehicle pretreated membranes (Figure 4C). This demonstrates that AM6544 bound irreversibly to hCB1 receptors at a concentration of 100 nM, such that [3H]‐SR141716A or [3H]‐CP55,940 were unable to engage AM6544‐occupied hCB1 receptors.
Under whole cell binding conditions, however, the pKd of [3H]‐SR141716A was right‐shifted, to 7.27 (SEM ± 0.06, n = 5), and similarly, the affinity of AM6544 was also reduced: pKi 6.25 (SEM ± 0.06, n = 5). AM6544 irreversible binding was also estimated using the whole cell binding method and was interpreted as being the concentration of ligand that was able to prevent [3H]‐SR141716A binding when applied as a 1 h pretreatment and subjected to washing prior to the [3H]‐SR141716A incubation. Under these conditions, the pEC50 of AM6544 knockdown was estimated to be 5.45 (SEM ± 0.11, n = 5).
Functional characterization of AM6544 under pretreatment conditions (as used in receptor knockdown studies, see below) in a cAMP signalling assay revealed inverse agonist behaviour, comparable with the effects of SR141716A. As shown in Supporting Information Figure S1, assay potency under these washout conditions was consistent with the aforementioned high pEC50 for receptor knockdown.
Systematic pharmacological receptor knockdown reverses pplss‐3HA‐hCB1 Gαs signalling
As further evidence to demonstrate the receptor number‐dependence of the CB1 receptor cAMP signalling phenotype, the irreversible CB1 antagonist, AM6544, was used to systematically reduce receptor number in both the 3HA‐hCB1 and pplss‐3HA‐hCB1 HEK stable cell lines. Following pharmacological knockdown, standard concentration–response curves were performed for CP55,940, WIN55,212‐2 and THC in the presence of 2.5 μM forskolin. The cAMP signalling of 3HA‐hCB1 HEKs showed decreases in agonist potency and efficacy following pretreatment with μM concentrations of AM6544 (Figure 5A, C, E). However, in pplss‐3HA‐hCB1 HEK cells, AM6544 pretreatment produced a concentration‐dependent loss of nett Gαs‐like signalling and progressive increase in Gαi‐like cAMP signalling. Pretreatment with intermediate concentrations of AM6544 (e.g. 1 μM) resulted in U‐shaped agonist concentration responses, as lower concentrations of agonist caused inhibition (e.g. 10 nM CP55,940, Figure 5B; 1 μM WIN55,212‐2, Figure 5D) but higher concentrations in the same agonist‐response curve resulted in cAMP increases. In cells stimulated with THC (Figure 5F), an agonist which does not cause Gαs‐like signalling at any concentration in the absence of PTX, inhibitory signalling was also restored following pretreatment with high‐concentrations of AM6544. Notably, pretreatment with concentrations of AM6544 that were sufficient to restore Gαi signalling resulted in lower potency agonist responses than the curves arising from signalling in the absence of antagonist, perhaps indicating some degree of competitive binding despite attempted washout of non‐irreversibly bound AM6544.
Figure 5.
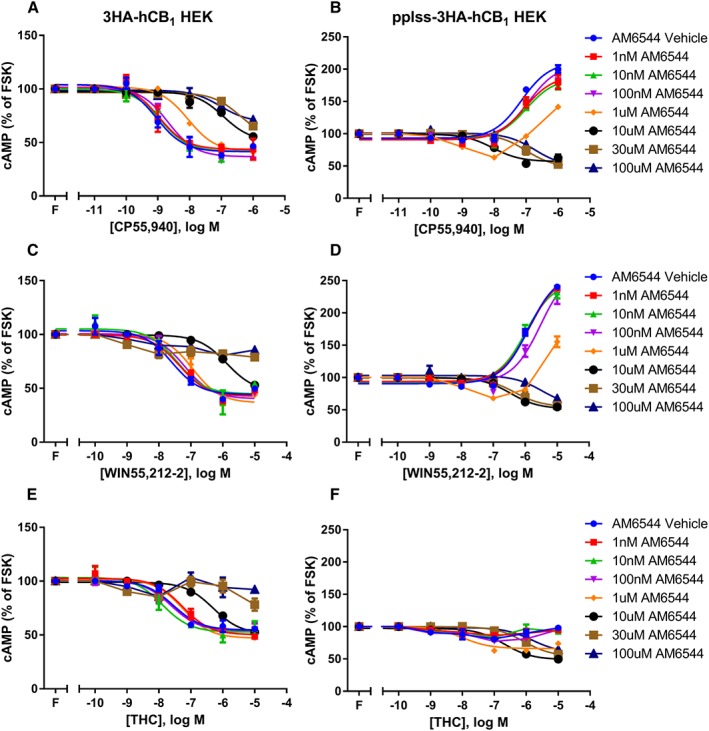
Concentration–response curves for cAMP formation showing 3HA‐hCB1 HEK cells (A, C, E) and pplss‐3HA‐hCB1 HEK cells (B, D, F) signalling on stimulation with 2.5 μM forskolin (FSK; ‘F’) and CP (A, B), WIN (C, D) or THC (E, F), following pretreatment in the presence of increasing concentrations of the irreversible CB1 receptor antagonist AM6544. Representative data are presented, demonstrating mean ± SEM of technical duplicates. Curves were created by AUC analysis of kinetic CAMYEL biosensor data, which were normalized to basal (0%) and forskolin (100%).
Supplementation of Gαi protein restores inhibitory CB1 receptor signalling
If the switch to Gαs signalling at high receptor levels is due to Gαi exhaustion, then it would follow that expression levels of either the receptor or the Gαi protein may drive the change in CB1 receptor signalling. CB1 receptors are known to interact with and signal through all three Gαi subtypes (Mukhopadhyay and Howlett, 2005); therefore, Gαi1 expression was supplemented by transfecting untagged GNAI1 (or matched empty vector) into the 3HA‐hCB1 and pplss‐3HA‐hCB1 HEK cell lines. cAMP assays revealed no obvious changes to agonist concentration‐responses in the 3HA‐hCB1 HEK cell line (whose nett cAMP signal is inhibitory under normal conditions, Figure 6A, C, E). However, in the pplss‐3HA‐hCB1 cells, increased Gαi1 expression resulted in substantially reduced CP55,940 and WIN55,212‐2‐dependent Gαs signalling efficacy, with inhibitory efficacy evident at some agonist concentrations (Figure 6B, D), and restoration of nett inhibitory signalling on stimulation with THC.
