

Abstract
Bifidobacteria are common gut commensals with purported health-promoting effects. This has encouraged scientific research into bifidobacteria, though recalcitrance to genetic manipulation and scarcity of molecular tools has hampered our knowledge on the precise molecular determinants of their health-promoting attributes and gut adaptation. To overcome this problem and facilitate functional genomic analyses in bifidobacteria, we created a large Tn5 transposon mutant library of the commensal Bifidobacterium breve UCC2003 that was further characterized by means of a Transposon Directed Insertion Sequencing (TraDIS) approach. Statistical analysis of transposon insertion distribution revealed a set of 453 genes that are essential for or markedly contribute to growth of this strain under laboratory conditions. These essential genes encode functions involved in the so-called bifid-shunt, most enzymes related to nucleotide biosynthesis and a range of housekeeping functions. Comparison to the Bifidobacterium and B. breve core genomes highlights a high degree of conservation of essential genes at the species and genus level, while comparison to essential gene datasets from other gut bacteria identified essential genes that appear specific to bifidobacteria. This work establishes a useful molecular tool for scientific discovery of bifidobacteria and identifies targets for further studies aimed at characterizing essential functions not previously examined in bifidobacteria.
Introduction
Bifidobacteria are Gram-positive commensal microorganisms, widely encountered within the gut ecosystem of mammals and (certain social) insects. Extensive research has attributed health-promoting traits to some bifidobacterial strains, such as vitamin production, amelioration of certain gastrointestinal disorders and inhibition of enteropathogens through production of a range of antimicrobial compounds1–3. The scarcity of molecular tools for bifidobacteria, however, has hampered progress in our understanding of the specific molecular mechanisms behind their adaptation to the intestinal ecosystem, interaction with their host and possible health-promoting activities.
In recent years, various comparative genomic analyses have shed light on the evolutionary adaptation of bifidobacteria to the intestinal environment4–7. Furthermore, during the past decade, a number of molecular tools have been developed or adapted to certain bifidobacterial strains, generating a basic molecular tool-box including cloning and expression vectors, reporter genes and a number of inducible promoters for the control of heterologous gene expression systems8, 9. Successful procedures for targeted mutagenesis have also been reported, providing essential tools to perform functional genomics, which is aimed at expanding our knowledge on host-colonization factors and metabolic capabilities of this group of gut commensals10–15. Mutagenesis represents the most accurate route to experimentally determine if a gene is essential for a specific attribute under particular environmental conditions. However, mutagenesis procedures in bifidobacteria are still tedious, their efficiencies are highly strain dependent and frequently focus on selected target genes. In a recent report by our research group, a random mutagenesis approach based on a customized Tn5 transposon was successfully applied to two different Bifidobacterium breve strains16, opening avenues to interrogate the functionality of bifidobacterial genes at genome level.
The genome-wide identification of essential genes, defined as those that cannot be mutated in vitro, or those in which a mutation reduces growth or viability under particular conditions, has been performed in other microorganisms, where screenings of mutant collections following growth under specific conditions are coupled to en mass genome-wide location of genomic disruptions. Such an approach allows the identification of the complete set of genes essential for a certain phenotype or bacterial attribute in a single experiment. Microarray-based approaches are used for mutant identification in TRAnsposon Site Hybridization (TraSH); while massive parallel sequencing is utilized in High-throughput Insertion Tracking Sequencing (HITS), Insertion Sequencing (In-Seq), Transposon Sequencing (Tn-seq) and Transposon Directed Insertion-Site Sequencing (TraDIS) technologies17–23.
Massive parallel sequencing has substantially increased the resolution power of genome-wide screening of high-density mutant collections21–23, thereby allowing the identification of essential genes, protein domains and intergenic regions with presumptive regulatory roles17, 23. Genome-wide random mutagenesis approaches are also very useful in assisting functional genome annotation and in complementing comparative genomic studies24–27. Such mutant library screening approaches have been used to characterize a variety of clinically relevant pathogenic microorganisms in search of novel antibacterial targets. However, so far only a small number of non-pathogenic gut commensals or potential probiotic bacteria, belonging to Bacteroides thetaiotaomicron and Lactobacillus casei, have been analyzed in this manner18, 28.
The aim of the current study was to further improve the applicability of the transposon mutagenesis method previously developed by our research group for B. breve UCC2003, by applying a genome-wide random mutagenesis approach coupled to transposon mapping by means of next generation sequencing. Analysis of transposon insertion distribution across the genome facilitated the identification of genes (designated here as essential genes) required for strain survival and normal growth on rich media under laboratory conditions. Comparative genomic analysis between the predicted essential genes and the Bifidobacterium core genome revealed a high level of conservation of essential genes across the genus Bifidobacterium and B. breve species, highlighting a set of highly conserved core metabolic functions, as well as a set of still uncharacterized essential functions in bifidobacteria.
Results and Discussion
Library preparation and TraDIS validation
In a previous publication by our research group Tn5 transposon-mediated mutagenesis was accomplished in B. breve UCC2003, enabling relatively high insertion frequencies and generating single and stable transposon insertions per cell16. In the current work, a tetracycline resistance-conferring, Tn5-based transposon was employed to create a large collection of insertion mutants comprised of approximately 58,000 tetracycline-resistant colonies which were pooled from 120 independent transformation experiments (See Materials and Methods). The size of this newly constructed mutant library outnumbers the predicted number of genes of B. breve UCC2003 genome (1,985 genes)29 by about 30-fold. Assuming an even distribution of transposon insertions in the B. breve UCC2003 genome (2.42 Mbp), the library would be expected to contain on average one insertion every 40 base pairs. Since the smallest identified Open Reading Frames (ORFs) in this microorganism are the (predicted) tRNA genes with a size of ~77 bp29, this library is expected to contain mutants in most non-essential genes. In order to confirm this expectation, a transposon directed insertion sequencing (TraDIS) approach was designed and implemented in this B. breve UCC2003-derived mutant collection in order to identify all (or at least the vast majority) transposon insertion points. This allowed us to experimentally confirm the transposition coverage of the library and to identify essential genes based on insertion distribution across the genome. An advantage of the TraDIS approach in comparison to other massive parallel sequencing-based tools, like Tn-seq, is that TraDIS can be applied to virtually any transposon library, without the requirement of any specific sequences within the transposon22.
A total of 2 million reads were obtained on a HiSeq2500 platform (Illumina) and deposited in the European Nucleotide Archive (ENA) under sample accession numbers ERR877647 and ERR877656. Analysis of the generated sequences allowed us to map a high percentage of the high quality reads (98%) to the B. breve UCC2003 reference genome (Genbank accesion number CP000303.1). High percentage of mapped reads (usually > 90%) has previously been described as a characteristic of the mapping approach used30, and allowed the identification of ~46,000 unique insertions with 1 estimated insertion in every 52 bp. Of these unique insertion points, approximately 36,000 were mapped within the deduced coding regions of predicted ORFs.
High-density insertion libraries, displaying insertions at every 10 bp or even less, provide the advantage of increasing the discriminatory resolution between essential and non-essential elements19, 31, 32. Based on the B. breve UCC2003 average gene length of 1099 bp29 and considering the estimated frequency of one insertion every 52 bp, every non-essential gene -including small genes (<200 bp)- is expected to have been targeted by the transposon in our mutant library. Furthermore, in the case of (apparently) non-essential genes, such as Bbr_0014 or Bbr_0055, the insertion frequency was even higher (Supplementary Table S1). Indeed, a rarefaction analysis showed that our library reached saturation level (Supplementary Figure S1). In total, 1,679 genes (84.6% of all ORFs identified on the UCC2003 genome) were found to harbor at least one transposon insertion, including representatives of all functional categories (Figs 1 and 2; see ins_count column in Supplementary Table S1).
Figure 1.
