

Abstract
Vascular Endothelial Growth Factors (VEGFs) are promising molecules for the treatment of ischemic diseases by pro-angiogenic therapy. Snake venom VEGFs are a novel subgroup with unique receptor binding profiles and as such are potential new therapeutic agents. We determined the ligand-receptor interactions, gene regulation and angiogenic properties of Vipera ammodytes venom VEGF, Vammin, and compared it to the canonical angiogenic factor VEGF-A to evaluate the use of Vammin for therapeutic angiogenesis. Vammin efficiently induced VEGFR-2 mediated proliferation and expression of genes associated with proliferation, migration and angiogenesis. VEGF-A165 and especially VEGF-A109 induced less pronounced effects. Vammin regulates a number of signaling pathways by inducing the expression of NR4A family nuclear receptors and regulators of calcium signaling and MAP kinase pathways. Interestingly, MARC1, which encodes an enzyme discovered to catalyze reduction of nitrate to NO, was identified as a novel VEGFR-2 regulated gene. In rabbit skeletal muscle adenoviral delivery of Vammin induced prominent angiogenic responses. Both the vector dose and the co-receptor binding of the ligand were critical parameters controlling the type of angiogenic response from sprouting angiogenesis to vessel enlargement. Vammin induced VEGFR-2/NRP-1 mediated signaling more effectively than VEGF-A, consequently it is a promising candidate for development of pro-angiogenic therapies.
Introduction
Vascular endothelial growth factors (VEGF) are key regulators of blood and lymphatic vessel formation and maintenance. They are potential gene candidates for therapeutic angiogenesis which aims to induce growth of new vasculature to ischemic tissues. To generate a sustained angiogenic response, delivery of the angiogenic factor by gene therapy vectors has been the favored approach. However, the completed phase II and III clinical trials have shown that a better understanding of VEGF biology and further development of the therapies are required for future clinical success1.
The VEGF family consists of five mammalian members VEGF-A, -B, -C, -D and placental growth factor (PlGF) as well as homologous proteins encoded by viruses (VEGF-E) and those found in snake venoms (VEGF-F). VEGFs mediate their signals through three VEGF receptor tyrosine kinases (VEGFR) -1, -2 and -3. The co-receptors, neuropilins (NRPs) -1 and -2 and heparan sulfate proteoglycans (HSPGs) modulate the functions of VEGFs. The VEGF family members are expressed as several isoforms, which differ in their HSPG and NRP binding properties2. The interactions of VEGFs with HSPGs mediate the formation of the VEGF concentration gradients required for proper vascular network formation during development3. VEGFR-1 binds VEGF-A with higher affinity than VEGFR-2, despite this VEGFR-2 is considered to be the main mediator of angiogenesis, as the kinase activity of VEGFR-1 is weak4. Nevertheless, VEGFR-1 has a crucial role during development as a non-signaling decoy receptor for VEGF-A5 and may also regulate the endothelial tip/stalk cell phenotype and sprouting by spatially sequestering VEGF from VEGFR-26, 7. NRPs bind specific forms of several VEGF family members8 and are required for the normal development of the cardiovascular system9. NRP-1 has been shown to be involved in the control of endothelial tip/stalk cell phenotype10 and guidance of tip cells11. The mechanism of action of NRPs is still largely unknown, but recent studies have highlighted the importance of short motifs with C-terminal arginine for binding to NRP b1 domain12 and implied that NRPs can modulate VEGFR-2 downstream signaling by multiple mechanisms2.
Snake venom VEGFs form the most recently identified VEGF-F subgroup with varying receptor specificities13. Previous studies have demonstrated that they share the common structure with mammalian VEGFs14 and are able to induce vascular permeability15 and lower blood pressure16. However, their capability to induce angiogenesis in vivo has not been previously studied, even though their unique receptor binding profiles may have therapeutic advantages over mammalian VEGFs. Vammin is a novel VEGF-F family member isolated from the venom of sand viper Vipera ammodytes 16 and is a highly potent VEGFR-2 ligand devoid of VEGFR-1 binding and binds also to NRPs and HSPGs17.
The normal growth of the vascular tree requires temporally and spatially precise guidance signals mediated by the interactions of the VEGFs with their multiple receptors and co-receptors, which the current gene transfer techniques cannot easily mimic. A better understanding of the (co-)receptor-ligand interactions and the subsequent biological outcomes are required for the development of improved novel pro-angiogenic therapies. In this study, we: 1) characterized the ligand-receptor interactions and biological functions of Vammin; 2) evaluated the angiogenic potency of Vammin in vivo in rabbit skeletal muscle using adenoviral gene transfer and; 3) studied the contribution of VEGFR-1 and co-receptor binding to VEGFR-2-mediated angiogenesis in vivo.
Results
Receptor binding properties
Competition binding assays were performed to characterize the binding of different VEGF-A forms and Vammin to VEGFR-1 and VEGFR-2. The VEGF-A forms were the only ligands binding to VEGFR-1 (Fig. 1B), whereas all tested factors bound to VEGFR-2 (Fig. 1C).
Figure 1.
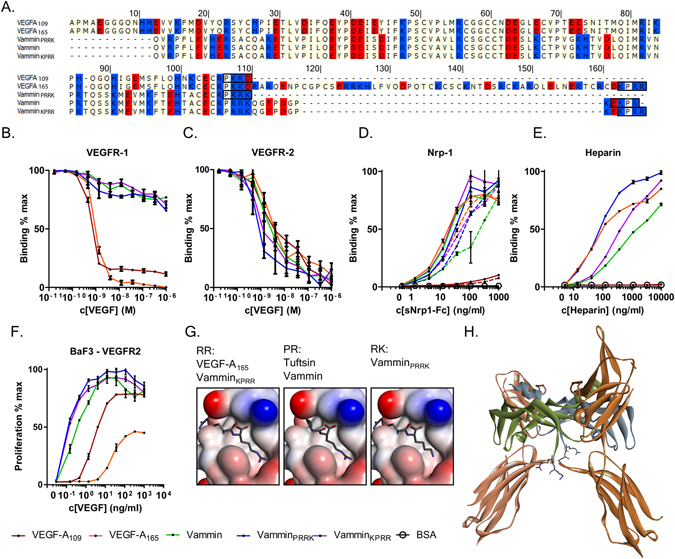
(A) Sequence alignment of recombinant VEGF-A and Vammin proteins. Basic amino acids are in blue and acidic in red background. The last four amino acids expected to be important for NRP-1 binding are in black boxes. (B) VEGFR-1 competition binding assay (C) VEGFR-2 competition binding assay (D) NRP-1 binding into VEGF coated plates the presence of heparin (solid lines) and without heparin (dotted lines) (E) Heparin binding in VEGF coated plates (F). BaF3-VEGFR-2 proliferation assay. In B to F the values are expressed as mean ± SD. (G) Binding of the two C-terminal amino acids into the NRP-1 binding pocket. RR and PR sequences are drawn based on VEGF-A165/NRP-1 and Tuftsin/NRP-1 complex structures. (PDB IDs 4DEQ and 2ORZ). RK sequence is modeled based on VEGF-A165/NRP-1 complex structure. Ligand amino acids are shown as sticks and NRP-1 as surface rendering colored by interpolated charge. (H) A model of location of the VamminPRRK C-terminus. Vammin crystal structure (PDB ID 1 WQ8) was superimposed onto VEGF-C/VEGFR-2 complex crystal structure (PDB ID 2X1W). The two VEGFR-2 monomers are seen with the brown colors and the two antiparallel dimer forming Vammin chains with green and blue. The C-terminal Arg97 and Lys98 of Vammin projecting outward from the complex are shown as sticks.
