

Abstract
Background and Purpose
Naturally occurring dysfunction of voltage‐gated sodium (NaV) channels results in complex disorders such as chronic pain, making these channels an attractive target for new therapies. In the pursuit of novel NaV modulators, we investigated spider venoms for new inhibitors of NaV channels.
Experimental Approach
We used high‐throughput screens to identify a NaV modulator in venom of the spider Davus fasciatus. Further characterization of this venom peptide was undertaken using fluorescent and electrophysiological assays, molecular modelling and a rodent pain model.
Key Results
We identified a potent NaV inhibitor named μ‐TRTX‐Df1a. This 34‐residue peptide fully inhibited responses mediated by NaV1.7 endogenously expressed in SH‐SY5Y cells. Df1a also inhibited voltage‐gated calcium (CaV3) currents but had no activity against the voltage‐gated potassium (KV2) channel. The modelled structure of Df1a, which contains an inhibitor cystine knot motif, is reminiscent of the NaV channel toxin ProTx‐I. Electrophysiology revealed that Df1a inhibits all NaV subtypes tested (hNaV1.1–1.7). Df1a also slowed fast inactivation of NaV1.1, NaV1.3 and NaV1.5 and modified the voltage‐dependence of activation and inactivation of most of the NaV subtypes. Df1a preferentially binds to the domain II voltage‐sensor and has additional interactions with the voltage sensors domains III and IV, which probably explains its modulatory features. Df1a was analgesic in vivo, reversing the spontaneous pain behaviours induced by the NaV activator OD1.
Conclusion and Implications
μ‐TRTX‐Df1a shows potential as a new molecule for the development of drugs to treat pain disorders mediated by voltage‐gated ion channels.
Abbreviations
- ACN
acetonitrile
- CaV
voltage‐gated calcium channel
- DIEA
N, N‐diisopropylethylamine
- DMF
N, N‐dimethylformamide
- HBTU
2‐(1H‐benzotriazol‐1‐yl)‐1,1,3,3‐tetramethyluronium hexafluorophosphate
- ICK
inhibitor cysteine knot
- KV
voltage‐gated potassium channel
- MALDI‐TOF
matrix‐assisted laser desorption/ionization time of flight
- NaV
voltage‐gated sodium channel
- RP‐HPLC
reversed‐phase high‐performance liquid chromatography
- TFA
trifluoroacetic acid
Introduction
Animal venoms have evolved into potent neurotoxins for predation and defence that cause harmful neurological alterations in insects and mammals (Klint et al., 2012; King and Hardy, 2013). These effects are induced by molecules acting on ion channels involved in the generation and transmission of electrical signals in neurons and muscles, which play a key role in several biological processes. The potency and ion channel selectivity of these neurotoxins have made them powerful pharmacological tools as well as therapeutic leads for treating channelopathy‐related disorders. These include conditions such as neuropathic pain, epilepsy and arrhythmia, which often involve dysfunction of voltage‐gated sodium (NaV) channels (Rogers et al., 2006; Catterall et al., 2010; Remme and Bezzina, 2010).
NaV channels are glycosylated transmembrane proteins involved in action potential generation and propagation in excitable cells. In mammals, the NaV channel family is composed of nine subtypes (NaV1.1–NaV1.9) that differ in their α‐subunit sequences and their pharmacological and functional properties. Alterations in NaV channel function and/or expression underlies a variety of disorders including chronic pain (Moldovan et al., 2013; Lauria et al., 2014). In particular, NaV1.3, NaV1.7, NaV1.8 and NaV1.9 channels have been strongly implicated in chronic pain (Liu and Wood, 2011). The expression of NaV1.3, NaV1.8 and NaV1.9 channels is altered during neuropathic pain (Dib‐Hajj et al., 1999), while individuals lacking functional NaV1.7 channels exhibit a congenital insensitivity to pain, with no other sensory abnormalities apart from anosmia (Cox et al., 2006). Several epileptic conditions are due to mutations in or altered expression of NaV1.1 and NaV1.2 channels (Meisler and Kearney, 2005), and numerous anticonvulsants act on NaV channels by stabilizing their fast inactivation or slow‐inactivated states (Ragsdale and Avoli, 1998; Errington et al., 2008). Other conditions such as congenital paramytonia and Brugada syndrome also present alterations in NaV channels (NaV1.4 and NaV1.5, respectively) (Amin et al., 2010; Zhao et al., 2012). Thus, NaV channels are common targets for the development of drugs to treat complex disorders.
Several neurotoxins sourced from animal venoms have been described as modulators of NaV channels (Herzig et al., 2011; Kaas et al., 2012). Most have high potency at NaV channels but lack selectivity for the relevant therapeutic targets and cause side effects and even death at low doses. Spider‐venom peptides have shown promising selectivity for the NaV channel subtypes involved in pain pathways (Klint et al., 2015) and they have been used as leads in the search for new analgesics. Using fluorescence assays and either endogenously or heterologously expressed NaV subtypes (Vetter et al., 2012; Cardoso et al., 2015), we have begun to systematically identify and characterize peptidic NaV channel modulators from spider crude venoms. In the present work, we describe the isolation and characterization of a new ion channel inhibitor (Df1a) from the tarantula Davus fasciatus using a high‐throughput fluorescence screen. Df1a (μ‐TRTX‐Df1a) potently inhibits hNaV and hCaV3 channels. Interestingly, Df1a displays a novel dual modulatory effect of specific hNaV channel subtypes, simultaneously inhibiting peak current and slowing fast inactivation, and it reversed pain behaviours induced by intraplantar OD1 activation of NaV in mice.
Methods
Animals
For behavioural assessment, we used adult male C57BL/6J mice aged 6–8 weeks weighing 20–25 g. Mice were housed in groups of three to four per cage, kept under a 12 h light–dark cycle, with standard rodent chow and water provided ad libitum.
Ethics statement
Ethical approval for in vivo experiments was obtained from The University of Queensland Animal Ethics Committee (AEC Approval Number IMB/PACE/326/15). Experiments involving animals were conducted in accordance with the Animal Care and Protection Regulation Qld (2012), the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes, 8th edition (2013) and the International Association for the Study of Pain Guidelines for the Use of Animals in Research. Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010; McGrath and Lilley, 2015).
Cell culture
The human neuroblastoma cell line SH‐SY5Y was maintained at 37°C in a humidified 5% CO2 incubator in Roswell Park Memorial Institute (RPMI) medium supplemented with 15% fetal bovine serum and 2 mM L‐glutamine. HEK 293 cells expressing recombinant hNaV channel subtypes co‐expressed with β1 auxiliary subunits (SB Drug Discovery, Glasgow, UK) were maintained at 37°C in a humidified 5% CO2 incubator in minimal essential medium supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin, 2 mM L‐glutamine and various concentrations of blasticidin, geneticin and zeocin according to the manufacturer's protocols. Replicating cells were sub‐cultured every 3–4 days in a 1:5 ratio using 0.25% trypsin/EDTA. CHO cells expressing recombinant hNaV1.6 channels (EZ cells, ChanTest Corp, OH, USA) were maintained at 37°C in a humidified 5% CO2 incubator in F‐12 medium supplemented with 10% FBS, 100 U·mL−1 penicillin and 100 μg·mL−1 streptomycin.
Venom fractionation
Venom from Davus fasciatus was obtained by electrical stimulation as previously described (Herzig and Hodgson, 2009). Dried venom (1 mg) was dissolved in 100 μL Milli‐Q water containing 0.05% trifluoroacetic acid (TFA) (Auspep, VIC, Australia) and 5% acetonitrile (ACN) and centrifuged at 11 952 × g for 10 min to remove particulates. Venom was fractionated by reversed‐phase HPLC (RP‐HPLC) using a C18 column (Vydac 4.6 × 250 mm, 5 μm, Grace Discovery Sciences, USA) with a gradient of solvent B (90% ACN in 0.045% TFA) in solvent A (0.05% TFA). The gradient was 5% B for 5 min, followed by 20 to 40% solvent B over 60 min at a flow rate 0.7 mL·min−1. Peaks were collected at 0.7 mL per well, and fractions were lyophilized before storage at −20°C.
Screening against the hNaV1.7 channel
Venom fractions were screened for inhibition of hNaV1.7 channels as previously described (Cardoso et al., 2015). Briefly, SH‐SY5Y cells were plated at 40 000 cells per well in 384‐well flat clear‐bottom black plates (Corning, NY, USA) and cultured at 37°C in a humidified 5% CO2 incubator for 48 h. Cells were loaded with 20 μL per well Calcium 4 dye (Molecular Devices, Sunnyvale, CA, USA) reconstituted in assay buffer containing (in mM) 140 NaCl, 11.5 glucose, 5.9 KCl, 1.4 MgCl2, 1.2 NaH2PO4, 5 NaHCO3, 1.8 CaCl2 and 10 HEPES pH 7.4 and incubated for 30 min at 37°C in a humidified 5% CO2 incubator. Fluorescence responses were recorded using excitation at 470–495 nm and emission at 515–575 nm for 10 s to set the baseline, then 600 s after addition of 10% venom fraction per well and for a further 300 s after co‐addition of 3 μM veratridine and 30 nM OD1.
Mass spectrometry and peptide sequencing
Peptide masses were determined by matrix‐assisted laser desorption ionization‐time of flight mass spectrometry (MS) (MALDI‐TOF MS) using a 4700 Proteomics Bioanalyser Model (Applied Biosystems, CA, USA). Df1a was dissolved in water mixed 1:1 (v.v‐1) with α‐cyano‐4‐hydroxy‐cinnamic acid matrix (7 mg·mL−1 in 50% ACN) and mass spectra acquired in positive reflector mode. The reported molecular weight of Df1a is for the monoisotopic M + H+ ion. N‐terminal sequencing was outsourced to the Australian Proteome Analysis Facility, Sydney, Australia. Briefly, the peptide was dissolved in urea (4 M) and ammonium bicarbonate (50 mM) and reduced with dithiothreitol (100 mM) at 56°C for 1 h under argon. The sample was then alkylated using acrylamide (220 mM) for 0.5 h in the dark. The reaction was quenched by the addition of excess dithiothreitol. After desalting by RP‐HPLC, the collected fraction was loaded onto pre‐cycled bioprene discs and subjected to 34 cycles of Edman degradation N‐terminal sequencing using an ABI 494 Procise Protein Sequencing System (Applied Biosystems).
Solid phase synthesis of Df1a
Solvents for RP‐HPLC consisted of 0.05% TFA/H2O (solvent A) and 90% ACN/0.043% TFA/H2O (solvent B). Analytical HPLC was performed on a Shimadzu LC20AT system using a Thermo Hypersil GOLD 2.1 × 100 mm C18 column heated at 40°C with a flow rate of 0.3 mL·min−1 and a gradient of 10 to 55% B over 30 min unless otherwise stated. The eluent was monitored at 214 nm unless otherwise stated. Preparative HPLC was performed on a Vydac 218TP1022 column running at a flow rate of 16 mL·min−1 using a gradient of 10 to 50% B over 40 min. MS was performed on an API2000 (ABI Sciex) mass spectrometer in positive ion mode. Df1a‐NH2 and Df1a‐OH were chain assembled on a Symphony (Protein Technologies Inc., AZ, USA) automated peptide synthesizer on Rink‐amide (loading 0.67 mmol·g−1) and Fmoc‐Phe‐Wang (loading 0.70 mmol·g−1) polystyrene resins, respectively, on a 0.1 mmol scale. Fmoc deprotections were achieved using 30% piperidine DMF (1 × 1.5 min, then 1 × 4 min). Couplings was performed in DMF using five equivalents of Fmoc‐amino acid/HBTU/DIEA (1:1:1) relative to resin loading for 2 × 20 min. Amino acid side chains were protected as follows: Asp(OtBu), Arg(Pbf), Cys(Trt), Gln(Trt), Glu(OtBu), His(Trt), Lys(Boc), Ser(tBu), Thr(tBu) and Trp(Boc). Cleavage from the resin and removal of side chain protecting groups was achieved by treatment with 95% TFA/2.5% triisopropylsilane/2.5% H2O at room temperature for 2 h. After most of the cleavage solution was evaporated under a stream of N2, the products were precipitated and washed with cold Et2O and lyophilized from 50% ACN/0.1% TFA/H2O. Df1a‐NH2: 192 mg; ESI‐MS (m/z): calc. (avg) 1362.6 Da [M + 3H]3+, found 1362.3 Da. The crude product was purified by preparative HPLC to give 50 mg of hexathiol Df1a‐NH2. Df1a‐OH: 204 mg; ESI‐MS (m/z): calc. (avg) 1362.9 Da [M + 3H]3+, found 1362.7 Da. The crude product was purified by preparative HPLC to give 50 mg of hexathiol Df1a‐OH.
