

Abstract
Background and purpose
Promoting energy metabolism is known to provide therapeutic effects for obesity and associated metabolic disorders. The present study evaluated the therapeutic effects of the newly identified bouchardatine (Bou) on obesity‐associated metabolic disorders and the molecular mechanisms of these effects.
Experimental approach
The molecular mode of action of Bou for its effects on lipid metabolism was first examined in 3T3‐L1 adipocytes and HepG2 cells. This was followed by an evaluation of its metabolic effects in mice fed a high‐fat diet for 16 weeks with Bou being administered in the last 5 weeks. Further mechanistic investigations were conducted in pertinent organs of the mice and relevant cell models.
Key results
In 3T3‐L1 adipocytes, Bou reduced lipid content and increased sirtuin 1 (SIRT1) activity to facilitate liver kinase B1 (LKB1) activation of AMPK. Chronic administration of Bou (50 mg∙kg−1 every other day) in mice significantly attenuated high‐fat diet‐induced increases in body weight gain, dyslipidaemia and fatty liver without affecting food intake and no adverse effects were detected. These metabolic effects were associated with activation of the SIRT1–LKB1–AMPK signalling pathway in adipose tissue and liver. Of particular note, UCP1 expression and mitochondrial biogenesis were increased in both white and brown adipose tissues of Bou‐treated mice. Incubation with Bou induced similar changes in primary brown adipocytes isolated from mice.
Conclusions and implications
Bou may have therapeutic potential for obesity‐related metabolic diseases by increasing the capacity of energy expenditure in adipose tissues and liver through a mechanism involving the SIRT1–LKB1–AMPK axis.
Abbreviations
- ACC
acetyl CoA carboxylase
- ACSL
long‐chain acyl‐CoA synthase
- AMPK
AMP‐activated protein kinase
- BAT
brown adipose tissue
- FAS
fatty acid synthase
- HF
high‐fat
- LKB1
liver kinase B‐1
- mtDNA
mitochondrial DNA
- nDNA
nuclear DNA
- PGC‐1α
PPAR γ coactivator‐1α
- SIRT1
sirtuin 1
- UCP1
uncoupling protein 1
- WAT
white adipose tissue
Introduction
Obesity is characterized by increased body fat mass, and this metabolic disorder results from interactions of multiple factors, which eventually favour a chronic positive energy balance (Goldzieher and Goldzieher, 1952; Carneiro et al., 2016). There is compelling evidence showing that obesity is a major cause of metabolic syndrome, which includes insulin resistance, dyslipidaemia, non‐alcoholic fatty liver, type 2 diabetes and cardiovascular disease (Kusminski et al., 2016; Sáez‐Lara et al., 2016). Current drugs for the treatment of obesity mainly include two classes: appetite inhibitors and gastrointestinal lipase inhibitors. However, they are still inadequate because of either a gradual loss of efficacy or associated adverse effects such as diarrhoea or even concerns of increased risk of mental illness (Bray et al., 2016). Apart from these treatments that act to inhibit the intake of energy, promoting energy expenditure is another approach for the treatment of obesity and associated diseases (Lebrasseur, 2012; Wang et al., 2013; Bi et al., 2014; Pyrzak et al., 2015). For example, various interventions to increase energy expenditure such as exercise are generally beneficial for the treatment of these metabolic disorders (Jeremic et al., 2016; Reddon et al., 2016). In a search for novel pharmacological agents that act by this mechanism, we have recently identified a small molecule bouchardatine (Bou, MW: 289.3) that displays a potent efficacy in decreasing lipid accumulation in 3T3‐L1 adipocytes (Rao et al., 2015a,b).
Bou is isolated from the plant Bouchardatia neurococca, and it is synthesized from rutaecarpine through oxidative fission of the 7/8 bond in 7,8‐dehydrorutaecarpine in plant (Wattanapiromsakul et al., 2003). The rutaecarpine class of compounds have been reported to reduce obesity (Kim et al., 2009), and they can also be used to treat inflammatory or cardiovascular disease (Moon et al., 1999; Jia and Hu, 2010). Interestingly, our previous studies demonstrated the potential efficacy of Bou for inhibiting lipid accumulation in cultured cells by activating the AMP kinase (AMPK) pathway (Rao et al., 2015a,b). However, how Bou induces the activation of AMPK remains elusive. Furthermore, the implicated therapeutic effects for metabolic disorders have not been tested in vivo in a model that closely mimics the pathogenesis of obesity in the general human population.
Therefore, this study aimed to investigate the molecular mechanism of how Bou activates AMPK and to characterize its effects on obesity and associated metabolic disorders in high‐fat diet (HF)‐fed mice, a well‐defined animal model that mimics the pathogenesis of obesity in the general population (Yoneda et al., 2010; Zeng et al., 2015; Kim et al., 2016a,b). At the same time, we intend to assess its safety profile by evaluating a wide range of parameters in plasma and key tissues. Our results showed that Bou may activate the AMPK pathway by promoting liver kinase B1 (LKB1) translocation from the nucleus to the cytoplasm through sirtuin 1 (SIRT1) in cells. In chronic HF diet‐fed mice, Bou attenuated obesity and related metabolic disorders by increasing the thermogenic capacity in white adipose tissue (WAT) as well as brown adipose tissue (BAT). These therapeutic effects occurred without detectable indications of toxicity or adverse effects.
Methods
Studies in cultured cells
Mouse 3T3‐L1 fibroblast and human hepatoma HepG2 cells were purchased from Type Culture Collection (ATCC, Rockefeller, Maryland, USA). Cells were maintained in DMEM supplemented with 10% FBS, penicillin (100 U∙mL−1) and streptomycin (100 μg∙mL−1). The effects of Bou on adipogenesis and lipogenesis were assessed in 3T3‐L1 cells based on our recent reports (Chen et al., 2015; Rao et al., 2015a,b), while the effects on lipid accumulation in HepG2 cells were examined in the presence of oleic acid (0.5 mM). Primary brown adipocytes were isolated from C57BL/6J mice as described by Jeong et al. (2015) in a mixture of DMEM and Ham's F‐12 (1:1) medium containing 10% FBS.
LKB1 plasmid and siRNA transfection
3T3‐L1 cells (3 × 105) were seeded on a six‐well plate (Corning, NewYork, USA). After the cell density reached 80%, cells were transfected with 2 μg LKB1 plasmid or 50 nM siRNA/SIRT1 siRNA for 24 h using Lipofectamine 3000 according to the manufacturer's instructions. The transfection of LKB1 plasmid, the siRNAs and their negative controls was conducted according to the manufacturer's instructions (Invitrogen, California, USA).
Assessment of lipid accumulation in cells
Cells were fixed by 4% paraformaldehyde for 1 h at room temperature and then subjected to oil red O staining as described before (Rao et al., 2015a,b). The levels of cellular triglyceride (TG) were extracted as recently described (Rao et al., 2015a,b) and determined by using commercial Peridochrom TG GPO‐PAP kit following the manufacturer's instructions.
Determination of NAD+/NADH, AMP/ATP and SIRT1 deacetylase activity
The NAD +/NADH ratio and AMP/ATP ratio in 3T3‐L1 adipocytes were calculated from their levels determined by using respective Quantification Kits according to the manufacturer's instructions. The deacetylase activity of SIRT1 in 3T3‐L1 adipocytes and liver was determined with a SIRT1 Fluorometric Drug Discovery Kit. Resveratrol and SRT1720 were used as SIRT1 activators.
RT‐qPCR and western blotting
Total RNA was extracted from cells or tissue using RNAiso Plus as reported before (Chen et al., 2015; Rao et al., 2015a,b). The sequences of oligonucleotide primers synthesized by Generay (Shanghai, China) are shown in Supporting Information Table S1. The relative levels of mRNA expression were normalized to the loading control actin by Quantity One Software (Bio‐Rad Laboratory, California, USA). For western blotting, protein lysates from cells and tissues were prepared as previously described (Chen et al., 2015; Rao et al., 2015a,b) for immunoblotting using specific antibodies as described in Supporting Information Table S2. Densitometry analysis was performed using Quantity One Software (Bio‐Rad Laboratory, California, USA) and quantified to the loading control GAPDH or β‐actin.