Ligand binding to NRP-1 b1 domain has been suggested to be dependent on C-terminal sequences resembling the VEGF-A exon 8 encoded C-terminus (CDKPRR) ending in free Arg. Previously, we showed that Vammin with a C-terminal sequence of PKEKPR binds to NRP-117. In this study, we further characterized the NRP-1 binding site. VamminKPRR, encoding 111 amino acids, was engineered to have an additional Arg residue in the C-terminus to mimic the last 4 amino acids (KPRR) in the C-terminus of VEGF-A. VamminPRRK, encoding 98 amino acids, was designed to correspond to the VEGF homology domain encoded by VEGF-A109. VamminPRRK was expected not to bind to NRP-1 as it encodes a C-terminal Lys, instead of Arg, which was previously found to be a required binding determinant for NRP-1. VEGF-A165 which has the C-terminal NRP-1/HSPG domain and VEGF-A109 totally lacking this domain were used as controls (Fig. 1A). All ligands were equally efficient in VEGFR-2 binding (Fig. 1C). As expected, VEGF-A109 did not bind to NRP-1, in contrast all Vammin proteins and VEGF-A165 bound NRP-1 (Fig. 1D). As was previously shown17, free heparin increased the binding to NRP-1 (Fig. 1D), and accordingly, all Vammin forms and VEGF-A165 bound heparin (Fig. 1E). The C-terminal Arg of NRP-1 ligands, forms salt bridges with the NRP-1 binding pocket with both its side chain and the free carboxyl group, as shown in complex structures of both VEGF-A165, VEGF-C and a small peptide Tuftsin with NRP-112, 18, 19. These bonds are modeled to be preserved also in VamminPRRK with the C-terminal Lys (Fig. 1G). In comparison, the C-terminal amino acid of VEGF-A109 is Asp with an opposite charge. Figure 1H shows a model of Vammin, containing amino acids up to the Lys 98, in complex with VEGFR-2. The Vammin C-terminus is located close to the VEGFR-2 domain 3 and projects outward from the VEGF/VEGFR complex. It is expected that VEGFR-2 and NRP-1 are able to bind simultaneously to form a signaling complex as shown previously17. The capability of the recombinant proteins to induce VEGFR-2 mediated cell proliferation was tested using VEGFR-2/EpoR expressing BaF3 cells (Fig. 1F). All three Vammin constructs were more potent and more effective in this assay than either of the VEGF-A forms.
Vammin induces angiogenesis in rabbit skeletal muscles
As Vammin was found to be a highly effective VEGFR-2 ligand, we generated an adenoviral vector encoding Vammin and compared its properties against vectors encoding either VEGF-A165, a VEGFR-1/VEGFR-2/NRP-1/HSPG ligand, or VEGF-A109 which is a VEGFR-1/VEGFR-2 ligand. Adenoviral vectors were injected into the rabbit hindlimb semimembranosus muscles in two doses, 1 × 109 and 1 × 1010 viral particles (vp). A control vector (AdCMV) was used in three doses 1 × 109, 1 × 1010 and 1 × 1011 vp. Blood perfusion in the target muscle was analyzed before and 6 days after the gene transfers using a contrast pulse sequence (CPS) ultrasound method, that is suitable for detecting blood flow in the capillary level vessels in addition to the larger vessels20. All three VEGF encoding vectors induced significant increases in blood perfusion (Fig. 2A and B). There were no significant differences in perfusion induced by the two different doses or between the different VEGF encoding vectors, but the higher doses were observed to cause increased perfusion also in the surrounding muscles, likely to be due to the spread of the growth factors and the vector. Edema was detected in the target muscles (Fig. 2A and C).
Figure 2.
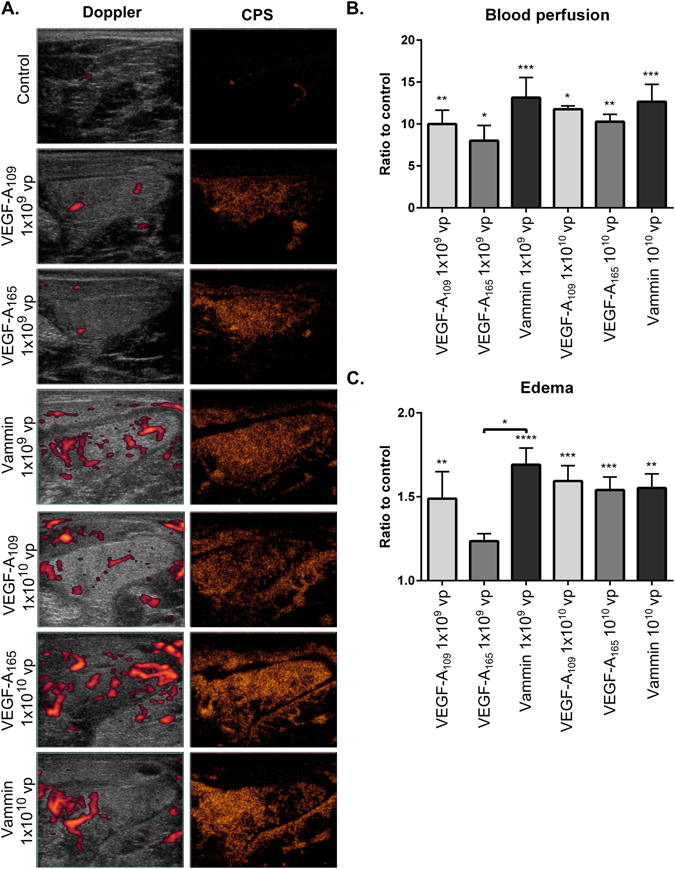
Vammin efficiently induces blood perfusion and edema in skeletal muscles. (A) Images with Doppler and CPS ultrasound from the semimembranosus muscles 6 days after the gene transfer. Increased perfusion is seen in all treated muscles. Edema is observed with both doses (B) Quantification of the blood perfusion from CPS ultrasound (n = 4–6 per group) (C) Quantification of edema from the ultrasound images (n = 4–6 per group). In B to C the data is presented as mean ± SEM. * P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Vector dose affects the vascular network morphology
The blood vessel network in the affected area was imaged using confocal and multiphoton microscopy techniques from 1 mm thick longitudinal sections of the muscle. The two different vector doses showed highly different capillary tree morphology (Fig. 3A). With 1 × 109 vp dose, the vessels were slightly enlarged and formed numerous connections in comparison to controls. With 1 × 1010 vp dose, all vessels were extensively enlarged (Fig. 3A,B and E) and fewer individual sprouts and connections were identified (Fig. 3A and F). VEGF-A109 induced uniform straight vessels running in a longitudinal direction through the muscle with very few connections. VEGF-A165 and Vammin caused a different vessel phenotype with enlarged vessels sending projections towards or fusing with adjacent vessels. At maximal affected areas, Vammin induced formation of CD31 positive vessels encircling the muscle fibers, whereas with VEGF-A165, individual sprouts were more clearly distinguished. Similar findings were seen in paraffin embedded cross-sections (Fig. 3B) where VEGF-A109-induced enlarged capillaries were homogenous in size and well separated from each other, whereas VEGF-A165 and Vammin induced highly enlarged vessels closely resembling the multiphoton imaging results.