Oxidative folding
Purified reduced peptide (10 mg of Df1a‐NH2 or Df1a‐OH), reduced glutathione (100 equiv.) and oxidized glutathione (10 equiv.) were dissolved in 6 M guanidine HCl (33 mL) then added to a solution of 0.1 M Tris (pH 8.0, 67 mL) and stirred at 4°C with exposure to air for 4 days. The major products were isolated by preparative HPLC. Df1a‐NH2: 2.9 mg; Electrospray ionization‐MS (EIS‐MS) (m/z): calc. (avg) 1360.6 Da [M + 3H]3+, found 1360.4 Da. Df1a‐OH: 2.5 mg; ESI‐MS (m/z): calc. (avg) 1360.9 Da [M + 3H]3+, found 1360.5 Da.
Patch‐clamp electrophysiology using mammalian cells
NaV channel experiments were performed in HEK 293 (SB Drug Discovery) cells expressing specific hNaV subtypes co‐expressed with the β1 auxiliary subunit or CHO expressing hNaV subtypes (EZ cells, ChanTest Corp) and Na+ currents measured by whole‐cell patch clamp using the automated QPatch 16X system (Biolin Scientific A/S, Ballerup, Denmark). The extracellular solution comprised (in mM) 1 CaCl2, 1 MgCl2, 5 HEPES, 3 KCl, 140 NaCl, 0.1 CdCl2 and 20 TEA‐Cl at pH 7.3 and 320 mOsm, and the intracellular solution comprised (in mM) 140 CsF, 1/5 EGTA/CsOH, 10 HEPES and 10 NaCl at pH 7.3 and 320 mOsm. The currents elicited were sampled at 25 kHz and filtered at 4 kHz. The average seal, whole‐cell and chip resistances values were (in MΩ) 3690, 997 and 2.08 respectively. Cells with less than 1 nA of peak Na+ current were not used in this study. Cells were maintained at a holding potential −80 mV and Na+ currents elicited by 20 ms voltage steps to 0 mV from a −120 mV conditioning pulse applied for 200 ms. To obtain concentration–response curves, cells were incubated for 5 min with increasing concentrations of Df1a. For on‐rate experiments, Na+ currents were measured at 15 s intervals over 15 min immediately following addition of Df1a at corresponding IC50 and 10× IC50 concentrations for the NaV subtypes analysed. For off‐rate measurements, cells were incubated for 10 min with Df1a at corresponding IC50 concentrations for the NaV subtypes analysed and Na+ currents assessed at 5 min intervals during saline washes. The K on, K off and K d were calculated using K d = K off/K on (nM), where K off = 1/τoff (s−1) and K on = (1/τon − K off)/[toxin] (nM−1·S−1). Voltage–activation relationships were obtained by measuring steady‐state Na+ currents elicited by step depolarizations from −110 to +80 mV using 10 mV increments. The peak conductance (G Na) was calculated from G = I/(V − V rev), where I, V and V rev represent the current value, membrane potential and reverse potential respectively. The voltage of steady‐state fast inactivation was estimated using a double‐pulse protocol with currents elicited by a 20 ms depolarizing potential of 0 mV following a 500 ms pre‐pulse to potentials from −130 to −10 mV using 10 mV increments. Voltage‐dependence of activation and inactivation relationships was examined either in the absence and presence of Df1a (5 min exposure) with the cells before application of the voltage protocols. Voltage‐gated calcium 3 (CaV3) channels experiments were recorded in CHO cells expressing hCaV3.1 and HEK 293 cells expressing hCaV3.2 and hCaV3.3 (these cell lines were kindly donated by Prof Emmanuel Bourinet from the Institute of Functional Genomics, Montpellier University, France and Prof Edward Perez‐Reyes from the School of Medicine, University of Virginia, USA) and Ca+2 currents measured by whole‐cell patch clamp using an automated QPatch 16X. The extracellular solution comprised (in mM) 5 CaCl2, 0.5 MgCl2, 10 HEPES and 157 TEA‐Cl at pH 7.3 and 320 mOsm, and the intracellular solution comprised (in mM) 140 CsF, 1 EGTA, 10 HEPES and 10 NaCl at pH 7.3 and 320 mOsm. The currents elicited were sampled at 25 kHz and filtered at 4 kHz. The average seal, whole‐cell and chip resistances values were (in MΩ) 5000, 1953 and 2.2 for CHO and 1536, 734 and 2.1 for HEK 293 respectively. Cells were maintained at a holding potential −90 mV and Ca2+ currents elicited by 60 ms voltage steps to −30 mV from a −120 mV conditioning pulse applied for 60 ms. To obtain concentration–response curves, cells were incubated for 5 min with increasing concentrations of Df1a. All experimental data were analysed using QPatch Assay Software v5.0 (Biolin Scientific A/S).
Determination of Df1a binding sites on hNaV1.7
hNaV1.7/rKV2.1 chimeras containing the extracellular S3‐S4 loop and helices of the hNaV1.7 paddle were generated previously (Klint et al., 2015). Xenopus laevis oocytes were injected with cRNA encoding hNaV1.7/rKV2.1 chimera or rKV2.1. Two‐electrode voltage clamp electrophysiology (Axoclamp 900A, Molecular Devices; 40 μL recording chamber) was used to measure currents 1–4 days after cRNA injection and incubation at 17°C in ND96 that contained (in mM) 96 NaCl, 2 KCl, 5 HEPES, 1 MgCl2, 1.8 CaCl2 and 50 μg·mL−1 gentamycin, pH 7.6. Data were filtered at 4 kHz and digitized at 20 kHz using pClamp software (Molecular Devices). Micro‐electrode resistances were 0.5–1 MΩ when filled with 3 M KCl. The external recording solution contained (in mM) 50 KCl, 50 NaCl, 5 HEPES, 1 MgCl2, 0.3 CaCl2 and pH 7.6 with NaOH. Experiments were performed at room temperature (∼22°C). Toxin samples were diluted in recording solution with 0.1% bovine serum albumin. Potassium currents were elicited by depolarization to +70 mV from a holding potential of −90 mV (−120 mV for the DIII chimera), with a tail voltage at −60 mV (−90 mV for the DIII chimera). Leak and background conductance, identified by blocking channels with agitoxin‐2, were subtracted for all experiments.
Activity of Df1a in vivo
The efficacy of Df1a to inhibit pain mediated by NaV channels in vivo was assessed in mice using the α‐scorpion toxin OD1 as previously described (Cardoso et al., 2015; Deuis et al., 2016). To induce spontaneous pain behaviours, the NaV activator OD1 (300 nM) ± Df1a‐OH (10 μM, 1 μM) or Df1a‐NH2 (10 μM, 1 μM) was administered by intraplantar injection into the left hind paw of mice in PBS containing 0.1% BSA in a volume of 40 μL under 2% isoflurane anaesthesia. Mice were then placed individually into Perspex boxes (10 × 10 × 10 cm), and the number of spontaneous pain behaviours (licks and flinches) was counted, by an observer unaware of the treatments received, for 10 min following injection of OD1 from video recordings.
Molecular model of Df1a
The three‐dimensional structure of Df1a was modelled using the NMR‐derived structure of β/ω‐theraphotoxin‐Tp1a (ProTx‐I, PDB code 2M9L) as a template (Gui et al., 2014). Backbone fitting and energy minimization were performed using the Swiss‐Model prediction algorithm (open source, http://swissmodel.expasy.org) (Arnold et al., 2006) and displayed using the PyMOL Software package (DeLano, 2002). The model was validated by inspection of the Ramachandran plot (Lovell et al., 2003).
Materials
Cell culture reagents were from Gibco, Life Technologies Corporation (Carlsbad, CA, USA), unless otherwise stated. For the RP‐HPLC, calcium influx assays, peptide synthesis and oxidative folding, electrophysiological assays and in vivo experiments, all reagents were from Sigma‐Aldrich (St Louis, MO, USA) unless otherwise stated.
Data analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology (Curtis et al., 2015). Curve fitting was achieved using GraphPad Prism Version 6 (GraphPad Software Inc, San Diego, CA, USA) using non‐linear regression with log[inhibitor] versus normalized response and variable Hill slope for dose–responses, Boltzmann sigmoidal equation for voltage‐dependence of activation and inactivation and exponential one‐phase association for on‐ and off‐rate analysis. Data are presented as mean ± SEM of at least five independent experiments, unless otherwise stated. For the in vitro experiments, statistical significance was determined by Student's paired t‐test assuming equal variance. For the in vivo experiments, statistical significance was determined by ANOVA with Dunnett's post test. P < 0.05 was considered significant.
Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Southan et al., 2016), and are permanently archived in the Concise Guide to PHARMACOLOGY 2015/16 (Alexander et al., 2015).
Results
Peptide toxin identification and chemical synthesis
Fractionation of Davus fasciatus venom (1 mg) using RP‐HPLC revealed eight dominant peaks eluting from 20 to 40% solvent B (Figure 1A). Testing aliquots (10%) of 1 min fractions for activity in SH‐SY5Y cells stimulated by co‐addition of OD1 and veratridine revealed three fractions that strongly inhibited hNaV1.7 (Figure 1A, grey circles). MALDI‐TOF MS analysis revealed that the peaks eluting at 30.8 and 32.2 min contained multiple peptides that proved difficult to separate and could not be definitively characterized (data not shown). The remaining fraction eluting at 36–37 min (33% B) contained a single major peptide with a monoisotopic mass of 4075.8 Da (Figure 1B). N‐terminal Edman sequencing disclosed a novel 34‐residue peptide that we named Df1a (μ‐TRTX‐Df1a). The peptide sequence had a calculated monoisotopic oxidized mass of 4076.8 Da, suggesting that it was C‐terminally amidated in its native form (Figure 1C). Sequence comparisons revealed that Df1a belonged to the Family 2 of NaV‐targeting spider toxins (NaSpTx), which is comprised of 33 to 41 residue peptides with hyper‐stable inhibitor cysteine knot (ICK) motifs that inhibit NaV, CaV and voltage‐gated potassium (KV) channels (Klint et al., 2012; Herzig and King, 2015). Sequence analysis revealed the highest identity with β/ω‐TRTX‐Tp1a (ProTx‐I, 73%), ω‐TRTX‐Hhn1a (64%) and TRTX‐Hh2a (61%) (Figure 1D).
Figure 1.