Immunofluorescence and confocal microscopy
Confocal microscopy was performed in an inverted microscope (Leica Microsystems, solmos, Germany). Cells were washed with ice‐cold PBS (at pH 4) and then fixed with 4% paraformaldehyde for 10 min at room temperature. The fixed cells were blocked by 5% goat serum (Bioss, Beijing, China) in PBS for 2 h at room temperature and then stained with a specific first antibody (LKB1 1:50, pLKB1 1:50 or SIRT1 1:100) at 4°C overnight. After staining with an appropriate AlexaFluor‐conjugated secondary antibody for 2 h at 37°C, the images were captured (magnification, ×400). Imaging analyses were performed using an LSM 510 laser confocal microscope (Zeiss, Jena, Germany).
Determination of LKB1 acetylation
LKB1 antibody was incubated with 400 μL of 50% protein G/A beads in PBS (pH 8.2) at 4°C for 2 h. After washing with PBS three times and 0.2 M triethanolamine, the beads were resuspended in 20 mM dimethylpimelimidate‐HCl in 0.2 M triethanolamine for 30 min at room temperature. The resuspended beads were transferred to 50 mM Tris buffer (pH 7.5) containing acetylation LKB1 antibody (1:100) for 15 min. The beads cross‐linked with the antibody were washed three times with Tris buffer containing 5% Tween‐20 (TBST, pH 8.0) and then resuspended with a 2× diluted blocking solution [5% BSA in TBST (m/v)] at 4°C until used. Approximately 1 mg of extracted protein lysates were incubated with cross‐linked LKB1 beads overnight. The immunoprecipitated beads were washed four times with a lysis buffer and then eluted with 30 μL of 2× SDS sample buffer for immunoblotting.
Immunoprecipitation
Approximately 500 μg of extracted proteins were used for immunoprecipitation by incubation with 20 μL Protein A/G and 10 μg primary antibodies for 2 h at 4°C under constant shaking according to the manufacturer's instructions. The immunoprecipitates were washed three to four times with a cold PBS, and the immunoprecipitated beads were washed four times with a immunoprecipitation (IP) lysis buffer and then eluted with 20 μL neutralized buffer; supernatants were separated and subjected to immunoblotting. Immunoprecipitates were separated on a 12% SDS‐PAGE followed by immunoblotting against specific antibodies with non‐specific IgG as a control.
Mitochondrial membrane potential
Mitochondrial inner membrane potential was measured using a Rodamine‐123 probe. Briefly, confluent 3T3‐L1 cells were exposed to an adipogenic cocktail in the presence or absence of Bou. Then, cells were collected and incubated with 1 μM Rhodamine 123 probe in the PBS solution at 37°C for 30 min in the dark. The fluorescence was measured with a fluorescence spectrophotometer using filter settings at 507 nm Ex and 529 nm Em.
Studies in animals
Male C57BL/6J mice (aged 7–8 weeks) bred at the Laboratory Animal Centre of Sun Yat‐sen University (Guangzhou, China) were used for the study. All animal procedures were approved by the Sun Yat‐sen University Committee on Ethics for the Use of Laboratory Animals in accordance with the Animal Welfare Legislation of China. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015). The mice were kept at 22 ± 1°C on a 12 h light/dark cycle with free access to food and water. After 1 week of acclimatization to the environment of this study, mice were randomly assigned to receive either a chow diet (CH, with 70% calories from starch) or a HF diet (with 60% calories from fat) ad libitum for up to 16 weeks. Both CH and HF groups were randomly divided into two subgroups at the beginning of week 12 to receive the treatment with Bou or its vehicle. Bou was dissolved in normal saline containing 5% DMSO and 10% castor oil and injected i.p. (50 mg∙kg−1 every other day) for 5 weeks. The control subgroups were administered the same volume of the vehicle. Body weight and food intake were monitored daily.
A glucose tolerance test (GTT) was performed after 2 weeks of treatment with Bou based on our previous reports (Chen et al., 2015). Briefly, mice were fasted for a period of 6 h and then injected with glucose (2 g.Kg‐1, i.p.). Insulin tolerance tests (ITTs; insulin load 0.6 U per mouse, i.p.) were performed, after 6 h fasting, after 4 weeks of treatment with Bou. Blood samples for both GTT and ITT were taken from the tail vein under local anaesthesia with saline containing 2% lidocaine to ensure animal well‐being and that it was not subjected to significant discomfort.
At the end of the study, mice were fasted for 8 h and anaesthetized by an i.p. injection of ketamine/xylazine. After the mice were fully anaesthetized, the eyeball was removed to collect blood samples in a tube containing 1 mM EDTA (Hoff, 2000) for the measurement of relevant plasma parameters. After the blood samples were collected, the anaesthetized mice were killed by cervical decapitation; the tissues of interest were weighed, freeze‐clamped or fixed in 4% formaldehyde solution. Plasma levels of TG, total cholesterol, LDL cholesterol, HDL cholesterol, alkaline phosphatase, glutamic‐pyruvic transaminase and aspartate aminotransferase, total protein, urea, blood urea nitrogen, creatinine, total bilirubin and albumin were determined by an Olympus AU 600 auto‐analyser (Olympus, Japan). Plasma levels of insulin were detected using an ultra‐sensitive insulin elisa kit. Liver TGs were extracted by the method of Folchand and determined by a Peridochrom Triglyceride GPO‐PAP kit as previously described (Zeng et al., 2015).
Determination of mitochondrial DNA copy numbers
Quantification of mitochondrial DNA (mtDNA) copy numbers was achieved by PCR. Briefly, DNA was extracted from inguinal WAT using a DNeasy Blood and Tissue kit. The copy numbers of nuclear DNA (nDNA) and mtDNA were assessed by PCR using primers (Supporting Information Table S1) targeted toward the cytochrome C gene (for mtDNA) and 18S rRNA (for nDNA).
Histological analysis
The tissues fixed in 4% formaldehyde solution were embedded in paraffin after dehydration in a graded ethanol series (70–100%). Embedded samples were sectioned (4 μm thick) with a rotary microtome and stained with haematoxylin and eosin (H&E) for microscopic examination. Sections were viewed with a light microscope (Olympus) and photographed at ×100 magnification. The section of immunohistochemistry image was first labelled by an identification number code without the information of the grouping. The numbers and sizes of adipocytes of each slide were calculated with imagej software (USA) by Servicebio Company (Beijing, China). Quantification analysis was performed in six randomly selected fields per sample in a blinded manner.
Statistical analysis
Results are expressed as the mean ± SEM. Data between two groups were analysed by Student's t‐test using Graphpad Prism (Graphpad Software Inc, California, USA). Statistical analysis for multiple groups was performed by one‐way ANOVA followed by Tukey's HSD post hoc tests. A P value of <0.05 was considered statistically significant. The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015).
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015a,b).
Results
Bou activates SIRT1 deacetylase during the stimulation of AMPK
Bou has been shown to decrease TG accumulation and activate AMPK in 3T3‐L1 adipocytes (Rao et al., 2015a,b). We considered it important to examine whether SIRT1 may be involved because this deacetylase is often activated during AMPK activation. Additionally, stimulation of SIRT1, such as with Res, can also reduce cellular lipid accumulation (Yoneda et al., 2010; Gu et al., 2014). As expected, Res dose‐dependently increased the activity of SIRT1 and decreased TG accumulation in 3T3‐L1 adipocytes while increasing the ratio of NAD+/NADH (Supporting Information Figure S1). Similar effects were also observed in HepG2 cells (Supporting Information Figure S1C, D). We therefore used 3T3‐L1 adipocytes to investigate the effects of Bou on NAD+/NADH ratio and SIRT1 activity in relation to its ability to reduce TG accumulation. As shown in Figure 1, Bou treatment increased the NAD+/NADH ratio and the deacetylase activity of SIRT1 along with its effect in reducing TG accumulation. Interestingly, the effects of Bou on TG levels and the phosphorylation of AMPKα as well as acetyl CoA carboxylase (ACC) were blunted when SIRT1 activity was inhibited with nicotinamide (NAM), a SIRT1 partial inhibitor (Li et al., 2015). These data suggest that Bou may stimulate the AMPK signalling pathway via activating the deacetylase SIRT1.
Figure 1.