Figure 3.
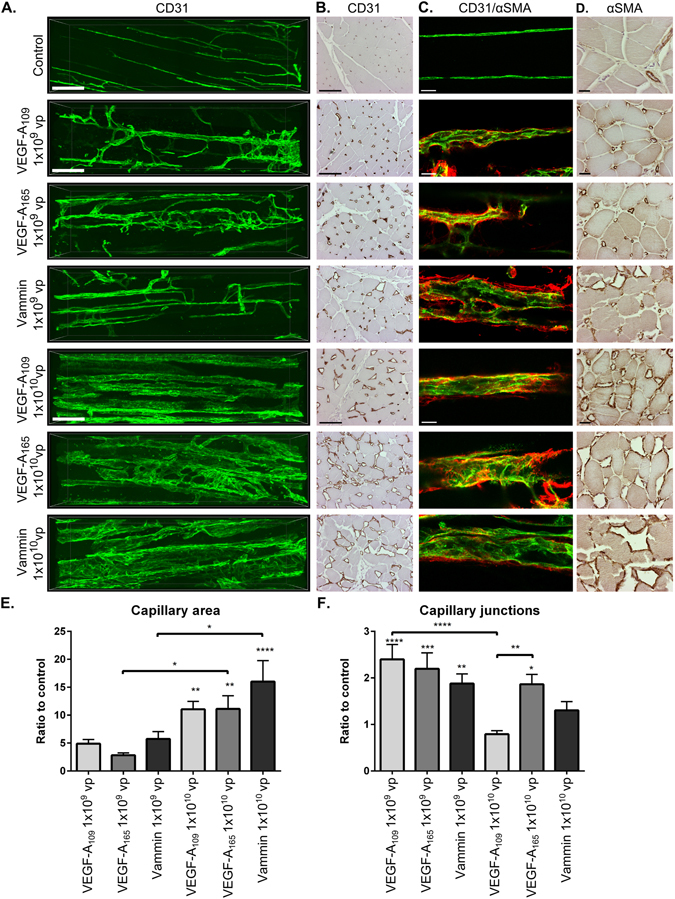
Both VEGF-A forms and Vammin induce dose and receptor binding profile specific changes in the vascular network (A). Multiphoton microscopy imaging of the blood vessel network by CD31 (green) labelled blood vessels from longitudinal sections of semimebranosus muscle. With 1 × 109 vp dose all tested factors induce sprouting angiogenesis and modest enlargement of capillaries. With 1 × 1010 vp dose, the normal organization of the blood vessel network is disturbed. VEGF-A109 induces enlarged capillaries that have notably few connections and sprouts. Scalebar 100 µm. (B) CD31 staining of paraffin embedded cross-sections of semimembranosus muscle. Scalebar 100 µm. (C) Confocal microscopy images from CD31 (green) and αSMA (red) double-stained muscles. αSMA positive staining surrounds the enlarged capillaries. Scalebar 25 µm. (D) αSMA staining of the cross-sections shows high coverage of αSMA positive cells in the enlarged capillaries. In control muscles, the staining is mainly seen in the arterioles and venules. Scalebar 25 µm (E) Quantification of the capillary area as ratio to the controls from paraffin embedded CD31 stained cross sections (n = 4–6 per group) (F) Quantification of the amount of capillary junctions as a ratio to the controls from confocal microscopy images of CD31 stained whole-immunomounts (n ≥ 9 images per group). In E to F the data is presented as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Cell recruitment after VEGF gene transfer and VEGF receptor expression
The recruitment of mural cells was determined by staining cross-sections for αSMA. Very few positive cells were detected in control muscle capillaries with αSMA staining, and these were limited mainly to arterioles and venules, whereas almost all enlarged capillaries in growth factor treated animals were covered with αSMA positive cells (Fig. 3D). When imaged with confocal microscope from the longitudinal sections, αSMA staining was mainly localized in the immediate vicinity of the CD31 stained endothelium (Fig. 3C). As detected from low magnification images of the muscle cross-sections, the localization of the CD31 positive cells and capillary enlargement in the target muscle were different between the two vector doses. The 1 × 1010 vp dose caused a uniform effect covering the majority of the muscle area. In contrast, in the lower 1 × 109 vp dose, the most dense CD31 positive staining was observed in the muscle perimysium, with CD31 positive cells packed around the edges of the muscle fascias (Fig. 4A).
Figure 4.
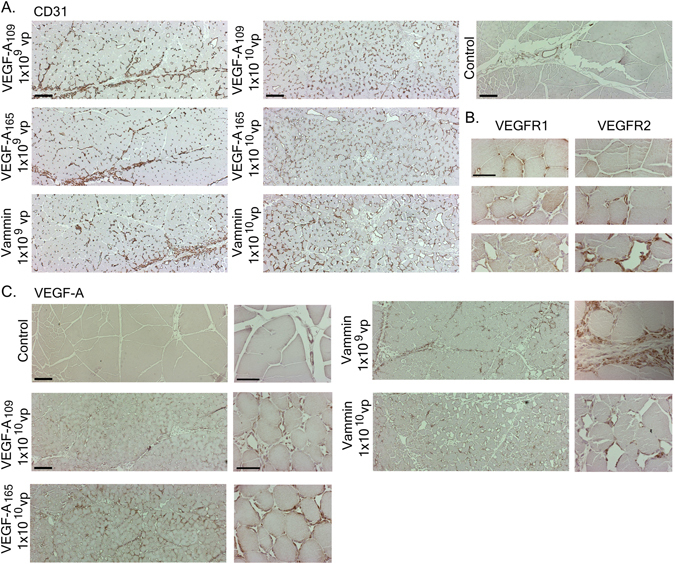
(A) Low magnification images from CD31 stained cross-sections of semimembranosus muscles show CD31 positive cells in the muscle perimysium in all 1 × 109 vp animals. With the 1 × 1010 vp dose, enlargement of capillaries is uniformly seen inside the muscle fascicles. Scalebar 200 µm. B. VEGFR-1 staining is intense in the small capillaries, but reduced in the heavily enlarged capillaries. VEGFR-2 staining is more intense in the enlarged capillaries. Pictures are from AdVammin 1 × 1010 vp treated muscle from areas of low, medium and high effect. Scalebar 50 µm. C. Diffuse VEGF-A staining is seen in 1 × 1010 vp AdVEGF-A transduced muscles surrounding the capillaries especially in VEGF-A165 muscles. The cells in the perimysium were strongly stained with VEGF-A also in AdVammin 1 × 109 vp transduced muscles suggesting that they may serve as a source of endogenous VEGF-A. VEGF-A is also detected around the highly enlarged capillaries in AdVammin 1 × 1010 vp muscle. Scalebar 200 µM in the low magnification (40x) images and 50 µm in the high magnification (400x) images.