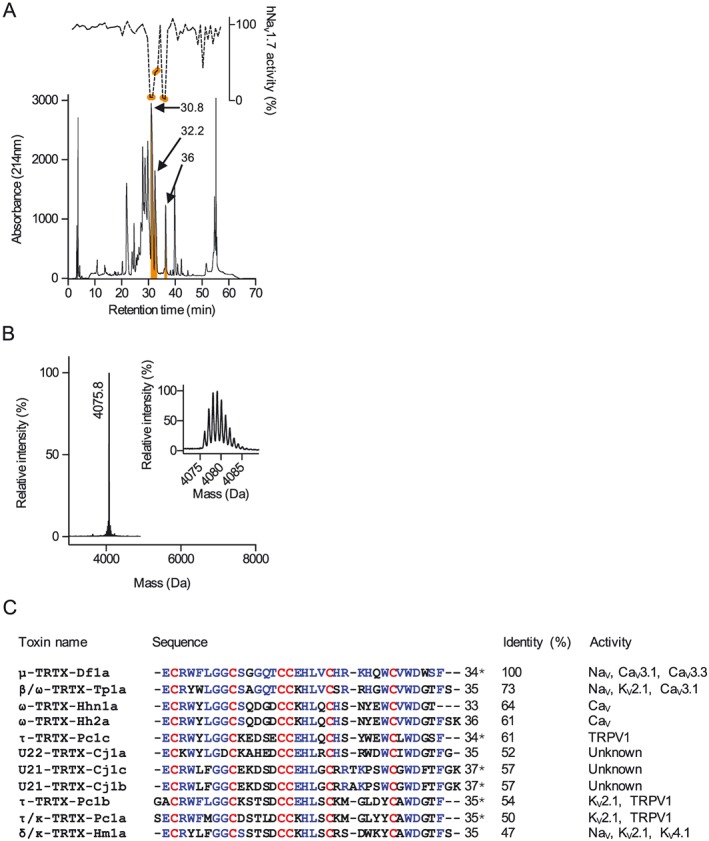
Venom fractionation, activity screening on NaV1.7, mass spectrometry and sequence determination of active fraction 36. (A) RP‐HPLC of Davus fasciatus crude venom (1 mg) was performed using a Vydac218TP C18 column using a three‐step gradient of acetonitrile/0.05% trifluoroacetic acid (5–10% B for 5 min, 20–40% B for 40 min and 40–80% B for 5 min). Fractions were collected at 0.7 mL·min−1 and screened for NaV1.7 inhibition using Calcium dye and a FLIPRTetra instrument. Fractions eluted at 30.8, 32.2 and 36 min showed strong NaV1.7 inhibition (orange shaded fractions). (B) MALDI‐TOF mass spectrometry of fraction 36 showing single predominant mass of 4075.8 Da. (C) Sequence identification and analysis of μ‐TRTX‐Df1a. Edman degradation analysis of the native toxin revealed a peptide with 34 residues containing six cysteines. The difference in the masses between the native Df1a and predicted mass of the amino acids sequence revealed by Edman degradation indicates the presence of a C‐terminal amidation in the native peptide. Sequence alignment of peptides toxins showing at least 47% identity in their amino acids sequence with Df1a. Identical residues are shown in blue and cysteine scaffold in red. The % identity is shown relative to Df1a, and the activity reported for each peptide toxin is presented in the far right column according to data sourced from the ArachnoServer database (Herzig et al., 2011). Asterisks denote C‐terminal amidation. Df1a showed highest identity with the toxin β/ω‐TRTX‐Tp1a (ProTx‐I) isolated from the tarantula Thrixopelma pruriens (Middleton et al., 2002).
Chemical synthesis was used to generate the C‐terminal amide and acid forms of Df1a (Figure 2A). Both native and synthetic Df1a‐NH2, as well as synthetic Df1a‐OH, eluted as a complex set of partially resolved peaks by analytical HPLC at 40°C (Figure 2A) but as two peaks at 23°C (Figure 2B). Comparable temperature‐dependent dynamics have been reported previously for human β‐defensins (Chino et al., 2006), while hanatoxin also elutes as two peaks, although temperature dependence was not reported (Takahashi et al., 2000). Importantly, all major peaks associated with synthetic Df1a‐NH2 co‐eluted with native Df1a, confirming the assigned sequence and that the native disulfide bond pairings had been achieved. In order to rapidly evaluate the activity of the synthetic Df1a over NaV channels, a fluorescence imaging plate reader (FLIPR)‐based fluorescence assay was performed. Synthetic Df1a‐NH2 was found to potently inhibit endogenous hNaV1.7 channels in SH‐SY5Y cells (Figure 2C). Given their structural identity and functional activity, synthetic Df1a (sDf1a‐NH2 and sDf1a‐OH) was used for all subsequent characterization studies.
Figure 2.
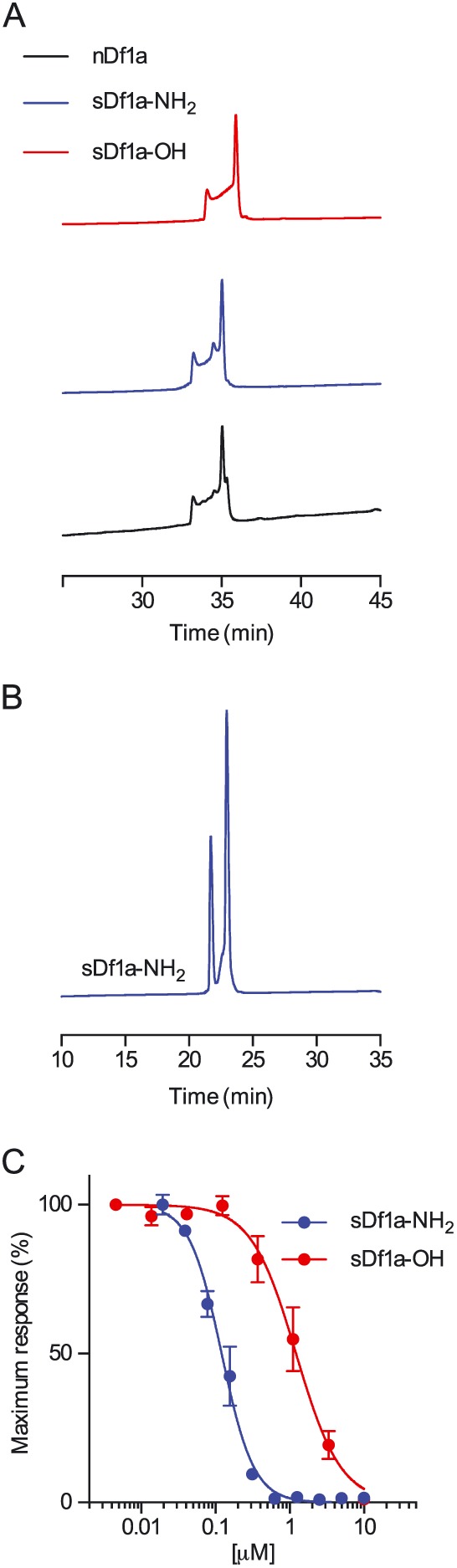
Comparison of the RP‐HPLC retention time of native μ‐TRTX‐Df1a and synthetic Df1a‐NH2 and Df1a‐OH. (A) Analytical RP‐HPLC chromatograms of native amidated Df1a, synthetic Df1a‐NH2 and synthetic Df1a‐OH. RP‐HPLC was performed on a Shimadzu LC20AT system using a Thermo Hypersil GOLD C18 column (2.1 × 100 mm) heated at 40°C. Peptides were eluted using a gradient of 5–50% B over 45 min with a flow rate of 0.3 mL·min−1. Native Df1a and synthetic Df1a‐NH2 eluted both at 33.2 (minor peak) and 35.1 (major peak) min, while synthetic Df1a‐OH eluted at 34.1 (minor peak) and 35.9 (major peak) min. (B) Analytical HPLC trace showing synthetic Df1a‐NH2 at 23°C. Lowering the temperature produced a deconvolution of the chromatogram resulting in only two peaks and disappearance of the unresolved portion between them observed at 40°C. (C) Activity of synthetic sDf1a‐NH2 and sDf1a‐OH over hNaV1.7 in SH‐SY5Y cells determined using a fluorescent assay. The IC50 for hNaV1.7 inhibitions were (in μM) 0.117 ± 0.006 and 1.24 ± 0.30 for sDf1a‐NH2 and sDf1a‐OH respectively. Data are presented as mean ± SEM, n = 9 independent experiments performed on three different days.
Effect of Df1a on NaV and CaV3 channels
We evaluated the effect of Df1a on a range of voltage‐gated ion channels by using automated whole‐cell patch clamp to examine its effect on hNaV1.1–1.5, hNaV1.7, hCaV3.2 and hCaV3.3 expressed in HEK 293 cells and hNaV1.6 and hCaV3.1 expressed in CHO cells (Figure 3A–J, Supporting Information Table S1). Df1a‐NH2 inhibited hNaV1.1–1.7 channels, with preference for hNaV1.2, hNaV1.3 and hNaV1.7. The C‐terminal acid form of Df1a (Df1a‐OH) lost potency at all NaV subtypes compared the C‐terminal amide form, especially at hNaV1.7, to be hNaV1.2, hNaV1.3 and hNaV1.6 preferring. The effect of Df1a was also investigated by two‐electrode voltage clamp in Xenopus laevis oocytes expressing hNaV1.7 showing Df1a‐NH2 inhibited this channel with more potency compared with Df1a‐OH and displayed IC50 values of up to 8.5‐fold difference compared with the automated whole‐cell patch clamp in mammalian cells (Supporting Information Figure S1A, C). Furthermore, the effect of ProTx‐I, a spider toxin also belonging to the NaSpTx Family 2, was investigated on hNaV1.7 by two‐electrode voltage clamp and automated whole‐cell patch clamp. ProTx‐I inhibited hNaV1.7 currents with similar IC50 values in both electrophysiology systems (Supporting Information Figures S1E and S2A). Df1a‐NH2 also inhibited the CaV3 channels responses, with preference for hCaV3.1 and hCaV3.3. This inhibition was less potent in the C‐terminal acid form of Df1a for all CaV3 subtypes. The IC50 values and Hill slope for each hNaV and hCaV3 subtypes in the presence of Df1a are described in Supporting Information Table S1.
Figure 3.
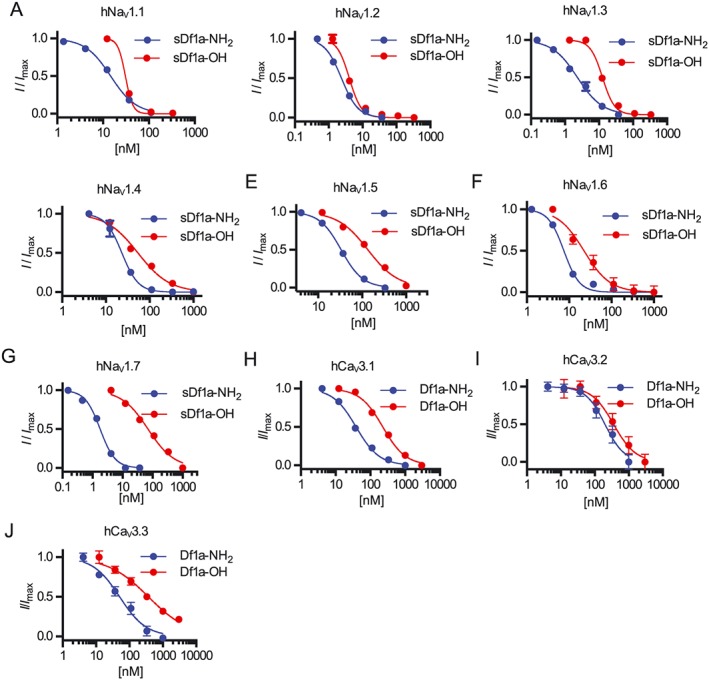
Inhibition of hNaV and hCaV3 subtypes by μ‐TRTX‐Df1a measured by automated patch clamp electrophysiology in QPatch 16X. Holding potential was −80 mV for hNaV and −90 mV for hCaV3. Na+ currents were elicited by 20 ms voltage steps to 0 mV from a −120 mV conditioning pulse applied for 200 ms, and Ca2+ currents were elicited by 60 ms voltage steps to −30 mV from a −120 mV conditioning pulse applied for 60 ms. Representative concentration–response curves for inhibition of (A) hNaV1.1, (B) hNaV1.2, (C) hNaV1.3, (D) hNaV1.4, (E) hNaV1.5, (F) hNaV1.6, (G) hNaV1.7, (H) hCaV3.1, (I) hCaV3.2 and (J) hCaV3.3. The IC50 values, calculated using I/I max values and non‐linear regression, were (in nM) 14.3 ± 0.1 (n = 5) and 30.7 ± 2.2 (n = 6) for hNaV1.1, 1.9 ± 0.5 (n = 5) and 3 ± 1.4 (n = 5) for hNaV1.2, 3 ± 0.7 (n = 5) and 10 ± 1 (n = 8) for hNaV1.3, 24 ± 1.8 (n = 5) and 53.6 ± 12 (n = 6) for hNaV1.4, 45.3 ± 6.8 (n = 5) and 125.6 ± 21 (n = 5) for hNaV1.5, 7.6 ± 0.4 (n = 5) and 23 ± 2.9 (n = 7) for hNaV1.6, 1.9 ± 0.08 (n = 6) and 60.5 ± 6.1 (n = 6) for hNaV1.7, 44.6 ± 5.8 (n = 8) and 216 ± 28.1 (n = 7) for hCaV3.1, 253 ± 45.7 (n = 6) and 371 ± 48.8 (n = 6) for hCaV3.2 and 48.4 ± 7.2 (n = 5) and 460 ± 43.7 (n = 7) for hCaV3.3, under application of sDf1a‐NH2 and sDf1a‐OH respectively. Data are presented as mean ± SEM from described n independent experiments; one cell was considered an independent experiment.