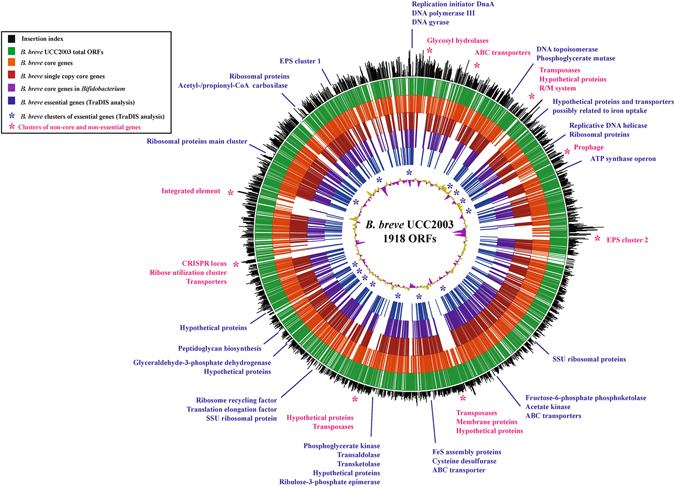
Genome-wide distribution of transposon insertions in B. breve UCC2003. Circular plot showing gene content, insertion distribution and gene comparison to Bifidobacterium and B. breve core genomes. Circular tracks are numbered from 1 (outer track) to 7 (inner track). Track 1 represents transposon insertion densities; track 2 indicates forward and reverse genes predicted in B. breve UCC2003 genome; track 3 indicates core B. breve genes; track 4 indicates B. breve core genes present in single copy; track 5 represents B. breve single copy core genes present in Bifidobacterium genus core genes; track 6 represents TraDIS predicted essential genes and track 7 represents GC bias, khaki indicates values > 1; purple < 1.
Figure 2.
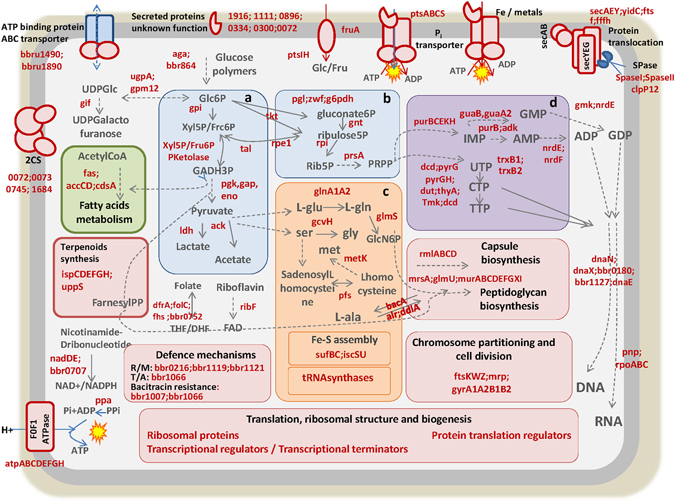
Reconstruction of essential metabolic pathways and functions, based on TraDIS-predicted essential genes in B. breve UCC2003. Gene names or locus tags are represented in red and metabolic intermediates in grey. Dashed arrows summarize multiple reactions. Panels A and B represent the essential central carbohydrate utilization pathways, bifid shunt and pentose phosphate pathway, respectively. Panel C represents essential steps within amino acid metabolism and biosynthesis pathways. Panel D highlights essential steps mapped onto nucleotide metabolic pathways.
One has to keep in mind that certain gene disruptions can only be retrieved under specific environmental conditions. For this reason, the transposon mutant collection was recovered in a rich laboratory medium containing multiple carbon sources (i.e. 0.5% ribose, 0.5% glucose and 0.1% starch). These sugars are known to be metabolized through independent metabolic pathways in B. breve UCC2003 and, accordingly, previous works from the research group have demonstrated that mutants unable to metabolize one of those carbohydrates are still able to survive and grow using one of the other two carbon sources10, 16, 33. Thus, by combining multiple carbohydrates in the recovery media, the number of genes able to tolerate transposon insertions in this library and thus classified as non-essential, was maximized. Accordingly, mutants in the amylopullulanase-encoding gene apuB and the neighbouring maltodextrin transport cluster, known not to grow on starch as the sole carbon source, are represented in our collection. Similarly, mutants in the ribose utilization cluster are also represented in the bank (Supplementary Table S1).
In agreement with other transposon library studies, the mapped insertions were well distributed across the whole genome, with transposon densities approximately 40% higher at regions close to the chromosomal origin of replication (Fig. 1). In fact, the division of B. breve UCC2003 chromosome in 8 sections showed how genes located around the ori region possess an average of 30 insertions per gene, which decreases to 12 insertions per gene in those genes located around the ter region (Supplementary Figure S2a). This has previously been reported as a common phenomenon in insertion mutant libraries and has been related to the presence of a high number of replication forks in the exponential phase of growth, which is when the transposon is introduced into the cell by electroporation34–36. Therefore, genes close to the origin of replication are encountered in multiple copies and are thus more likely to be targeted by transposon mutagenesis. Another possible explanation for this phenomenon is related to the configuration of the genomic DNA, which would be more relaxed and accessible for transposon insertion to occur in regions close to the replication fork17, 19. Consistent with the latter notion, we also observed that our library shows a bias towards increased transposon frequencies in A + T rich regions, where genes display transposon densities up to 46% higher as compared to the rest of the chromosome (Supplementary Table S1, Supplementary Figs S3a and S3b), an observation in agreement with previous reports17, 19.
Prediction of essential genes from TraDIS analysis
Analysis of mapped reads revealed that 1,670 protein-encoding genes (87%), which includes 7 out of the 50 identified ribosomal protein-specifying genes, 2 out of 6 rRNA-encoding genes and 7 out of the 54 identified tRNA-encoding genes harbored at least one transposon insertion. Furthermore, 255 protein-specifying genes, 4 rRNA-encoding genes and 47 tRNA-encoding genes were found not to harbour any insertion whatsoever. Several bioinformatic approaches and statistical models have been described that predict essential genes in genome wide transposon mapping experiments23, 30, 35, 37–39. In the current work, candidate essential genes – i.e. those that are shown not to harbour transposon insertions - in the B. breve UCC2003 mutant library, were predicted from the transposon insertion distribution through calculation of an insertion index per gene as previously described30 (Supplementary Fig. S4). Cut-off criteria to classify genes as essential were established based on the bimodal distribution of insertion indexes as previously described30 and indicated in the Materials and Methods section (Supplementary Fig. S4). Additional manual curation and inspection of each gene was performed in order to exclude situations of insertions not evenly distributed across the whole gene length, and thus not affecting gene function. In addition, our mapping parameters do not allow for repeat mapping and therefore, no insertions were mapped in transposons as these are expected to represent false positives, and are for this reason excluded from the essential gene list. Overall, our analysis resulted in a compiled list of (what we designate here as) essential genes, which includes 453 genes (22.8% of the total number of identified genes on the genome), including 50 that encode ribosomal proteins, 49 that represent tRNA-encoding genes and 4 being rRNA-encoding genes. The predicted percentage of essential genes is within the range reported from genome-wide surveys of essential genes in other microorganisms, which has been shown to range from 5 to 35% of the overall gene content30–32, 40–42.