VEGFR-1 staining was intense in the quiescent small capillaries of the controls and in areas with little capillary enlargement in VEGF samples (Fig. 4B). However, staining was notably weak in the strongly enlarged capillaries. On the contrary, VEGFR-2 staining was weak in the small capillaries, but intense in the enlarged capillaries. VEGF-A protein expression was detected in both VEGF-A109 and VEGF-A165 samples and surprisingly some also in Vammin samples (Fig. 4C), but not in the controls. The antibody used was verified to be able to detect VEGF-A109 and VEGF-A165 equally in both direct ELISA and western blotting, but showed no cross-reactivity towards Vammin in these assays. In the 1 × 109 vp dose groups, the majority of the VEGF-A staining localized with the cells of the perimysium as shown with Vammin, whereas in samples from the 1 × 1010 vp dose, intense VEGF-A staining was found between the muscle fibers in the VEGF-A165 samples and less intense in the VEGF-A109 samples. Some VEGF-A staining was also detected in the strongly enlarged capillaries of the Vammin samples.
Gene regulation in endothelial cells
To investigate the molecular mechanisms behind the observed differences in biological functions of the different tested factors, we stimulated HUVECs with adenoviral vectors encoding the tested factors and quantitated differential gene expression by RNA-sequencing. In general, the gene expression profiles were similar between all tested factors (Fig. 5A). We found 410 significantly differentially regulated genes with Vammin, 65 with VEGF-A165 and 28 with VEGF-A109 when compared to the control AdCMV vector (Fig. 5B and Supplementary data). In gene ontology analysis, Vammin and both forms of VEGF-A differentially regulated genes that were significantly associated with proliferation of cells, migration of cells and angiogenesis (Fig. 5B and C). When compared to VEGF-A165, Vammin induced significant differences in the expression of genes associated with proliferation and angiogenesis, but not migration (Fig. 5D). 14 genes were significantly differentially expressed between VEGF-A165 and VEGF-A109 (Fig. 5E), most notably NR4A1, NR4A2 and NR4A3 transcription factors, previously associated with VEGF-A165 signaling21.
Figure 5.
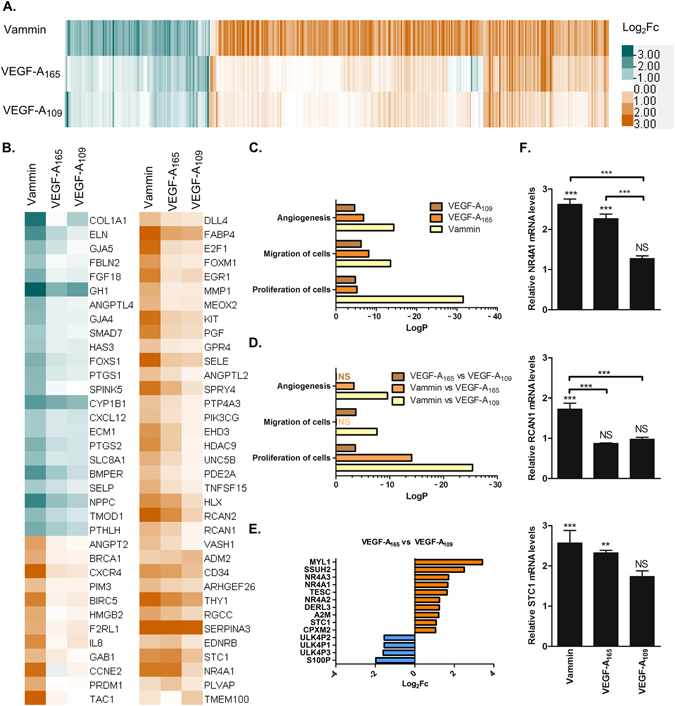
(A) Heatmap of fold changes (Log2) in gene expression levels between vectors encoding the tested factors and the AdCMV control vector. (B) Heatmap of fold changed (Log2) of the angiogenesis associated genes identified in the gene ontology analysis. (C and D). Gene ontology analysis using the Ingenuity Pathway Analysis: The top 3 most significantly enriched physiological functions are presented. (E) The genes differentially expressed between VEGF-A165 and VEGF-A109. F. qRT-PCR analysis of the expression of NR4A1, RCAN1 and STC1 in human dermal blood endothelial cells. The data is presented as mean ± SEM. **P < 0.01, ***P < 0.001.
Vammin regulated the expression of several genes encoding major angiogenesis contributing factors. ANGPT2 (Angiopoietin II), ANGPTL2 (Angiopoietin like II), DLL4 (Delta like ligand 4), PGF (PlGF) and VASH (Vasohibin 1) were upregulated whereas ANGPTL4 (Angiopoietin like IV), BMPER (BMP binding endothelial regulator) and CXCL12 (Stromal cell derived factor 1) were downregulated. Vammin also upregulated cell surface receptor encoding genes KIT, UNC5B and F2RL1 that has recently been shown to function as a switch between angiogenesis and vessel maturation depending on the cellular localization22. Of potential controllers of cellular signaling, GAB1 and four Sprouty family members controlling MAPK pathway signaling and Ca2+- Calcineurin-NFAT pathway signaling regulators RCAN1 and -2 and TESC were upregulated. Other potential new angiogenesis controlling signaling mediators included PALD1 phosphatase which was recently shown to be expressed in developing vasculature23 and kinase PBK which was found to phosphorylate p38MAPK in proliferating cells24. Interestingly, MARC1 gene encoding an enzyme catalyzing nitric oxide (NO) formation by nitrite reduction25 was upregulated by both VEGF-A165 and Vammin. To verify the high efficacy of Vammin to induce angiogenic signaling we stimulated human dermal blood endothelial cells (HDBEC) with the adenoviral vectors and using qPCR analyzed the expression of known VEGF regulated genes STC126, RCAN127 and NR4A121. These genes were all significantly upregulated by Vammin (Fig. 5F).
Discussion
The purpose of this study was to characterize the properties and angiogenic potential of a novel snake venom derived factor Vammin. To put the effects into context, we compared Vammin to two forms of a well characterized pro-angiogenic molecule VEGF-A. Additionally, we were interested in how the differential binding of the tested factors to VEGFR-1, NRP-1 and HSPGs affects the angiogenic responses.
NRPs have emerged as important contributors to angiogenesis, but their mechanism of action has remained elusive. A common feature of most NRP binding proteins is a C-terminal free Arg residue. However, the C-terminally modified Vammins showed that NRP-1 binding does not strictly depend on the C-terminal Arg residue since a related basic residue Lys also facilitated binding and is compatible with the NRP binding pocket. This is supported by earlier findings demonstrating that peptides with a C-terminal Lys could compete with VEGF-A165 for NRP-1 binding, albeit with lower efficacy compared to the ones with C-terminal Arg28, 29. In contrast, VEGF-A109 with C-terminal Asp is not capable of binding NRP-1 due to the opposite charge. The relatively relaxed binding determinants, binding motif which resembles the pro-protein convertase recognition site (R/K)Xn(R/K) and the ability of several unrelated factors to bind12, 30 suggest that NRPs may have a role as modulators of many different signaling pathways. The use of both VEGF-A109 and VEGF-A165 in this study allowed us to evaluate potential genes specific for NRP in the context of VEGFR signaling. Notably, in comparison to VEGF-A165, VEGF-A109 failed to induce expression of NR4A1, NR4A2 and NR4A3 orphan nuclear receptors, which are regulated by the Ca-Calcineurin-NFAT signaling21. Additionally, other genes involved in calcium signaling including MYL1, TESC, STC1 and S100P were differentially regulated by these factors. This suggests that modulation of VEGFR-2 induced calcium signaling may be one important function of NRPs.