Effect of Df1a on activation and inactivation of hNaV channels
Most spider toxins that inhibit NaV channels are gating modifiers that alter the voltage‐dependence of channel gating (Catterall et al., 2007). In order to investigate the mode of action of Df1a, we examined its effect on the voltage‐dependence of NaV channel activation and steady‐state inactivation using automated whole‐cell patch clamp using HEK293 cells expressing the hNaV1.1–1.5 and 1.7 and CHO cells expressing hNaV1.6. We found that both Df1a‐NH2 and Df1a‐OH shifted the voltage‐dependence of activation and of steady‐state fast inactivation of hNaV subtypes to more hyperpolarizing or depolarizing potentials when applied at respective IC50 concentration for each NaV channel subtype (Figure 4). More specifically, Df1a considerably altered the voltage‐dependence of activation of hNaV1.2 (ΔV50 (mV) –6.9 ± 1.3 for Df1a‐OH), hNaV1.3 (ΔV50 (mV) 10.6 ± 1.6 and 5.6 ± 1.3 for Df1a‐NH2 and OH respectively), hNaV1.5 (ΔV50 (mV) –7.2 ± 2.1 for Df1a‐OH) and hNaV1.7 (ΔV50 (mV) 10.7 ± 1 and 5.3 ± 1.5 for Df1a‐NH2 and OH respectively) (Figure 4B, C, E, G). Moreover, Df1a shifted the voltage‐dependence of steady‐state fast inactivation of hNaV1.1 (ΔV50 (mV) –19.1 ± 2.6 for Df1a‐NH2), hNaV1.2 (ΔV50 (mV) –9.1 ± 2.2 and −5.3 ± 1.3 for Df1a‐NH2 and OH respectively), hNaV1.6 (ΔV50 (mV) –6.5 ± 0.98 and −8 ± 2.6 for Df1a‐NH2 and OH respectively) and hNaV1.7 (ΔV50 (mV) –16.9 ± 2.9 and −17.5 ± 2.3 for Df1a‐NH2 and OH respectively) (Figure 4A, B, D, F). In striking contrast, Df1a shifted the steady‐state inactivation for hNaV1.3 to more depolarizing potentials (ΔV50 (mV) 9.6 ± 1.5 and 7.9 ± 2.3 for Df1a‐NH2 and OH respectively) (Figure 4C). ΔV50 values not reported were between −5 and +5 mV. There was a general trend for Df1a to shift both voltage‐dependence of activation and of steady‐state fast inactivation of the hNaV subtypes to more hyperpolarizing potentials, with exceptions found for the subtypes hNaV1.3 and hNaV1.7 (Figure 4H). The effect of Df1a on activation and inactivation of hNaV1.7 was also investigated by two‐electrode voltage clamp in Xenopus laevis oocytes (Supporting Information Figure S1B, D). No effect on the voltage‐dependence of activation and inactivation of hNaV1.7 was observed in the presence of Df1a‐NH2 and Df1a‐OH when two‐electrode voltage clamp in oocytes was used. A similar effect in hNaV1.7 was observed in the presence of ProTx‐I, where there were no significant changes in the voltage‐dependence of activation and inactivation using both two‐electrode voltage clamp and automated whole‐cell patch clamp electrophysiology systems (Supporting Information Figures S1F and S2B).
Figure 4.
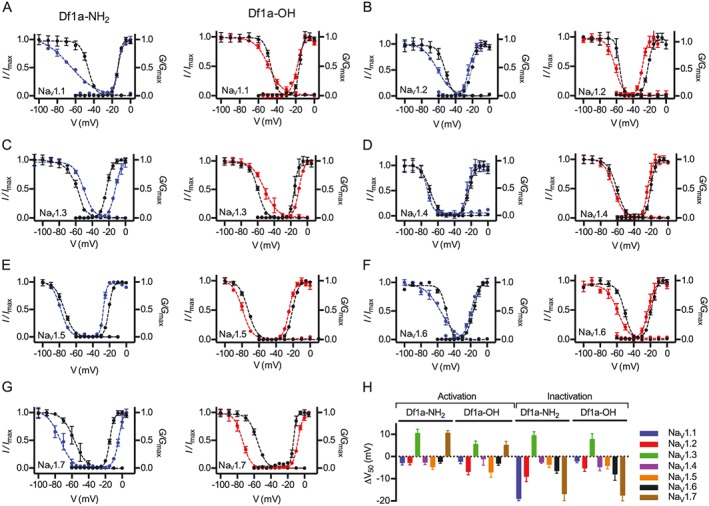
Modulation of the voltage‐dependence of hNaV channel activation and inactivation gating in the presence of μ‐TRTX‐Df1a. Data (mean ± SEM, n = 5, one cell was considered an independent experiment) for (A) hNaV1.1, (B) hNaV1.2, (C) hNaV1.3, (D) hNaV1.4, (E) hNaV1.5, (F) hNaV1.6 and (G) hNaV1.7 are plotted as G/G max or I/I max. Cells were held at −80 mV. μ‐TRTX‐Df1a C‐terminal amide and acid were applied at respective IC50 concentration for each NaV channel subtype as described in Figure 3 and Supporting Information Table S1. Steady state kinetics were estimated by currents elicited at 5 mV increment steps ranging from −110 to +80 mV. Conductance was calculated using G = I/(V–V rev) in which I, V and V rev are the current value, membrane potential and reverse potential respectively. The voltage‐dependence of fast inactivation was estimated using a double‐pulse protocol where currents were elicited by a 20 ms depolarizing potential of 0 mV following a 500 ms pre‐pulse at potentials ranging from −130 to −10 mV with 10 mV increments. Steady‐state activation and inactivation V50 were determined by the Boltzmann equation. Both C‐terminal acid and amide forms of sDf1a applied at respective IC50 concentrations modified the gating properties of hNaV channels by shifting the voltage‐dependence of activation and steady‐state inactivation to more hyperpolarizing or depolarizing potentials. The ΔV50 was calculated, showing most of the voltage shifts towards more hyperpolarizing potentials, except for hNaV1.3 and hNaV1.7 that had some of these shifts to more hyperpolarizing potentials (H).
A slowing in the fast inactivation of NaV channels was observed for hNaV1.1, hNaV1.3 and hNaV1.5 in the presence of Df1a‐NH2 and Df1a‐OH (Figure 5). Representative traces show slowed fast inactivation occurring simultaneously with peak current inhibition in the presence of Df1a at the respective IC50 concentrations for each channel subtype (Figure 5A). The slowing in fast inactivation was fully inhibited in the presence of 1 μM Df1a, along with peak current. The inactivation decay time constant (τ) was calculated, revealing this toxin slows the inactivation of NaV1.1 by 2.1‐ and 2.3‐fold, NaV1.3 by 12‐ and 9‐fold and NaV1.5 by 2.1‐ and 1.8‐fold in the presence of Df1a‐NH2 and Df1a‐OH respectively. The remaining fraction of currents was calculated at 20 ms after 0 mV application, showing NaV1.3 with the highest fraction of persistent currents, followed by NaV1.5 and NaV1.1. (Figure 5B–D).
Figure 5.
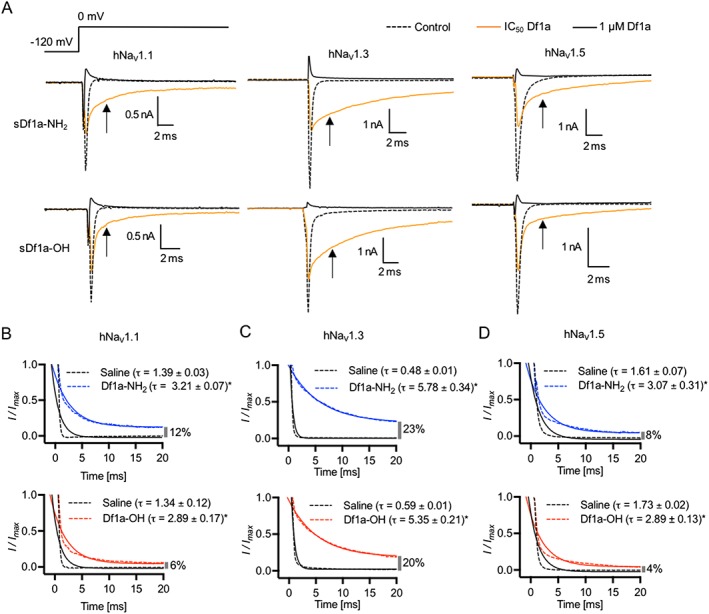
μ‐TRTX‐Df1a slows the fast inactivation of hNaV1.1, hNaV1.3 and hNaV1.5 along with peak current reduction after the application of sDf1a‐NH2 or sDf1a‐OH. The same effect is not observed in other hNaV subtypes tested, which presented only peak current reduction after the application of sDf1a‐NH2 or sDf1a‐OH (data not shown). Cells were held at −80 mV and Na+ currents were elicited by 20 ms voltage steps to 0 mV from a −120 mV conditioning pulse applied for 200 ms. (A) Representative traces of hNaV1.1, hNaV1.3 and hNaV1.5 in the presence of sDf1a‐NH2 and sDf1a‐OH. Cells were applied with the correspondent IC50 (black traces) and 1 μM sDf1a (grey trances) for each NaV subtype and incubated for 5 min before depolarization at 0 mV. No toxin controls are presented as dashed traces. Current traces showing the slowing in fast inactivation are featured by arrows. The slowing of fast inactivation at 5 ms after application of 0 mV was plotted against the log scale of various concentrations of Df1a, and maximum slowing in inactivation evaluated. (B–D) hNaV1.1, hNaV1.3 and hNaV1.5 inactivation decay time constant (τ) and percentage of remaining currents were calculated in the presence of respective IC50 concentrations of Df1a for each channel subtype. Blue traces represent the time constant (τ) in the presence of Df1a‐NH2 and red traces in the presence of Df1a‐OH. Remaining currents (%) are presented in the far right of the X‐axis at each graph. A marked slowing of fast inactivation was observed for the NaV1.3 channel subtype in the presence of Df1a, which also displayed the highest percentage of remaining currents at 20 ms. Data are presented as mean ± SEM from n ≥ 5 independent experiments for each ion channel assayed; one cell was considered as an independent experiment (see Supporting Information Table S1).