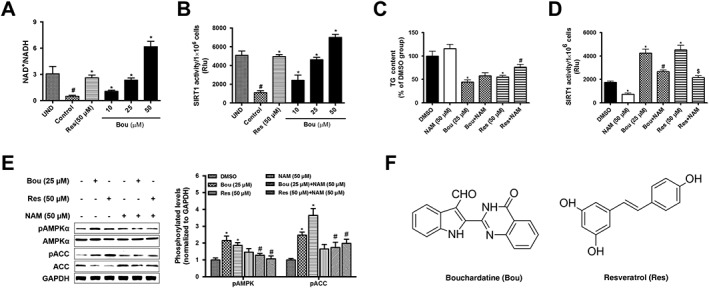
Bouchardatine increases the activity of SIRT1 and activates the AMPK pathway in 3T3‐L1 adipocytes. Confluent 3T3‐L1 pre‐adipocytes were incubated in an adipogenic cocktail (IBMX, dexamethasone, and insulin) for 9 days in the presence or absence of Bou and Res respectively. After the treatment, NAD+/NADH (A), deacetylase activity of SIRT1 (B) and TG levels (C) were determined. * P < 0.05 compared with UND (undifferentiated) group; #P < 0.05 compared with DMSO treatment group. The activity of SIRT1 (D) was determined in the presence or absence of NAM (SIRT1 inhibitor). * P < 0.05 compared with DMSO group; #P < 0.05 compared with Bou group; $ P < 0.05 compared with Res group. The AMPK signalling pathway was assessed by determining the levels of key‐related proteins (E). *P < 0.05 compared with DMSO group; #P < 0.05 compared with Bou‐ or Res‐treated group. (F) Chemical structures of Res and Bou. Data are expressed as means ± SEM from five independent experiments.
Bou phosphorylates LKB1 during the stimulation of AMPK
As LKB1 is a key kinase controlling AMPK activity (Woods et al., 2003), we next examined the effects of Bou on this AMPK upstream kinase. As shown in Figure 2, Bou dose‐ and time‐dependently increased both the phosphorylation and total levels of LKB1 during the activation of AMPK. These effects were abolished by the SIRT1 inhibitor NAM. In contrast, overexpression of LKB1 enhanced the effects of Bou on the activation of AMPK and TG content. These results suggest that the activation of AMPK by Bou is mediated by LKB1.
Figure 2.
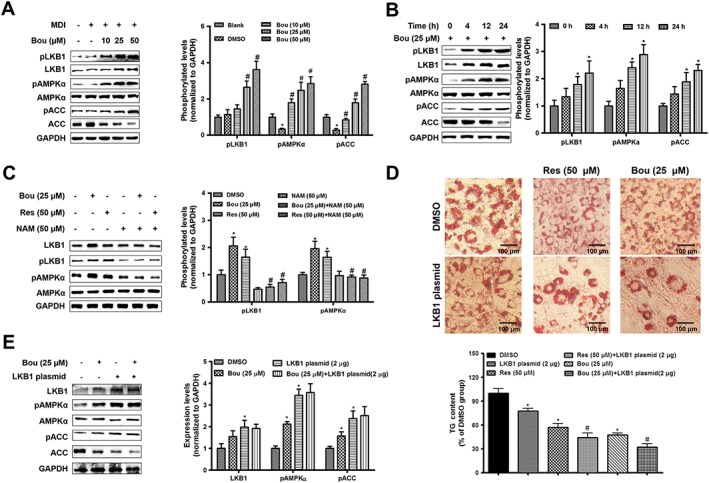
Bouchardatine activates the AMPK signalling pathway via LKB1 in 3T3‐L1 adipocytes. Confluent 3T3‐L1 pre‐adipocytes were exposed to an adipogenic cocktail (IBMX, dexamethasone, and insulin) for 9 days in the presence or absence of Bou. (A) Dose‐dependent effect of Bou on the LKB1–AMPK pathway. *P < 0.05 compared with undifferentiated group; #P < 0.05 compared with DMSO group. (B) Time‐dependent effect of Bou on the LKB1–AMPK pathway. *P < 0.05 compared with DMSO group. (C) Effect of Bou in the presence or absence of NAM on the expression of the LKB1–AMPK pathway. *P < 0.05 compared with DMSO group; #P < 0.05 compared with Res or Bou group. (D) Effect of LKB1 on the TG levels. LKB1 plasmid transfection as described in the Methods section. Original magnification, 100×. Scale bar, 100 μm. * P < 0.05 compared with DMSO group; #P < 0.05 compared with Bou or Res group. (E) Effect of LKB1 transfection on the AMPK pathway. *P < 0.05 compared with DMSO group. Data were quantified from six independent experiments.
Bou stimulates the AMPK signalling pathway via the SIRT1–LKB1 complex
It has been suggested that deacetylation of LKB1 by SIRT1 promotes the phosphorylation of AMPK via facilitating the translocation of LKB1 from the nucleus to the cytosol (Hou et al., 2008; Yang et al., 2014). We analysed the distribution of LKB1 between the cytosolic and nucleus using their respective loading markers actin and lamin B (Luo, et al., 2015; Moreno, et al., 2016). Consistent with this notion, Bou significantly reduced the acetylation of LKB1 and increased its levels in the cytosol in a similar pattern as Res (Figure 3A, B). Immunofluorescence assay also showed that Bou induced LKB1 translocation to the cytosol and increased its phosphorylation (Figure 3C). Res is a non‐specific activator of SIRT1 (Borra, et al., 2005); we further treated cells with SRT1720, a specific activator of SIRT1. As shown in Supporting Information Figure S2, similar to Bou, SRT1720 treatment increased the levels of LKB1 predominantly in the cytosol relative to the nucleus. IP assay showed an increase in the protein levels of LKB1 co‐precipitated with SIRT1 antibody after treatment with Bou. Similarly, IP with LKB1 antibody also showed an increase in SIRT1 levels after the incubation with Bou (Figure 3D). Furthermore, a knockdown of SIRT1 by siRNA decreased the acetylation of LKB1 and phosphorylation of both LKB1 and AMPKα (Figure 3E). These results suggest that Bou induces the translocation of LKB1 from the nucleus to the cytosol as a result of its deacetylation via activating SIRT1.
Figure 3.
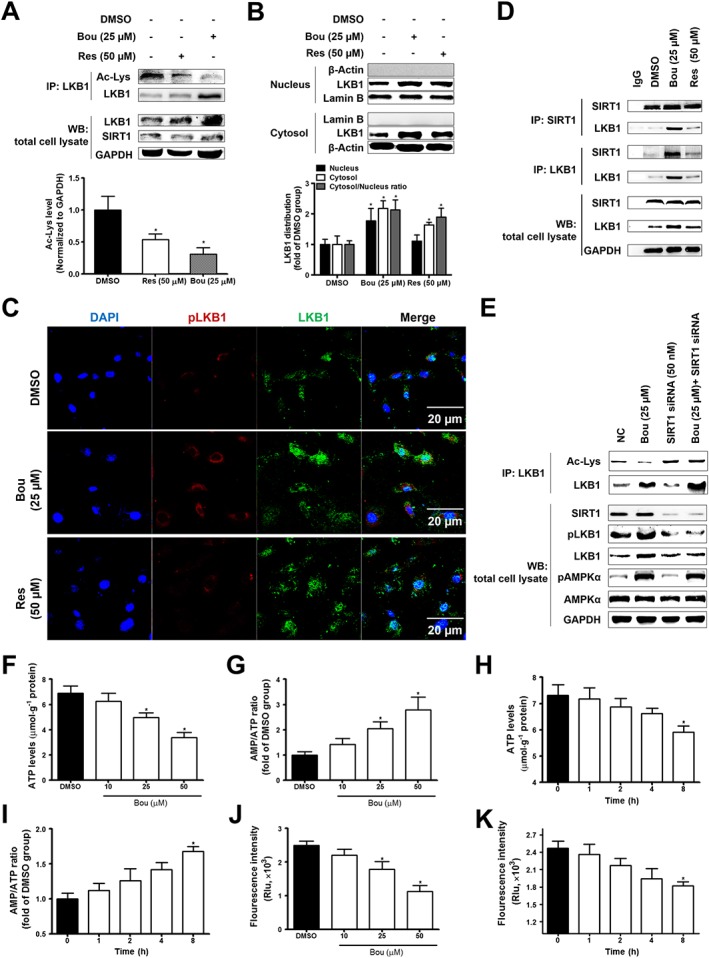
Bouchardatine induces LKB1 translocation via modulating SIRT1 activity in 3T3‐L1 adipocytes. Confluent 3T3‐L1 pre‐adipocytes were incubated in MDI (a mixture of IBMX, dexamethasone, and insulin) for 9 days in the presence or absence of Bou. After the treatment, LKB1 acetylation (A) and distribution (B, C) were determined. *P < 0.05 compared with DMSO group. Interaction between SIRT1 and LKB1 were assayed by co‐immunoprecipitation (D). (E) Protein levels of SIRT1–LKB1–AMPK pathway after SIRT1 siRNA (50 nM) transfection. The Bou‐treated 3T3‐L1 were collected for ATP levels, AMP/ATP ratio determined (F–I) and mitochondrial membrane potential assay (J, K) as described in the Methods section. *P < 0.05 compared with DMSO group. Data were quantified from five independent experiments. WB, western blot; NC, negative control group.