Figure 6.
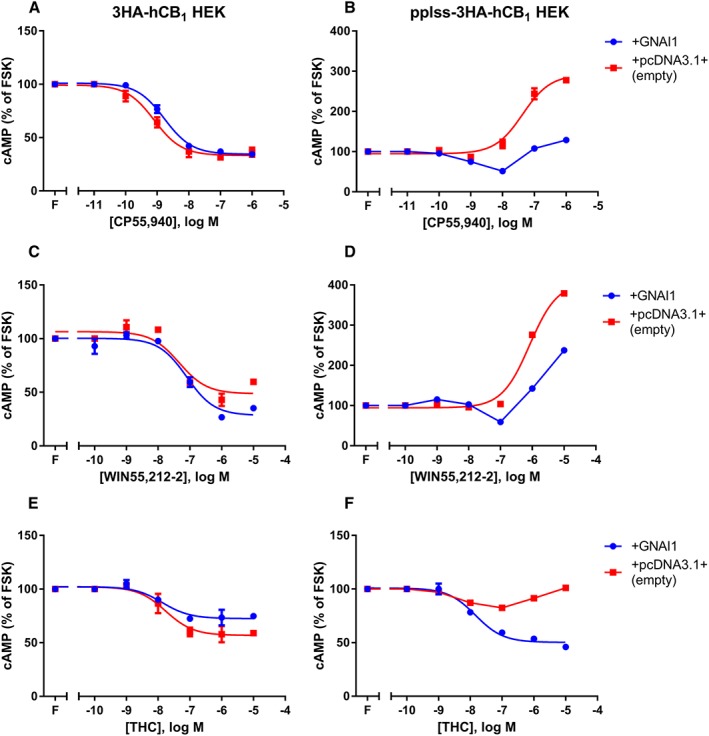
Concentration–response curves for cAMP formation showing 3HA‐hCB1 HEK cells (A, C, E) and pplss‐3HA‐hCB1 HEK cells (B, D, F) signalling on stimulation with 2.5 μM forskolin (FSK; ‘F’) and CP (A, B), WIN (C, D) or THC (E, F), following transfection either with supplementary GNAI1 (encoding Gαi1) or empty vector. Representative data are presented, demonstrating mean ± SEM of technical duplicates. Curves were created by AUC analysis of kinetic CAMYEL biosensor data, which were normalized to basal (0%) and forskolin (100%).
Constitutive cAMP signalling in stable 3HA‐hCB1 and pplss‐3HA‐hCB1 HEK cells is Gαi‐linked
The divergence of agonist‐induced signalling for CB1 receptors at different levels of expression led us to investigate the nature of CB1 receptor constitutive signalling in each cell line. As expected, in 3HA‐hCB1 cells SR141716A concentration‐dependently inhibited constitutive Gαi‐mediated inhibition of cAMP, resulting in an increase in cAMP. Following PTX pretreatment, this effect was lost (Figure 7A), indicating that the effect SR141716A inhibited was indeed Gαi‐linked. Interestingly, SR141716A treatment of pplss‐3HA‐hCB1 HEK cells resulted in a much more pronounced inhibition of Gαi activity, as demonstrated by the much larger increase in cAMP. This effect was wholly PTX‐sensitive (Figure 7B).
Figure 7.
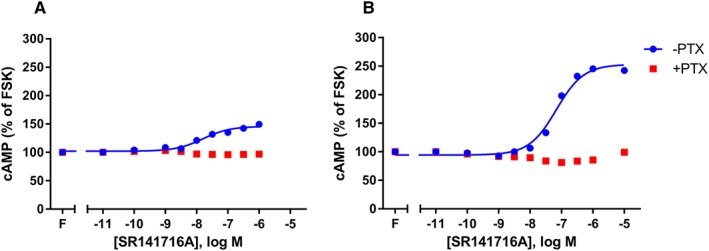
Concentration–response curves showing inhibition by the inverse agonist SR141716A of cAMP signals derived from CB1 receptor constitutive activity, in both 3HA‐hCB1 (A) and pplss‐3HA‐hCB1 (B) HEK cells. SR141716A concentration–response curves were performed each in the presence and absence of forskolin (‘F’) and PTX. Representative data are presented, demonstrating mean ± SEM of technical duplicates. Curves were created by AUC analysis of kinetic CAMYEL biosensor data, which were normalized to basal (0%) and forskolin (100%).
pERK, a pathway mediated through multiple G protein subtypes, provides further evidence of Gαi‐independent signalling
Time course experiments were performed for activation (phosphorylation) of ERK in both cell lines. In the lower‐expressing 3HA‐hCB1 cells, high concentrations of the agonists CP55,940 and WIN55,212‐2 elicited similar pERK maxima of approximately 40% of a 5 min 100 nM PMA stimulation (Figure 8A, C), while 10 μM THC elicited a partially efficacious response of approximately 20% of the PMA response (Figure 8E). In each instance, this effect was essentially wholly PTX‐sensitive, indicating that the pERK responses observed were Gαi‐mediated. Interestingly, in the pplss‐3HA‐hCB1 cells, the same stimulation conditions elicited larger efficacy responses that were predominantly not PTX‐sensitive. Specifically, stimulation with either CP55,940 or WIN55,212‐2 (Figure 8B, D) resulted in pERK signals of approximately 60% of the PMA response in the absence of PTX, and this effect was attenuated by just 10–20% when Gαi was inhibited with PTX. Under these conditions, THC stimulation (Figure 8F) demonstrated low efficacy in the 3HA‐hCB1 cells, and a relatively greater proportion of the pERK signal (approximately half) was sensitive to PTX. Time course differences are also apparent in these data: 3HA‐hCB1 HEK cells showed pERK maxima at approximately 3–4 min stimulation times, and this signal decayed rapidly to basal within 10 min, while the pplss‐3HA‐hCB1 cells showed an earlier increase in signal (pERK levels at 1 min stimulation already tended to be above basal), a peak at 3–4 min stimulation, but then a delayed decay, as pERK levels tended to plateau at approximately 6–7 min stimulation and gradually returning toward basal pERK state over a period longer than the 30 min of the assay.