Kinetics of Df1a inhibition and current recovery in hNaV1.3 and hNaV1.7 channels
Association/dissociation rates and reversibility can impact considerably on the therapeutic potential of ion channel modulators. Thus, we used automated whole‐cell patch clamp electrophysiology to measure on‐ and off‐rates for Df1a inhibition of hNaV1.3 and hNaV1.7. Current inhibition and recovery were estimated following application of the amidated and acid forms of Df1a (Figure 6A–H, Table 1). The association rates for hNaV1.3 (K on) were slower for Df1a‐NH2 compared with Df1a‐OH at IC50 values (τ(min) 3.92 and 1.51 for Df1a‐NH2 and Df1a‐OH respectively) (Figure 6A), while for hNaV1.7 the association rates (K on) were faster for Df1a‐NH2 compared with Df1a‐OH at IC50 values (τ(min) 0.64 and 3.06 for Df1a‐NH2 and Df1a‐OH respectively) (Figure 6B). When applied at concentration 10 times the respective IC50 value, Df1a‐OH showed faster association rates for both hNaV1.3 and hNaV1.7. Current traces after 2.5 min incubation with Df1a showed a persistent slowing in fast inactivation for hNaV1.3, which was absent in hNaV1.7 (Figure 6C–F). The inhibition of hNaV1.3 and hNaV1.7 was quasi‐irreversible for Df1a‐NH2, while for Df1a‐OH, the inhibition was quasi‐irreversible for hNaV1.7 but reversible for hNaV1.3 (Figure 6G, H). The irreversibility of Df1a was tested using a − 80 mV holding potential, and more hyperpolarized holding potentials should be tested in the near future to further evaluate irreversibility of this toxin over NaV channels.
Figure 6.
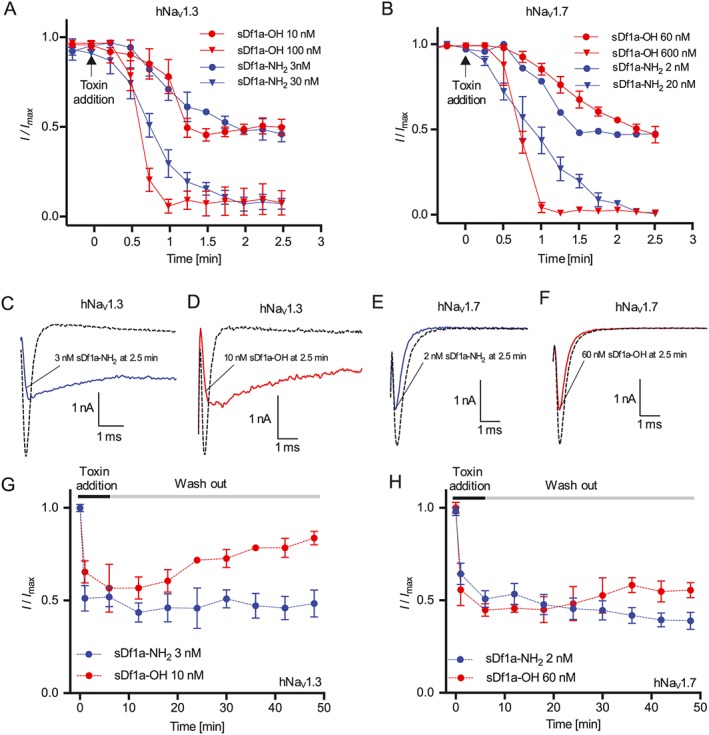
Kinetics of hNaV current inhibition and recovery in the presence of μ‐TRTX‐Df1a. Cells were maintained at a holding potential −80 mV and Na+ currents elicited by 20 ms voltage steps to 0 mV from a −120 mV conditioning pulse applied for 200 ms. (A) For on‐rates, Na+ currents were recorded every 15 s for 15 min after toxin addition. The on‐rates for hNaV1.3 were 4.34 and 2.03 min at 3 and 30 nM sDf1a‐NH2, respectively, and 1.14 and 1.13 min at 10 and 100 nM sDf1a‐OH respectively. (B) For hNaV1.7, the on‐rates were 0.64 and 1.32 min at 2 and 20 nM sDf1a‐NH2 and 3.06 and 1.32 min at 60 and 600 M sDf1a‐OH respectively. (C–F) Representative Na+ current traces after 2.5 min incubation with Df1a along to consecutive pulses of 0 mV with 15 s intervals. A persistent slowing in fast inactivation associated with peak current reduction was observed for hNaV1.3 in the presence of (C) 3 nM sDf1a‐NH2 and (D) 10 nM sDf1a‐OH, while for hNaV1.7, only a peak reduction was observed in the presence of (E) 2 nM sDf1a‐NH2 and (F) 60 nM sDf1a‐OH. (G–H) For the wash‐out of sDf1a‐NH2 and sDf1a‐OH over hNaV1.3 and hNaV1.7, cells were incubated for 10 min with Df1a and Na+ currents assessed at 5 min intervals during saline washes. The inhibition by sDf1a‐OH at the IC50 concentration was reversible only in the hNaV1.3 subtype, while for sDf1a‐NH2 over hNaV1.3 and hNaV1.7, and sDf1a‐OH over hNaV1.7 the inhibition remained quasi‐irreversible under the experimental conditions applied at up 50 min of recording. The K on, K off and K d were calculated using K d = K off/K on (nM), where K off = 1/τoff (s−1) and K on = (1/τon − K off)/[toxin] (nM−1·S−1). Data are presented as mean ± SEM, n = 5 independent experiments for each condition assayed; one cell was considered as an independent experiment.
Table 1.
Kinetics of current inhibition and recovery of hNaV1.3 and hNaV1.7 channels after application of μ‐TRTX‐Df1a
| Channel | Toxin | Concentration | K on | K off | K d |
|---|---|---|---|---|---|
| nM | (nM−1·s−1) | (s−1) | (nM) | ||
| hNaV1.3 | sDf1a‐NH2 | 3 | 1.4 ± 0.1 × 10−3 | 0 | 0 |
| 30 | 2.9 ± 0.7 × 10−4 | ND | ND | ||
| sDf1a‐OH | 10 | 1.5 ± 0.2 × 10−3 | 8.8 ± 1.5 x 10−3 | 12.5 ± 1.3 | |
| 100 | 1.6 ± 0.3 × 10−4 | ND | ND | ||
| hNaV1.7 | sDf1a‐NH2 | 2 | 1.3 ± 0.2 × 10−2 | 0 | 0 |
| 20 | 6.3 ± 1.3 × 10−4 | ND | ND | ||
| sDf1a‐OH | 60 | 9.0 ± 1.2 × 10−5 | 0 | 0 | |
| 600 | 2.1 ± 0.2 × 10−5 | ND | ND |
sDf1a‐NH2 was applied at 3 and 30 nM for hNaV1.3 and 2 and 20 nM for hNaV1.7, while sDf1a‐OH was applied at 10 and 100 nM for hNaV1.3 and 60 and 600 for hNaV1.7, and sodium currents measured. The kinetics of inhibition and recovery of inhibition were determined from the I/Imax as a function of time from traces shown in Figure 7A–H fitted to a single exponential. Values are from n = 5 independent experiments (mean ± SEM). ND, not determined.
NaV1.7 sites interacting with Df1a
We used a panel of previously described hNaV1.7/rKv2.1 S3–S4 paddle chimeras (Klint et al., 2015) to map the binding site for Df1a on hNaV1.7 (Figure 7). The acid and amide forms of Df1a had no effect on the DI chimera or wild‐type rKV2.1 at 1 μM concentration, and they only partially inhibited currents from the DIII and DIV chimeras. In contrast, 1 μM Df1a‐NH2 and Df1a‐OH significantly inhibited currents from the DII chimera. Thus, our data indicate that Df1a primarily interacts with the DII voltage sensor of hNaV1.7, with weaker interactions with VSD III and IV.
Figure 7.
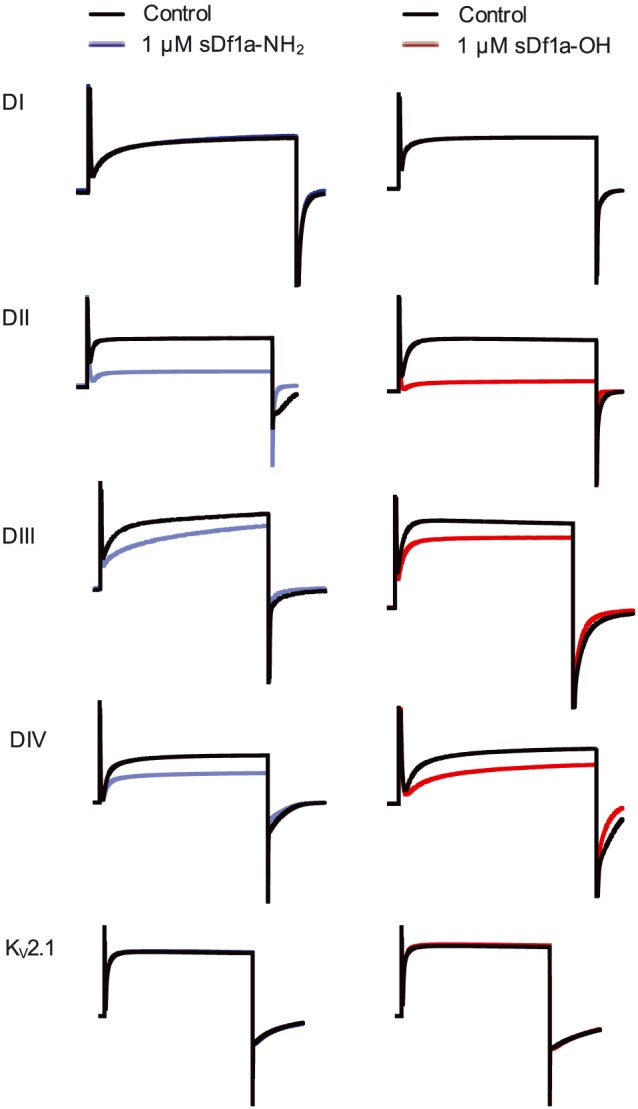
Binding sites of μ‐TRTX‐Df1a over hNaV.7. Chimeras hNaV1.7/rKV2.1 containing the paddles S3‐S4 from DI‐DIV from NaV1.7 were used to explore the binding site of Df1a over NaV1.7. Potassium currents were elicited by depolarization to +70 mV. The currents are shown before and after addition of 1 μM Df1a toxin. sDf1a (both C‐terminally acid and amide) preferentially binds to S3‐S4 loop region in DII of NaV1.7, followed by DIII and DIV. Df1a (C‐terminally acid and amide) had no effect on wild‐type rKV2.1 at up to 1 μM. Data are from n = 5 independent experiments for each condition assayed; one oocyte was considered for each independent experiment.
Df1a is analgesic in a mouse model of pain
In order to evaluate the potential of Df1a to reverse peripheral pain in vivo, we use an OD1‐induced pain model. Intraplantar injection of OD1, a scorpion toxin that preferentially potentiates the activity of NaV1.4, NaV1.6 and NaV1.7 channels (Durek et al., 2013), causes rapid development of spontaneous pain in mice as evidenced by flinching and licking of the affected hind paw (Cardoso et al., 2015; Deuis et al., 2016). Intraplantar injection of 10 μM Df1a‐NH2 or Df1a‐OH (400 pmol in a 40 μL injection) significantly reduced spontaneous pain behaviour (Figure 8; OD1, 102 ± 4 flinches 10 min−1; 10 μM Df1a‐NH2, 56 ± 7 flinches 10 min−1; 1 μM Df1a‐OH, 73 ± 8 flinches 10 min−1; 10 μM Df1a‐OH, 30 ± 6 flinches 10 min−1, P < 0.05). No significant reduction in the pain behaviour was observed when Df1a‐NH2 or Df1a‐OH was administered at 1 μM (40 pmol in a 40 μL injection). Data are presented as mean ± SEM of n = 5 mice per group treated with Df1a and n = 12 mice in the control group.
Figure 8.