Bou functions as an uncoupler
To elucidate the mechanism of the activation of the SIRT1–LKB1–AMPK axis by Bou, we examined the effect of Bou on the ATP levels and AMP/ATP ratio in 3T3‐L1 adipocytes. As expected, treatment with Bou decreased ATP levels, thus an increase in the AMP/ATP ratio in a dose‐ and time‐dependent manner (Figure 3F–I). Moreover, Bou decreased mitochondrial membrane potential (Figure 3J, K). These results suggest that Bou may act as an uncoupler of mitochondrial oxidative phosphorylation to inhibit ATP synthesis.
Bou attenuates metabolic disorders in chronic HF‐fed mice
To assess whether Bou exerts in vivo therapeutic effects on the metabolic syndrome devoid of the influence on calorie intake, we administered Bou (50 mg∙kg−1, i.p.) to HF‐fed mice with matched caloric intake to the vehicle controlled HF group. As expected, HF‐induced mice gained more significantly body weight, but this was progressively returned to the level of CH‐fed mice after 5 weeks of administration with Bou (Figure 4A, B). The increased plasma levels of free fatty acid, carbohydrate, TG, LDL, glucose and insulin were also significantly reduced (Table 1). Both GTT and ITT were significantly improved in HF mice after the treatment with Bou (Figure 4C, D). No major effect was observed on these parameters in CH‐fed mice after Bou administration, except for a mild increase in plasma levels of glucose.
Figure 4.
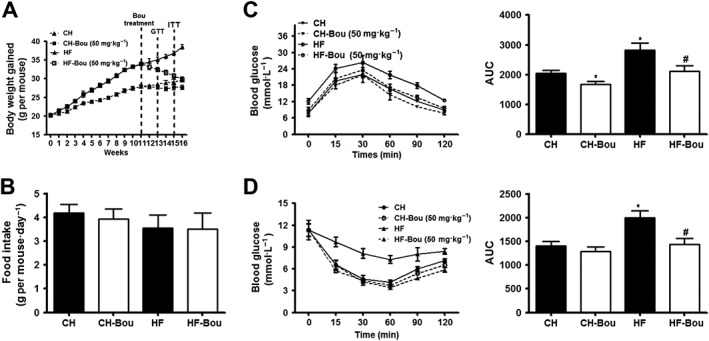
Bouchardatine reduces body weight gain and dyslipidaemia in high‐fat diet‐fed mice. Mice were fed a HF diet for 16 weeks, and Bou was administered in the last 5 weeks at a dose of 50 mg·kg−1 each other day by i.p. injection. (A) Body weight changes over time. (B) Food intake. (C) Glucose tolerance test. (D) Insulin resistance test. * P < 0.05 compared with CH group; # P < 0.05 compared with HF group. (n = 8 mice in each group).
Table 1.
Chronic effects of Bou on metabolic parameters in mice
| CH | CH‐Bou | HF | HF‐Bou | |
|---|---|---|---|---|
| Plasma FFA (mM) | 1.1 ± 0.06 | 0.9 ± 0.03 | 2.1 ± 0.15* | 1.3 ± 0.16‡ |
| Plasma TG (mM) | 1.2 ± 0.37 | 1.1 ± 0.22 | 2.5 ± 0.2* | 1.5 ± 0.3‡ |
| Plasma CHO (mM) | 2.1 ± 0.1 | 1.6 ± 0.1* | 2.2 ± 0.2 | 2.0 ± 0.4 |
| Plasma HDL (mM) | 1.7 ± 0.1 | 1.9 ± 0.3 | 1.47 ± 0.14* | 1.6 ± 0.2 |
| Plasma LDL (mM) | 0.15 ± 0.02 | 0.14 ± 0.03 | 0.3 ± 0.04* | 0.2 ± 0.04 |
| Plasma glucose (mM) | 4.4 ± 0.3 | 4.7 ± 0.5 | 10.8 ± 1.4* | 6.8 ± 1.4‡ |
| Plasma insulin (ng·mL−1) | 2.2 ± 0.3 | 2.0 ± 0.4 | 3.6 ± 0.2* | 2.7 ± 0.4‡ |
| Plasma AST (U·L−1) | 101 ± 13 | 110 ± 11 | 135 ± 6* | 125 ± 8 |
| Plasma ALT (U·L−1) | 25.8 ± 2.9 | 25.90 ± 5.2 | 32.2 ± 5.3 | 28.8 ± 6.9 |
| Plasma ALP (U·L−1) | 49.7 ± 7.0 | 42.2 ± 4.3 | 62.8 ± 8.3 | 54.4 ± 3.3‡ |
| Plasma UREA (mM) | 12.7 ± 1.2 | 11.2 ± 1.7 | 13.1 ± 2.2 | 13.0 ± 2.7 |
| Plasma BUN (mM) | 5.3 ± 0.6 | 4.8 ± 0.9 | 6.9 ± 1.5 | 6.1 ± 0.7 |
| Plasma CR (μM) | 78.8 ± 7.9 | 47.5 ± 7.9 | 99.8 ± 6.1 | 74.6 ± 11.6‡ |
| Plasma TBIL (mM) | 1.2 ± 0.3 | 1.2 ± 0.5 | 1.1 ± 0.4 | 1.3 ± 0.3 |
| Plasma TP (mM) | 49.6 ± 1.8 | 52.1 ± 4.2 | 62.9 ± 4.4 | 54.2 ± 5.2 |
| Plasma ALB (mM) | 19.7 ± 0.6 | 19.7 ± 1.5 | 19.1 ± 1.8 | 18.5 ± 1.6 |
| Liver weight (g) | 1.5 ± 0.5 | 1.5 ± 0.2 | 1.9 ± 0.2* | 1.6 ± 0.2‡ |
| eWAT (g) | 0.32 ± 0.05 | 0.27 ± 0.05 | 0.82 ± 0.17 | 0.51 ± 0.15 |
| sWAT (g) | 0.24 ± 0.03 | 0.22 ± 0.08 | 0.60 ± 0.09 | 0.34 ± 0.06 |
| Total fat mass (g) | 0.56 ± 0.005 | 0.49 ± 0.03 | 1.42 ± 0.13* | 0.85 ± 0.09‡ |
Plasma samples were collected after 5 weeks of treatment with Bou and analysed by automatic biochemical analyser.
P < 0.05 compared with CH group.
P < 0.05 compared with HF mice (n = 8 per group).
TC, total cholesterol; LDL‐c, LDL‐cholesterol; HDL‐c, HDL‐cholesterol; ALP, alkaline phosphatase; ALT, glutamic‐pyruvic transaminase; AST, aspartate aminotransferase; TP, total protein; BUN, blood urea nitrogen; CR, creatinine; TBIL, total bilirubin; ALB, albumin; FFA, free fatty acid; CHO, carbohydrate.
Next, we measured relevant plasma parameters and tissue weights to examine whether there was any indication of toxicity. As shown in Table 1, Bou treatment did not affect any of the parameters indicative of toxicity in the liver or kidney. There was no major difference in Bou–treated mice regarding the size or appearance of the tissues (Supporting Information Figure S3A) or the ratios of relevant tissues to body weight in Bou‐treated mice (Supporting Information Figure S3B). Histological examination of brain, heart, kidney and pancreas with H&E staining in Bou‐treated mice also showed similar patterns to those of the control mice (Supporting Information Figure S3C).