Figure 8.
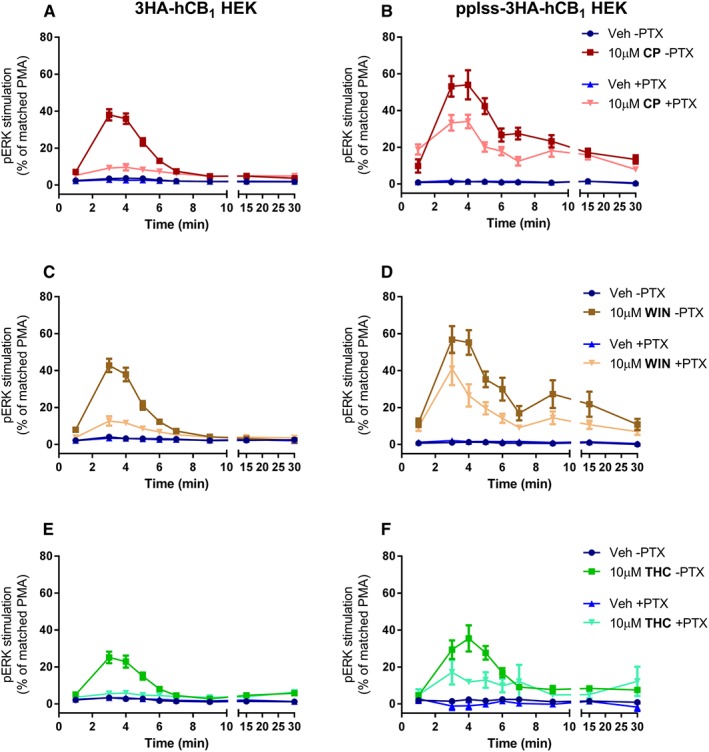
Time course of pERK activation in 3HA‐hCB1 HEK cells (A, C, E) and pplss‐3HA‐hCB1 HEK cells (B, D, F) on stimulation with CP (A, B), WIN (C, D) or THC (E, F), following >16 h pretreatment in the absence or presence of PTX. Combined data (n = 5) were normalized to the level of pERK induced by treatment with PMA for 5 min (100%) and U0126 for 30 min (0%).
Discussion and conclusions
In this study, CB1 receptor expression level is shown to be a novel determinant of signalling outcome, and this effect was found to be dependent on the cannabinoid agonist used and the expression level of Gαi protein. Assays for cAMP signalling revealed that some ‘full’ agonists of the CB1 receptor, particularly CP55,940, elicit maximal signalling in just one of the two pathways characterized, an observation consistent with the findings of Bonhaus et al. (1998). Additionally, the two CB1 partial agonists included in the cAMP screen in this study, THC and BAY, showed limited ability to stimulate cAMP, requiring both PTX pretreatment and high CB1 receptor expression levels for partial Gαs‐like efficacy to be unmasked. The receptor number‐driven switch from Gαi to Gαs signalling was shown to be specific to CB1 receptors and could be reversed both by systematic pharmacological knockdown with the novel irreversible antagonist AM6544 (Patent US8084451, 2011) and by increasing the expression level of Gαi1 protein. Finally, characterization of pERK activation was undertaken to reveal the relative contributions of Gαi activation and alternative pathways to a downstream pathway. These data showed that most (but not all) of the signalling in the lower expressing 3HA‐hCB1 HEK cells is Gαi‐mediated and that most of the signalling in the higher expressing pplss‐3HA‐hCB1 HEK cells is not Gαi‐mediated.
To date, the signalling phenotype of the CB1 receptor has been viewed as almost entirely Gαi‐mediated under normal circumstances. At best, this is an oversimplification that has arisen because the easiest, and by far most common, means for assaying Gαi and Gαs protein activity is changes in forskolin‐stimulated cAMP. An obvious but inherent limitation of this endpoint, however, is that it is not equipped to distinguish contributions of the different G protein subtypes to the total signal, a problem that originates from the mutually antagonistic effects that Gαi and Gαs activity exert on the cAMP pathway. Indeed, recent work by Eldeeb et al. (2016) would suggest that nett cAMP signalling does not necessarily reflect CB1 receptor‐mediated G protein activity, as Gαs protein cycling was elicited concentration‐dependently following agonist stimulation under conditions where the nett cAMP effect was inhibitory. The use of PTX to irreversibly and specifically inactivate Gαi proteins is a widely used approach that aids in cAMP phenotype characterization. However, this approach is of limited utility in the context of CB1 receptor characterization because both the circumstances under which Gαs activity occurs, and the extent to which it exists within canonical signalling, remain uncharacterized. Without a reliable means to eliminate Gαs activity, it is also imponderable as to whether PTX unmasks ‘secondary’ signalling pathways with magnitude equal to that in the absence of PTX, or whether it effectively drives greater efficacy in the non‐preferred pathway simply because of reduced competition of non‐preferred effectors for active receptors. The levels of CB1 receptors present in these cell lines are not dissimilar to that observed in native tissue. Where the pplss‐3HA‐hCB1 cells used in this study contained approximately 2700 fmol·mg−1 hCB1 receptor protein (vs. approximately 1000 fmol·mg−1 for the lower‐expressing cell line), whole rat brain ‘sausage’ homogenate sections have been shown to contain approximately 1158 fmol·mg−1 of rCB1 receptor protein (Herkenham et al., 1991), and membranes prepared from mouse brain contained 1810 fmol·mg−1 of mCB1 receptor protein (Abood et al., 1997), where neither study attempted to account for differences in density across different brain regions. Experiments to quantify cannabinoid binding in guinea pig forebrain homogenates revealed the presence of as much as 5658 fmol·mg−1 of CB1 receptor protein, as determined by binding of the CB1‐selective radioligand [3H]‐SR141716A, although estimates using nonselective radioligands ([3H]‐CP55,940 and [3H]‐WIN55,212‐2) reported 4281 and 2032 fmol·mg−1 protein respectively (Ross et al., 1998).