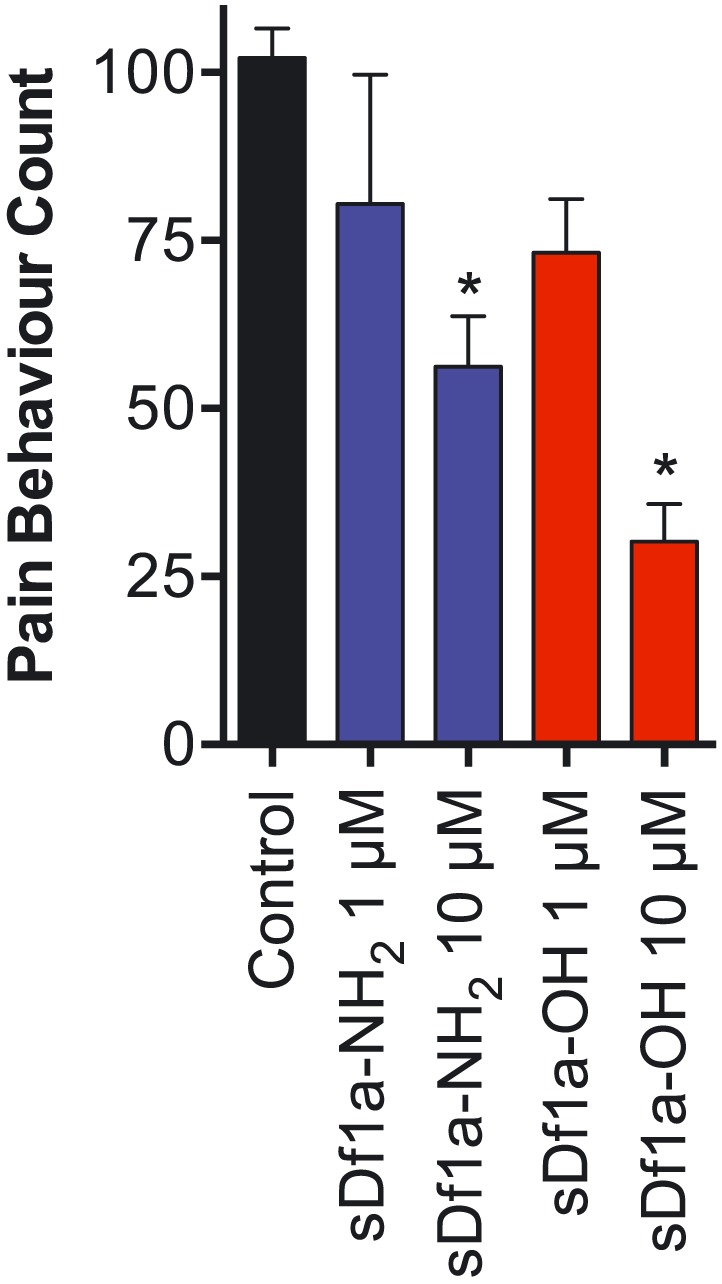
Antinociceptive effects of μ‐TRTX‐Df1a. (A) Intraplantar injection of the NaV1.7 activator OD1 (300 nM) led to rapid development of nocifensive behaviour in mice. This spontaneous pain behaviour, measured by the number of paw licks and flinches, was attenuated in a concentration‐dependent manner by co‐administration of sDf1a‐OH at 1 and 10 μM, and sDf1a‐NH2 at 10 μM but not at 1 μM. Data are presented as mean ± SEM of n = 5 mice per group treated with Df1a and n = 12 mice in the control group. *P < 0.05.
Structure analysis of Df1a
The 3D structure of μ‐TRTX‐Df1a was modelled using the structure of β/ω‐theraphotoxin‐Tp1a (ProTx‐I, PDB code 2M9L) as a template (Gui et al., 2014) (Figure 9A). The model obtained had a GMQE score of 0.91, with 82% of residues in the favoured region of the Ramachandran plot. The structural model revealed Df1a adopts an ICK fold that is organized into two distinct faces; a hydrophobic patch surrounded by charged residues lies opposite a face comprised primarily of neutral hydrophilic residues face (Figure 9B). The hydrophobic patch is dominated by aromatic residues in Df1a, including the central W4, F5, W27 and W30 residues and the peripheral W32 and F34 residues. This is reminiscent of the hydrophobic patch present in the ProTx‐I (Y4, W5, W27, W30 and F34), although there are significant differences including E17K, Q26G and W32G (Figure 9C). Nevertheless, the structural data suggest that these toxins share similar binding properties to NaV1.7.
Figure 9.
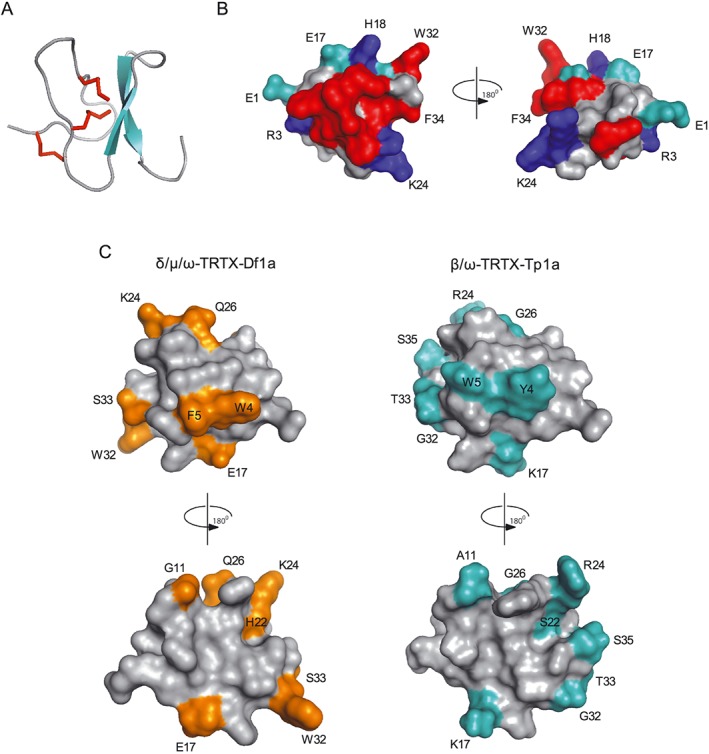
Molecular modelling and structural features of μ‐TRTX‐Df1a. The three dimensional structure of Df1a was modelled using the NMR structure of β/ω‐theraphotoxin‐Tp1a (ProTx‐I) (Gui et al., 2014). (A) Ribbon representation showing β‐sheet (cyan) and Cys‐bridges (red) of a typical ICK peptide. (B) Surface representation of Df1a structure with 180° rotation shown in cyan: negatively charged, blue: positively charged and red: hydrophobic residues (aromatics). Residues present in these regions are labelled (E1, R3, E17, H18, K24, W32 and F34). (C) Comparison of the structures of Df1a and ProTx‐I. The differences in amino acids residues between these toxins are highlighted in orange and cyan respectively. These residues are W4Y, F5W, G11A, E17K, H22S, K24R, Q26G, W32G, S33T for Df1a and ProTx‐I, respectively, and S35 is present only in ProTx‐I. Structures are shown in two orientations, rotated by 180°.
Discussion
Spider venoms have proven to be a rich source of peptide ligands that modulate NaV channels (King, 2011). Their ability to target ion channels at allosteric binding sites often enhances target selectivity and opens up opportunities for new ion channel therapeutics. In the present work, we report the discovery and characterization of a new NaV and CaV3 channel inhibitor named Df1a from the tarantula Davus fasciatus. Df1a belongs to NaSpTx Family 2, whose members contain an ICK motif and highly conserved N‐ and C‐termini. The closest orthologue of Df1a is the NaV and CaV3 channel inhibitor ProTx‐I (Middleton et al., 2002). However, ProTxI causes a reduction in peak current without altering channel inactivation, whereas Df1a has the dual effect of reducing peak current and slowing fast inactivation of some NaV channel subtypes.
Electrophysiology using mammalian cells revealed that Df1a inhibits hNaV channels with a rank order of potency 1.7 > 1.2 > 1.3 > 1.6 > 1.1 > 1.4 > 1.5 and hCaV3 channels with a rank order 3.1 > 3.3 > 3.2. Interestingly, C‐terminal amidation of Df1a increased potency by 1.7‐ to 4‐fold for hNaV1.1 to hNaV1.6, 1.2‐ to 8.7‐fold for hCaV3.1 to hCaV3.3 and remarkably by 32‐fold for hNaV1.7. The effect on hNaV1.7 was also observed using two‐electrode voltage clamp in oocytes; in this system, C‐terminal amidation increased potency against NaV1.7 by 22‐fold. C‐terminal amidation has been reported to significantly increase the potency of other NaSpTx peptides. The amidated form of huwentoxin‐IV, a toxin isolated from the spider Haplopelma schmidti, is 42‐fold more potent against NaV1.7 (Sermadiras et al., 2013). The C‐terminally amidated form of ProTx‐III, a toxin that we recently isolated from the spider Thrixopelma pruriens, is 4.6‐fold more potent against NaV1.7 and 8.9‐ and 3.5‐fold more potent against NaV1.1 and NaV1.3 respectively (Cardoso et al., 2015). Although these peptides do not display favourable selectivity for key ion channels involved in complex disorders, the remarkable increase in potency of their C‐terminal amide forms is important for the rational development of peptide drugs that target voltage‐gated ion channels.
Among the ion channels studied in this work, hNaV1.3, hNaV1.7 and hCaV3.2 are known to be involved in pain disorders. hNaV1.3 is up‐regulated in dorsal root ganglion neurons during chronic constriction nerve injury and following axotomy (Waxman et al., 1994; Dib‐Hajj et al., 1999). A lack of NaV1.7 function induces insensitivity to pain (Cox et al., 2006), and a monoclonal antibody against NaV1.7 suppresses inflammatory and neuropathic pain in mice (Lee et al., 2014). Altered T‐type Ca2+ currents are involved in somatic and visceral pain signalling, and the hyperalgesia induced by L‐cysteine was reported to be present in wild‐type but not CaV3.2‐knockout mice (Nelson et al., 2007; Maeda et al., 2009). These studies emphasize the potential of Df1a as a tool peptide to help guiding the development of novel drugs that simultaneously target NaV and CaV channels involved in chronic pain states.
Patch‐clamp studies revealed that Df1a alters the voltage‐dependence of activation and inactivation of hNaV channels. Other spider toxins belonging to NaSpTx2 and which alter the gating properties of NaV channels are β‐TRTX‐Cm2a and μ‐TRTX‐Cj1a (Bosmans et al., 2006; Chen et al., 2009). Curiously, Df1a also slowed fast inactivation of hNaV1.1, hNaV1.3 and hNaV1.5. A similar effect was observed for the spider toxin JZTX‐XI, which slowed fast inactivation of TTX‐R and TTX‐S sodium channels (Liao et al., 2006; Tao et al., 2016), and for the spider toxin ProTx‐II at NaV1.2 to NaV1.5 and NaV1.7 channels (Xiao et al., 2010). However, ProTx‐II induces persistent currents in hNaV1.7 at saturating concentrations, while Df1a is a full inhibitor of NaV channel currents irrespective of subtype. Interestingly, Df1a showed no changes in the voltage‐dependence of activation and inactivation of hNaV1.7 using two‐electrode voltage clamp in oocytes. Differences in membrane composition between mammalian cells and oocytes that potentially influence how spider toxins bind to the NaV1.7 channel could explain the effects observed (Henriques et al., 2016). Furthermore, for the two‐electrode voltage clamp studies reported here, no β subunit was co‐expressed with NaV1.7, which could alter gating properties as well as toxin affinity to the channel. It was previously demonstrated that in oocytes, in the absence of the β1 subunit, fast inactivation of NaV is slowed and steady‐state inactivation altered by a ΔV50 of 6 mV (Shcherbatko et al., 1999). In this present study, the ΔV50 of NaV1.7 steady‐state inactivation observed in the presence and absence of the β1 subunit was 12 mV, which confirms the slowing in inactivation previously observed. Furthermore, in studies in which NaV1.2 was expressed in oocytes, the spider toxin ProTx‐II completely lost affinity to this channel in the presence of the β4 subunit, while the scorpion toxin TsVII showed potent activity over NaV1.2 only in the absence of β4 (Gilchrist et al., 2013).