Functional annotation of essential genes in B. breve UCC2003
Functional analysis of predicted essential protein-encoding genes in B. breve UCC2003 was performed. The full list of essential genes predicted in our B. breve UCC2003 library, along with corresponding COG families, KEGG pathways and functional classification, are presented in Supplementary Table S2. This analysis revealed entire metabolic pathways or specific steps in particular pathways that are critical to support B. breve UCC2003 growth under the applied conditions. It is worth noting that our prediction of essential genes was performed straight away following the mutagenesis procedure, without any prior enrichment step recovering the mutants in liquid media. Therefore, essential genes are estimated based exclusively on the inability to recover viable mutants. Accordingly, a substantial proportion of house-keeping functions, including proteins responsible for DNA replication and transcription, manufacture of cell envelope components, such as cell wall and membrane, the Sec-dependent protein translocation pathway, and central routes for energy acquisition and conversion are abundant among TraDIS-predicted essential functions. Among the cell envelope components, it is worth highlighting that, from the two exopolysaccharide clusters encoded by B. breve UCC2003, only the genes encoding for the EPS cluster 1 (Bbr_1798 to Bbr_1793) were deemed essential (Fig. 1). This represents a novel finding since previous work had only inactivated and proved functionality of EPS cluster 2 (Bbr_0434 to Bbr_0451) which was not essential for in vitro growth, yet is required for in vivo colonization43. In addition, most genes involved in central glycolytic routes, the specific bifid-shunt44 and the pentose phosphate pathway, crucial for production of energy and precursors for other metabolic routes, were found to be essential (Fig. 2). Furthermore, all elements involved in ATP synthase assembly (atpA, atpB, atpC, atpD, atpE, atpF, atpG and atpH) appeared essential. In fact, ATP synthase which couples H+ extrusion to ATP synthesis is necessary to maintain cellular pH homeostasis, required as bifidobacteria growth leads to production of organic acids45 (Fig. 2, Table 1, Supplementary Table S2).
Table 1.
COG and KEGG classification of B. breve essential genes.
| COG Family | Pathway (KEGG) | Genes | %* |
|---|---|---|---|
| Amino acid transport and metabolism | Alanine aspartate glutamate | 2 | 0.5% |
| Amino acid related functions | 3 | 0.8% | |
| Glycine serine threonine | 1 | 0.3% | |
| Selenocompounds | 1 | 0.3% | |
| Carbohydrate transport and metabolism | Transporters | 4 | 1.0% |
| Amino sugar metabolism | 1 | 0.3% | |
| Bifid shunt | 3 | 0.8% | |
| Carbohydrate related functions | 5 | 1.3% | |
| Carbohydrate transport and metabolism | 2 | 0.5% | |
| Glycolysis/gluconeogenesis | 2 | 0.5% | |
| Glycosyl hydrolases | 2 | 0.5% | |
| Pentose phosphate | 7 | 1.7% | |
| Cell cycle control, cell division, chromosome partitioning | Cell cycle control, cell division, chromosome partitioning | 6 | 1.5% |
| Cell wall/membrane/envelope biogenesis | Alanine aspartate glutamate | 1 | 0.3% |
| Amino sugar metabolism | 1 | 0.3% | |
| Capsule biosynthesis | 6 | 1.5% | |
| Cell wall associated functions | 8 | 2.0% | |
| D-alanine | 2 | 0.5% | |
| Peptidoglycan biosynthesis | 9 | 2.3% | |
| Coenzyme transport and metabolism | Coenzyme related functions | 4 | 1.0% |
| Cysteine methionine | 1 | 0.3% | |
| Folate | 4 | 1.0% | |
| Nicotinate nicotinamide metabolism | 3 | 0.8% | |
| Panthotenate coA | 1 | 0.3% | |
| Riboflavin utilization (energy metabolism) | 1 | 0.3% | |
| Thiamine | 1 | 0.3% | |
| Defence mechanism | Transporters | 3 | 0.8% |
| Defence mechanism | 4 | 1.0% | |
| DNA, replication, recombination and repair | DNA related functions | 3 | 0.8% |
| Recombination | 4 | 1.0% | |
| Repair | 5 | 1.3% | |
| Replication | 15 | 3.8% | |
| Energy production and conversion | Citrate cycle | 2 | 0.5% |
| Energy production conversion related functions | 13 | 3.3% | |
| Pyruvate metabolism | 1 | 0.3% | |
| Inorganic ion transport and metabolism | Transporters | 12 | 3.0% |
| Inositol phosphate | 1 | 0.3% | |
| Intracellular trafficking, secretion, and vesicular transport | Secretion | 1 | 0.3% |
| Secretion (sec pathway) | 7 | 1.8% | |
| Lipid transport and metabolism | Fatty acids biosynthesis | 3 | 0.8% |
| Lipids metabolism | 5 | 1.3% | |
| Panthotenate coA | 1 | 0.3% | |
| Terpenoid | 7 | 1.8% | |
| Nucleotide transport and metabolism | Cysteine methionine | 1 | 0.3% |
| Nucleotide related functions | 1 | 0.3% | |
| Purine metabolism | 9 | 2.3% | |
| Pyrimidine metabolism | 11 | 2.8% | |
| Nucleotide transport and metabolism | 2 | 0.5% | |
| Posttranslational modification, protein turnover, chaperones | Posttranslational modification, protein turnover, chaperones | 12 | 3.0% |
| RNA processing and modification | RNA processing and modification | 3 | 0.8% |
| Signal transduction mechanisms | Signal transduction mechanisms | 3 | 0.8% |
| Signal transduction related functions | 6 | 1.5% | |
| Transcription | RNA polymerase | 4 | 1.0% |
| Transcription | 1 | 0.3% | |
| Transcriptional regulator | 10 | 2.5% | |
| Translation, ribosomal structure and biogenesis | Ribosome | 52 | 13.0% |
| Translation | 18 | 4.5% | |
| tRNA processing and modification | 2 | 0.5% | |
| tRNA synthetase | 22 | 5.5% | |
| tRNA-dihydrouridine synthase | 4 | 1.0% | |
| General function prediction only | General function prediction only | 15 | 3.8% |
| Unassigned function | Unassigned function | 56 | 14.0% |
| TOTAL | 400 | 100.0% |
*Percentage of the protein encoding predicted essential genes from B. breve UCC2003, mapped into each COG and KEGG functional category.
With regards to specific anabolic routes, the majority of the genes involved in purine and pyrimidine synthesis were found to be essential, suggesting that B. breve UCC2003 relies primarily on de novo nucleotide synthesis. Furthermore, essential genes related to amino acid metabolism map into the biosynthesis of L-glutamate and L-glutamine which are precursors for the aminosugar metabolic pathway. Among them, the genes glmS (locus tag Bbr_0524) and glnA (locus tags Bbr_1278 and Bbr_0670) were retrieved as essential in our analysis. The product encoded by glmS is known to play a central regulatory role in nitrogen metabolism, catalyzes key steps for the synthesis of cell wall precursors, and has also been found essential in Bacteroides 18, 32, 46. Interestingly, our data suggests that also the presumptive transcriptional regulator of glmS, encoded by glnR (locus tag Bbr_0292), is essential. Another essential gene related to amino acid metabolism is gcvH (locus tag Bbr_0481) whose encoded protein participates in glycine detoxification47. The limited presence of essential genes in other amino acid biosynthetic routes suggests that B. breve UCC2003 possesses the capacity to import most required amino acids from the environment, although specific amino acid transporters have not yet been functionally characterized in this strain.
Putative inorganic ion transporters were also retrieved as essential functions, including predicted import systems for manganese, phosphate, potassium and iron. These ions are essential cofactors for many cellular enzymes and some import systems have previously been found to be essential for intestinal colonization of enteropathogens48, 49. Furthermore, Mn2+ uptake systems have been associated with oxidative stress protection in oral bacteria50. In fact, functions involved in the maintenance of redox homeostasis, such as glutaredoxin production and thioredoxin reductase (predicted to be encoded by Bbr_0039, Bbr_1901 and Bbr_1919 locus tags), were also found essential in our analysis. Bifidobacteria are anaerobic bacteria and are known to be exquisitely sensitive to oxygen, therefore defence mechanisms against oxidative damage are crucial. Besides, redox homeostasis regulates key metabolic functions including signal transduction mechanisms and central carbon metabolism51. Elements predicted to be involved in assembly of iron-sulfur clusters (IscS, IscU, sufB, sufC, fdx, represented by locus tags Bbr_0910, Bbr_0911, Bbr_0907, Bbr_0909, Bbr_1664), which have not been previously characterized in bifidobacteria, were also found essential in our analysis. The corresponding protein complexes are known to sense oxygen and metal availability and regulate gene expression profiles allowing rapid adaptation to environmental changes52, although they have mainly been studied in pathogenic bacteria. Also two genes encoding predicted orphan ATP binding proteins (Bbr_1490 and Bbr_1890), not located in proximity to any ABC transporter-encoding genes, were found to be essential in our analysis therefore suggesting they might energize crucial transport systems in the cell.