The concept of functional selectivity adds further complexity to the characterization of CB1 receptor signalling (see Urban et al., 2007 for a review). Qualitative means of estimating bias are frequently confounded by variables such as cell background (e.g. different Gαi and Gαs expression levels) or, in cAMP signalling, concentrations of forskolin (higher concentrations will overstate Gαi activity but understate Gαs activity, and vice versa). An empirical method of analysis that purports to objectively describe functional selectivity (not be influenced by the aforementioned variables) is the Operational model. This approach produces parameters for affinity, KA, and efficacy, τ, based on the classical principle that the two levels of interaction that they represent (agonist‐receptor interactions and receptor‐signalling proteins interactions, respectively) are contingent for profiling a given signalling response (Black and Leff, 1983). A value obtained from these variables, termed the transduction coefficient, Δlog(τ/KA), may therefore be considered an agglomeration of both interactions to allow approximation of an agonist's intrinsic efficacy (‘stimulus per receptor’) (Kenakin et al., 2012). Comparisons of the transduction coefficients of different pathways (for example, Gαi and Gαs signalling) are subsequently represented as single values in the form of a bias factor, ΔΔlog(τ/KA). Operational analysis was performed on the cAMP data included in this study, for 3HA‐hCB1 HEK cells in the absence and presence of PTX (Table 2). These data did not reveal significant bias for any agonist. However, it must be noted that neither THC nor BAY elicited any cAMP effects when cells had been pretreated with PTX, and therefore, concentration–response curves could not be fitted, nor operational parameters obtained. A simple EC50 comparative analysis suggests that if THC and BAY underwent the same potency shift as the other ligands (approximately 100‐fold), then responses would still be detectable within this assay. It would follow that the apparent inability of these ligands to signal in the Gαs‐like pathway must indicate either a much larger shift in potency or an actual lack of Gαs signalling efficacy. Either way, these explanations indicate that THC and BAY are both biased toward the inhibitory pathway and against the stimulatory pathway.
In this study, experiments of two different designs were performed to demonstrate the role of receptor number in the reported signalling switch for CB1 receptors. The first, sorting of HEK cells transiently transfected with a construct for pplss‐3HA‐hCB1 by FACS (Figure 3), revealed in ‘very low’ to ‘medium’‐expressing cells a progressive increase in inhibitory cAMP signalling, then (with continued increases in receptor expression) a gradual loss of efficacy in the inhibitory pathway, and ultimately nett stimulatory cAMP signalling. The reduction in Gαi signalling at ‘intermediate’ receptor expression probably indicates the stimulatory signalling component acting competitively with Gαi for receptors – as more receptors are activated, Gαi protein saturation may be occurring as Gαs recruitment increases. Significantly, this suggests that the switch in CB1 receptor signalling from Gαi to Gαs is dependent on the expression of all three proteins, and potentially other accessory proteins. We hypothesize that the CB1 receptor possesses high affinity for Gαi, but that this is a relatively low‐efficacy signalling pathway. Conversely, the CB1 receptor has lower affinity for Gαs, but this pathway has higher intrinsic activity and is therefore capable of overwhelming the effects of Gαi. Certainly, the availability and abundance of G protein effectors has already been shown to contribute to alterations in signalling for other GPCRs (Nasman et al., 2001). It is also possible that cAMP increases results from increased activation of Gαi‐insensitive adenylate cyclase (AC) isoforms. HEK cells express the transcripts of multiple isoforms of AC (Atwood et al., 2011). Different AC isoforms are modulated by different Gα subtypes: all nine AC isoforms are sensitive to stimulation by Gαs/Olf family proteins, but some isoforms (e.g. AC2, 3, 4 and 7) are not directly regulated by Gαi (Dessauer, 2009).
The second assay type that demonstrated the CB1 receptor specificity of the switch involved systematic pharmacological knockdown with the novel antagonist AM6544. In order to determine compound affinity, homogenate binding competition‐displacement assays were performed and revealed low nanomolar‐range affinity. However, in live cell functional assays, micromolar concentrations of AM6544 were required in order for receptor knockdown to be observed (Figure 5) – a difference in concentration that is incongruent with simple occupancy. Further affinity characterization involved whole cell binding assays, which were performed under matched conditions to functional assays (Figure 5). As reported, this assay type revealed a right‐shifted micromolar‐range pKi and a similar right‐shift in SR141716A affinity, emphasizing the dependence of ligand binding on assay conditions. AM6544 was found to concentration‐dependently cause a switch of nett stimulatory cAMP signalling back to canonical inhibitory signalling in the pplss‐3HA‐hCB1 cells (Figure 5B, D, F), although pretreatment with even very high concentrations (100 μM) failed to completely abolish signalling, probably indicating both that only a portion of receptors were AM6544‐occupied and therefore irreversibly blocked under these assay conditions, and that the cAMP pathway has a substantial degree of receptor reserve.