Bou reduces body fat associated with inhibition of lipid synthesis and browning in WAT
Consistent with the reduction in body weight, the HF‐fed mice treated with Bou displayed marked reduction in total WAT (~40%) (Table 1) and the size of adipocytes WAT (Figure 5A). These effects were associated with increased phosphorylation of protein LKB1, AMPKα and ACC (Figure 5B) and decreased the levels of protein SREBP‐1c (Sterol regulatory element binding protein‐1c), ACC, fatty acid synthase (FAS) and stearoyl CoA dehydrogenase‐1 (SCD‐1; Figure 5C). Furthermore, chronic treatment with Bou increased the copy numbers of mtDNA (Figure 5D and Supporting Information Figure S4A), mitochondrial transcription factor [PPAR γ coactivator‐1 α (PGC‐1α)] and fatty acid oxidation‐related enzymes long‐chain acyl‐CoA synthase (ACSL) and carnitine palmitoyl transferase‐1b (Figure 5E). There was a robust increase in the expression levels of uncoupling protein 1 (UCP1) at both transcriptional and translational levels (Figure 5F, G). These results suggest that the inhibition of fatty acid synthesis and browning in WAT are involved in the anti‐obesity effect of Bou via activation of the SIRT1–LKB1–AMPK pathway.
Figure 5.
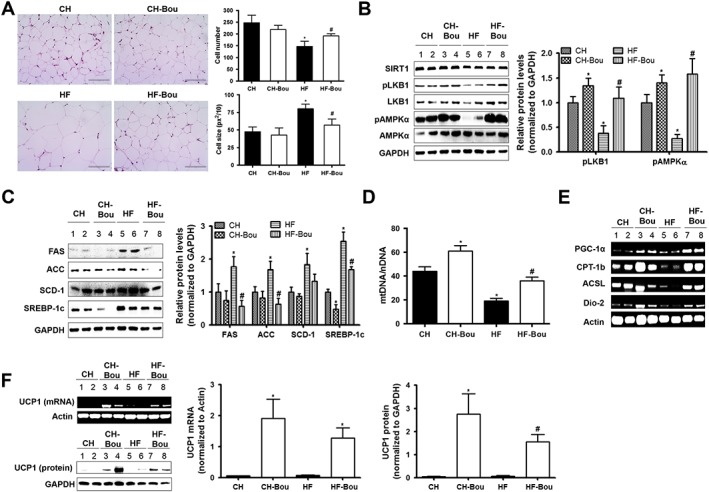
Bouchardatine reduces fat mass associated with the browning in WAT. (A) Representative images of H&E staining in epididymal WAT after 5 weeks of treatment with Bou. Original magnification, 400×. Scale bar, 400 μm. (B) Effects on the protein levels of SIRT1, LKB1 and AMPK. (C) Effects on the expression levels of fatty acid synthesis‐related proteins: FAS, ACC, SCD‐1 and SREBP‐1c. (D) Effects on the ratio of mtDNA/nDNA. (E) Effects on the expression of proteins related to the browning. (F) Effects on the levels of mRNA and protein of UCP1. * P < 0.05 compared with CH group; #P < 0.05 compared with HF group (n = 8 mice in each group). CPT‐1b, carnitine palmitoyl transferase‐1b.
Bou reverses the reduced thermogenic capacity of BAT in HF mice
In HF‐fed mice, brown adipocytes was full of enlarged lipid droplets and showed reductions in the markers of thermogenic capacity PGC‐1α, UCP1 and mtDNA content (Figure 6). Bou administration reversed the size of brown adipocytes in HF mice to the similar levels of CH mice and increased the number of mtDNA copies (Figure 6B and Supporting Information Figure S4B). Along with these changes, the phosphorylation of AMPKα and the contents of both PGC‐1α and UCP1 were increased (Figure 6C–E). In isolated primary brown adipocytes, incubation with Bou also increased the levels of PGC‐1α and UCP1 and the phosphorylation of AMPKα (Figure 6F–G).
Figure 6.
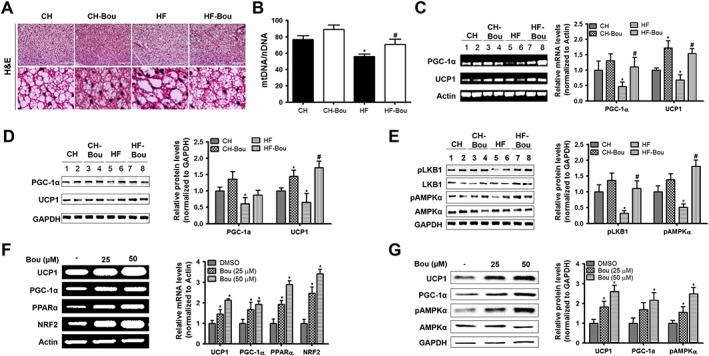
Bouchardatine ameliorates HF‐induced suppression of thermogenic capacity in BAT. (A) Representative images of H&E staining in interscapular BAT after 5 weeks of treatment with Bou. Original magnification, 400×. Scale bar, 400 μm. (B) Effects on the ratio of mtDNA/nDNA. (C, D) Effects on the expressions of PGC‐1α and UCP1. (E) Effects on the protein levels of SIRT1, LKB1 and AMPK. * P < 0.05 compared with CH group; #P < 0.05 compared with HF group (n = 8 mice in each group). (F) Expression of browning markers in cultured primary brown adipocytes after incubation with Bou for 24 h. (G) Expression of UCP1 and AMPK pathway‐related proteins in cultured primary brown adipocytes. *P < 0.05 compared with DMSO group. Data were quantified from five independent experiments. NRF2, nuclear factor 2.
Bou ameliorates hepatic steatosis and activates the SIRT1–LKB1–AMPK pathway
Bou treatment markedly reversed the increased hepatic TG content in HF mice (Figure 7A). The ratio of NAD+/NADH was increased after administered with Bou (Figure 7B) associated with increased deacetylation activity of SIRT1 (Figure 7C). Moreover, Bou treatment decreased ATP content (thus increased the AMP/ATP ratio) (Figure 7D–E) and increased the phosphorylated and total LKB1 as well as the phosphorylation of AMPKα and ACC (Figure 7F). These results demonstrate that Bou ameliorates HF‐induced hepatic steatosis in association with the activation of the SIRT–AMPK pathway.
Figure 7.
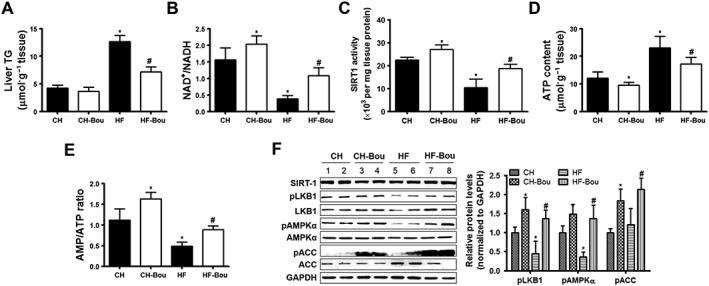
Bouchardatine ameliorates HF‐induced lipid accumulation and activates the LKB1–AMPK pathway in the liver. Liver TG levels and phosphorylated levels of LKB1–AMPK–ACC were measured after Bou treatment. (A) TG levels. (B) NAD+/NADH ratio. (C) SIRT1 activity. (D, E) ATP levels and AMP/ATP ratio. (F) Expression of LKB1–AMPK signalling pathway. * P < 0.05, compared with CH group; # P < 0.05 compared with HF group (n = 8 mice in each group).
Discussion
Repurposing therapeutic agents is an attractive strategy for the identification of new drugs which are more applicable to metabolic disease, as advantages can be taken from the existing knowledge about the safety and molecular mode of actions (Zhang et al., 2014; Turner et al., 2016). Using this approach, the present study has identified Bou as a potential novel agent for the treatment of obesity and associated metabolic disorders through a distinctive mechanism. Bou promotes energy metabolism in cells as indicated by increasing the ratio of AMP/ATP and particularly the ratio of NAD+/NADH. The latter activates SIRT1, which deacetylates LKB1 to facilitate the translocation of LKB1 into the cytoplasm. This stimulates the AMPK pathway, which reduces lipid accumulation by inhibiting lipid synthesis and promoting fatty acid oxidation. In HF mice, Bou is able to reduce body weight gain, plasma levels of lipids, fat mass, hepatic steatosis and glucose intolerance. These effects were associated with an activation of the SIRT1–LKB1–AMPK axis in adipose tissues and liver. Finally, our assessment of the parameters for liver and kidney functions suggest that Bou treatment is safe without organ toxicity or other detectable adverse effects.