Remarkably, 53 essential genes found in this work encode hypothetical proteins with unknown functions, highlighting the limited knowledge we have on associated fundamental aspects of bifidobacterial metabolism and physiology. These findings also highlight the need for research on essential hypothetical proteins in order to define their biological function. Moreover, 11 out of the identified 453 essential genes in B. breve UCC2003 correspond to transcriptional regulators that have not been functionally characterized in bifidobacteria, of which nine are predicted to be involved in signal sensing and transduction, therefore representing interesting targets for future studies.
Conserved essential genes in the Bifidobacterium and B. breve core genomes
Comparative genomics has led to important insights with regards to evolutionary drivers and pathways of bacterial taxa, allowing the identification of genes encoding functions that can be lost or acquired through horizontal gene transfer, as well as those that are seemingly very important as they are maintained across a particular taxonomic group. In this context, the concept of a core genome, consisting of the complete set of orthologous genes present in single or multiple copy shared among a group of bacteria, provides a framework to discern and understand the conserved basic biology of bifidobacteria4–7. Core genomes are the result of long-term natural selection, as determined by environmental conditions that facilitate the loss of non-essential genes, while at the same time preserve those functions that are critical for bacterial survival, competitiveness and persistence in their natural niche6, 53.
Comparative genomics coupled to transposon mutagenesis mapping has indeed been used to better define the minimal essential core genome and to reconstruct bacterial evolution, selective adaptive trait acquisition and evolutionary conservation in a number of microorganisms25, 42, 54, 55. For these reasons, substantial overlap between the in silico computed Bifidobacterium and B. breve core-genomes on one hand, and the identified essential genes from our TraDIS approach on the other, was expected. In order to test this expectation, the TraDIS-deduced essential genes of B. breve UCC2003 were compared to the core genomes of B. breve (1514 genes) and Bifidobacterium (929 genes), which were predicted based on 8 complete B. breve genomes5 and the genomes of 47 bifidobacterial type strains6, respectively. This analysis revealed that 377 out of 400 TraDIS-predicted essential genes in B. breve UCC2003 (94%) were also part of the B. breve core genome (Fig. 3a; Supplementary Table S4). This overlap is expected to occur within single-copy core genes, as they are most likely to include essential core functions evolved from a common ancestor and stably retained by selective pressure56. In concordance with this expectation, 75% of the B. breve core genome (1141 out of 1514 genes) is represented by genes present in single copy (orthologues). Remarkably, 85% of the TraDIS-determined essential genes (343 out of 400 genes) indeed belong to this B. breve single-copy core-genome (Supplementary Table S4). Since essential functions should not only be conserved at species level, but may also be retained at genus level, the candidate essential genes were also compared with single-copy core genes of the genus Bifidobacterium. This analysis revealed that indeed a large proportion of B. breve single-copy core genes (65%) are present in the Bifidobacterium core-genome, including the majority of essential genes identified by transposon insertion mapping in B. breve UCC2003 (81%) and representatives of all functional categories (Fig. 3a and b; and Supplementary Table 4). As the mutant library was tested under in vitro conditions we estimate that 43% of core genome functions in the Bifidobacterium genus (326 out of 748), as predicted in this work, are essential for cell viability under laboratory conditions (Supplementary Table S4). This set of genes comprises functions belonging to most COG functional categories, though in different proportions, yet is particularly enriched in house-keeping functions, such as those involved in DNA maintenance, replication, transcription and translation, in the biosynthesis of cell envelope components, and in central metabolic pathways for energy generation and metabolite conversion functions (Table 1, Fig. 3b; and Supplementary Table S4). This is in agreement with data from other microorganisms and supports the high level of conservation of a range of house-keeping functions41, 42.
Figure 3.
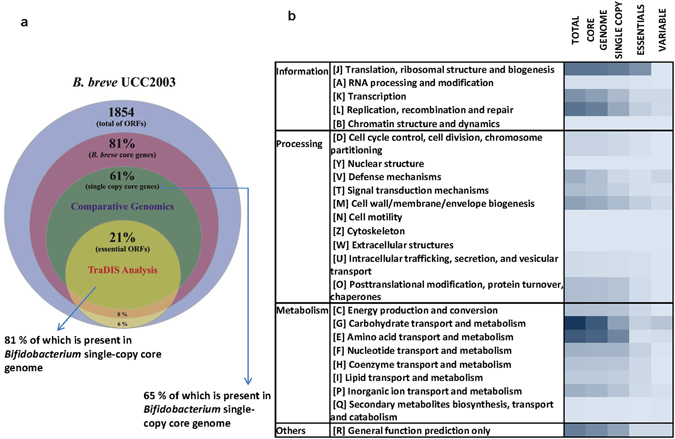
(a) Venn Diagram representing the overlap between the essential genes determined by TraDIS analysis for B. breve UCC2003, the Bifidobacterium and B. breve core genomes, as well as B. breve core genes present in single copy. (b) Heatmap representing in colour gradient the frequency of COG categories across all the different type of gene families resulting from comparative analysis of B. breve UCC2003 total gene content and TraDIS predicted essential genes, against the available Bifidobacterium genomes.
Bifidobacteria are specialized towards carbohydrate utilization as reflected by the fact that ~10% of the protein-encoding genes in the B. breve UCC2003 genome falls into the functional category devoted to carbohydrate transport and metabolism29, 53, 57–60. Among the carbohydrate utilization capabilities identified as essential in B. breve UCC2003 and shared with the Bifidobacterium core genome, a significant proportion is dedicated to central carbon metabolism (i.e. the bifid shunt and the partially present citric acid cycle) and associated energy metabolism (e.g. pyruvate and pentose phosphate pathways), which also generates important intermediates for biosynthetic purposes (e.g. for the biosynthesis of peptidoglycan, amino acids, fatty acids and nucleotides) (Table 1 and Supplementary Table S2).
Differential niche-adaptation of particular strains to specific environments, may also account for the existence of strain-specific essential genes, not shared in a broader core-genome context53, 61. For instance, among the B. breve UCC2003 essential genes that are not part of either the B. breve or Bifidobacterium core genome, various predicted defence mechanisms are included such as DNA methylases and toxin-antitoxin systems. DNA methylases in B. breve UCC2003 (encoded by genes BbrI, BbrII and BbrIII) have previously been identified as part of restriction-modification systems, which represent one of the defence barriers against invading DNA, such as bacteriophages11. These three methylase-encoding genes were found to be essential in our analysis, thereby confirming that the corresponding restriction enzymes are active in B. breve UCC2003. This represents a novel finding since previous experiments by our research team could only prove activity of one of these endonucleases (BbrIII) encoded by this strain11. This was most likely due to limited knowledge on the target sequences or on specific mechanisms of reaction on the endonucleases encoded by BbrI and BbrII11, 62. Regarding toxin-antitoxin systems, they have not been thoroughly characterized in either B. breve or Bifidobacterium, however, they are typically associated with mobile genetic elements. Interestingly, in the particular case of this study, we were able to localize as non-core, yet essential, the antitoxin component of a presumptive toxin-antitoxin system (encoded by Bbr_1066 and Bbr_1067), therefore suggesting the corresponding toxin component is active. Indeed, another chromosomal region (located between chromosome positions 907740 and 907450), which had escaped previous ORF detection and annotation efforts, and displaying homology to a toxin-antitoxin module harbours uneven transposon insertions, pointing towards the existence of a second active toxin-antitoxin system in the chromosome of this strain.