Experiments to increase Gαi expression by transfection were performed to investigate the influence of G protein expression level on the signalling switch (Figure 6). No substantial differences were observed in the signalling of the 3HA‐hCB1 cells, which signal through Gαi under usual circumstances (Figure 6A, C, E). In the pplss‐3HA‐hCB1 cells, supplementary Gαi1 expression dramatically altered the concentration–response curves, producing U‐shaped concentration‐response curves for CP55,940 and WIN55,212‐2 and resulting in an inhibitory curve for THC (Figure 6B, D, F). The incomplete reversal of signalling with CP55,940 and WIN55,212‐2 probably arises due to our assay design, wherein partial transfection efficiency resulted in only a proportion of cells receiving the GNAI1 construct by transfection, and therefore, a range of Gαi1 expression levels were assayed, including a proportion of cells with no additional Gαi1 expression. Interestingly, however, in these assays, agonist efficacies align with Gα pathway bias: CP55,940 exhibits partial efficacy in the Gαs‐like pathway in the absence of PTX and demonstrates almost no nett signalling at 1 μM CP55,940 following GNAI1 transfection (Figure 6B); WIN55,212‐2 exhibits full efficacy in the Gαs‐like pathway and demonstrates less pronounced Gαi rescue in cells overexpressing Gαi (Figure 6D); and THC exhibits no efficacy in the Gαs‐like pathway in the absence of PTX, while also being most amenable to demonstrating nett Gαi rescue following GNAI1 transfection, being the only agonist for which an inhibitory concentration–response curve could be fitted (Figure 6F). Similarly, it is worth noting that at the ratios at which CB1 receptors and its signalling effectors are expressed following GNAI1 transfection in this assay, functional bias is genuinely druggable: for example, 10 μM WIN55,212‐2 (Figure 6D) results in a nett stimulatory cAMP signalling response, while 1 μM THC (Figure 6F) results in a nett inhibitory signal.
Characterization of the differential constitutive signalling of the 3HA‐hCB1 and pplss‐3HA‐hCB1 HEK cells was based on the Gαi exhaustion hypothesis that under unstimulated conditions, irrespective of receptor expression level, receptors are less likely to be limited for their preferred G protein effector because a minority of receptors are in an active conformation at any moment. The inverse agonist SR141716A induced concentration‐dependent stimulation of cAMP in both cell lines (Figure 7), and these effects were PTX‐sensitive, indicating that these were completely mediated by Gαi. Intriguingly, this results in a situation in pplss‐3HA‐hCB1 cells where stimulation with both agonists and inverse agonists has the same effect on signalling, to increase cAMP, albeit by different mechanisms.
Although cAMP assays facilitated substantial characterization of the interplay between receptor number and signalling outcome, the mutually antagonistic consequences of Gαi and Gαs activation on cAMP levels limit ability to discern activation states for each distinct pathway. Characterization of pERK activation in 3HA‐hCB1 and pplss‐3HA‐hCB1 HEK cells was undertaken in order to better observe an effect of concurrent activity in the Gαi pathway and alternative Gαs‐like pathway and also to examine a downstream consequence of the CB1 receptor signalling switch. In a result which further extends understanding of the mechanism of the signalling switch, the pERK signal in 3HA‐hCB1 cells was PTX sensitive in every condition (Figure 8A, C, E), but in pplss‐3HA‐hCB1 cells, it was predominantly PTX‐independent, suggesting that the alternative signalling pathway may not simply exist additively with Gαi signalling (as required by the Gαi exhaustion hypothesis) but may replace it, in agreement with a pleiotropic switch mechanism (Maudsley et al., 2005). Significantly, however, it is possible that the pathway mediating the non‐PTX‐sensitive pERK signal in the pplss‐3HA‐hCB1 cells is not the same as the Gαs‐like signal that gives rise to stimulatory cAMP signalling: CB1 receptor coupling to Gαq has also been reported previously (Lauckner et al., 2005), and this pathway has also been shown to elicit ERK phosphorylation responses (Asimaki and Mangoura, 2011).
Taken together, the data we present support more than one mechanism for the switch from the canonical Gαi pathway to the alternative Gαs‐like pathway: the central idea of receptor number driving the switch and the fact that it can be reversed competitively by altering G protein expression strongly support receptor coupling promiscuity and affinity/efficacy interactions that underpin the Gαi hypothesis; however, the apparent change in signalling balance between Gαi and other effectors in pERK characterization (as opposed to the alternative effectors simply adding to the total signal) better supports a pleiotropic switch mechanism. The potential for more than one signalling switch mechanism is significant from a biomedical perspective. This is because only the pleiotropic mechanism is directly targetable pharmacologically by design of a biased agonist (it requires that stabilization of a particular receptor conformation is sufficient to drive differential signalling), whereas the Gαi exhaustion mechanism requires both receptor agonism and a secondary means to either promote coupling of the favoured G protein or diminish the activity of the disfavoured one. In development of CB1 receptor‐targeted interventions, the ability to target one Gα pathway over another will be of interest therapeutically. For example, in an in vitro model of Huntington's disease, CB1 receptor Gαs‐mediated increases in cAMP have been found to exacerbate cell death by promoting aggregation of mutant huntingtin (Scotter et al., 2010). Similarly, in a different in vitro Huntington's disease model, CB1 receptor‐mediated Gαi activity has recently been suggested to be therapeutically beneficial, as Gαi‐mediated pERK is substantially reduced in diseased cells compared with matched controls, and agonists that demonstrated Gαi bias in those assays were shown to improve cell viability (Laprairie et al., 2016).