To further investigate the site of action of Df1a, we used a panel of hNaV1.7/KV2.1 chimeras (Klint et al., 2015). These experiments revealed that Df1a preferentially interacts with the DII voltage sensor of hNaV1.7, and to a less extent with the DIII and DIV voltage sensors. The data also revealed that Df1a has no effect on KV2.1, although its closest orthologue ProTx‐I potent inhibits this potassium channel (Middleton et al., 2002). Previous reports have shown that inhibition of NaV1.7 by spider toxins is mediated through interactions with the DII voltage sensor (Klint et al., 2015), whereas toxins that slow fast inactivation primarily interact with the DIV voltage sensor (Rogers et al., 1996; Mitrovic et al., 2000; Campos et al., 2008; Xiao et al., 2010; Tao et al., 2016). Similar studies performed with ProTx‐II demonstrated interactions of this toxin with the DI, DII and DIV voltage sensors of NaV1.2 (Bosmans et al., 2008), suggesting that DII and DIV interactions mediate the dual modulatory effects observed for both Df1a and ProTx‐II. Df1a does not affect fast inactivation of hNaV1.7, consistent with its more robust binding to the DII voltage sensor of this channel. In contrast, since Df1a does slow fast inactivation of NaV1.1, NaV1.3 and NaV1.5, we speculate that the toxin interacts more avidly with the DIV voltage sensor in these NaV channel subtypes. Although the chimeric channels provide useful insights into the binding sites for Df1a on NaV channels, these interactions might differ among NaV channel subtypes and/or in the complete NaV voltage‐sensor constructs.
Modelling of the Df1a structure revealed an ICK motif typical of spider toxins gating modifiers (Klint et al., 2012). Furthermore, the structure of Df1a displayed surface similarities with the structure of ProTx‐I (Gui et al., 2014) and three other spider toxins belonging to the NaSpTx Family 2 and inhibitors of KV (κ‐TRTX‐Gr1a and κ‐TRTX‐Scg1a) and CaV (ω‐TRXT‐Gr1a) (Takahashi et al., 2000; Takeuchi et al., 2002; Wang et al., 2004). These displayed a conserved large hydrophobic patch surrounded by positively charged residues which is potentially involved in the interactions with the hydrophobic core of the cell membrane and the S3–S4 linker regions of the voltage‐gated ion channels. The pharmacophore for ProTx‐I interaction with hNaV1.2 was established by alanine scan (Gui et al., 2014), from which the key residues identified were found to be identical in Df1a, suggesting that these toxins possibly share a similar pharmacophore for NaV channels. Further studies are required to elucidate pharmacophores for Df1a as well as identify residues that might enhance selectivity towards key channels subtypes and its potential as a lead for treating NaV‐ and CaV3‐related disorders.
Finally, given the important role of NaV channels in nociception, we tested the analgesic efficacy of Df1a in a rodent pain model in which nocifensive behaviour is elicited by intraplantar administration of the NaV activator OD1 (Durek et al., 2013; Cardoso et al., 2015; Deuis et al., 2016). Intraplantar administration of Df1a‐NH2 and Df1a‐OH reduced nocifensive behaviour by 42 and 71%, respectively, suggesting that Df1a can effectively inhibit OD1‐induced pain at peripheral sensory nerve endings in the skin. Although we have evidence Df1a inhibits NaV and CaV3 channels involved in peripheral sensory pain, the exact targets of the observed analgesic effect remain to be fully elucidated. Interestingly, the Df1a C‐terminal acid was less potent than the amide form at the human NaV isoform co‐expressed with β1 but more effective at reducing pain behaviours in a rodent pain model. This effect could be related to differences between the human and rodent ion channel isoforms and/or due to distinct expression and combinations of auxiliary β subunits in vivo, which could lead to altered sensitivities to Df1a (Isom, 2001; Gilchrist et al., 2013). Assessment of the analgesic potential of Df1a in disease‐specific pain models will provide additional insight into the clinical potential of this peptide for the treatment of chronic pain.
In conclusion, we have characterized a new spider toxin named μ‐TRTX‐Df1a. Df1a is a potent NaV and CaV3 channel inhibitor that in addition to shifting the voltage‐dependence of activation and inactivation produces a prominent slowing in fast inactivation at specific NaV channels subtypes. Channel inhibition in vitro was enhanced by C‐terminal amidation of Df1a, but the C‐terminal acid form showed better analgesic properties in vivo. The in vivo analgesic efficacy of Df1a suggests it might be a useful lead for the development of analgesics targeting NaV‐mediated pain.
Author contributions
F.C.C., R.J.L. and G.F.K. were responsible for the research conception and design. F.C.C., Z.D., J.J.S., I.V., J.R.D. and V.H. did the conduction of experiments, analysis and interpretation of data. F.C.C. and R.J.L. wrote the manuscript. F.C.C., Z.D., J.R.D., V.H., P.F.A., G.F.K. and R.J.L. revised the manuscript.
Conflict of interest
The authors declare no conflicts of interest.
Declaration of transparency and scientific rigour
This Declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research recommended by funding agencies, publishers and other organisations engaged with supporting research.
Supporting information
Figure S1 ProTx‐I and Df1a effects on hNav1.7 expressed in oocytes. Briefly, Xenopus laevis oocytes were injected with cRNA encoding hNaV α subunit hNaV1.7 and two‐electrode voltage clamp electrophysiology was performed as per described in the Methods section of the main text, however the external recording solution contained (in mM) 96 NaCl, 2 KCl, 5 HEPES, 1 MgCl2, 1.8 CaCl2, pH 7.45 with NaOH. Leak and background conductance, identified by blocking channels with tetrodotoxin, were subtracted for all experiments. Dose–response curves were generated using a non‐linear fit function (log(inhibitor) versus. response (three parameters) in Prism version 6.0 software. (A, C and E) Inhibition of Na+ currents in the presence of Df1a‐NH2, Df1a‐OH and ProTx‐I showed a IC50 values of (in nM) 17 (95% CI 11 to 28), 369 (95% CI 257 to 530) and 64 (95% CI 43 to 97) respectively. (B, D and F) No significant changes in the voltage‐dependence of activation and inactivation were observed after application of 30 nM Df1a‐NH2, 300 nM Df1a‐OH and 60 nM ProTx‐I. The V50 values for the voltage‐dependence of activation and inactivation controls were −15.2 ± 0.4 and −45.7 ± 1.71 mV respectively. Data are representedasmean ± SEM, n ≥ 5.
Figure S2 ProTx‐I effect on hNav1.7/β1 expressed in HEK 293 cells measured by automated patch clamp electrophysiology in QPatch 16X. Exper iments and data analysis were performed as per descr ibed in the Methods section of the main text. (A) ProTx‐I inhibited hNaV1.7 currents with a IC50 value of 43.5 (95% CI 40.7 to 45.08). (B) No signif icant changes in the voltagedependence of activation and inactivation were observed in the presence of ProTx‐I, with maximum change of −3.8 mV in the steady‐state inactivation and 0.5 mV in the voltage of activation of hNaV1.7. Data are represented as mean ± SEM, n = 5.
Table S1 IC50 and Hill slope values calculated using I/I max values and non‐linear regression for the μ‐TRTX‐Df1a activity over hNaV and hCaV3 channels evaluated in automated whole cell patch‐clamp and mammalian recombinant cell lines. The decay in potency observed in the acid form of Df1awas estimatedas the ratio of the IC50 values against theDf1a amide.
Acknowledgements
This work was supported by the Australian Research Council (linkage Grant to G.F.K., R.J.L. and P.F.A. and Future Fellowship to I.V.) and Australian National Health & Medical Research Council (programme grant APP1072113 to R.J.L., G.F.K. and P.F.A. and Principal Research Fellowships to R.J.L., G.F.K. and P.F.A.). We thank Dr Frank Bosmans (School of Medicine, Johns Hopkins University, MD, USA) for hNaV1.7/rKV2.1 chimeras and Prof Emmanuel Bourinet (Institute of Functional Genomics, Montpellier University, France) and Prof Edward Perez‐Reyes (School of Medicine, University of Virginia, USA) for Cav3 cell lines. We thank Dr Alun Jones (Institute for Molecular Bioscience, The University of Queensland, QLD, Australia) for help with mass spectrometry. We thank Frank and Claudia Schneider (Ludwigshafen, Germany) for providing the spider for venom extraction. Access to the Australian Proteome Analysis Facility is facilitated by support from the Australian Government's National Collaborative Research Infrastructure Strategy.
Cardoso, F. C. , Dekan, Z. , Smith, J. J. , Deuis, J. R. , Vetter, I. , Herzig, V. , Alewood, P. F. , King, G. F. , and Lewis, R. J. (2017) Modulatory features of the novel spider toxin μ‐TRTX‐Df1a isolated from the venom of the spider Davus fasciatus . British Journal of Pharmacology, 174: 2528–2544. doi: 10.1111/bph.13865.
Contributor Information
Fernanda C Cardoso, Email: f.caldascardoso@uq.edu.au.
Richard J Lewis, Email: r.lewis@uq.edu.au.
References
- Alexander SPH, Catterall WA, Kelly E, Marrion N, Peters JA, Benson HE et al. (2015). The Concise Guide to PHARMACOLOGY 2015/16: Voltage‐gated ion channels. Br J Pharmacol 172: 5904–5941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin AS, Klemens CA, Verkerk AO, Meregalli PG, Asghari‐Roodsari A, de Bakker JM et al. (2010). Fever‐triggered ventricular arrhythmias in Brugada syndrome and type 2 long‐QT syndrome. Neth Heart J 18: 165–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K, Bordoli L, Kopp J, Schwede T (2006). The SWISS‐MODEL workspace: a web‐based environment for protein structure homology modelling. Bioinformatics 22: 195–201. [DOI] [PubMed] [Google Scholar]
- Bosmans F, Rash L, Zhu S, Diochot S, Lazdunski M, Escoubas P et al. (2006). Four novel tarantula toxins as selective modulators of voltage‐gated sodium channel subtypes. Mol Pharmacol 69: 419–429. [DOI] [PubMed] [Google Scholar]
- Bosmans F, Martin‐Eauclaire MF, Swartz KJ (2008). Deconstructing voltage sensor function and pharmacology in sodium channels. Nature 456: 202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campos FV, Chanda B, Beirao PS, Bezanilla F (2008). Alpha‐scorpion toxin impairs a conformational change that leads to fast inactivation of muscle sodium channels. J Gen Physiol 132: 251–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso FC, Dekan Z, Rosengren KJ, Erickson A, Vetter I, Deuis J et al. (2015). Identification and characterization of ProTx‐III [μ‐TRTX‐Tp1a], a new voltage‐gated sodium channel inhibitor from venom of the tarantula Thrixopelma pruriens . Mol Pharmacol 88: 291–303. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Cestele S, Yarov‐Yarovoy V, Yu FH, Konoki K, Scheuer T (2007). Voltage‐gated ion channels and gating modifier toxins. Toxicon 49: 124–141. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Kalume F, Oakley JC (2010). NaV1.1 channels and epilepsy. J Physiol 588: 1849–1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang Y, Rong M, Zhao L, Jiang L, Zhang D et al. (2009). Expression and characterization of jingzhaotoxin‐34, a novel neurotoxin from the venom of the tarantula Chilobrachys jingzhao. Peptides 30: 1042–1048. [DOI] [PubMed] [Google Scholar]
- Chino N, Kubo S, Nishio H, Nishiuchi Y, Nakazato M, Kimura T (2006). Chemical synthesis of human β‐defensin (hBD)‐1,‐2,‐3 and ‐4: optimization of the oxidative folding reaction. Int J Pept Res Ther 12: 203–209. [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K et al. (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444: 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis MJ, Bond RA, Spina D, Ahluwalia A, Alexander SP, Giembycz MA et al. (2015). Experimental design and analysis and their reporting: new guidance for publication in BJP. Br J Pharmacol 172: 3461–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLano WL (2002). The PyMOL Molecular Graphics System, Version 1.5.0.4 Schrödinger, LLC.