In addition, genes essential for bifidobacterial persistence in their natural environment, represented by the gastrointestinal tract, but not required for in vitro survival are expected to be present across all bifidobacterial representatives, but may not be among the candidate essential genes delineated in this work. Examples of this are the genes encoding Tad pili or those responsible for autoinducer-2 production29, 63, mutants of which were present in the library. Mutations in other B. breve UCC2003 functions essential for intestinal colonization in mice, like the non-core gene cluster involved in the biosynthesis of exopolysaccharide (EPS cluster 2)43, were also present in the library. Indeed, further research is required to discern the genes that are required for successful gut colonization of B. breve UCC2003 (as opposed to being essential for growth under laboratory conditions).
Comparison between B. breve UCC2003 essential functions and those found for other (gut) bacteria
In order to obtain further information on essential metabolic pathways specific for bifidobacteria and to understand their specific in vitro growth requirements, the 400 deduced proteins encoded by the identified essential genes of B. breve UCC2003 were compared to proteins corresponding to essential gene surveys reported for other bacteria. Bacteroides thetaiotaomicron and Bacteroides fragilis are the closest bacteria to bifidobacteria, that have previously been surveyed for essential genes by means of transposon sequencing approaches18, 32 (Supplementary Table S5). Although being phylogenetically distant from bifidobacteria, they share the same environmental niche as inhabitants of the human gut. Thus blast-based analysis were used to establish gene orthology between Bacteroides and Bifidobacterium, and to compare the essential gene compilation detected using the TraDIS approach for these gut bacteria. Despite the limitations of blast-based alignments to identify orthologs accross phyla, as compared to other alignment tools (eg Hidden Markov Model based tools), this analysis revealed that a core set of 106 genes was found essential for these three bacteria, including many housekeeping functions (e.g. DNA maintenance and replication, cell wall and membrane biogenesis and ribosomal and transcriptional machinery). Furthermore the analysis revealed that 223 predicted essential genes in B. breve UCC2003 were not detected by TraDIS analysis in Bacteroides representatives (Fig. 4a; Supplementary Table S5).
Figure 4.

(a) Venn Diagram representing the overlap of essential genes determined by transposon sequencing in B. breve UCC2003, Bacteroides fragilis 638 R and Bacteroides thetaiotaomicron VPI-5482. (b) Distribution per COG functional classification of essential genes in the three compared taxa.
A comparison of the COG classification distribution of essential genes in the three compared taxa (Fig. 4b), showed that Bifidobacterium essential genes are particularly enriched in carbohydrate utilization capabilities, nucleotide transport and metabolism, and energy production and conversion. Bifidobacteria are specialized in the utilization of a range of plant- and host-derived carbohydrates, and can rapidly switch their metabolism to adjust to fluctuations in nutrient availability in their natural environment, thus it is not surprising that they have adopted an intricate network of carbohydrate utilization pathways53, 64.
Since the three compared TraDIS analyses were performed on libraries with very different coverages, it may be that some essential genes escaped the TraDIS-based predictions. Therefore, a comparative genomic analysis was performed to compare the Bifidobacterium predicted essential genes and the two Bacteroides genomes. This analysis showed that 126 essential genes of B. breve UCC2003 do not exhibit sequence homology to any gene in the two analysed Bacteroides genomes, thus constituting bifidobacterial unique genes (Supplementary Table S6). These genes are also part of the Bifidobacterium single copy core genome and include functions related to central carbon metabolism, such as the Bifidobacterium-specific enzyme fructose-6-phosphoketolase (Bbr_0776) (Supplementary Table S6). A cluster encoding a predicted iron-transport system was found among the B. breve UCC2003 specific essential functions (Bbr_0221 to Bbr_0223)65. While iron is an essential cofactor for all living bacteria specially anaerobes, the fact that orthologous genes do not appear essential in Bacteroides despite their key function in the maintenance of cell homeostasis might either indicate that Bacteroides encode additional genes that can compensate the absence of one of the genes or that this systems performs other still undefined essential functions in B. breve UCC2003.
Moreover, the essential genes of B. breve UCC2003 that do not exhibit homology with the two Bacteroides essential gene sets include a relatively high proportion of genes, which encode proteins of unknown functions or for which only general functional predictions can be obtained (46%) (NB. the overall B. breve UCC2003 essential gene set contains 15% genes whose products are predicted to have either an unknown or very general function). This highlights that the biological functions of a substantial proportion of essential bifidobacterial genes are still unknown. Indeed, a recent report on the synthetic construction of a minimum Mycoplasma genome required the inclusion of 149 (32%) genes of unknown function41. These findings and the high proportion of essential hypothetical functions in bifidobacteria emphasize the importance of studying genes encoding hypothetical proteins, particularly because the conclusions of many (microbiome) studies heavily rely on functional predictions.
Finally, it should be mentioned that 29% of the 126 B. breve UCC2003 essential genes that do not have homologs in the two analysed Bacteroides genomes, represent putative secreted, cell surface-associated proteins or functions that have a direct effect on bacterial surface structure and properties (Supplementary Table S6). These may be crucial in environmental sensing and adaptation, and/or microbe-host interactions, and therefore further investigation into the functionality of hypothetical secreted proteins in bifidobacteria is warranted66.
Conclusions
We identified the genes essential for growth of a commensal Bifidobacterium strain under in vitro growth conditions, based on a random mutagenesis approach coupled to simultaneous mapping of the disruption point for ~46,000 different insertion sites. Comparison to Bifidobacterium and B. breve core-genomes revealed significant overlap with our identified candidate essential gene set, especially at single-copy core genes. Essential genes not shared with the Bifidobacterium core genome may either represent strain-specific adaptive traits or functions required under specific environmental conditions such as those mimicking food or intestinal environments. In addition, comparison between essential gene sets in other gut commensals, revealed B. breve UCC2003-specific essential functions, which upon further study will contribute to a better comprehension of bifidobacterial physiology and evolution. The current work provides a fundamental framework to gain functional insights into bifidobacterial molecular traits at a genome-wide level. Expanding identification and tracking of disrupted genes in collections of bifidobacterial mutants under specific environmental conditions will assist functional annotation in this genus and contribute to a better definition of the minimal bifidobacterial genome. Such knowledge in turn will help to depict a more accurate definition of the bifidobacterial lineage. Furthermore, this work allowed identifying a set of essential functions not previously characterized in bifidobacteria including a range of hypothetical and secreted proteins, the EPS cluster 1 and several orphan ATPases, among others, which represent interesting targets for further studies.
Methods
Bacterial strains and culture conditions
Bifidobacterium breve UCC2003 was routinely grown anaerobically at 37 °C on RCM (Oxoid) or De Man Rogosa Sharpe (mMRS) made from first principles67, and supplemented with 0.05% L-cysteine and appropriate carbon sources. Escherichia coli EC101 cells were grown on LB supplemented with tetracycline (10 μg ml−1) or kanamycin (50 μg ml−1) for selection of plasmid-containing cells. B. breve UCC2003 transposon mutants were routinely grown on RCM supplemented with 0.5% ribose and 10 μg ml−1 tetracycline, or mMRS supplemented with 0.5% L-cysteine-HCl and appropriate carbon sources as indicated. D-ribose, D-glucose, starch (potato-derived) and D-lactose were purchased from Sigma.
Creation of second generation Tn5 based transposon, transposome assembly and library construction
A second generation tetracycline resistant transposon was created essentially as previously described16. In order to make the transposon library compatible with Transposon Mediated Differential Hybridization (TMDH) approaches, outwards facing T7 promoters were included at the ends of a tetracycline-resistant Tn-5 transposon. For this purpose, Fw-Tet-pMOD2T7 and Rev-Tet-pMOD2T7 primers (Table 2) were used to amplify a tetracycline resistance cassette, tetW, from pAM5 plasmid DNA68. This PCR product was digested with XbaI and SphI, and subsequently ligated to similarly digested pMOD2 (Epicentre Biotechnology) to yield pMOD2-tetT7.
Table 2.