Bou is a natural small molecule isolated from the fruits of Evodia rutaecarpa. This plant was selected using the approach of drug repurposing (Turner et al., 2016) based on its traditional use for the treatment of inflammatory and cardiovascular diseases (Zhao et al., 2015). We selected Bou as a leading compound for the present study because our previous studies showed its potent efficacy in reducing lipid accumulation in 3T3‐L1 cells by stimulating AMPK (Rao et al., 2015a,b).
As AMPK can be activated by increased AMP upon energy depletion (Russo et al., 2015; Evans et al., 2016), we first examined the energy status of the cell. Our results suggest that Bou acts like an uncoupler to decrease ATP levels. This leads to an increase in the AMP/ATP ratio, which activates AMPK. One of the crucial upstream kinases controlling the activity of AMPK is LKB1, which is mainly localized in the nucleus under basal conditions (Lee et al., 2012). This AMPK upstream kinase is translocated to the cytoplasm by interacting with two adaptor proteins STE20‐related adaptor and MO25 to phosphorylate AMPK during energy stress. Thus, the LKB1–AMPK pathway is coupled with cellular energy status (Woods et al., 2003; Wang et al., 2012; Ritho et al., 2015; Hu and Liu, 2016). The present study found that Bou activates LKB1–AMPK pathway involving the translocation of LKB1 from the nucleus to the cytoplasm as a result of the increased AMP/ATP ratio.
We further investigated the relationship between SIRT1 and AMPK during the treatment with Bou because SIRT1 can modulate the activity of LKB1 by deacetylating at the lysine residue (K48) (Lan et al., 2008). SIRT1 is a highly conserved NAD+‐dependent deacetylases, and its expression and enzymatic activity are enhanced during an increase in the NAD+/NADH ratio (Tong and Kraus, 2010). Similar to Res (Yoneda et al., 2010; Andrade et al., 2014; Li et al., 2015), Bou activates SIRT1 to acetylate LKB1 via increased NAD+/NADH ratio. This interpretation is supported by the inhibition of LKB1 by the SIRT1‐specific inhibitor SRT1720 and the knockdown of SIRT1 using siRNA. Consistent with these data in cultured cells, administration of Bou to mice was found to increase the NAD+/NADH ratio and SIRT1 activity and lead to the AMPK pathway activation in both adipose tissues and the liver. Collectively, these results suggest that Bou activates the SIRT1–LKB1–AMPK signalling pathway.
It has been well known that activation of the AMPK pathway inhibits lipid synthesis but promotes mitochondrial oxidation of fatty acids (Russo et al., 2015; Kim et al., 2016a,b; Lin et al., 2015). Consistent with these dual effects on lipid metabolism, Bou were found to reduce lipid accumulation (indicated by the decrease in TG content) in 3T3‐L1 adipocytes, HepG2 cells and the liver of HF mice. Because chronic administration of Bou ameliorated obesity without inhibition of calorie intake, it is likely that this metabolic effect results from an increase in energy expenditure. As activated AMPK pathway has been suggested to induce the transformation of WAT to BAT (termed as ‘browning’) to develop the thermogenic function (Zhang et al., 2015), we further examined the chronic effects of Bou on the expression of UCP1 in both WAT and BAT in mice.
UCP1 is a mitochondrial protein that dissipates energy produced from the oxidation of NADH as heat rather than driving the synthesis of ATP to store the energy (Poher et al., 2015; Poekes et al., 2015). Indeed, the expression of UCP1 in WAT of HF mice was increased following the treatment with Bou. UCP1 is a hallmark of BAT, and recent studies have revealed that this protein is induced along with increase in the content of mitochondria in WAT during browning. This transformation is mediated by PGC‐1α and ACSL under the regulation of the SIRT1–AMPK axis (Shan et al., 2013; Wang et al., 2015; Yoneshiro and Saito, 2015; Zhang et al., 2015; Baskaran et al., 2016). Our results also showed significant increases in mtDNA copies and the key regulators (including PGC‐1α and nuclear factor 2) for UCP1 and mtDNA expression. Therefore, the increased expression of UCP1 and mtDNA indicates that the browning of WAT is likely to be involved in the effects of Bou in reducing obesity and lipid accumulation.
In addition, Bou reversed the reductions in UCP1 and mtDNA in the BAT of HF mice by activating the SIRT1–AMPK axis to maintain its thermogenic capacity. The results from isolated primary cultured brown adipocytes confirm the direct effects of Bou on expression of UCP1 via the SIRT1–AMPK pathway. These findings indicate that both WAT and BAT are likely to be important sites for the effects of Bou to reduce obesity in vivo. Increased thermogenic capacity may also occur in the liver following chronic administration of Bou as suggested by its effects on the SIRT1–AMPK pathway as well as hepatic steatosis. As a result of reduced obesity, the associated dyslipidemia and glucose tolerance were ameliorated in the HF mice after the treatment with Bou.
Liver and kidney are sensitive to drug toxicity as they are important organs for drug deposition and metabolism. In the past, several anti‐obesity and anti‐diabetic drugs were withdrawn from the clinical use due to the concerns about their potential toxicity in these organs (Padwal and Majumdar, 2007). To examine whether or not Bou may have these undesirable effects, we analysed the plasma parameters indicative of the function of the liver and kidney. These results showed no significant changes in any of these parameters, suggesting that Bou is safe at the dose which produces these metabolic effects.
In summary, the present study shows that Bou is a promising therapeutic agent for obesity and associated metabolic disorders through a mechanism different from the current anti‐diabetic drug metformin which activates AMPK by inhibiting mitochondrial respiration (Luengo et al., 2014). Our findings suggest the SIRT1–LKB1–AMPK browning mechanism is likely to underpin the observed therapeutic effects of Bou against obesity and associated metabolic disorders (as depicted in Figure 8). These findings together with its safety profile suggest that Bou may be repurposed for the treatment of obesity and associated metabolic disorders including hepatic steatosis and type 2 diabetes.
Figure 8.
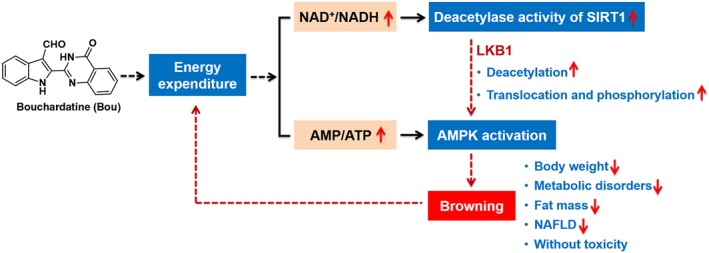
Proposed mechanism by which bouchardatine (Bou) ameliorates obesity associated metabolic disorders.
Author contribution
Z.‐S.H., L.‐Q.G. and J.‐M.Y. conceived and designed the study. Y.R. and Y.‐T.L. performed the screening and mechanism study of Bou in vitro and in vivo. H.Y., L.G., Z.X. and H.L. performed compound synthesis. Z.‐S.H. and J.‐M.Y. provided reagents, materials and analysis tools. Y.R., J.‐M.Y. and Z.‐S.H. wrote the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Table S1 Primers sequences used for PCR.
Table S2 Information of Antibody.
Figure S1 Effect of resveratrol (Res) on the TG levels and SIRT1 activity in cells. (A) Effect of Res on the ratio of NAD+/NADH in 3 T3‐L1 adipocytes. *P < 0.05, **P < 0.01, ***P < 0.001 compared with UND group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with DMSO treatment group. (B) Effect of Res on the deacetylase activity of SIRT1 in 3 T3‐L1 adipocytes. *P < 0.05, **P < 0.01, ***P < 0.001 compared with UND group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with DMSO treatment group. (C‐D) TG content analysis by TG assays and Oil‐Red O staining. *P < 0.05, **P < 0.01, ***P < 0.001 compared with DMSO group. Data are expressed as means ± standard errors from 5 independent experiments.