In the BaF3-VEGFR-2/EpoR proliferation assay, Vammin was a highly potent VEGFR-2 ligand when compared to either of the VEGF-A forms. Surprisingly, also VEGF-A109 was both a more potent and more efficient stimulator of these cells than VEGF-A165. Since the affinities of these ligands towards VEGFR-2 are comparable, this indicates that there are also other factors involved in determining the efficacy of signaling via VEGFR-2. A similar finding was previously shown for VEGFR-1 activation by VEGF-B and PlGF, which was affected by the differential orientation of the subunits of the dimerized receptor31. A similar phenomenon could also explain the observed differences of these factors in this assay. In HUVECs, Vammin induced a gene expression profile in line with a VEGFR-2 ligand activation. However, the changes were more prominent than with VEGF-A, especially Vammin induced the expression of genes linked to cellular proliferation. It is likely, that this is due to both the highly efficient VEGFR-2 activation and avoidance of VEGFR-1 binding, which limits VEGFR-2 mediated endothelial proliferation during embryonic angiogenesis5 and controls the amount of free VEGF-A available to signal through VEGFR-27. However, it cannot completely be ruled out that some of the actions of Vammin could be mediated by yet unknown receptors on endothelial cells. Vammin induced expression of several genes known to contribute to angiogenesis including VEGF signaling targets NR4A1, RCAN1 and STC1. Additionally, several of the identified genes were involved in the regulation of MAP kinase pathway and Ca2+-Calcineurin pathway, both of which are important mediators of the VEGFR-2 signaling. Interestingly, MARC1, a gene encoding an enzyme recently discovered to catalyze NO reduction from nitrite25 was also upregulated. Given the importance of NO in VEGF signaling and the vascular system function, this might represent an alternative NO synthesis pathway to eNOS mediated NO generation from L-Arginine and may contribute to VEGF induced angiogenesis.
Adenoviral gene delivery to rabbit hindlimb verified that Vammin is a highly efficient angiogenic factor in vivo, greatly inducing blood perfusion in the target tissue. Whereas, the other VEGFR-2 specific ligand, VEGF-E, has been shown to induce less vascular permeability than VEGF-A32, Vammin induced at least as much edema as VEGF-A. The difference may reflect the lower VEGFR-2 binding affinity of VEGF-E17. Importantly, we found that even constant potent stimulation of VEGFR-2 over several days in vivo by Vammin does not lead to clearance of the receptor from the endothelium. The angiogenic effect of VEGFs can be divided into sprouting angiogenesis and capillary enlargement and the microenviromental concentrations of VEGFs are important for the type of angiogenic response33. Low doses of VEGF-A165 induce mainly sprouting angiogenesis and higher doses cause capillary enlargement and even formation of aberrant vascular structures34. This strict dose dependent effect of VEGFs applies also to both VEGF-A109 and Vammin. The tendency for sprouting angiogenesis with the low dose may be due to the gene transfer causing local hotspots of VEGF production, thereby generating intramuscular growth factor gradients. With the higher dose, the more efficient transduction causes more uniform production of VEGF around the muscle leading to saturated stimulus and mainly enlargement of the vessels. Accordingly, the high VEGF concentrations have recently been shown to synchronize endothelial cell function through Notch and VE-Cadherin mediated mechanism and to prevent sprouting35.
The major difference between the three ligands was the tendency of high dose VEGF-A109 to induce uniform formation of straight enlarged vessels with very few connections. This is in line with previous findings, showing that the interactions of VEGF-A with heparan sulphates and NRPs are required for sprouting angiogenesis during development11, 36 and may also reflect the attenuated response of VEGF-A109 seen in gene expression analysis. As both VEGF-A165 and Vammin are dual NRP-1/HSPG ligands, we can’t speculate which one of the interactions caused the difference in comparison to VEGF-A109. However, previous publications suggested that both may contribute37. In addition to the cell signaling mediated differences, the observed differential deposition and microenviromental concentrations of the heparin binding and non-binding factors are likely to be important. In addition to heparan sulphates, VEGFs also bind to several other extracellular matrix components38. These interactions and the difference in VEGFR-1 binding likely cause the subtle differences observed in vessel morphology with Vammin and VEGF-A165. The results suggest that modification of the ECM binding properties of VEGFs could be beneficial for developing VEGF based pro-angiogenic therapies. For example, fully soluble VEGF forms may be useful therapeutic agents to induce collateral growth by arteriogenesis with lesser disturbance of the capillary tree structure.
VEGFs are also known to be capable of recruiting bone marrow derived circulating cells that produce other angiogenic mediators and that may incorporate into the newly forming vessels39. A prominent feature after gene transfers was the presence of large amounts of CD31 positive cells in the muscle perimysium. These cells were determined to be a potential source for endogenous VEGF-A, which may have in part contributed to the angiogenic effect seen especially with the lower viral dose. Additionally recruitment of mural cells to the newly developed blood vessels is a critical step in the formation of stable vasculature. All three tested factors were efficient at recruiting αSMA positive cells into the enlarged vessels as was previously seen with VEGF-A165 40. The lack of staining in the control muscle capillaries may reflect findings that αSMA is not expressed in quiescent pericytes and, therefore, its expression is induced only after angiogenic stimulus41. In the RNA-seq experiment, the tested factors did not directly upregulate genes linked to pericyte recruitment such as PDGF. Therefore, the stimulus may arise in vivo through increased blood flow and shear stress, rather than direct changes in VEGF induced gene expression.
This study sheds light into the interactions of NRP-1 with its VEGF ligands demonstrating the flexible binding determinants to NRP-1 and suggests that the NR4A nuclear receptors and calcium signaling may be important mediators of NRP-1 biological functions. Importantly, it demonstrates that the VEGFR-2 specific ligand Vammin is a highly potent molecule capable of inducing angiogenesis in vivo and warrants further studies in ischemic animal models of therapeutic angiogenesis. Binding to VEGFR-1 does not enhance the angiogenic response, but may rather limit the efficacy. This is in line with VEGF-DΔNΔC which despite its fairly low VEGFR-2 binding affinity is a potent angiogenic growth factor in vivo 42. The use of high doses of VEGF encoding gene transfer vectors for clinical applications comes with a risk of side effects due to vector escape into distant organs34. The high potency of Vammin to specifically stimulate VEGFR-2/NRP-1 signaling allows the use of very low doses of gene transfer vectors, and may therefore help to prevent systemic side effects. Alternatively, it may enable the use of different gene transfer vectors, with lower in vivo transduction efficacy, or cell type specific, but weaker, promoters, than the commonly used strong viral promoters.
Methods
Recombinant proteins and in vitro assays
The recombinant VEGF-A109, VEGF-A165, VamminKPRR and VamminPRRK proteins were produced in adherent HEK293T cells by calcium phosphate transfection with plasmids encoding N-terminally His-tagged proteins. On the following day, DMEM (Sigma-Aldrich) supplemented with 10% FBS was replaced with serum free media and cells were incubated for 2 days. The proteins were purified using HisTrapExcel columns with ÄktaAvant (GE Healthcare) using gradient elution from 0 to 500 mM Imidazole in PBS. The collected fractions were analyzed by SDS-PAGE. Pure fractions were pooled, diluted to PBS and concentrated using Amicon Ultra-15 3 K Centrifugal Filter Units and final buffer exchange to PBS was performed using HiTrap Desalting columns (GE Healthcare). Protein concentration was assessed using a BCA Protein Assay Kit (Thermo Scientific) and verified by SDS-PAGE. Recombinant Vammin, sVEGFR2-Fc and sNRP1-Fc were produced as described earlier17.