Oligonucleotides and plasmids used in this work.
| Name | Sequence | Reference |
|---|---|---|
| Transposon generation | ||
| pMOD2 | Source of Tn5 terminal ends, AmpR | Epicentre Biotechnology |
| pMOD2-tetT7 | tetR pMOD2 derivative, containing outwards facing T7 promoters at both transposon ends | This work |
| Fw-Tet-pMOD2T7 | CGCTAGTCTAGACCCTATAGTGAGTCGTATTAGCTCATGTACGGTAAGGAG | This work |
| Rev-Tet-pMOD2T7 | CGCTAGGCATGCCCCTATAGTGAGTCGTATTAGCAAAACCCTCGGTCGGTCTGACCGGGGGTTTTGATTACATTACCTTCTGAAACATATGGC | This work |
| pMOD < MCS > Fw | ATTCAGGCTGCGCAACTGT | Epicentre Biotechnology |
| pMOD < MCS > Rev | GTCAGTGAGCGAGGAAGCGGAAG | Epicentre Biotechnology |
| TraDIS sequencing approach | ||
| 5′ PCR enrichment primer sequence | AATGATACGGCGACCACCGAGATCTACACAATTCGAGCCAATATGCGAGAACACCCG | This work |
| 3′ PCR enrichment primer sequence | AATGATACGGCGACCACCGAGATCTACACATGCAAGCTTGCCAACGACTACGCACTAGC | This work |
| 5′ Sequencing primer sequence | ACCCGAGAAAATTCATCGATGATGGTTGAGATGTGTA | This work |
| 3′ Sequencing primer sequence | ACCCGAGAAAATTCATCGATGATGGTTGAGATGTGTA | This work |
aRestriction sites are underlined.
The Tn5-derived transposon section was then PCR amplified from pMOD2-tetT7 by employing primer combination pMOD < MCS > Fw and pMOD < MCS > Rev (Table 2). The generated PCR product was purified using High pure PCR purification columns (Roche), and transposon ends were pruned by restriction with PshAI. Then DNA was purified and concentrated to 400 ng μl−1 before being assembled with purified Tn5 transposase according to manufacturer’s instructions (Epicentre Biotechnology). About 120 independent electroporation reactions were performed using B. breve UCC2003 competent cells, freshly prepared for transformation purposes as previously described16. Transformation mixes were spread plated onto RCA agar supplemented with 0.5% ribose and 10 μg ml−1 tetracycline, and plates were incubated for 2–3 days anaerobically at 37 °C. In order to create a pool of mutants, plates were flooded with 1 ml of RCM supplemented with 0.5% ribose, 30% glycerol and 10 μg ml−1 tetracycline, and clones were scraped from the plates using disposable sterile spreaders. These bacterial suspensions were stored at −80 °C. A master stock was created by pooling the mutants originated from multiple independent experiments.
DNA isolation and adaptation of TraDIS sequencing and gene essentiality prediction
DNA isolation from the pool of mutants was performed according to previously described procedures69. 2 μg DNA was sequenced on a HiSeq2500 platform (Illumina) as previously described30, using TraDIS-specific primers designed in this study (see Table 2). Briefly, a read length cut-off of 42 bp after trimming 10 bp off the Tn was used before mapping. Mapping was performed using SMALT v 0.7.6 implemented in Bio-Tradis pipeline using a base PHRED quality threshold of 30 (threshold of 96% of exactly matching nucleotides as a fraction of the read length). No mismatches of reads were allowed in the mapping. Then, gene essentiality was established following a previously described method30. Then, transposon insertions located in the 3′ end of a given gene (representing 10% of the gene sequence length) were first excluded, since they do not necessarily inactivate the encoded protein. Then, the insertion index (number of insertion sites per gene length) was calculated for each gene and the distribution of insertion indexes was assessed as being bimodal. Gamma distributions of such indexes were then fitted to the putative essential (mode = 0) and non-essential peaks of the empirical distribution. Log2-likelihood ratios (LLR) were also retrieved between the fitted distributions. Gene essentiality was established based on LLR < −2, with essentiality cut-off set at an insertion index of 0.0015. Genes were classified as non-essential with LLR > 2 and an insertion index cut-off of 0.0021. Insertion indexes falling between these two values were classified as “ambiguous” (Supplementary Figure S4).
Comparative analysis and core genome predictions
Comparative analyses at species and genus level were conducted using an all-against-all, bi-directional BLASTP alignment70 (cut-off: E-value 0.0001, with at least 50% identity across at least 50% of either protein sequence). The BLAST output was then clustered into functionally-related protein families using the Markov Cluster Algorithm (MCL) implemented in the mclblastline pipeline v12–067871. Members of the core-genome were selected based on their presence in all strains and a filter to distinguish orthologues (single-copy) from paralogues (multiple copy) was applied. Members of the core-genome were defined as genes present in each B. breve representative. For the comparative analyses extended at genus level, since the fully sequenced B. breve UCC2003 was compared with draft genome sequences, we decided to apply an extended core-genome that included genes present in at least 80% of the sequences (in order to exclude the possibility that genes not sequenced were considered absent).
The same BLAST-based comparative analysis was used to compare the Bifidobacterium breve UCC2003 essential genes with those found in other (gut) bacteria (e.g. Bacteroides). Coding sequences of B. breve UCC2003, Bacteroides fragilis 638 R and Bacteroides thetaiotaomicron VPI-5482 were compared using reciprocal BLASTP alignment70 and homologous genes were selected using at least 20% identity across at least 50% of either protein sequence of significant BLAST hits (cut-off: E-value 0.0001) using the MCL algorithm. The information obtained by this clustering was employed to determine the overlap between the essential genes in each TraDIS library and visualized in a Venn diagram and heatmap.
Functional classification of essential genes and metabolic pathway reconstruction
All B. breve UCC2003 genes were functionally classified using a combination of COG category assignment72 based on significant BLASTP70 alignment against COG database (cut-off: E-value 0.0001, with at least 50% identity across at least 50% of either protein sequence) and refined with the online available information on B. breve UCC2003 KEGG pathways (http://www.genome.jp/kegg). The results of this functional classification assignment were then summarized in heatmap (Fig. 3b). The same BLAST-based approach was used to retrieve the COG classification of the essential genes described in other bacteria (belonging to the phylum Bacteroides) to be compared with B. breve UCC2003 (see above).
Electronic supplementary material
Acknowledgements
DvS, FB, LR and MOCM are members of The APC Microbiome Institute, which is a research centre funded by Science Foundation Ireland (SFI), through the Irish Government’s National Development Plan. The authors and their work were supported by SFI (Grant SFI/12/RC/2273) and HRB (Grant No. 513 PDTM/20011/9). Sequencing was supported by the Wellcome Trust, grant number WT098051. CJB and AKC were supported by the Medical Research Council, grant number G1100100/1.
Author Contributions
L.R., D.v.S., M.O.C.M. and T.L. designed the research. L.R., C.B., A.C. performed the experiments. L.R., F.B., C.B. and D.v.S. analyzed the data. L.R., F.B. created the figures and drafted the manuscript. All authors read and approved the manuscript.
Competing Interests
The authors declare that they have no competing interests.
Footnotes
Lorena Ruiz and Francesca Bottacini contributed equally to this work.