- Deuis JR, Wingerd JS, Winter Z, Durek T, Dekan Z, Zimmermann K et al. (2016). Analgesic effects of GpTx‐1, PF‐04856264 and CNV1014802 in a mouse model of NaV1.7‐mediated pain. Toxins 8: 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Fjell J, Cummins TR, Zheng Z, Fried K, LaMotte R et al. (1999). Plasticity of sodium channel expression in DRG neurons in the chronic constriction injury model of neuropathic pain. Pain 83: 591–600. [DOI] [PubMed] [Google Scholar]
- Durek T, Vetter I, Wang CI, Motin L, Knapp O, Adams DJ et al. (2013). Chemical engineering and structural and pharmacological characterization of the alpha‐scorpion toxin OD1. ACS Chem Biol 8: 1215–1222. [DOI] [PubMed] [Google Scholar]
- Errington AC, Stohr T, Heers C, Lees G (2008). The investigational anticonvulsant lacosamide selectively enhances slow inactivation of voltage‐gated sodium channels. Mol Pharmacol 73: 157–169. [DOI] [PubMed] [Google Scholar]
- Gilchrist J, Das S, Van Petegem F, Bosmans F (2013). Crystallographic insights into sodium‐channel modulation by the beta 4 subunit. Proc Natl Acad Sci U S A 110: E5016–E5024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui J, Liu B, Cao G, Lipchik AM, Perez M, Dekan Z et al. (2014). A tarantula‐venom peptide antagonizes the TRPA1 nociceptor ion channel by binding to the S1‐S4 gating domain. Curr Biol 24: 473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques ST, Deplazes E, Lawrence N, Cheneval O, Chaousis S, Inserra M et al. (2016). Interaction of tarantula venom peptide protx‐ii with lipid membranes is a prerequisite for its inhibition of human voltage‐gated sodium channel NaV1.7. J Biol Chem 291: 17049–17065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig V, Hodgson WC (2009). Intersexual variations in the pharmacological properties of Coremiocnemis tropix (Araneae, Theraphosidae) spider venom. Toxicon 53: 196–205. [DOI] [PubMed] [Google Scholar]
- Herzig V, King GF (2015). The cystine knot is responsible for the exceptional stability of the insecticidal spider toxin ω‐Hexatoxin‐Hv1a. Toxins (Basel) 7: 4366–4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig V, Wood DL, Newell F, Chaumeil PA, Kaas Q, Binford GJ et al. (2011). ArachnoServer 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res 39: D653–D657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL (2001). Sodium channel beta subunits: anything but auxiliary. Neuroscientist 7: 42–54. [DOI] [PubMed] [Google Scholar]
- Kaas Q, Yu R, Jin AH, Dutertre S, Craik DJ (2012). ConoServer: updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res 40: D325–D330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG (2010). Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160: 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King GF (2011). Venoms as a platform for human drugs: translating toxins into therapeutics. Expert Opin Biol Ther 11: 1469–1484. [DOI] [PubMed] [Google Scholar]
- King GF, Hardy MC (2013). Spider‐venom peptides: structure, pharmacology, and potential for control of insect pests. Annu Rev Entomol 58: 475–496. [DOI] [PubMed] [Google Scholar]
- Klint JK, Senff S, Rupasinghe DB, Er SY, Herzig V, Nicholson GM et al. (2012). Spider‐venom peptides that target voltage‐gated sodium channels: pharmacological tools and potential therapeutic leads. Toxicon 60: 478–491. [DOI] [PubMed] [Google Scholar]
- Klint JK, Smith JJ, Vetter I, Rupasinghe DB, Er SY, Senff S et al. (2015). Seven novel modulators of the analgesic target NaV1.7 uncovered using a high‐throughput venom‐based discovery approach. Br J Pharmacol 172: 2445–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria G, Ziegler D, Malik R, Merkies IS, Waxman SG, Faber CG et al. (2014). The role of sodium channels in painful diabetic and idiopathic neuropathy. Curr Diab Rep 14: 538. [DOI] [PubMed] [Google Scholar]
- Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T et al. (2014). A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell 157: 1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Liao Z, Yuan C, Deng M, Li J, Chen J, Yang Y et al. (2006). Solution structure and functional characterization of jingzhaotoxin‐XI: a novel gating modifier of both potassium and sodium channels. Biochemistry 45: 15591–15600. [DOI] [PubMed] [Google Scholar]
- Liu M, Wood JN (2011). The roles of sodium channels in nociception: implications for mechanisms of neuropathic pain. Pain Med 12 (Suppl 3): S93–S99. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG et al. (2003). Structure validation by Cα geometry: Φ, Ψ and Cβ deviation. Proteins 50: 437–450. [DOI] [PubMed] [Google Scholar]
- Maeda Y, Aoki Y, Sekiguchi F, Matsunami M, Takahashi T, Nishikawa H et al. (2009). Hyperalgesia induced by spinal and peripheral hydrogen sulfide: evidence for involvement of CaV3.2 T‐type calcium channels. Pain 142: 127–132. [DOI] [PubMed] [Google Scholar]
- McGrath JC, Lilley E (2015). Implementing guidelines on reporting research using animals (ARRIVE etc.): new requirements for publication in BJP. Br J Pharmacol 172: 3189–3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meisler MH, Kearney JA (2005). Sodium channel mutations in epilepsy and other neurological disorders. J Clin Invest 115: 2010–2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton RE, Warren VA, Kraus RL, Hwang JC, Liu CJ, Dai G et al. (2002). Two tarantula peptides inhibit activation of multiple sodium channels. Biochemistry 41: 14734–14747. [DOI] [PubMed] [Google Scholar]
- Mitrovic N, George AL Jr, Horn R (2000). Role of domain 4 in sodium channel slow inactivation. J Gen Physiol 115: 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldovan M, Alvarez S, Romer Rosberg M, Krarup C (2013). Axonal voltage‐gated ion channels as pharmacological targets for pain. Eur J Pharmacol 708: 105–112. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Woo J, Kang HW, Vitko I, Barrett PQ, Perez‐Reyes E et al. (2007). Reducing agents sensitize C‐type nociceptors by relieving high‐affinity zinc inhibition of T‐type calcium channels. J Neurosci 27: 8250–8260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragsdale DS, Avoli M (1998). Sodium channels as molecular targets for antiepileptic drugs. Brain Res Brain Res Rev 26: 16–28. [DOI] [PubMed] [Google Scholar]
- Remme CA, Bezzina CR (2010). Sodium channel (dys)function and cardiac arrhythmias. Cardiovasc Ther 28: 287–294. [DOI] [PubMed] [Google Scholar]
- Rogers JC, Qu Y, Tanada TN, Scheuer T, Catterall WA (1996). Molecular determinants of high affinity binding of α‐scorpion toxin and sea anemone toxin in the S3‐S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J Biol Chem 271: 15950–15962. [DOI] [PubMed] [Google Scholar]
- Rogers M, Tang L, Madge DJ, Stevens EB (2006). The role of sodium channels in neuropathic pain. Semin Cell Dev Biol 17: 571–581. [DOI] [PubMed] [Google Scholar]
- Sermadiras I, Revell J, Linley JE, Sandercock A, Ravn P (2013). Recombinant expression and in vitro characterisation of active Huwentoxin‐IV. PLoS One 8: e83202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shcherbatko A, Ono F, Mandel G, Brehm P (1999). Voltage‐dependent sodium channel function is regulated through membrane mechanics. Biophys J 77: 1945–1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Southan C, Sharman JL, Benson HE, Faccenda E, Pawson AJ, Alexander SP et al. (2016). The IUPHAR/BPS Guide to PHARMACOLOGY in 2016: towards curated quantitative interactions between 1300 protein targets and 6000 ligands. Nucleic Acids Res 44: D1054–D1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Kim JI, Min HJ, Sato K, Swartz KJ, Shimada I (2000). Solution structure of hanatoxin1, a gating modifier of voltage‐dependent K(+) channels: common surface features of gating modifier toxins. J Mol Biol 297: 771–780. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Park E, Lee C, Kim J, Takahashi H, Swartz K et al. (2002). Solution structure of omega‐grammotoxin SIA, a gating modifier of P/Q and N‐type Ca(2+) channel. J Mol Biol 321: 517–526. [DOI] [PubMed] [Google Scholar]
- Tao H, Chen X, Lu M, Wu Y, Deng M, Zeng X et al. (2016). Molecular determinant for the tarantula toxin Jingzhaotoxin‐I slowing the fast inactivation of voltage‐gated sodium channels. Toxicon 111: 13–21. [DOI] [PubMed] [Google Scholar]
- Vetter I, Mozar CA, Durek T, Wingerd JS, Alewood PF, Christie MJ et al. (2012). Characterisation of NaV types endogenously expressed in human SH‐SY5Y neuroblastoma cells. Biochem Pharmacol 83: 1562–1571. [DOI] [PubMed] [Google Scholar]
- Wang JM, Roh SH, Kim S, Lee CW, Kim JI, Swartz KJ (2004). Molecular surface of tarantula toxins interacting with voltage sensors in KV channels. J Gen Physiol 123: 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Kocsis JD, Black JA (1994). Type III sodium channel mRNA is expressed in embryonic but not adult spinal sensory neurons, and is reexpressed following axotomy. J Neurophysiol 72: 466–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y, Blumenthal K, Jackson JO 2nd, Liang S, Cummins TR (2010). The tarantula toxins ProTx‐II and Huwentoxin‐IV differentially interact with human NaV1.7 voltage sensors to inhibit channel activation and inactivation. Mol Pharmacol 78: 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Dupre N, Puymirat J, Chahine M (2012). Biophysical characterization of M1476I, a sodium channel founder mutation associated with cold‐induced myotonia in French Canadians. J Physiol 590: 2629–2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 ProTx‐I and Df1a effects on hNav1.7 expressed in oocytes. Briefly, Xenopus laevis oocytes were injected with cRNA encoding hNaV α subunit hNaV1.7 and two‐electrode voltage clamp electrophysiology was performed as per described in the Methods section of the main text, however the external recording solution contained (in mM) 96 NaCl, 2 KCl, 5 HEPES, 1 MgCl2, 1.8 CaCl2, pH 7.45 with NaOH. Leak and background conductance, identified by blocking channels with tetrodotoxin, were subtracted for all experiments. Dose–response curves were generated using a non‐linear fit function (log(inhibitor) versus. response (three parameters) in Prism version 6.0 software. (A, C and E) Inhibition of Na+ currents in the presence of Df1a‐NH2, Df1a‐OH and ProTx‐I showed a IC50 values of (in nM) 17 (95% CI 11 to 28), 369 (95% CI 257 to 530) and 64 (95% CI 43 to 97) respectively. (B, D and F) No significant changes in the voltage‐dependence of activation and inactivation were observed after application of 30 nM Df1a‐NH2, 300 nM Df1a‐OH and 60 nM ProTx‐I. The V50 values for the voltage‐dependence of activation and inactivation controls were −15.2 ± 0.4 and −45.7 ± 1.71 mV respectively. Data are representedasmean ± SEM, n ≥ 5.
Figure S2 ProTx‐I effect on hNav1.7/β1 expressed in HEK 293 cells measured by automated patch clamp electrophysiology in QPatch 16X. Exper iments and data analysis were performed as per descr ibed in the Methods section of the main text. (A) ProTx‐I inhibited hNaV1.7 currents with a IC50 value of 43.5 (95% CI 40.7 to 45.08). (B) No signif icant changes in the voltagedependence of activation and inactivation were observed in the presence of ProTx‐I, with maximum change of −3.8 mV in the steady‐state inactivation and 0.5 mV in the voltage of activation of hNaV1.7. Data are represented as mean ± SEM, n = 5.
Table S1 IC50 and Hill slope values calculated using I/I max values and non‐linear regression for the μ‐TRTX‐Df1a activity over hNaV and hCaV3 channels evaluated in automated whole cell patch‐clamp and mammalian recombinant cell lines. The decay in potency observed in the acid form of Df1awas estimatedas the ratio of the IC50 values against theDf1a amide.