Figure S2 SRT1720 increases LKB1 translocation to cytosol in 3 T3‐L1 adipocytes. Confluent 3 T3‐L1 pre‐adipocytes were exposed to adipogenic cocktail (MDI) for consecutive 9 days in the presence or absence of SRT1720 treatment. After treatment, LKB1 in cytosol and nucleus were extracted and determined by western blot, GAPDH and Lamin B were loaded as loading control. *P < 0.05, compared with DMSO group.
Figure S3 Toxicity analysis of bouchardatine in vivo. (A) Appearance of tissue was captured. (B‐C) Tissue/body weight ratio. (C) Representative H&E staining from tissue sections after 5 weeks of Bou treatment. Original magnification, 100×. Scale bar, 100 μm.
Figure S4 Expression levels of Cytochrome C gene (for mtDNA) and 18S rRNA (for nuclear DNA) in eWAT (A) and BAT (B) of mice.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81330077, 91213302 and 81273433). Special Fund was also from Science and Technology Development in Guangdong Province (2016A020217004) and Guangdong Provincial Key Laboratory of Construction Foundation (2011A060901014). Y. R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Rao, Y. , Yu, H. , Gao, L. , Lu, Y.‐T. , Xu, Z. , Liu, H. , Gu, L.‐Q. , Ye, J.‐M. , and Huang, Z.‐S. (2017) Natural alkaloid bouchardatine ameliorates metabolic disorders in high‐fat diet‐fed mice by stimulating the sirtuin 1/liver kinase B‐1/AMPK axis. British Journal of Pharmacology, 174: 2457–2470. doi: 10.1111/bph.13855.
Contributor Information
Ji‐Ming Ye, Email: jiming.ye@rmit.edu.au.
Zhi‐Shu Huang, Email: ceshzs@mail.sysu.edu.cn.
References
- Alexander SPH, Fabbro D, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015a). The Concise Guide to PHARMACOLOGY 2015/16: Enzymes. Br J Pharmacol 172: 6024–6109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander SPH, Kelly E, Marrion N, Peters JA, Benson HE, Faccenda E et al. (2015b). The Concise Guide to PHARMACOLOGY 2015/16: Transporters. Br J Pharmacol 172: 6110–6202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade JM, Frade AC, Guimarães JB, Freitas KM, Lopes MT, Guimarães AL et al. (2014). Resveratrol increases brown adipose tissue thermogenesis markers by increasing SIRT1 and energy expenditure and decreasing fat accumulation in adipose tissue of mice fed a standard diet. Eur J Nutr 53: 1503–1510. [DOI] [PubMed] [Google Scholar]
- Baskaran P, Krishnan V, Ren J, Thyagarajan B (2016). Capsaicin induces browning of white adipose tissue and counters obesity by activating TRPV1 dependent mechanism. Br J Pharmacol 173: 2369–2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi P, Shan T, Liu W, Yue F, Yang X, Liang XR et al. (2014). Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med 20: 911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borra MT, Smith BC, Denu JM (2005). Mechanism of human SIRT1 activation by resveratrol. J Biol Chem 280: 17187–17195. [DOI] [PubMed] [Google Scholar]
- Bray GA, Fruhbeck G, Ryan DH, Wilding JP (2016). Management of obesity. Lancet 387: 1947–1956. [DOI] [PubMed] [Google Scholar]
- Carneiro IP, Elliott SA, Siervo M, Padwal R, Bertoli S, Battezzati A et al. (2016). Is obesity associated with altered energy expenditure? Adv Nutr 7: 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YC, Zeng XY, He Y, Wang B, Zhou H, Chen JW et al. (2015). Rutaecarpine analogues reduce lipid accumulation in adipocytes via inhibiting adipogenesis/lipogenesis with AMPK activation and UPR suppression. ACS Chem Biol 10: 1570–1571. [DOI] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans RM, Wall CE, Yu RT, Atkins AR, Downes M, Evans RM (2016). Nuclear receptors and AMPK: can exercise mimetics cure diabetes? J Mol Endocrinol 57: R49–R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldzieher MA, Goldzieher JW (1952). Past and present concepts of energy metabolism and obesity. Am J Dig Dis 19: 69–72. [DOI] [PubMed] [Google Scholar]
- Gu XS, Wang ZB, Ye Z, Lei JP, Li L, Su DF et al. (2014). Resveratrol, an activator of SIRT1, upregulates AMPK and improves cardiac function in heart failure. Genet Mol Res 13: 323–335. [DOI] [PubMed] [Google Scholar]
- Hoff J (2000). Methods of blood collection in the mouse. Lab Anim 29: 47–53. [Google Scholar]
- Hou XY, Xu SQ, Maitland‐Toolan KA, Sato K, Jiang B, Ido Y et al. (2008). SIRT1 regulates hepatocyte lipid metabolism through activating AMP‐activated protein kinase. J Biol Chem 283: 20015–20026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M, Liu B (2016). Resveratrol via activation of LKB1‐AMPK signaling suppresses oxidative stress to prevent endothelial dysfunction in diabetic mice. Clin Exp Hypertens 38: 381–387. [DOI] [PubMed] [Google Scholar]
- Jeremic N, Chatuverdi P, Tyagi SC (2016). Browning of white fat: novel insight into factors, mechanisms and therapeutics. J Cell Physiol 232: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong MY, Kim HL, Park J, Jung Y, Youn DH, Lee JH et al. (2015). Rubi Fructus (Rubus coreanus) activates the expression of thermogenic genes in vivo and in vitro . Int J Obes (Lond) 39: 456–464. [DOI] [PubMed] [Google Scholar]
- Jia S, Hu C (2010). Pharmacological effects of rutaecarpine as a cardiovascular protective agent. Molecules 15: 1873–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Park M, Jeong J, Kim H, Lee SK, Lee E et al. (2016a). Cholinesterase inhibitor donepezil increases mitochondrial biogenesis through AMP‐activated protein kinase in the hippocampus. Neuropsychobiology 73: 81–91. [DOI] [PubMed] [Google Scholar]
- Kim J, Kim MH, Choi YY, Hong J, Yang WM (2016b). Inhibitory effects of Leonurus sibiricus on weight gain after menopause in ovariectomized and high‐fat diet‐fed mice. J Nat Med 70: 522–530. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Lee SJ, Lee S, Chae S, Han MD, Mar W et al. (2009). Rutecarpine ameliorates bodyweight gain through the inhibition of orexigenic neuropeptides NPY and AgRP in mice. Biochem Biophys Res Commun 389: 437–442. [DOI] [PubMed] [Google Scholar]
- Kusminski CM, Bickel PE, Scherer PE (2016). Targeting adipose tissue in the treatment of obesity‐associated diabetes. Nat Rev Drug Discov 15: 639–660. [DOI] [PubMed] [Google Scholar]
- Lan F, Cacicedo JM, Ruderman N, Ido Y (2008). SIRT1 Modulation of the acetylation status, cytosolic localization, and activity of LKB1. J Biol Chem 283: 27628–27635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebrasseur NK (2012). Building muscle, browning fat and preventing obesity by inhibiting myostatin. Diabetologia 55: 13–17. [DOI] [PubMed] [Google Scholar]
- Lee J, Hong S‐W, Rhee E‐J, Lee W‐Y (2012). GLP‐1 receptor agonist and non‐alcoholic fatty liver disease. Diabetes Metab J 36: 262–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Dou X, Li S, Zhang X, Zeng Y, Song Z (2015). NAMtinamide ameliorates palmitate‐induced ER stress in hepatocytes via cAMP/PKA/CREB pathway‐dependent Sirt1 upregulation. Biochim Biophys Acta 1853: 2929–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Elf S, Shan C, Kang HB, Ji Q, Zhou L et al. (2015). 6‐Phosphogluconate dehydrogenase links oxidative PPP, lipogenesis and tumor growth by inhibiting LKB1–AMPK signaling. Nat Cell Biol 17: 1484–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo A, Sullivan LB, Heiden MG (2014). Understanding the complex‐I‐ty of metformin action: limiting mitochondrial respiration to improve cancer therapy. BMC Biol 12: 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ML, Gong C, Chen CH, Hu H, Huang PY, Zheng M et al. (2015). The Rab2A GTPase promotes breast cancer stem cells and tumorigenesis via ERK signaling activation. Cell Rep 11: 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno A, Carrington JT, Albergante L, Al Mamun M, Haagensen EJ, Komseli ES et al. (2016). Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. Proc Natl Acad Sci U S A 113: E5757–E5764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moon TC, Murakami M, Kudo I, Son KH, Kim HP, Kang SS et al. (1999). A new class of COX‐2 inhibitor, rutaecarpine from Evodia rutaecarpa . Inflamm Res 48: 621–625. [DOI] [PubMed] [Google Scholar]