Receptor binding assays
For VEGFR-1 and VEGFR-2 binding assays sVEGFR1-Fc (R&D Systems) and sVEGFR2-Fc proteins were pre-incubated for 1 h with dilution series of VEGF proteins in the presence of 100 ng/ml heparin (Sigma-Aldrich), the mixture was transferred to VEGF-A165 coated plates, blocked with 1% BSA in TBS, pH 7.5, for 2 h, and incubated for 1 h. For NRP-1 and Heparin binding assays 96-well plates were coated with the VEGF ligands overnight at pH 9.5 in bicarbonate buffer. Plates were blocked with 1% BSA in TBS, pH 7.5, for 2 h. sNRP1-Fc fusion protein and biotinylated heparin (Sigma-Aldrich) were incubated for 1 h at room temperature. The bound receptor-Fc fusion proteins and heparin-biotin were detected accordingly with anti-human IgG peroxidase (Sigma-Aldrich) or Streptavidin peroxidase and TMB (Sigma-Aldrich), respectively. Absorbances of stopped reactions were measured at 450 nm.
Proliferation assay
BaF3-VEGFR-2 cells43 were cultured in DMEM supplemented with 10% FBS, 1% Penicillin-Streptomycin, 500 µg/ml G418 (InvivoGen) and 2 ng/ml rmIL3 (Calbiochem). Proliferation assay was performed by plating the cells in culture medium without rmIL3 at 18 000 cells per well and adding the indicated amounts of the ligands. Cells were incubated for 2 days and proliferation was measured using Cell Titer 96 Aqueuous One Solution Assay System (Promega).
Adenovirus generation
Replication-deficient E1-E3 deleted clinical GMP-grade adenoviruses (serotype 5) were produced in 293 cells. Empty vector containing the CMV promoter and poly(A) tail was used as a control virus. All viruses used in the present study were tested for sterility, mycoplasma and endotoxin, and functionality in cell cultures before the animal studies.
Gene transfers
All animal experiments and procedures used in this study were approved by the National Animal Experiment Board in Finland and carried out in accordance with the EU Act on the Protection of Animals Used for Scientific or Educational Purposes (497/2013) and The Finnish Act on Animal Experimentation. Animals were kept in standard housing conditions in The National Laboratory Animal Center of The University of Eastern Finland. Diet and water were provided ad libitum. New Zealand White rabbits received intramuscular injections of adenoviral vectors encoding Vammin (1 × 109 and 1 × 10 × 1010 viral particles [vp]), VEGF-A109 (1 × 109 and 1 × 1010 vp) or VEGF-A165 (1 × 109 and 1 × 1010 vp) or CMV promoter (1 × 109 and 1 × 1010 and 1 × 1011 vp) into the semimembranosus muscle of the thigh (n = 34). Rabbits were anesthetized with medetomidine (Domitor, 0.3 mg/kg, Orion) and ketamine (Ketalar, 20 mg/kg, Pfizer) and intramuscular gene transfers were performed in ten x 100 µl aliquots. One rabbit receiving VEGF-A109 1 × 1010 vp gene transfer died at day 6 before ultrasound analysis and was excluded from analysis.
Ultrasound analysis
Contrast pulse sequence (CPS) ultrasound imaging was performed before and at d6 after gene transfer for the analysis of changes in perfusion in the treated muscles as described previously 20. Acuson Sequoia 512 and 15 L8 transducer (Siemens AG, Erlangen, Germany) with Sonovue (Bracco, Milano, Italy) contrast agent were used to measure the perfusion in the muscles and the peak intensities were analyzed. One rabbit receiving VEGF-A109 1 × 1010 vp gene transfer did not generate reliable contrast pulse sequence (CPS) response due to technical error and was excluded from CPS analysis. Other analyses were normally performed from this animal. Edema was determined as ratios of the area of the semimembranosus muscle before and at d6 after the gene transfer in the ultrasound figures.
Immunohistochemistry
Tissues were fixed in 4% PFA, embedded in paraffin, and sectioned at 4–7 µm thickness. Immunostainings were performed using the following antibodies: α-SMA (1:500, Sigma-Aldrich, Cat. no A5228), CD31 (1:50, Dako, Cat. no M0823), VEGFR-1 (1:250, Santa Cruz Biotechnology, Cat no sc-31173), VEGFR-2 (1:250, Santa Cruz Biotechnology, Cat. no sc-6251) and VEGF-A (1:500, Santa Cruz Biotechnology, Cat no sc-7269). Photographs were taken with Olympus AX70 microscope (Olympus Optical, Tokyo, Japan). For whole-mount tissue imaging, 1 mm thick longitudinal sections were cut and stained with CD31 (1:100, Dako, Cat. no M0823) and α-SMA conjugated with Cy3 (1:500, Sigma-Aldrich, Cat. no C6198). For CD31, goat anti-mouse Alexa 488 (1:500; Invitrogen, Cat. no A-11001) was used. Imaging was performed by Nikon A1R multiphoton microscope (MPLSM) and LSM700 Zeiss confocal microscope (LSM).
Analysis of gene expression
Human umbilical vein endothelial cells (HUVECs) were isolated from umbilical cords obtained from the maternity ward of Kuopio University Hospital. Informed consent was obtained from all the umbilical cord donors. All experiments were carried out in accordance with the European Union guidelines and regulations with the approval by the Kuopio University Hospital Ethics Committee (license numbers 58/2012 and 341/2015). Human dermal blood endothelial cells (HDBECs) were obtained from PromoCell (Heidelberg, Germany). Cells were used at early (I-V) passages and grown on plastic surface coated with 0.05% gelatin/10 µg/ml fibronectin (Sigma) in EBM Endothelial Cell Basal Medium supplied with EGM SingleQuots (Lonza). After 24 h of adenoviral transductions, starvation medium (EBM + 0.5% FBS) was changed to the cells. After another 24 h, RNA was extracted with RNeasy Mini Kit (Qiagen) accrording to the manufacturer’s instructions.
For RNA sequencing RNA was enriched for Poly(A)-RNA with MicroPoly(A) Purist Kit (Life Technologies). RNA was fragmented using RNA Fragmentation Reagents (Life Technologies) and purified by running through P-30 column (Bio-Rad, Hercules, CA, USA). Fragmented RNA was dephosphorylated with PNK (New England Biolabs, Ipswich, MA, USA) followed by heat-inactivation. Dephosphorylation reactions were purified using RNA Clean & Concentrator™-5 kit (Zymo Research). Poly(A)-tailing and cDNA synthesis was performed as described44. However, for reverse transcription an oligo allowing custom barcoding during final amplification was used: /5Phos/GATCGTCGGACTGTAGAACTCTGAAC/iSp18/TCAGACGTGTGCTCTTCCGATCTTTTTTTTTTTTTTTTTTTTVN (IDTDNA). After cDNA synthesis, Exonuclease I (New England Biolabs) was used to catalyze the removal of excess oligos. The DNA-RNA hybrid was purified using ChIP DNA clean & Concentrator Kit (Zymo Research Corporation, Irvine, CA, USA), RNaseH treated and circularized using CircLigase (EpiBio). The libraries were amplified for 11 cycles using the following primers:
5′-AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGACG-3′ and 5′-CAAGCAGAAGACGGCATACGAGATXXXXXGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT (barcode XXXXXX-underlined). The final product was ran on Novex 10%-TBE gel, purified and cleaned as above. The libraries were sequenced on HiSeq 2000 for 50 cycles according to the manufacturer’s instructions.