Electronic supplementary material
Supplementary information accompanies this paper at doi:10.1038/s41598-017-05795-y
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Fukuda S, Toh H, Taylor TD, Ohno H, Hattori M. Acetate-producing bifidobacteria protect the host from enteropathogenic infection via carbohydrate transporters. Gut Microbes. 2012;3(5):449–454. doi: 10.4161/gmic.21214. [DOI] [PubMed] [Google Scholar]
- 2.Tojo R, et al. Intestinal microbiota in health and disease: Role of bifidobacteria in gut homeostasis. World Journal of Gastroenterology. 2014;20(41):15163–15176. doi: 10.3748/wjg.v20.i41.15163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Russell DA, Ross RP, Fitzgerald GF, Stanton C. Metabolic activities and probiotic potential of bifidobacteria. International Journal of Food Microbiology. 2011;149(1):88–105. doi: 10.1016/j.ijfoodmicro.2011.06.003. [DOI] [PubMed] [Google Scholar]
- 4.Bottacini F, et al. Comparative genomics of the genus Bifidobacterium. Microbiology. 2010;156(Pt 11):3243–54. doi: 10.1099/mic.0.039545-0. [DOI] [PubMed] [Google Scholar]
- 5.Bottacini F, et al. Comparative genomics of the Bifidobacterium breve taxon. BMC Genomics. 2014;15:170–188. doi: 10.1186/1471-2164-15-170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Milani C, et al. Genomic encyclopedia of type strains of the genus Bifidobacterium. Applied Environmental Microbiology. 2014;80(20):6290:302. doi: 10.1128/AEM.02308-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun Z, et al. Comparative genomic analysis of 45 type strains of the genus Bifidobacterium: a snapshot of its genetic diversity and evolution. PLoS One. 2015;10(2):e0117912. doi: 10.1371/journal.pone.0117912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun Z, Baur A, Zhurina D, Yuan J, Riedel CU. Accessing the inaccessible: molecular tools for bifidobacteria. Applied Environmental Microbiology. 2012;78(15):5035–42. doi: 10.1128/AEM.00551-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fukiya S, Hirayama Y, Sakanaka M, Kano Y, Yokota A. Technological advances in Bifidobacterial molecular genetics: application to functional genomics and medical treatments. Biosciences of Microbiota, Food and Health. 2012;31(2):15–25. doi: 10.12938/bmfh.31.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Connell-Motherway M, et al. Characterization of ApuB, an extracellular type II amylopullulanase from Bifidobacterium breve UCC2003. Applied and Environmental Microbiology. 2008;74(20):6271–9. doi: 10.1128/AEM.01169-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.O’Connell-Motherway M, O’Driscoll J, Fitzgerald GF, Van Sinderen D. Overcoming the restriction barrier to plasmid transformation and targeted mutagenesis in Bifidobacterium breve UCC2003. Microbial Biotechnology. 2009;2(3):321–32. doi: 10.1111/j.1751-7915.2008.00071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hirayama Y, et al. Development of a double-crossover markerless gene deletion system in Bifidobacterium longum: functional analysis of the α-galactosidase gene for raffinose assimilation. Applied and Environmental Microbiology. 2012;78(14):4984–94. doi: 10.1128/AEM.00588-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sakaguchi K, et al. The pyrE gene as a bidirectional selection marker in Bifidobacterium longum 105-A. Bioscience Microbiota and Food Health. 2013;32(2):59–68. doi: 10.12938/bmfh.32.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hidalgo-Cantabrana C, et al. A single mutation in the gene responsible for the mucoid phenotype of Bifidobacterium animalis subsp. lactis confers surface and functional characteristics. Applied and Environmental Microbiology. 2015;81(23):7960–8. doi: 10.1128/AEM.02095-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Callaghan A, Bottacini F, O’Connell-Motherway M, van Sinderen D. Pangenome analysis of Bifidobacterium longum and site-directed mutagenesis through by-pass of restriction modification systems. BMC Genomics. 2015;16(1):832. doi: 10.1186/s12864-015-1968-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruiz L, O’Connell-Motherway M, Lanigan N, van Sinderen D. Transposon mutagenesis in Bifidobacterium breve: Construction and characterization of a Tn5 transposon mutant library for Bifidobacterium breve UCC2003. PLOS One. 2013;8(5):e64699. doi: 10.1371/journal.pone.0064699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barquist L, et al. A comparison of dense transposon insertion libraries in the Salmonella serovars Thyphi and Typhimurium. Nucleic Acids Research. 2013;41(8):4549–64. doi: 10.1093/nar/gkt148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goodman AL, et al. Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe. 2009;6:279–89. doi: 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Langridge GC. Simultaneous assay of every Salmonella Typhi gene using one million transposon mutants. Genome Research. 2009;19:2308–16. doi: 10.1101/gr.097097.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gawronski JD, Wong SM, Giannoukos G, Ward DV, Akerley BJ. Tracking insertion mutants within libraries by deep sequencing and a genome-wide screen for Haemophilus genes required in the lung. Proceedings National Academy of Sciences USA. 2009;106:16422–7. doi: 10.1073/pnas.0906627106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.van Opijnen T, Bodi KL, Camilli A. Tn-seq: high-throughput parallel sequencing for fitness and genetic interaction studies in microorganisms. Nat Methods. 2009;6(10):767–72. doi: 10.1038/nmeth.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Opijnen T, Camilli A. Transposon insertion sequencing: a new tool for systems-level analysis of microorganisms. Nature Reviews in Microbiology. 2013;11(7):435–42. doi: 10.1038/nrmicro3033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeJesus MA, et al. Bayesian analysis of gene essentiality based on sequencing of transposon insertion libraries. Bioinformatics. 2013;29(6):695–703. doi: 10.1093/bioinformatics/btt043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Deutschbauer A, et al. Evidence-based annotation of gene function in Shewanella oneidensis MR-1 using genome-wide fitness profiling across 121 conditions. PLoS Genetics. 2004;7(11):e1002385. doi: 10.1371/journal.pgen.1002385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu W, et al. Comparative genomics of Mycoplasma: analysis of conserved essential genes and diversity of the pan-genome. PLoS One. 2012;7(4):e35698. doi: 10.1371/journal.pone.0035698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuehl JV, et al. Functional genomics with a comprehensive library of transposon mutants for the sulfate-reducing bacterium Desulfovibrio alaskensis G20. Molecular Biology. 2014;5(3):e01041. doi: 10.1128/mBio.01041-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hooven TA, et al. The essential genome of Streptococcus agalactiae. BMC Genomics. 2016;17(1):406–418. doi: 10.1186/s12864-016-2741-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Licandro-Seraut H, Scornec H, Pédron T, Cavin JF, Sansonetti PJ. Functional genomics of Lactobacillus casei establishment in the gut. Proceedings National Academy of Sciences USA. 2014;11(30):E3101–3109. doi: 10.1073/pnas.1411883111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.O’Connell-Motherway M, et al. Functional genome analysis of Bifidobacterium breve UCC2003 reveals type IVb tight adherence (Tad) pili as an essential and conserved host-colonization factor. Proceedings National Academy of Sciences USA. 2011;108(27):11217–22. doi: 10.1073/pnas.1105380108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barquist, L. et al. The TraDIS toolkit: sequencing and analysis for dense transposon mutant libraries. Bioinformatics 32(7), 1109–11. [DOI] [PMC free article] [PubMed]