One of the greatest remaining obstacles in biomedical sciences generally is the difficulty in relating easily characterized near‐signalling phenotypes with whole cell‐level endpoints, as mid‐stream signalling pathways are relatively poorly characterized and diverge greatly between different cell types and disease states – indeed, a recent review by Velasco et al. (2016) observed that oncogenically transformed cells may exhibit different signalling phenotypes to non‐transformed cells and that the molecular reasons for this are unknown. An early study into the effects of cannabinoids on cancer growth revealed that THC treatment inhibited growth of C6 rat glioma cells and induced cell death (Sanchez et al., 1998), a finding now supported by other studies (Gomez del Pulgar et al., 2002; Salazar et al., 2009). Confusingly, however, more recent data suggests that blockade of CB1 receptors can also lead to apoptosis in glioma cells and primary human cells (Ciaglia et al., 2015), meaning that agonism and antagonism of CB1 receptors may not confer opposite effects on cell fate, even in related cell types. Additionally, high expression of CB1 receptors has been shown to correlate with poor prognosis in prostate cancer (Chung et al., 2009; Fowler et al., 2010) as well as stage two microsatellite stable colorectal cancer (Gustafsson et al., 2011). These endpoints have not been associated with near‐signalling phenotypes, but one study to date has revealed that astrocytoma subclones signal through the pro‐survival Akt pathway in a manner dependent on high expression of CB1 receptors (Cudaback et al., 2010), a finding that was replicated in prostate cancer tissue (Cipriano et al., 2013). Relatedly, an up‐regulation of CB1 receptors has been reported in a variety of other cancers, including Hodgkin lymphoma (Benz et al., 2013), human epithelial ovarian tumours (Messalli et al., 2014) and stage four colorectal cancer (Jung et al., 2013) and has been shown to correlate with disease severity in the latter two cases. Thus, substantial conflicts remain in the literature and emphasize the complex interconnections between CB1 receptor near signalling phenotypes and downstream endpoints such as cell fate. Our study may help to provide a basis for deconvoluting the conflicting data regarding CB1 receptor near‐signalling and cell fate, though much more research is required in order to understand the roles that the signalling switch plays in normal physiology and disease and the ways in which downstream endpoints are affected.
Author contributions
D.B.F. designed and performed experiments, analysed the data and wrote the paper. E.E.C., M.R.H. and A.K. designed and performed experiments. N.L.G. analysed the data and wrote the paper. V.K.V. designed and performed experiments, wrote the paper and generated the AM6544 compound. A.M. generated the AM6544 compound. M.G. designed experiments, analysed the data and wrote the paper.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 Cyclic AMP concentration–response curves showing 3HA‐hCB1 HEK cell signalling on stimulation with vehicle or 2.5 μM FSK, following pretreatment in the presence of increasing concentrations of the CB1 irreversible antagonist AM6544. Representative data. Curves were created by area‐under‐the‐curve analysis of kinetic CAMYEL biosensor data which was normalized to basal (0%) and FSK (100%).
Acknowledgements
The authors gratefully acknowledge grants to MG from the Marsden Fund of the Royal Society of New Zealand and the University of Auckland Faculty Research Development Fund, and National Institutes of Health grants R01DA041435 and P01DA009158 to AM. We also thank Stephen Edgar (University of Auckland, Department of Molecular Medicine and Pathology) for his assistance with flow cytometry.
Finlay, D. B. , Cawston, E. E. , Grimsey, N. L. , Hunter, M. R. , Korde, A. , Vemuri, V. K. , Makriyannis, A. , and Glass, M. (2017) Gαs signalling of the CB1 receptor and the influence of receptor number. British Journal of Pharmacology, 174: 2545–2562. doi: 10.1111/bph.13866.
Contributor Information
Alexandros Makriyannis, Email: a.makriyannis@northeastern.edu.
Michelle Glass, Email: m.glass@auckland.ac.nz.
References
- Abadji V, Lucas‐Lenard JM, Chin C, Kendall DA (1999). Involvement of the carboxyl terminus of the third intracellular loop of the cannabinoid CB1 receptor in constitutive activation of Gs. J Neurochem 72: 2032–2038. [DOI] [PubMed] [Google Scholar]
- Abood ME, Ditto KE, Noel MA, Showalter VM, Tao Q (1997). Isolation and expression of a mouse CB1 cannabinoid receptor gene. Comparison of binding properties with those of native CB1 receptors in mouse brain and N18TG2 neuroblastoma cells. Biochem Pharmacol 53: 207–214. [DOI] [PubMed] [Google Scholar]
- Alexander SP, Davenport AP, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The concise guide to PHARMACOLOGY 2015/16: G protein‐coupled receptors. Br J Pharmacol 172: 5744–5869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asimaki O, Mangoura D (2011). Cannabinoid receptor 1 induces a biphasic ERK activation via multiprotein signaling complex formation of proximal kinases PKCepsilon, Src, and Fyn in primary neurons. Neurochem Int 58: 135–144. [DOI] [PubMed] [Google Scholar]
- Atwood BK, Lopez J, Wager‐Miller J, Mackie K, Straiker A (2011). Expression of G protein‐coupled receptors and related proteins in HEK293, AtT20, BV2, and N18 cell lines as revealed by microarray analysis. BMC Genomics 12: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagher AM, Laprairie RB, Kelly ME, Denovan‐Wright EM (2016). Antagonism of dopamine receptor 2 long (D2L) affects cannabinoid receptor 1 (CB1) signaling in a cell culture model of striatal medium spiny projection neurons. Mol Pharmacol 89: 652–666. [DOI] [PubMed] [Google Scholar]
- Bash R, Rubovitch V, Gafni M, Sarne Y (2003). The stimulatory effect of cannabinoids on calcium uptake is mediated by Gs GTP‐binding proteins and cAMP formation. Neurosignals 12: 39–44. [DOI] [PubMed] [Google Scholar]
- Belin D, Bost S, Vassalli JD, Strub K (1996). A two‐step recognition of signal sequences determines the translocation efficiency of proteins. EMBO J 15: 468–478. [PMC free article] [PubMed] [Google Scholar]