Two independent experiments were performed with the 1st consisting of AdCMV and AdVammin, AdVEGF-A109 and AdVEGF-A165 as two replicates and the 2nd consisting of AdCMV, AdVammin, AdVEGF-A109 and AdVEGF-A165 and AdVammin + AdCMV as two replicates. Data analysis was performed using HOMER 4.3 and the detailed instructions for analysis can be found at http://homer.salk.edu/homer 45. RNA-Seq was mapped using tophat allowing up to two mismatches and reporting only one alignment for each read. Poor quality reads were filtered out (minimum 97% of bp over quality cutoff 10). Each sequencing experiment was normalized to a total of 107 uniquely mapped tags and visualized by preparing custom tracks for the UCSC Genome browser. Differentially expressed genes were identified using edgeR46, thresholds of FDR <0.05, reads per kb per million reads >0.5 and fold change >2 were used. Hierarchical clustering of the log2 fold-changes was performed by Cluster 3.047 using Pearson correlation as distance method and average linkage as clustering method. The output from clustering was viewed using Java Treeview8. The gene ontology analysis was performed by Qiagen’s Ingenuity Pathway Analysis (IPA, Qiagen Redwood City, www.qiagen.com/ingenuity).
For the qRT-PCR the amount and purity of total RNA were measured with NanoDrop ND-1000 Spectrophotometer (NanoDrop Technologies Inc, Wilmington, DE, USA). One microgram of total RNA was reverse transcribed into cDNA using random hexamers (Thermo Scientific) and Revert Aid Reverse Transcriptase (Thermo Scientific). Quantitative measurements of mRNA levels were done using the Assays-on-Demand gene expression products (Life Technologies) with the StepOnePlus Real-Time PCR System (Life Technologies). Alternatively, Power SYBR Green PCR Master Mix (Life Technologies) and 10 pmol of primers were used (Supplemental Table I). Cycling parameters were 95 °C for 10 min, 40 cycles of 95 °C (15 s) and 60 °C (6fFor whole tissue lysate, respectively.
The RNA-seq data can be found under GEO accession number GSE68535.
Statistical analysis
Results are expressed as the mean ± SEM. Statistical significance was evaluated with One-way ANOVA followed by Bonferroni’s corrected t-test. P < 0.05 was considered statistically significant.
Electronic supplementary material
Acknowledgements
We thank Joonas Malinen, Mervi Nieminen, Maarit Mähönen, Sari Järveläinen, Tiina Koponen and Svetlana Laidinen for excellent technical assistance and Nihay Laham-Karam and Nicholas Downes for the linguistic review. This study was funded by The Academy of Finland, ERC Advanced Grant, The Finnish Foundation for Cardiovascular Research, The Instrumentarium Science Foundation, The Finnish Medical Foundation, The Alfred Kordelin Foundation, The Finnish Cultural Foundation and The Paulo Foundation.
Author Contributions
Pyry I.Toivanen and Seppo Ylä-Herttuala designed research and wrote the manuscript. Pyry I.Toivanen, Tiina Nieminen, Johanna P.Laakkonen, Tommi Heikura and Minna U.Kaikkonen performed research and analyzed data. Data and materials availability: AdVEGF-A109, AdVEGF-A165 and AdVammin need MTA from A.I.Virtanen Institute for Molecular Sciences, University of Eastern Finland.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05876-y
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ylä-Herttuala, S., Bridges, C., Katz, M. G. & Korpisalo, P. Angiogenic gene therapy in cardiovascular diseases: dream or vision? Eur. Heart J. 38, 1365–1371 (2017). [DOI] [PMC free article] [PubMed]
- 2.Simons M, Gordon E, Claesson-Welsh L. Mechanisms and regulation of endothelial VEGF receptor signalling. Nat. Rev. Mol. Cell Biol. 2016;17:611–625. doi: 10.1038/nrm.2016.87. [DOI] [PubMed] [Google Scholar]
- 3.Ferrara N. Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol. Biol. Cell. 2010;21:687–90. doi: 10.1091/mbc.E09-07-0590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M, Heldin CH. Different signal transduction properties of KDR and Flt1, two receptors for vascular endothelial growth factor. J. Biol. Chem. 1994;269:26988–26995. [PubMed] [Google Scholar]
- 5.Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M. Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc. Natl. Acad. Sci. USA. 1998;95:9349. doi: 10.1073/pnas.95.16.9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hellström M, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445:776–80. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 7.Kappas NC, et al. The VEGF receptor Flt-1 spatially modulates Flk-1 signaling and blood vessel branching. J. Cell Biol. 2008;181:847–58. doi: 10.1083/jcb.200709114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pellet-Many C, Frankel P, Jia H, Zachary I. Neuropilins: structure, function and role in disease. Biochem. J. 2008;411:211–26. doi: 10.1042/BJ20071639. [DOI] [PubMed] [Google Scholar]
- 9.Takashima S, et al. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc. Natl. Acad. Sci. USA. 2002;99:3657–62. doi: 10.1073/pnas.022017899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fantin A, et al. NRP1 acts cell autonomously in endothelium to promote tip cell function during sprouting angiogenesis. Blood. 2013;121:2352–62. doi: 10.1182/blood-2012-05-424713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gerhardt H, et al. Neuropilin-1 is required for endothelial tip cell guidance in the developing central nervous system. Dev. Dyn. 2004;231:503–9. doi: 10.1002/dvdy.20148. [DOI] [PubMed] [Google Scholar]
- 12.Parker MW, et al. Structural Basis for VEGF-C Binding to Neuropilin-2 and Sequestration by a Soluble Splice Form. Structure. 2015;23:677–687. doi: 10.1016/j.str.2015.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamazaki Y, et al. Snake venom Vascular Endothelial Growth Factors (VEGF-Fs) exclusively vary their structures and functions among species. J. Biol. Chem. 2009;284:9885–91. doi: 10.1074/jbc.M809071200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Suto K, Yamazaki Y, Morita T, Mizuno H. Crystal structures of novel vascular endothelial growth factors (VEGF) from snake venoms: insight into selective VEGF binding to kinase insert domain-containing receptor but not to fms-like tyrosine kinase-1. J. Biol. Chem. 2005;280:2126–31. doi: 10.1074/jbc.M411395200. [DOI] [PubMed] [Google Scholar]
- 15.Matsunaga Y, Yamazaki Y, Suzuki H, Morita T. VEGF-A and VEGF-F evoke distinct changes in vascular ultrastructure. Biochem. Biophys. Res. Commun. 2009;379:872–5. doi: 10.1016/j.bbrc.2008.12.129. [DOI] [PubMed] [Google Scholar]
- 16.Yamazaki Y, Takani K, Atoda H, Morita T. Snake venom vascular endothelial growth factors (VEGFs) exhibit potent activity through their specific recognition of KDR (VEGF receptor 2) J. Biol. Chem. 2003;278:51985–51988. doi: 10.1074/jbc.C300454200. [DOI] [PubMed] [Google Scholar]