- 31.Santiago M, et al. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics. 2015;16:252–270. doi: 10.1186/s12864-015-1361-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veeranagouda Y, Husain F, Tenorio EL, Wexler HM. Identification of genes required for the survival of B. fragilis using massive parallel sequencing of a saturated transposon mutant library. BMC Genomics. 2014;15:429–440. doi: 10.1186/1471-2164-15-429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pokusaeva K, et al. Ribose utilization by the human commensal Bifidobacterium breve UCC2003. Microbial. Biotechnology. 2010;3(3):311–323. doi: 10.1111/j.1751-7915.2009.00152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhong J, Kargerg M, Lambowithz AM. Targeted random bacterial gene disruption using a group II intron (targetron) vector containing a retrotransposition-activated selectable marker. Nucleic Acids Research. 2003;31(6):1656–64. doi: 10.1093/nar/gkg248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zomer A, Burghout P, Bootsma HJ, Hermans PWM, van Hijum SAFT. ESSENTIALS: software for rapid analysis of high throughput transposon insertion sequencing data. PLoS One. 2012;7(8):e43012. doi: 10.1371/journal.pone.0043012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller CA, et al. The dynamics of genome replication using deep sequencing. Nucleic Acids Research. 2014;42(1):e3. doi: 10.1093/nar/gkt878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pritchard JR, et al. ARTIST: high-resolution genome-wide assessment of fitness using transposon-insertion sequencing. PLoS Genetics. 2014;10(11):e1004782. doi: 10.1371/journal.pgen.1004782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.DeJesus MA, Ioerger TR. A Hidden Markov model for identifying essential and growth-defect regions in bacterial genomes from transposon insertion sequencing data. BMC Bioniformatics. 2013;14:303–315. doi: 10.1186/1471-2105-14-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Deng J, Su S, Lin X, Hassett DJ, Lu LJ. A statistical framework for improving genomic annotations of prokaryotic essential genes. PLoS One. 2013;8(3):e58178. doi: 10.1371/journal.pone.0058178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dembek M, et al. High-throughput analysis of gene essentiality and sporulation in Clostridium difficile. Molecular Biology. 2015;24(6):e02383–14. doi: 10.1128/mBio.02383-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hutchison, C.A. et al. Design and synthesis of a minimal bacterial genome. Science351(6280) (2016). [DOI] [PubMed]
- 42.LeBreton Y, et al. Essential genes in the core genome of the human pathogen Streptococcus pyogenes. Scientific Reports. 2015;5:9838–9851. doi: 10.1038/srep09838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fanning S, et al. Bifidobacterial surface-exopolysaccharide facilitates commensal-host interaction through immune modulation and pathogen protection. Proceedings of the National Academy of Sciences USA. 2012;109(6):2108–13. doi: 10.1073/pnas.1115621109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fushinobu S. Unique sugar metabolic pathways of bifidobacteria. Bioscience Biotechnology and Biochemistry. 2010;74(12):2374–84. doi: 10.1271/bbb.100494. [DOI] [PubMed] [Google Scholar]
- 45.Jin J, et al. Investigation of growth phase-dependent acid tolerance in Bifidobacterium longum BBMN68. Current Microbiology. 2016;73(5):660–667. doi: 10.1007/s00284-016-1111-z. [DOI] [PubMed] [Google Scholar]
- 46.Harth G, Maslesa-Galić S, Tullius MV, Horwitz MA. All four Mycobacterium tuberculosis glnA genes encode glutamine synthetase activities but only GlnA1 is abundantly expressed and essential for bacterial homeostasis. Molecular Microbiology. 2005;58(4):1157–72. doi: 10.1111/j.1365-2958.2005.04899.x. [DOI] [PubMed] [Google Scholar]
- 47.Tezuka T, Ohnishi Y. Two glycine riboswitches activate the glycine cleavage system essential for glycine detoxification in Streptomyces griseus. Journal of Bacteriology. 2014;196(7):1369–1376. doi: 10.1128/JB.01480-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sheng Y, et al. Dual Zinc transporter systems in Vibrio cholerae promote competitive advantages over gut microbiome. Infection and Immunity. 2015;83(10):3902–8. doi: 10.1128/IAI.00447-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Naikare H, Palyada K, Panciera R, Marlow D, Stintzi A. Major role for FeoB in Campylobacter jejuni ferrous iron acquisition, gut colonization, and intracellular survival. Infection and Immunity. 2006;74(10):5433–5444. doi: 10.1128/IAI.00052-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang X, Tong H, Dong X. PerR-regulated manganese ion uptake contributes to oxidative stress defence in an oral Streptococcus. Applied and Environmental Microbiology. 2014;80(8):2351–9. doi: 10.1128/AEM.00064-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Green J, Paget MS. Bacterial redox sensors. Nature Reviews Microbiology. 2004;2:954–966. doi: 10.1038/nrmicro1022. [DOI] [PubMed] [Google Scholar]
- 52.Miller HK, Auerbuch V. Bacterial iron-sulfur cluster sensors in mammalian pathogens. Metallomics. 2015;7(6):943–56. doi: 10.1039/C5MT00012B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Milani, C. et al. Genomics of the genus Bifidobacteirum reveals species-specific adaptation to the glycan-rich gut environment. Applied and Environmental Microbiology pii: AEM.03500–15 (2015). [DOI] [PMC free article] [PubMed]
- 54.Canals R, et al. High-throughput comparison of gene fitness among related bacteria. BMC Genomics. 2012;13:212–225. doi: 10.1186/1471-2164-13-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glass JI, et al. Essential genes of a minimal bacterium. Proceedings National Academy of Sciences USA. 2006;103(2):425–30. doi: 10.1073/pnas.0510013103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Charlebois RL, Doolittle WF. Computing prokaryotic gene ubiquity: rescuing the core from extinction. Genome Research. 2004;14(12):2469–77. doi: 10.1101/gr.3024704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Egan M, O’Connell-Motherway M, Ventura M, van Sinderen D. Metabolism of sialic acid by Bifidobacterium breve UCC2003. Applied and Environmental Microbiology. 2014;80(14):4414–26. doi: 10.1128/AEM.01114-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ruiz-Moyano S, et al. Variation in consumption of human milk oligosaccharides by infant gut-associated strains of Bifidobacterium breve. Applied and Environmental Microbiology. 2013;79(19):6040–9. doi: 10.1128/AEM.01843-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McLaughlin HP, et al. Carbohydrate catabolic diversity of bifidobacteria and lactobacilli of human origin. International Journal of Food Microbiology. 2015;203:109–21. doi: 10.1016/j.ijfoodmicro.2015.03.008. [DOI] [PubMed] [Google Scholar]
- 60.O’Connell KJ, et al. Metabolism of four α-glycosidic linkage-containing oligosaccharides by Bifidobacterium breve UCC2003. Applied and Environmental Microbiology. 2013;79(20):6280–92. doi: 10.1128/AEM.01775-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma S, Markham PF, Browning GF. Genes found essential in other mycoplasmas are dispensable in Mycoplasma bovis. PLoS One. 2014;9(6):e97100. doi: 10.1371/journal.pone.0097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gowers DM, Bellamy SR, Halford SE. One recognition sequence, seven restriction enzymes, five reaction mechanisms. Nucleic Acids Research. 2004;32(11):3469–79. doi: 10.1093/nar/gkh685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Christiaen SE, et al. Autoinducer-2 plays a crucial role in gut colonization and probiotic functionlity of Bifidobacterium breve UCC2003. PLoS One. 2014;9(5):e98111. doi: 10.1371/journal.pone.0098111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Milani C, et al. Bifidobacteria exhibit social behaviour through carbohydrate resource sharing in the gut. Scientific Reports. 2015;5:15782. doi: 10.1038/srep15782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cronin M, Zomer A, Fitzgerald GF, van Sinderen D. Identification of iron-regulated genes of Bifidobacterium breve UCC2003 as a basis for controlled gene expression. Bioengineered Bugs. 2012;3(3):157–167. doi: 10.4161/bbug.18985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sánchez B, Urdaci MC, Margolles A. Extracellular proteins secreted by probiotic bacteria as mediators of effects that promote mucosa-bacteria interactions. Microbiology. 2010;156(Pt 11):3232–42. doi: 10.1099/mic.0.044057-0. [DOI] [PubMed] [Google Scholar]
- 67.De Man JC, Rogosa M, Sharpe E. A medium for the cultivation of lactobacilli. Journal of Applied Bacteriology. 1960;23(1):130–135. doi: 10.1111/j.1365-2672.1960.tb00188.x. [DOI] [Google Scholar]
- 68.Alvarez-Martin P, Flórez AB, Margolles A, del Solar G, Mayo B. Improved cloning vectors for bifidobacteria, based on the Bifidobacterium catenulatum pBC1 replicon. Applied and Environmental Microbiology. 2008;74(15):4656–4665. doi: 10.1128/AEM.00074-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.MacConaill LE, Fitzgerald GF, van Sinderen D. Investigation of protein export in Bifidobacterium breve UCC2003. Applied and Environmental Microbiology. 2003;69:6994–7001. doi: 10.1128/AEM.69.12.6994-7001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Altschul SF, et al. Basic local alignment search tool. Journal of Molecular Biology. 1990;215(3):403–10. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 71.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleid Acids Research. 2002;30(7):1575–84. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tatusov RL, Galperin MY, Natale DA, Koonin EV. The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Research. 2000;28(1):33–6. doi: 10.1093/nar/28.1.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.