- Benz AH, Renne C, Maronde E, Koch M, Grabiec U, Kallendrusch S et al. (2013). Expression and functional relevance of cannabinoid receptor 1 in Hodgkin lymphoma. PLoS One 8: e81675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black JW, Leff P (1983). Operational models of pharmacological agonism. Proc R Soc Lond Ser B Biol Sci 220: 141–162. [DOI] [PubMed] [Google Scholar]
- Bonhaus DW, Chang LK, Kwan J, Martin GR (1998). Dual activation and inhibition of adenylyl cyclase by cannabinoid receptor agonists: evidence for agonist‐specific trafficking of intracellular responses. J Pharmacol Exp Ther 287: 884–888. [PubMed] [Google Scholar]
- Cawston EE, Redmond WJ, Breen CM, Grimsey NL, Connor M, Glass M (2013). Real‐time characterization of cannabinoid receptor 1 (CB1) allosteric modulators reveals novel mechanism of action. Br J Pharmacol 170: 893–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung SC, Hammarsten P, Josefsson A, Stattin P, Granfors T, Egevad L et al. (2009). A high cannabinoid CB(1) receptor immunoreactivity is associated with disease severity and outcome in prostate cancer. Eur J Cancer 45: 174–182. [DOI] [PubMed] [Google Scholar]
- Ciaglia E, Torelli G, Pisanti S, Picardi P, D'Alessandro A, Laezza C et al. (2015). Cannabinoid receptor CB1 regulates STAT3 activity and its expression dictates the responsiveness to SR141716 treatment in human glioma patients' cells. Oncotarget 6: 15464–15481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipriano M, Haggstrom J, Hammarsten P, Fowler CJ (2013). Association between cannabinoid CB(1) receptor expression and Akt signalling in prostate cancer. PLoS One 8: e65798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeaux Y, Briddon SJ, Megson AE, McDonnell J, Dickenson JM, Hill SJ (2000). Influence of receptor number on functional responses elicited by agonists acting at the human adenosine A(1) receptor: evidence for signaling pathway‐dependent changes in agonist potency and relative intrinsic activity. Mol Pharmacol 58: 1075–1084. [DOI] [PubMed] [Google Scholar]
- Cudaback E, Marrs W, Moeller T, Stella N (2010). The expression level of CB1 and CB2 receptors determines their efficacy at inducing apoptosis in astrocytomas. PLoS One 5: e8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Vry J, Denzer D, Reissmueller E, Eijckenboom M, Heil M, Meier H et al. (2004). 3‐[2‐cyano‐3‐(trifluoromethyl)phenoxy]phenyl‐4,4,4‐trifluoro‐1‐butanesulfonate (BAY 59‐3074): a novel cannabinoid Cb1/Cb2 receptor partial agonist with antihyperalgesic and antiallodynic effects. J Pharmacol Exp Ther 310: 620–632. [DOI] [PubMed] [Google Scholar]
- Dessauer CW (2009). Adenylyl cyclase – A‐kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol 76: 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldeeb K, Leone‐Kabler S, Howlett AC (2016). CB1 cannabinoid receptor‐mediated increases in cyclic AMP accumulation are correlated with reduced Gi/o function. J Basic Clin Physiol Pharmacol 27: 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferre S (2015). The GPCR heterotetramer: challenging classical pharmacology. Trends Pharmacol Sci 36: 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finlay DB, Joseph WR, Grimsey NL, Glass M (2016). GPR18 undergoes a high degree of constitutive trafficking but is unresponsive to N‐Arachidonoyl Glycine. PeerJ 4: e1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Hammarsten P, Bergh A (2010). Tumour cannabinoid CB(1) receptor and phosphorylated epidermal growth factor receptor expression are additive prognostic markers for prostate cancer. PLoS One 5: e15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass M, Felder CC (1997). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors augments cAMP accumulation in striatal neurons: evidence for a Gs linkage to the CB1 receptor. J Neurosci 17: 5327–5333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez del Pulgar T, Velasco G, Sanchez C, Haro A, Guzman M (2002). De novo‐synthesized ceramide is involved in cannabinoid‐induced apoptosis. Biochem J 363 (Pt 1): 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimsey NL, Goodfellow CE, Dragunow M, Glass M (2011). Cannabinoid receptor 2 undergoes Rab5‐mediated internalization and recycles via a Rab11‐dependent pathway. Biochim Biophys Acta 1813: 1554–1560. [DOI] [PubMed] [Google Scholar]
- Gustafsson SB, Palmqvist R, Henriksson ML, Dahlin AM, Edin S, Jacobsson SO et al. (2011). High tumour cannabinoid CB1 receptor immunoreactivity negatively impacts disease‐specific survival in stage II microsatellite stable colorectal cancer. PLoS One 6: e23003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall DA, Langmead CJ (2010). Matching models to data: a receptor pharmacologist's guide. Br J Pharmacol 161: 1276–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC (1991). Characterization and localization of cannabinoid receptors in rat brain: a quantitative in vitro autoradiographic study. J Neurosci 11: 563–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua T, Vemuri K, Pu M, Qu L, Han GW, Wu Y et al. (2016). Crystal structure of the human cannabinoid receptor CB1. Cell 167: 750–762 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter MR, Grimsey NL, Glass M (2016). Sulfation of the FLAG epitope is affected by co‐expression of G protein‐coupled receptors in a mammalian cell model. Sci Rep 6: 27316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter MR, Finlay DB, Macdonald CE, Cawston EE, Grimsey NL, Glass M (2017). Real time measurement of cannabinoid receptor mediated cAMP signalling. In Cannabinoids and Their Receptors, Methods in Enzymology. 593, In press. [DOI] [PubMed]
- Janero DR, Yaddanapudi S, Zvonok N, Subramanian KV, Shukla VG, Stahl E et al. (2015). Molecular‐interaction and signaling profiles of AM3677, a novel covalent agonist selective for the cannabinoid 1 receptor. ACS Chem Nerosci 6: 1400–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrahian A, Watts VJ, Barker EL (2004). D2 dopamine receptors modulate Galpha‐subunit coupling of the CB1 cannabinoid receptor. J Pharmacol Exp Ther 308: 880–886. [DOI] [PubMed] [Google Scholar]