- 17.Nieminen T, et al. The impact of the receptor binding profiles of the vascular endothelial growth factors on their angiogenic features. Biochim. Biophys. Acta. 2014;1840:454–63. doi: 10.1016/j.bbagen.2013.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Parker MW, Xu P, Li X, Vander Kooi CW. Structural basis for selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J. Biol. Chem. 2012;287:11082–9. doi: 10.1074/jbc.M111.331140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vander Kooi CW, et al. Structural basis for ligand and heparin binding to neuropilin B domains. Proc. Natl. Acad. Sci. USA. 2007;104:6152–7. doi: 10.1073/pnas.0700043104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rissanen TT, et al. High-resolution ultrasound perfusion imaging of therapeutic angiogenesis. JACC. Cardiovasc. Imaging. 2008;1:83–91. doi: 10.1016/j.jcmg.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 21.Liu D, Jia H, Holmes DIR, Stannard A, Zachary I. Vascular endothelial growth factor-regulated gene expression in endothelial cells: KDR-mediated induction of Egr3 and the related nuclear receptors Nur77, Nurr1, and Nor1. Arterioscler. Thromb. Vasc. Biol. 2003;23:2002–7. doi: 10.1161/01.ATV.0000098644.03153.6F. [DOI] [PubMed] [Google Scholar]
- 22.Joyal J-S, et al. Subcellular localization of coagulation factor II receptor-like 1 in neurons governs angiogenesis. Nat. Med. 2014;20:1165–73. doi: 10.1038/nm.3669. [DOI] [PubMed] [Google Scholar]
- 23.Wallgard E, et al. Paladin (X99384) is expressed in the vasculature and shifts from endothelial to vascular smooth muscle cells during mouse development. Dev. Dyn. 2012;241:770–86. doi: 10.1002/dvdy.23753. [DOI] [PubMed] [Google Scholar]
- 24.Dougherty JD, et al. PBK/TOPK, a proliferating neural progenitor-specific mitogen-activated protein kinase kinase. J. Neurosci. 2005;25:10773–85. doi: 10.1523/JNEUROSCI.3207-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sparacino-Watkins CE, et al. Nitrite reductase and nitric-oxide synthase activity of the mitochondrial molybdopterin enzymes mARC1 and mARC2. J. Biol. Chem. 2014;289:10345–58. doi: 10.1074/jbc.M114.555177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holmes DIR, Zachary IC. Vascular endothelial growth factor regulates stanniocalcin-1 expression via neuropilin-1-dependent regulation of KDR and synergism with fibroblast growth factor-2. Cell. Signal. 2008;20:569–79. doi: 10.1016/j.cellsig.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 27.Hesser BA, et al. Down syndrome critical region protein 1 (DSCR1), a novel VEGF target gene that regulates expression of inflammatory markers on activated endothelial cells. Blood. 2004;104:149–58. doi: 10.1182/blood-2004-01-0273. [DOI] [PubMed] [Google Scholar]
- 28.Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc. Natl. Acad. Sci. USA. 2009;106:16157–16162. doi: 10.1073/pnas.0908201106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Starzec A, et al. Structure-function analysis of the antiangiogenic ATWLPPR peptide inhibiting VEGF165 binding to neuropilin-1 and molecular dynamics simulations of the ATWLPPR/neuropilin-1 complex. Peptides. 2007;28:2397–2402. doi: 10.1016/j.peptides.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 30.Zachary I. Neuropilins: role in signalling, angiogenesis and disease. Chem. Immunol. Allergy. 2014;99:37–70. doi: 10.1159/000354169. [DOI] [PubMed] [Google Scholar]
- 31.Anisimov A, et al. The basis for the distinct biological activities of vascular endothelial growth factor receptor-1 ligands. Sci. Signal. 2013;6:ra52. doi: 10.1126/scisignal.2003905. [DOI] [PubMed] [Google Scholar]
- 32.Kiba A, Sagara H, Hara T, Shibuya M. VEGFR-2-specific ligand VEGF-E induces non-edematous hyper-vascularization in mice. Biochem. Biophys. Res. Commun. 2003;301:371–7. doi: 10.1016/S0006-291X(02)03033-4. [DOI] [PubMed] [Google Scholar]
- 33.Ozawa CR, et al. Microenvironmental VEGF concentration, not total dose, determines a threshold between normal and aberrant angiogenesis. J. Clin. Invest. 2004;113:516–27. doi: 10.1172/JCI18420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Korpisalo P, et al. Capillary enlargement, not sprouting angiogenesis, determines beneficial therapeutic effects and side effects of angiogenic gene therapy. Eur. Heart J. 2011;32:1664–1672. doi: 10.1093/eurheartj/ehq433. [DOI] [PubMed] [Google Scholar]
- 35.Bentley K, et al. The role of differential VE-cadherin dynamics in cell rearrangement during angiogenesis. Nat. Cell Biol. 2014;16:309–21. doi: 10.1038/ncb2926. [DOI] [PubMed] [Google Scholar]
- 36.Ruhrberg C, et al. Spatially restricted patterning cues provided by heparin-binding VEGF-A control blood vessel branching morphogenesis. Genes Dev. 2002;16:2684–98. doi: 10.1101/gad.242002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kawamura H, et al. Neuropilin-1 in regulation of VEGF-induced activation of p38MAPK and endothelial cell organization. Blood. 2008;112:3638–49. doi: 10.1182/blood-2007-12-125856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martino MM, et al. Growth Factors Engineered for Super-Affinity to the Extracellular Matrix Enhance Tissue Healing. Science (80−) 2014;343:885–888. doi: 10.1126/science.1247663. [DOI] [PubMed] [Google Scholar]
- 39.Grunewald M, et al. VEGF-induced adult neovascularization: Recruitment, retention, and role of accessory cells. Cell. 2006;124:175–189. doi: 10.1016/j.cell.2005.10.036. [DOI] [PubMed] [Google Scholar]
- 40.Korpisalo P, et al. Vascular endothelial growth factor-A and platelet-derived growth factor-B combination gene therapy prolongs angiogenic effects via recruitment of interstitial mononuclear cells and paracrine effects rather than improved pericyte coverage of angiogenic ves. Circ. Res. 2008;103:1092–9. doi: 10.1161/CIRCRESAHA.108.182287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Armulik A, Genové G, Betsholtz C. Pericytes: developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell. 2011;21:193–215. doi: 10.1016/j.devcel.2011.07.001. [DOI] [PubMed] [Google Scholar]
- 42.Rissanen TT, et al. VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ. Res. 2003;92:1098–1106. doi: 10.1161/01.RES.0000073584.46059.E3. [DOI] [PubMed] [Google Scholar]
- 43.Achen MG, et al. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4) Proc. Natl. Acad. Sci. USA. 1998;95:548–53. doi: 10.1073/pnas.95.2.548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang D, et al. Reprogramming transcription by distinct classes of enhancers functionally defined by eRNA. Nature. 2011;474:390–4. doi: 10.1038/nature10006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 2010;38:576–89. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DeHoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–4. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.