- Padwal RS, Majumdar SR (2007). Drug treatments for obesity: orlistat, sibutramine, and rimonabant. Lancet 369: 71–77. [DOI] [PubMed] [Google Scholar]
- Poekes L, Lanthier N, Leclercq IA (2015). Brown adipose tissue: a potential target in the fight against obesity and the metabolic syndrome. Clin Sci 129: 933–949. [DOI] [PubMed] [Google Scholar]
- Poher AL, Altirriba J, Veyrat‐Durebex C, Rohner‐Jeanrenaud F (2015). Brown adipose tissue activity as a target for the treatment of obesity/insulin resistance. Front Physiol 6: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyrzak B, Demkow U, Kucharska AM (2015). Brown adipose tissue and browning agents: irisin and FGF21 in the development of obesity in children and adolescents. Adv Exp Med Biol 866: 25–34. [DOI] [PubMed] [Google Scholar]
- Rao Y, Liu H, Gao L, Yu H, Ou TM, Tan JH et al. (2015a). Synthesis and biological evaluation of novel bouchardatine derivatives as potential adipogenesis/lipogenesis inhibitors for antiobesity treatment. J Med Chem 58: 9395–9413. [DOI] [PubMed] [Google Scholar]
- Rao Y, Liu H, Gao L, Yu H, Tan JH, Ou TM et al. (2015b). Discovery of natural alkaloid bouchardatine as a novel inhibitor of adipogenesis/lipogenesis in 3T3‐L1 adipocytes. Bioorg Med Chem 23: 4719–4727. [DOI] [PubMed] [Google Scholar]
- Reddon H, Guéant JL, Meyre D (2016). The importance of gene‐environment interactions in human obesity. Clin Sci 130: 1571–1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritho J, Arold ST, Yeh ET (2015). A critical SUMO1 modification of LKB1 regulates AMPK activity during energy stress. Cell Rep 12: 734–742. [DOI] [PubMed] [Google Scholar]
- Russo GL, Russo M, Ungaro P (2015). AMP‐activated protein kinase: a target for old drugs against diabetes and cancer. Biochem Pharmacol 86: 339–350. [DOI] [PubMed] [Google Scholar]
- Sáez‐Lara MJ, Robles‐Sanchez C, Ruiz‐Ojeda FJ, Plaza‐Diaz J, Gil A (2016). Effects of probiotics and pynbiotics on obesity, insulin resistance syndrome, type 2 diabetes and non‐alcoholic fatty liver disease: a review of human clinical trials. Int J Mol Sci 17: 928–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shan TZ, Liang XR, Bi PP, Kuang SH (2013). Myostatin knockout drives browning of white adipose tissue through activating the AMPK–PGC1 alpha‐Fndc5 pathway in muscle. FASEB J 27: 1981–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 proteins targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Z, Kraus WL (2010). SIRT1‐dependent regulation of chromatin and transcription: linking NAD+ metabolism and signaling to the control of cellular functions. Biochim Biophys Acta 1804: 1666–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner N, Zeng XY, Osborne B, Rogers S, Ye JM (2016). Repurposing drugs to target the diabetes epidemic. Trends Pharmacol Sci 37: 379–389. [DOI] [PubMed] [Google Scholar]
- Wang F, Tian DR, Tso P, Han JS (2012). Diet‐induced obese rats exhibit impaired LKB1–AMPK signaling in hypothalamus and adipose tissue. Peptides 35: 23–30. [DOI] [PubMed] [Google Scholar]
- Wang L, Teng R, Di L, Rogers H, Wu H, Kopp JB et al. (2013). PPARalpha and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes 62: 4122–4131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Liang X, Yang Q, Fu X, Rogers CJ, Zhu M et al. (2015). Resveratrol induces brown‐like adipocyte formation in white fat through activation of AMP‐activated protein kinase (AMPK) α1. Int J Obes (Lond) 39: 967–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wattanapiromsakul C, Forster PI, Waterman PG (2003). Alkaloids and limonoids from Bouchardatia neurococca: systematic significance. Phytochemistry 64: 609–615. [DOI] [PubMed] [Google Scholar]
- Woods A, Johnstone SR, Dickerson K, Leiper FC, Fryer LG, Neumann D et al. (2003). LKB1 is the upstream kinase in the AMP‐activated protein kinase cascade. Curr Biol 13: 2004–2008. [DOI] [PubMed] [Google Scholar]
- Yang Y, Li W, Liu Y, Sun Y, Li Y, Yao Q et al. (2014). Alpha‐lipoic acid improves high‐fat diet‐induced hepatic steatosis by modulating the transcription factors SREBP‐1, FoxO1 and Nrf2 via the SIRT1/LKB1/AMPK pathway. J Nutr Biochem 283: 20015–20026. [DOI] [PubMed] [Google Scholar]
- Yoneda M, Guo Y, Ono H, Nakatsu Y, Zhang J, Cui X et al. (2010). Decreased SIRT1 expression and LKB1 phosphorylation occur with long‐term high‐fat diet feeding, in addition to AMPK phosphorylation impairment in the early phase. Obes Res Clin Pract 4: e163–e246. [DOI] [PubMed] [Google Scholar]
- Yoneshiro T, Saito M (2015). Activation and recruitment of brown adipose tissue as anti‐obesity regimens in humans. Ann Med 47: 133–141. [DOI] [PubMed] [Google Scholar]
- Zeng XY, Wang H, Bai F, Zhou X, Li SP, Ren LP et al. (2015). Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target. Br J Pharmacol 172: 4303–4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Guan M, Townsend KL, Huang TL, An D, Yan X et al. (2015). MicroRNA‐455 regulates brown adipogenesis via a novel HIF1a–AMPK–PGC1α signaling network. EMBO Rep 10: 1378–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang H, Li B, Meng X, Wang J, Zhang Y et al. (2014). Berberine activates thermogenesis in white and brown adipose tissue. Nat Commun 5: 5493–5507. [DOI] [PubMed] [Google Scholar]
- Zhao N, Li ZL, Li DH, Sun DT, Bai J, Pei YH et al. (2015). Quinolone and indole alkaloids from the fuits of Euodia retaecarpa and their cytotoxicity against two human cancer cell lines. Phytochemistry 109: 133–139. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Primers sequences used for PCR.
Table S2 Information of Antibody.
Figure S1 Effect of resveratrol (Res) on the TG levels and SIRT1 activity in cells. (A) Effect of Res on the ratio of NAD+/NADH in 3 T3‐L1 adipocytes. *P < 0.05, **P < 0.01, ***P < 0.001 compared with UND group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with DMSO treatment group. (B) Effect of Res on the deacetylase activity of SIRT1 in 3 T3‐L1 adipocytes. *P < 0.05, **P < 0.01, ***P < 0.001 compared with UND group; #P < 0.05, ##P < 0.01, ###P < 0.001 compared with DMSO treatment group. (C‐D) TG content analysis by TG assays and Oil‐Red O staining. *P < 0.05, **P < 0.01, ***P < 0.001 compared with DMSO group. Data are expressed as means ± standard errors from 5 independent experiments.
Figure S2 SRT1720 increases LKB1 translocation to cytosol in 3 T3‐L1 adipocytes. Confluent 3 T3‐L1 pre‐adipocytes were exposed to adipogenic cocktail (MDI) for consecutive 9 days in the presence or absence of SRT1720 treatment. After treatment, LKB1 in cytosol and nucleus were extracted and determined by western blot, GAPDH and Lamin B were loaded as loading control. *P < 0.05, compared with DMSO group.
Figure S3 Toxicity analysis of bouchardatine in vivo. (A) Appearance of tissue was captured. (B‐C) Tissue/body weight ratio. (C) Representative H&E staining from tissue sections after 5 weeks of Bou treatment. Original magnification, 100×. Scale bar, 100 μm.
Figure S4 Expression levels of Cytochrome C gene (for mtDNA) and 18S rRNA (for nuclear DNA) in eWAT (A) and BAT (B) of mice.