- Jiang LI, Collins J, Davis R, Lin KM, DeCamp D, Roach T et al. (2007). Use of a cAMP BRET sensor to characterize a novel regulation of cAMP by the sphingosine 1‐phosphate/G13 pathway. J Biol Chem 282: 10576–10584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin LQ, Wang HY, Friedman E (2001). Stimulated D(1) dopamine receptors couple to multiple Galpha proteins in different brain regions. J Neurochem 78: 981–990. [DOI] [PubMed] [Google Scholar]
- Jung CK, Kang WK, Park JM, Ahn HJ, Kim SW, Taek Oh S et al. (2013). Expression of the cannabinoid type I receptor and prognosis following surgery in colorectal cancer. Oncol Lett 5: 870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearn CS, Blake‐Palmer K, Daniel E, Mackie K, Glass M (2005). Concurrent stimulation of cannabinoid CB1 and dopamine D2 receptors enhances heterodimer formation: a mechanism for receptor cross‐talk? Mol Pharmacol 67: 1697–1704. [DOI] [PubMed] [Google Scholar]
- Kenakin T, Watson C, Muniz‐Medina V, Christopoulos A, Novick S (2012). A simple method for quantifying functional selectivity and agonist bias. ACS Chem Nerosci 3: 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurzchalia TV, Wiedmann M, Girshovich AS, Bochkareva ES, Bielka H, Rapoport TA (1986). The signal sequence of nascent preprolactin interacts with the 54K polypeptide of the signal recognition particle. Nature 320: 634–636. [DOI] [PubMed] [Google Scholar]
- Laprairie RB, Bagher AM, Kelly ME, Denovan‐Wright EM (2016). Biased type 1 cannabinoid receptor signaling influences neuronal viability in a cell culture model of Huntington disease. Mol Pharmacol 89: 364–375. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Hille B, Mackie K (2005). The cannabinoid agonist WIN55,212‐2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc Natl Acad Sci U S A 102: 19144–19149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laugwitz KL, Allgeier A, Offermanns S, Spicher K, Van Sande J, Dumont JE et al. (1996). The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families. Proc Natl Acad Sci U S A 93: 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcellino D, Carriba P, Filip M, Borgkvist A, Frankowska M, Bellido I et al. (2008). Antagonistic cannabinoid CB1/dopamine D2 receptor interactions in striatal CB1/D2 heteromers. A combined neurochemical and behavioral analysis. Neuropharmacology 54: 815–823. [DOI] [PubMed] [Google Scholar]
- Maudsley S, Martin B, Luttrell LM (2005). The origins of diversity and specificity in g protein‐coupled receptor signaling. J Pharmacol Exp Ther 314: 485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh BT, Hudson B, Yegorova S, Jollimore CA, Kelly ME (2007). Agonist‐dependent cannabinoid receptor signalling in human trabecular meshwork cells. Br J Pharmacol 152: 1111–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messalli EM, Grauso F, Luise R, Angelini A, Rossiello R (2014). Cannabinoid receptor type 1 immunoreactivity and disease severity in human epithelial ovarian tumors. Am J Obstet Gynecol 211: 234 e231–234 e236. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay S, Howlett AC (2005). Chemically distinct ligands promote differential CB1 cannabinoid receptor‐Gi protein interactions. Mol Pharmacol 67: 2016–2024. [DOI] [PubMed] [Google Scholar]
- Nasman J, Kukkonen JP, Ammoun S, Akerman KE (2001). Role of G‐protein availability in differential signaling by alpha 2‐adrenoceptors. Biochem Pharmacol 62: 913–922. [DOI] [PubMed] [Google Scholar]
- Pertwee RG (2008). The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9‐tetrahydrocannabinol, cannabidiol and delta9‐tetrahydrocannabivarin. Br J Pharmacol 153: 199–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przybyla JA, Watts VJ (2010). Ligand‐induced regulation and localization of cannabinoid CB1 and dopamine D2L receptor heterodimers. J Pharmacol Exp Ther 332: 710–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Brockie HC, Fernando SR, Saha B, Razdan RK, Pertwee RG (1998). Comparison of cannabinoid binding sites in guinea‐pig forebrain and small intestine. Br J Pharmacol 125: 1345–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar M, Carracedo A, Salanueva IJ, Hernandez‐Tiedra S, Lorente M, Egia A et al. (2009). Cannabinoid action induces autophagy‐mediated cell death through stimulation of ER stress in human glioma cells. J Clin Invest 119: 1359–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C, Galve‐Roperh I, Canova C, Brachet P, Guzman M (1998). Delta9‐tetrahydrocannabinol induces apoptosis in C6 glioma cells. FEBS Lett 436: 6–10. [DOI] [PubMed] [Google Scholar]
- Scotter EL, Goodfellow CE, Graham ES, Dragunow M, Glass M (2010). Neuroprotective potential of CB1 receptor agonists in an in vitro model of Huntington's disease. Br J Pharmacol 160: 747–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44 (D1): D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban JD, Clarke WP, von Zastrow M, Nichols DE, Kobilka B, Weinstein H et al. (2007). Functional selectivity and classical concepts of quantitative pharmacology. J Pharmacol Exp Ther 320: 1–13. [DOI] [PubMed] [Google Scholar]
- Velasco G, Hernandez‐Tiedra S, Davila D, Lorente M (2016). The use of cannabinoids as anticancer agents. Prog Neuropsychopharmacol Biol Psychiatry 64: 259–266.26071989 [Google Scholar]
- van der Westhuizen ET, Breton B, Christopoulos A, Bouvier M (2014). Quantification of ligand bias for clinically relevant beta2‐adrenergic receptor ligands: implications for drug taxonomy. Mol Pharmacol 85: 492–509. [DOI] [PubMed] [Google Scholar]
- Xu W, Filppula SA, Mercier R, Yaddanapudi S, Pavlopoulos S, Cai J et al. (2005). Purification and mass spectroscopic analysis of human CB1 cannabinoid receptor functionally expressed using the baculovirus system. J Pept Res 66: 138–150. [DOI] [PubMed] [Google Scholar]
- Zhu X, Gilbert S, Birnbaumer M, Birnbaumer L (1994). Dual signaling potential is common among Gs‐coupled receptors and dependent on receptor density. Mol Pharmacol 46: 460–469. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Cyclic AMP concentration–response curves showing 3HA‐hCB1 HEK cell signalling on stimulation with vehicle or 2.5 μM FSK, following pretreatment in the presence of increasing concentrations of the CB1 irreversible antagonist AM6544. Representative data. Curves were created by area‐under‐the‐curve analysis of kinetic CAMYEL biosensor data which was normalized to basal (0%) and FSK (100%).